Back to Archived Journals » Metalloproteinases In Medicine » Volume 6
The role of tissue inhibitors of metalloproteinases in microvascular endothelial cell barrier dysfunction during sepsis
Authors Jayawardena DP ![]() , Kulkarni NP
, Kulkarni NP ![]() , Gill SE
, Gill SE ![]()
Received 2 October 2018
Accepted for publication 29 November 2018
Published 7 May 2019 Volume 2019:6 Pages 1—12
DOI https://doi.org/10.2147/MNM.S156245
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yoshifumi Itoh
Devika P Jayawardena,1,2 Nidhi P Kulkarni,1,2 Sean E Gill1–4
1Centre for Critical Illness Research, Lawson Health Research Institute, London, ON, Canada; 2Department of Physiology and Pharmacology, Western University, London, ON, Canada; 3Division of Respirology, Western University, London, ON, Canada; 4Department of Medicine, Western University, London, ON, Canada
Abstract: Sepsis is defined as a dysregulated host response to an infection leading to organ dysfunction and failure. During sepsis circulating proinflammatory mediators, such as cytokines and bacterial products, as well as activated inflammatory cells cause dysfunction of the microvasculature, specifically the microvascular endothelial cells (MVECs), leading to loss of MVEC barrier function and increased permeability. While multiple mechanisms are known to initiate septic MVEC barrier dysfunction, our understanding of mechanisms that promote MVEC barrier stability, especially under septic conditions, is limited. Formation and maintenance of the MVEC barrier requires stability of MVEC cell–cell junctions, which control the passage of fluid and macromolecules across the endothelium, as well as the extracellular matrix (ECM), which provides critical signaling cues to MVECs through MVEC–ECM interactions. Importantly, disruption of either MVEC intercellular junctions or of the ECM surrounding MVECs can lead to barrier dysfunction. Metalloproteinases, including the matrix metalloproteinases (MMPs) as well as the closely related a disintegrin and metalloproteinases (ADAMs) and ADAMs with thrombospondin repeats (ADAMTSs), are a family of enzymes capable of proteolytically processing the ECM as well as other proteins associated with MVECs, including cell–cell junctional proteins and leukocyte adhesion receptors. Collectively, this suggests that metalloproteinases may be critical mediators of MVEC barrier function. While metalloproteinases are regulated at multiple levels, the tissue inhibitors of metalloproteinases (TIMPs) are known to be the primary inhibitors of metalloproteinase activity. Moreover, in addition to inhibiting metalloproteinases, TIMPs have also been found to have metalloproteinase-independent functions. Specifically, TIMPs have been found to regulate vascular endothelial growth factor signaling, focal adhesion kinase-dependent cell survival, and leukocyte–MVEC interaction. Thus, TIMPs have the potential to promote MVEC barrier stability and potentially restrict septic MVEC barrier dysfunction through both metalloproteinase-dependent and metalloproteinase-independent mechanisms.
Keywords: sepsis, microvascular permeability, inflammation, TIMPs
Sepsis
Sepsis, which is defined as life-threatening organ dysfunction due to a dysregulated host response, is an increasingly common and serious human disease with significant morbidity and mortality.1 Specifically, sepsis is one of the leading causes of mortality in intensive care units2 and the primary cause of death from infection, generally due to multiple organ dysfunction (MOD).1 MOD is the result of septic disturbances in cardiovascular function, especially of the microvasculature, which is critically driven by activation, injury, and dysfunction of microvascular endothelial cells (MVECs). Importantly, even those that survive sepsis often suffer from long-term morbidities associated with septic organ damage, which contributes to the extensive healthcare costs associated with the disease.3,4
Pathophysiology of sepsis
The pathogenesis of sepsis occurs via multiple mechanisms, all of which are associated with the establishment of a dysregulated and often overexuberant inflammatory response leading to MVEC dysfunction.5 During infection, release of foreign toxins, such as lipopolysaccharide (LPS), which are recognized by pattern recognition receptors, such as the toll-like receptors, results in activation of the host’s immune system and the release of inflammatory mediators (proinflammatory and anti-inflammatory).5
Proinflammatory mediators, including cytokines such as tumor necrosis factor alpha (TNFα), interferon γ (INFγ), IL 1β, and IL6 have numerous, sometimes overlapping, functions. For example, they promote endothelial cell (EC)–leukocyte adhesion through increased expression of adhesion molecules on ECs, such as intercellular adhesion molecule 1 (ICAM1), endothelial (E)-selectin, and vascular cell adhesion molecule 1 (VCAM1).6 Moreover, they are also known to induce a loss of MVEC barrier function leading to increased vascular permeability.6 Importantly, levels of TNFα, as well as IL1β and IL6, are well documented to increase under septic conditions, and these changes in cytokine levels appear to be associated with patient survival.7
Inflammation is an absolutely required host response to tissue damage and/or to an infection; however, this inflammatory response must be tightly regulated.8 In general, this regulation requires the action of proinflammatory cytokines to be modulated by the action of anti-inflammatory molecules as a form of regulatory immunosuppression.5 In sepsis, however, the proinflammatory response is augmented and the compensatory anti-inflammatory reaction is unable to suppress it, allowing for ongoing systemic inflammation leading to MVEC dysfunction and MOD.5 Moreover, the anti-inflammatory response, which is characterized by activation of anti-inflammatory cytokines (eg, IL10, IL1 receptor antagonist, transforming growth factor beta [TGFβ]), may be associated with immunosuppression resulting in attenuated host responses to pathogens and potentially, a higher risk of secondary, opportunistic infections.9,10
ECs and barrier formation
Septic MOD is largely due to dysfunction of the vasculature, especially the microvasculature or capillaries – the smallest vessels that feed each organ.11 This microvascular dysfunction is the consequence of injury and dysfunction to the ECs lining the microvasculature, the MVECs. MVECs are arranged as a monolayer with cell–cell junctions, such as the adherens and tight junctions, between them.12 These cell–cell junctions control the passage of substances across MVECs.12 For instance, the paracellular pathway, one of the major filtration routes, consists of adherens and tight junctions that mediate the movement of fluid, ions (mainly uncharged but some charged molecules), and smaller substances, while limiting larger proteins, such as albumin.13 Adherens junctions are comprised of vascular endothelial cadherin (VE-cadherin), a calcium-dependent adhesion protein that is involved in cell stability, monolayer integrity, and cell signaling.14 Tight junctions are formed through the association of claudin with occludin and are crucial in regulating the exchange of ions and solutes between MVECs.15 Junctional adhesion molecules, a family of transmembrane proteins, also contribute to the formation and maintenance of tight junctions, through their role in leukocyte adhesion and transmigration across the endothelium.16
In addition to cell–cell junctions, which join MVECs laterally, MVECs interact with the underlying extracellular matrix (ECM) within the basement membrane via integrins.17 Integrin activation and binding to the ECM leads to the recruitment of signaling proteins such as focal adhesion kinase (FAK) and the tyrosine kinase Fyn, leading to the formation of complexes referred to as focal adhesions.18 The formation of focal adhesions and the interaction of MVECs with the ECM further promotes MVEC barrier function and serves to restrict microvascular permeability.18
Further, the surrounding ECM, which is largely comprised of collagen, fibronectin, and glycosaminoglycans (GAGs), adds to the restrictive property of ECs that is required for normal tissue functioning.17 Collagen is the main structural component and helps regulate cell adhesion, cell migration, and elasticity of the tissue by providing tensile strength.19 Heparan sulfate, one of the most abundant GAGs, has numerous biological functions, including cell–matrix interactions, chemokine activation, and regulation of degradative enzymes.20 Fibronectin is important not only for organization of the ECM components but also plays a role in cell adhesion and migration, and as a mechano-regulator.19 These ECM components are intertwined in a complex network that maintains the structural integrity of the matrix while allowing it to resist tensile stresses and limit movement of molecules under healthy conditions.
Thus, the interaction between adjacent MVECs (cell–cell junctions) as well as between MVECs and the ECM (cell–ECM interactions) is crucial for MVEC barrier function, and disruption of these interactions under pathological conditions, such as sepsis, leads to MVEC barrier dysfunction and increased permeability.
EC dysfunction in sepsis
The homeostatic function of the microvasculature in each organ is locally regulated by MVECs. Importantly, healthy MVECs form a semi-selective permeability barrier, permitting paracellular (between cells) flux of small molecules (eg, ions, urea, glucose) into tissues and organs, but largely excluding movement of larger macromolecules and cells.21 Moreover, while MVECs are also active participants in immune surveillance, under basal conditions MVECs generally act to restrict the inflammatory response.22
Under pathological conditions, such as sepsis, the release of cytokines by activated immune cells (tissue macrophages and circulating leukocytes), direct leukocyte–MVEC interaction, and stimulation of MVECs with bacterial products (eg, LPS) result in MVEC activation and subsequent dysfunction. MVEC activation subsequently leads to increased MVEC-dependent expression of proinflammatory cytokines, cell surface leukocyte receptors, such as ICAM1 and VCAM1, and of extracellular enzymes known to regulate inflammatory signaling pathways, such as the matrix metalloproteinases (MMPs) and closely related a disintegrin and metalloproteinases (ADAMs).22,23 Moreover, this MVEC dysfunction is associated with a loss of MVEC barrier function leading to extravascular leak and accumulation of protein-rich edema fluid, which is a critical pathophysiologic feature of MOD.
There are multiple potential mechanisms mediating the loss of MVEC barrier function under septic conditions. For example, MVEC retraction due to actin cytoskeleton rearrangement and microtubule-dependent MVEC contraction is thought to promote gap formation and subsequent barrier dysfunction.21 Additional mechanisms, such as transcytosis, which occurs via active, homeostatic, cell surface receptor-dependent transcellular transport of specific macromolecules, as well as MVEC apoptosis, are known to occur during sepsis; however, the direct impact of these mechanisms on loss of MVEC barrier function is unclear.21,24,25
Loss of MVEC intercellular junctions, including both adherens and tight junctions, has also been found to occur under septic conditions and is thought to be a critical driver of MVEC barrier dysfunction.14,21,26 This septic disruption of MVEC intercellular junctions is initiated by multiple mechanisms and is characterized by the loss of key junctional proteins, such as VE-cadherin. For example, loss of membrane VE-cadherin can be due to phosphorylation of VE-cadherin, direct cleavage by extracellular proteases, including MMPs and ADAMs, and loss of attachment to submembrane cytoplasmic adaptor proteins (eg, β-catenin), which link VE-cadherin to the actin cytoskeleton.27–30 Moreover, it has also been suggested that alterations to the basement membrane leading to altered MVEC–ECM interactions may also lead to disruption of intercellular junctions and MVEC barrier dysfunction.31 Collectively, these lines of evidence highlight the variety, and resulting complexity, of mechanisms implicated in MVEC barrier dysfunction. Furthermore, they also highlight the potential role for metalloproteinases and thus the importance of the tissue inhibitors of metalloproteinases (TIMPs).
Tissue inhibitors of metalloproteinases
The integrity of the microvasculature is tightly controlled by various endogenous regulators. One such family of regulators is the TIMPs, which, as the name implies, are the endogenous inhibitors of the metalloproteinase family.32,33 When activated, ECs express multiple metalloproteinases, including MMPs and ADAMs.34–36 Excess metalloproteinase activity, due to increased expression and/or a lack of inhibition, corresponds with uncontrolled ECM degradation, disruption of intercellular junctions, and in many instances, the propagation of a proinflammatory microenvironment, all of which have been linked with several pathologies, including sepsis, and are required for septic MVEC barrier dysfunction and MOD.37,38 As such, TIMPs are important players in regulating ECM turnover, stability of intercellular junctions, and endothelial signaling (Figure 1).37
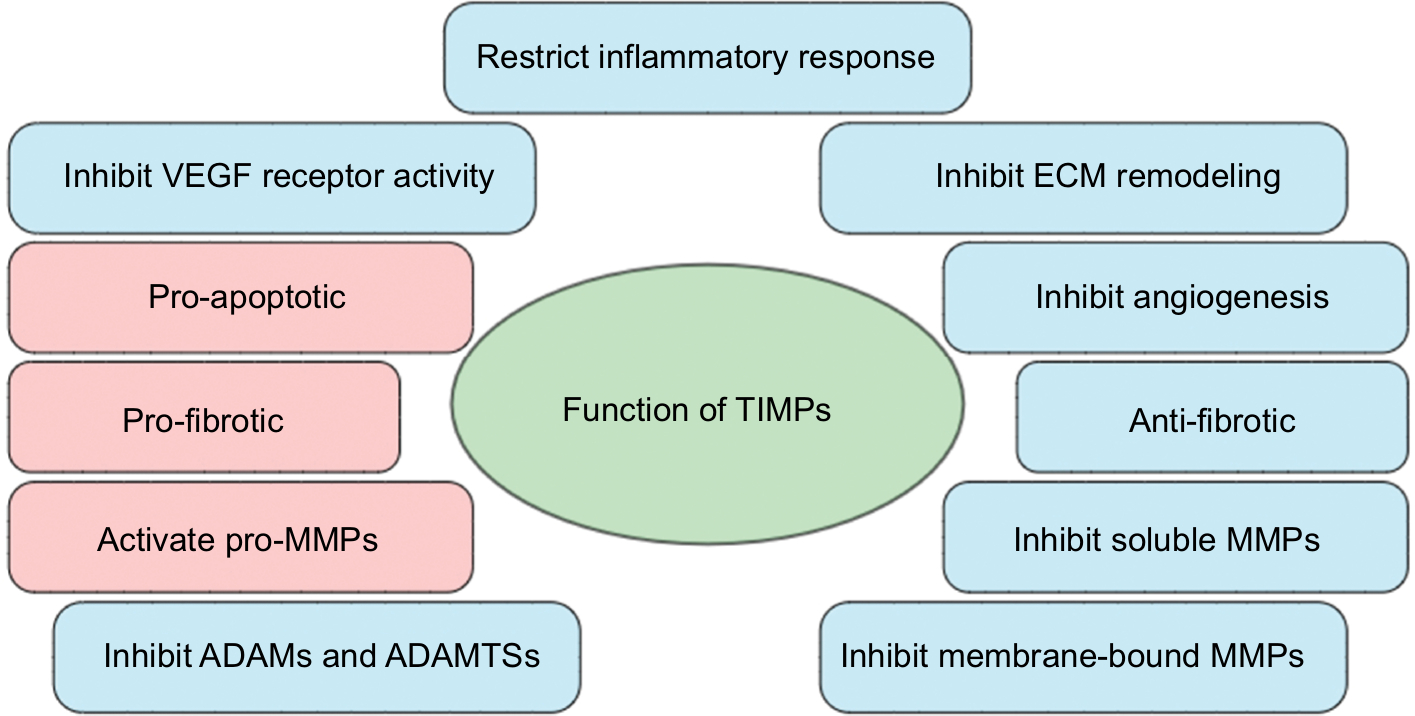 | Figure 1 Common functions of tissue inhibitors of metalloproteinases (TIMPs) in microvascular endothelial cell (MVEC) barrier function. Notes: In general, TIMPs promote MVEC barrier function and restrict inflammation, which occurs through a variety of metalloproteinase-dependent and metalloproteinase-independent mechanisms (blue bubbles). For example, TIMPs are involved in regulating the composition and structure of the extracellular matrix (ECM), largely through their ability to inhibit matrix metalloproteinases (MMPs), a disintegrin and metalloproteases (ADAMs), and ADAMs with thrombospondin repeats (ADAMTSs). Regulation of metalloproteinases also enables TIMPs to restrict the proinflammatory response by inhibiting shedding and/or activation of proinflammatory cytokines (eg, tumor necrosis factor α). Additionally, TIMPs appear to inhibit angiogenesis by binding to the VEGF receptor through metalloproteinase-independent mechanisms. While TIMPs appear to primarily function to stabilize the MVEC barrier, there is some limited, indirect evidence of TIMPs promoting barrier dysfunction (red bubbles). These mechanisms that have the potential to promote MVEC barrier dysfunction include promoting endothelial cell apoptosis, activation of latent MMPs, and the initiation of ECM accumulation leading to fibrosis. |
TIMP structure and function
There are four TIMP isoforms in mammals (TIMP1, -2,-3, -4), all of which are constitutively expressed in many tissues.37,39 Expression of individual TIMPs can also be differentially induced or inhibited within specific tissues and under certain conditions (ie, during development, following injury or infection, etc), with these changes in TIMP expression regulated at the transcriptional level by cytokines, growth factors, and miRNAs.39 For instance, there is a significant increase in TIMP1 and -3 in keratinocytes during wound healing following acute injury (3–5 days postinjury), whereas there is no evidence of TIMP1 and -3 expression in the epidermis following chronic or persistent injury.40 While TIMP1 and -2 are constitutively active under healthy conditions at low levels, their expression increases significantly during diseased states and is highest during early stages of cutaneous wound repair.41 Collectively, these studies demonstrate that TIMP expression appears to be highly regulated, both temporally and spatially.40
All TIMPs contain an N- and C-terminal domain connected through six conserved disulfide bonds giving the TIMPs a wedge-like appearance.37 The N-terminal domain is responsible for the inhibition of most metalloproteinases through formation of a ridge that slots into the metalloproteinase catalytic site, blocking the active site Zn2+ molecule.37 The C-terminal domain is critical for protein–protein interactions and is thought to have a limited role in metalloproteinase inhibition. There is evidence, however, that the C-terminal domain of TIMP1 and -2 binds latent MMP9 and -2, respectively, and this interaction leads to MMP activation, which highlights the complexity of the MMP–TIMP relationship.39 The C-terminus of TIMP1, -2, and -3 has also been found to play a significant role in the metalloproteinase-independent functions of TIMPs.42
Metalloproteinase structure and function
One of the primary and best characterized roles for TIMPs is the regulation of metalloproteinase activity, including the MMPs, ADAMs, and ADAMs with thrombospondin repeats (ADAMTSs) (Figure 1). MMPs are Zn2+-dependent metalloendopeptidases that have various functions, including remodeling the ECM and cleaving cell surface proteins.43 MMP expression is tightly regulated by proinflammatory cytokines, growth factors, and hormones, as well as cell–cell and cell–matrix interactions.43 Most MMPs share a common underlying domain structure, which includes a signal peptide, a pro-peptide, a catalytic domain, a hinge region, and a C-terminal domain. A small subset of MMPs contains additional structures, such as a transmembrane domain and a cytoplasmic tail.
Other members of the metalloproteinase family regulated by TIMPs include the ADAMs and ADAMTSs.44 ADAMs are largely involved in the cleavage and shedding of cell surface molecules, including those involved in inflammation.44 ADAMTSs play an important role in tissue morphology and the pathological remodeling of the endothelium during inflammation.45
Metalloproteinase-dependent functions of TIMPs
As already mentioned, the traditional function of TIMPs has always been thought to be inhibition of metalloproteinase activity. The action of individual TIMPs, however, depends on three factors: the specific TIMP being examined, the cellular environment and tissue type, and the specific metalloproteinase. Moreover, each TIMP has distinct affinities for specific MMPs. For instance, TIMP1 has a greater affinity for MMP3 than the other TIMPs; however, TIMP1 also appears to have a limited role in regulation of the membrane-type MMPs (MMP14, -15,-16, -17, -24, and -26).39
Further, while there are structural similarities between the individual TIMPs, the physiological functions in most cases are unique, and in some cases are only now being identified. For example, the ability of TIMPs to inhibit MMPs led to the belief by many that an increase in TIMP expression leads to ECM accumulation and fibrosis, and a decrease in TIMP expression leads to ECM degradation (Figure 1).46 Importantly, there is certainly evidence to support this idea. For instance, increased expression of TIMP2 has been implicated in idiopathic pulmonary fibrosis due to its role in MMP inhibition and the consequent ECM accumulation.47 Moreover, in patients with COPD, overexpression of MMP1 and -9 was associated with destruction of the pulmonary ECM and subsequently, loss of the normal pulmonary architecture.48
Analysis of the varied phenotypes present in mice lacking individual or multiple TIMPs, however, revealed a differential effect on ECM turnover. For example, mice lacking TIMP3 have significantly increased collagen accumulation and fibrosis following bleomycin-induced lung injury compared to wild-type (WT) mice, likely due to an overexuberant and persistent inflammatory response.49 Collectively, these studies suggest divergent, sometimes opposing, roles of TIMPs as regulators of ECM turnover (Figure 1).46
TIMP regulation of MMP activity
Metalloproteinase inhibition by TIMPs impacts cell function through multiple pathways. For example, TIMP1 has been found to regulate cell migration. Specifically, TIMP1 can inhibit MVEC migration by restricting the shedding of the cell surface proteins, VE-cadherin, and platelet endothelial cell adhesion molecule 1 (PECAM1), likely through inhibition of MMP9.50 Further, TIMP1 also has been implicated in restricting epithelial cell migration specifically through inhibition of MMP7-dependent syndecan-1 shedding.51 Thus, while TIMP1 has been found to be a critical mediator of cell migration in multiple cell types, this function is not mediated through the same MMPs, which again highlights the importance of the microenvironment and the cell-type-specific nature of the metalloproteinase–TIMP interaction.
Some TIMPs also have what appear to be roles that are contradictory to their function as inhibitors of metalloproteinases. Namely, multiple TIMPs have been linked to activation of latent MMPs. For example, latent MMP2 can bind TIMP2, -3, and -4, and latent MMP9 can bind TIMP1 and -3.39 The best characterized role of these is the interaction between latent MMP2 and TIMP2.52 The N-terminal domain of TIMP2 binds to MMP14 while the C-terminal domain binds to latent MMP2. This interaction brings the latent MMP2 into close proximity with a second MMP14 leading to full activation of MMP2.52,53 This highlights the importance of the specific MMP in dictating TIMP function.
TIMP regulation of ADAM and ADAMTS activity
TIMPs are also known to inhibit both ADAMs and ADAMTSs. Of the four TIMP family members, TIMP3 is considered to have the broadest range of action as, along with inhibiting MMPs, it inhibits many ADAMs and ADAMTSs, including ADAM10, -12, -17, and -28, as well as ADAMTS1, -2, -4, and -5.37 TIMP3-dependent inhibition of ADAM17, which is also known as TNFα converting enzyme (TACE), is critical to ensure an appropriate inflammatory response as ADAM17 catalyzes the release of membrane-bound TNFα and other members of the TNF superfamily (Figure 2).54,55 ADAM17 is also involved in shedding membrane-bound TGFα (Figure 2).56 Soluble TGFα is a ligand for EGF receptors and when cleaved from the membrane surfaces of ECs, activates EGF receptors and the downstream pathways responsible for cell proliferation56 and has been implicated to an increased expression of IL8 and neutrophil migration.57 Loss of TIMP3 leads to increased ADAM17 activity, augmented TNFα and TGFα release, and consequently overexuberant inflammation (Figure 2).55 Moreover, TIMP3-dependent inhibition of ADAMTS4 and -5 appears to inhibit aggrecan degradation and thereby mediate ECM turnover.58,59
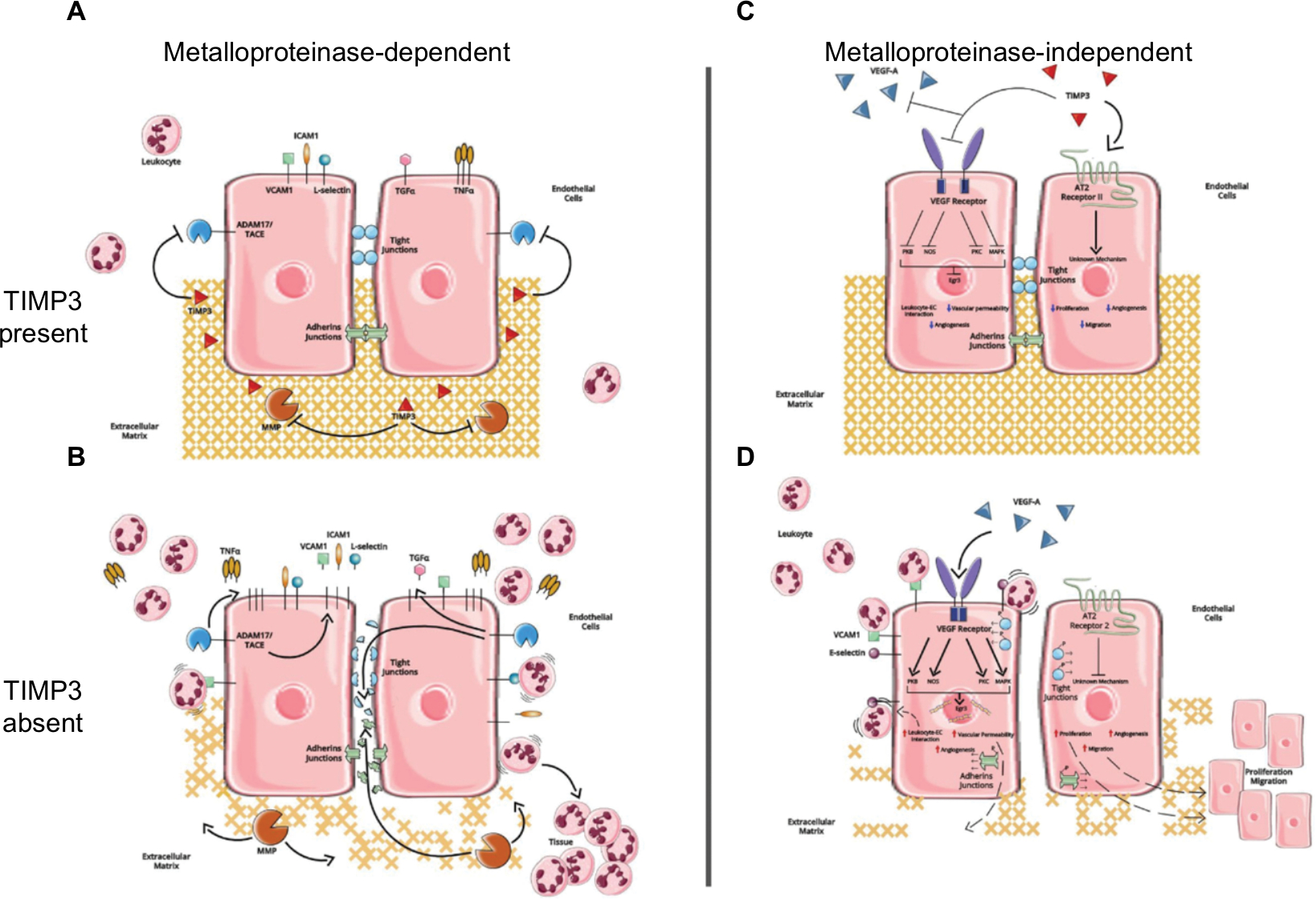 | Figure 2 Predicted metalloproteinase-dependent and metalloproteinase-independent functions of TIMP3 in MVECs. Notes: (A) In the presence of TIMP3, MMPs and ADAM17/ tumor necrosis factor alpha converting enzyme (TACE) are inhibited. The structure and composition of the ECM as well as the MVEC intercellular junctions are maintained. Moreover, leukocyte adhesion and extravasation are limited. (B) In the absence of TIMP3, increased MMP activity leads to degradation of the ECM and proteins associated with intercellular junctions. Additionally, increased ADAM17 activity leads to increased release of TNFα, which propagates the proinflammatory response. Expression of leukocyte receptors, such as intercellular adhesion molecule 1 and vascular cell adhesion molecule 1, is increased on the surface of MVECs. Moreover, shedding of these receptors by metalloproteinases is thought to promote leukocyte extravasation into the underlying tissue. Membrane-bound transforming growth factor α is also shed from the cell surface by ADAM17, resulting in binding to and activation of EGF receptors to promote cell proliferation, cell migration, and angiogenesis. (C) TIMP3 binds the VEGF receptor, inhibiting its downstream cascade by preventing interaction with and binding of its ligand, VEGF-A. TIMP3 also binds to and activates the angiotensin type II (AT2) receptor II. The combined effects of VEGF receptor inhibition and AT2 receptor II activation decrease cell proliferation and migration, inhibit angiogenesis, reduce vascular permeability, and inhibit leukocyte–EC interaction. (D) The absence of TIMP3 allows VEGF to bind to its receptor, activating the MAPK, protein kinase B, protein kinase C, and nitric oxide synthase pathways. This ultimately leads to increased expression of the early growth response protein 3 (Egr3) and phosphorylation of proteins associated with MVEC intercellular junctions. Moreover, the AT2 receptor II does not get activated due to the lack of TIMP3 binding. Overall, the combined effects of VEGF activation and AT2 receptor II inactivation result in increased cell proliferation and migration, increased angiogenesis, increased expression of cell surface adhesion molecules leading to leukocyte–EC interaction, and vascular permeability. Abbreviations: TIMP3, tissue inhibitors of metalloproteinases; MVECs, microvascular endothelial cells; MMP, matrix metalloproteinases; ECM, extracellular matrix; ADAM, a disintegrin and metalloproteinase; TNFα, tumor necrosis factor alpha. |
Metalloproteinase-independent functions of TIMPs
TIMPs are multifunctional proteins involved in numerous biological processes, such as regulation of cell proliferation, migration and invasion, angiogenesis, and apoptosis.60–62 While some of these functions may be a result of the ability of TIMPs to inhibit metalloproteinases, recent studies have shown that many of the TIMPs elicit downstream responses independent of metalloproteinase inhibition. For instance, TIMP1 was implicated in regulating CD4+ T-cell migration across the blood–brain barrier (BBB) during viral encephalitis.63 This was demonstrated using mice deficient for TIMP1 infected with a neurotropic coronavirus and quantifying the accumulation of CD4+ T cells in the perivasculature of the BBB.63 Specifically, Savarin et al found that in mice lacking TIMP1, accumulation of CD4+ T cells in the perivasculature was increased following viral encephalomyelitis, and that this increase was independent of any altered chemokines, MMP activity, or other TIMP involvement.63 This suggests TIMP1 controls the leukocyte access to the central nervous system through the BBB via a metalloproteinase-independent mechanism that may involve oligodendrocyte progenitor differentiation and remyelination.63
TIMP regulation of cell signaling
TIMP1 and -2 also have pro-mitogenic activity, which has been shown to be through metalloproteinase-independent mechanisms. Specifically, TIMP1 and -2 bind to Ras receptors on mesenchymal and epithelial cells, including keratinocytes and fibroblasts, as well as some cancer cells to activate the MAPK pathway and the protein kinase A (PKA) pathway and thereby induce proliferation.42
The effect of TIMP1 and -2 on proliferation, however, appears to be cell-type dependent. For example, in human breast epithelial cells (MCF10A), TIMP1 downregulates the expression of Cyclin D1 through upregulation of P27 (a cycline-dependent kinase inhibitor).37 This downregulation of Cyclin D1 suppresses cell growth; however, the cells remain viable suggesting that TIMP1 promotes cell survival.37 TIMP2 has been found to suppress EC proliferation by binding the α3β1 integrin and activating SH2-protein tyrosine phosphatase 1 (SHP1). This activation of SHP1 leads to dephosphorylation of the VEGF receptors and fibroblast growth factor receptors and inhibits MAPK signaling.42
TIMP3 also has multiple metalloproteinase-independent functions, many of which mediate EC proliferation and survival. For example, TIMP3 has been shown to bind the VEGF receptor, block VEGF–VEGFR2 interaction, and thereby restrict downstream VEGF receptor signaling and subsequent EC migration and proliferation (Figure 2).64 TIMP3 has also been suggested to inhibit EC migration and proliferation by binding to the angiotensin II type 2 receptor and preventing Akt activation (Figure 2). However, the underlying mechanisms are still unknown and require further research.
TIMP regulation of apoptosis
TIMP1 has also been found to have significant anti-apoptotic activity and to promote cell survival. Specifically, TIMP1 has been found to activate the focal adhesion kinase (FAK)-phosphoinositol-3 kinase (PI3K) pathway, potentially through interaction with CD63, a protein in the tetraspanin family, and protect epithelial cells from apoptotic death induced in one of two ways – by culturing cells in ployHEMA-coated wells, which prevents cell adhesion and causes anoikis, and through growth factor withdrawal by culturing cells in serum-free medium.62 Human breast epithelial cells were treated with a mutant TIMP1 that had limited affinity for MMPs, and the treated cells showed a greater survival rate than other groups treated with TIMP2 or a synthetic metalloproteinase inhibitor, suggesting that TIMP1 has potent, metalloproteinase-independent mechanisms for inhibiting apoptosis.62
Interestingly, in addition to restricting EC migration and proliferation, TIMP3 has also been implicated in promoting EC apoptosis. Specifically, overexpression of TIMP3 in pulmonary artery ECs was associated with induction of apoptosis and this appeared to be due to inhibition of a FAK-dependent survival pathway.65 Collectively, these studies provide direct evidence of the ability of TIMPs to mediate MVEC function (ie, regulation of VEGF signaling, of FAK-dependent survival, and of leukocyte recruitment) through mechanisms independent of their ability to inhibit metalloproteinases.
TIMPs and sepsis
Septic MOD is largely thought to be due to dysfunction of the microvasculature, specifically of the MVECs, leading to loss of MVEC barrier function and tissue edema.66 There are many potential regulators of septic MVEC dysfunction, and TIMPs, through their ability to inhibit metalloproteinases as well as through their metalloproteinase-independent functions, are one such regulator.
Expression of TIMPs and metalloproteinases
The expression of TIMPs, as well as MMPs, has been found to be dysregulated in septic patients. Specifically, patients with severe sepsis showed elevated plasma levels of MMP3, -7, -8, and -9 as well as TIMP1, -2, and -4 upon admission to the intensive care unit.67 Importantly, while TIMP1 serum levels tend to be high in septic patients throughout the duration of disease, increased TIMP1 concentration also appears to be associated with increased severity of sepsis.68,69
The ratio between TIMP1 and MMP9 may also be predictive of disease severity and patient survival as nonsurviving septic patients showed a significant increase in TIMP1/MMP9 ratio during the first week of diagnosis.70 This ratio may also function as a potential biomarker to predict the progression, severity, and outcome of sepsis in regard to tissue injury, specifically kidney disease. For example, patients with sepsis-associated acute kidney injury were found to have higher serum TIMP1 levels, lower MMP9 levels, and a lower MMP9/TIMP1 ratio than those patients that did not develop kidney injury.71
In addition to changes in TIMP1 levels, the levels of TIMP2 in serum were lower in septic patients compared to healthy controls.72 Moreover, the MMP2/TIMP2 ratio was found to be elevated in the serum of septic patients.72 Collectively, these studies provide multiple lines of evidence that TIMP and metalloproteinase, specifically MMP, expression becomes dysregulated during sepsis. The cell source, however, and whether these observed septic changes are due to alterations in MVECs remains to be determined.
There is certainly evidence of TIMP expression within different vascular beds throughout the body, including microvascular beds. For example, TIMP1, -3, and -4 are expressed by brain MVECs.73,74 Interestingly, TIMP3 is also expressed by pulmonary MVECs, whereas TIMP2 and -4 do not appear to be.75,76 Expression of TIMPs by cells associated with the vasculature has also been found to be altered following vascular injury. Specifically, expression of TIMP4 (both RNA and protein) was observed in the carotid artery within 24 hours of balloon injury and increased expression over the course of 2 weeks.77 TIMPs are also often expressed by cells other than EC associated with the vasculature. For example, TIMP3 is expressed by pericytes and appears to be critical for the ability of pericytes to promote MVEC barrier function.78 Additionally, this expression of TIMP3 by human pericytes is augmented under shear stress.79 Importantly, this expression of TIMPs often appears to be altered following injury and inflammation. For example, stimulation of brain MVECs with IL1β and TNFα increases Timp1 expression and decreases Timp3 expression.73 Thus, the expression of TIMPs by the vasculature, especially MVECs, as well as the changes following injury and inflammation, provides further support of a critical role for TIMPs in regulating MVEC function under both healthy and pathological conditions.
TIMP regulation of endothelial cell dysfunction
The potential for TIMPs to mediate MVEC function is without question but surprisingly, there is limited direct evidence of a role for MVEC-derived TIMPs in endothelial dysfunction. However, analysis of studies utilizing animal models from mice globally deficient for individual TIMPs as well as those using recombinant TIMPs has provided convincing data to suggest that TIMPs may in fact be critical mediators of endothelial dysfunction under pathological conditions. For example, disruption of the BBB is augmented in mice lacking TIMP1 compared to WT mice following cerebral ischemia, and this appears to be due to increased MMP9 activity.80 Additionally, MMP9 has been shown to degrade the tight junction proteins, claudin 5 and occludin, in brain MVECs leading to a loss of MVEC barrier function, and upregulation of TIMP1 in these brain MVECs reversed this degradation and restored the MVEC barrier.81
TIMP2 and TIMP3 have also been found to promote stability of the BBB; however, these changes appear to be mediated through metalloproteinase-independent functions. Specifically, BBB disruption was found to be augmented in Timp2−/− mice following cerebral ischemia.80 However, unlike in Timp1−/− mice, the augmented BBB disruption in Timp2−/− mice was not associated with increased MMP activity.80 Additionally, injection of recombinant TIMP3 was found to diminish BBB disruption following traumatic brain injury, and treatment of mice with a synthetic metalloproteinase inhibitor was unable to recapitulate the effect of recombinant TIMP3.82
Use of Timp3−/− mice has provided further support for a critical role of TIMP3 in regulating MVEC barrier function. The role identified using mice lacking TIMP3, however, appears to depend on the ability of TIMP3 to inhibit metalloproteinases. For example, mice lacking TIMP3 have increased pulmonary microvascular permeability under basal conditions and this is likely due to the absence of MVEC-derived TIMP3 as pulmonary MVECs isolated from Timp3−/− mice have significantly increased permeability under both basal and septic conditions vs WT mice.75 Moreover, this effect was likely due to dysregulated metalloproteinase activity as treatment of Timp3−/− pulmonary MVECs in vitro with GM6001 rescued the augmented permeability and restored MVEC barrier function under basal conditions.75 Timp3−/− mice have increased permeability under basal conditions in their snout and kidneys, as demonstrated by increased Evans blue-labeled albumin leak.78 Interestingly, this augmented leak was suggested to be due to loss of pericyte-derived TIMP3 and a subsequent increase in metalloproteinase activity.78 Taken together, these studies highlight the important role of TIMP3 in promoting MVEC barrier function, as well as the importance of TIMPs expressed by both MVECs and the supporting cells within the vascular microenvironment. Interestingly, while TIMP4 is expressed by cells associated with the vasculature and has been implicated in various pathologies such as cardiovascular disease83 and tumorigenesis,84 there is limited evidence suggesting a role for TIMP4 in regulating MVEC barrier function.
Metalloproteinase-dependent disruption of intercellular junctions
Additionally, although indirect, evidence of a potential role for TIMPs in MVEC barrier function is that multiple MMPs and ADAMs have been shown to cleave MVEC intercellular junctional proteins, including those associated with adherens and tight junctions, thereby promoting vascular permeability. For example, MMP7, ADAM10, and ADAM12 have been shown to cleave VE-cadherin (adherens junction), and MMP2 and -9 as well as ADAM17 have been shown to cleave occludin, zona occludens 1, and JAMs (tight junctions).35,38,85–87
Further indirect evidence is provided by several studies utilizing synthetic metalloproteinase inhibitors, such as GM6001. For example, stimulation of HUVEC with TNFα leads to increased metalloproteinase activity, which is associated with increased permeability. Treatment with GM6001 or with GI254023X, an ADAM10-specific inhibitor, reduced the TNFα-dependent increase in permeability, at least partly, through inhibiting VE-cadherin cleavage.88,89 Moreover, GM6001 treatment of brain MVECs was also found to inhibit microvascular permeability, and this reduction was found to be due to inhibition of MMP9 leading to increased stability of proteins associated with tight junctions.81,90 Collectively, these studies illustrate the importance of metalloproteinase inhibition in the regulation of MVEC barrier dysfunction and highlight the potential role of TIMPs.
Metalloproteinase-dependent ECM degradation
MMPs and ADAMTSs are also involved in degradation and remodeling of the ECM surrounding the vasculature, which can also modify EC function through disrupted endothelial–ECM interactions. For example, degradation of the basement membrane proteins fibronectin, laminin, and type IV collagen by MMP2 and -9 is associated with increased MVEC permeability in vitro as well as in vivo.91,92 These studies demonstrate that metalloproteinases can affect MVEC barrier function through multiple mechanisms, including through ECM remodeling. As such, the inhibition of these enzymes by TIMPs is a critical checkpoint required to maintain MVEC barrier function.
TIMP regulation of VEGF signaling
Finally, another additional mechanism through which TIMPs may mediate MVEC barrier function is through regulation of VEGF and activation of the VEGF signaling pathways. VEGF, which is critical for angiogenesis, is also known to be a potent mediator of increased endothelial permeability through promoting phosphorylation and internalization of VE-cadherin and subsequent disassembly of cell–cell adhesive contacts.93 Importantly, multiple TIMPs have been found to restrict VEGF signaling. For example, TIMP1 has been found to inhibit VEGF-stimulated EC proliferation leading to reduced vessel growth in ovaries potentially through increased release of sVEGFR1 from ECs, thereby reducing the bioavailability of VEGF.94 Moreover, both TIMP2 and -3 have also been found to inhibit EC proliferation and migration specifically through inhibition of VEGF signaling.64,95,96 However, unlike TIMP1, TIMP3-dependent inhibition of VEGF signaling occurs through direct interaction between TIMP3 and VEGFR2, which subsequently blocks VEGF–VEGFR2 interaction.64,95,96 Thus, these studies demonstrate the potential of TIMPs to regulate MVEC barrier function through direct control of VEGF signaling independent of their ability to inhibit metalloproteinase activity.
TIMP regulation of inflammation
Dysregulated inflammation is a key pathological feature of septic MOD. Moreover, this dysregulated inflammation ultimately results in increased leukocyte–MVEC interaction, which has been associated with a loss of MVEC barrier function. Importantly, TIMPs have been shown to regulate leukocyte–MVEC interaction, largely through their ability to regulate metalloproteinase activity.
While multiple TIMPs have been implicated in inflammation, TIMP3 appears to have the most significant role. Specifically, TIMP3 is best known as the physiological inhibitor of ADAM17 or TACE (Figure 2). For example, ADAM17/TACE activity was increased following a partial hepatectomy in mice lacking TIMP3 leading to increased release of TNFα in the liver.55 TIMP3 also inhibits shedding of VCAM1 from aortic EC following treatment with IL1β and TNFα.97 Further evidence of this was provided by Timp3−/− mice, as aortic ECs isolated from Timp3−/− mice had increased VCAM1 shedding vs those from WT mice. Moreover, the augmented shedding of VCAM1 in the absence of TIMP3 was due to increased ADAM17 activity as knockdown of ADAM17 protected against increased VCAM1 shedding.97
In addition to ADAM17, MMPs are also capable of mediating leukocyte–MVEC interaction through shedding of leukocyte receptors, such as ICAM and VCAM. For example, MMP14 and ADAM10 have been found to cleave ICAM1 from the cell surface of MVEC.98,99 Importantly, shedding of both ICAM1 and VCAM1 by metalloproteinases reduces MVEC surface expression of both ICAM1 and VCAM1, resulting in diminished leukocyte–MVEC interaction.100 Taken together, these studies strongly support a role for TIMPs in modulating the inflammatory response through control of proinflammatory cytokine release as well as stabilization of leukocyte receptors on the MVEC surface.
Conclusion
Septic organ dysfunction is primarily thought to be due to activation and subsequent dysfunction of MVECs, leading to a loss of MVEC barrier function and increased vascular permeability. There are multiple mechanisms known to promote MVEC dysfunction, including interaction with circulating proinflammatory mediators, such as cytokines and bacterial products, as well as with activated leukocytes. TIMPs appear to have a critical role in MVEC barrier function. This role for TIMPs occurs through both inhibition of metalloproteinases, which promotes stability of MVEC intercellular junctions and inhibits ECM remodeling, as well as through metalloproteinase-independent functions. These metalloproteinase-independent functions include inhibition of VEGF signaling and FAK-dependent cell survival, as well as leukocyte–MVEC interaction. The role of individual TIMPs, especially with respect to metalloproteinase-independent functions as well as leukocyte–MVEC interaction and the specific impact this has on septic MVEC barrier dysfunction, is unclear at this time and as such is the subject of ongoing studies. However, in general, the primary function of TIMPs appears to be to promote MVEC barrier function, and as such TIMPS may be critically important in restoring barrier function following infection and injury.
Disclosure
The authors report no conflicts of interest in this work.
References
Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801. | ||
Genga KR, Russell JA. Update of sepsis in the intensive care unit. J Innate Immun. 2017;9(5):441–455. | ||
Yende S, Austin S, Rhodes A, et al. Long-term quality of life among survivors of severe sepsis: analyses of two international trials. Crit Care Med. 2016;44(8):1461–1467. | ||
Barichello T, Sayana P, Giridharan VV, et al. Long-term cognitive outcomes after sepsis: a translational systematic review. Mol Neurobiol. Epub 2018 Apr 23. | ||
Sagy M, Al-Qaqaa Y, Kim P. Definitions and pathophysiology of sepsis. Curr Probl Pediatr Adolesc Health Care. 2013;43(10):260–263. | ||
6.Duque GA, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. | ||
Sullivan JS, Kilpatrick L, Costarino AT, Lee SC, Harris MC. Correlation of plasma cytokine elevations with mortality rate in children with sepsis. J Pediatr. 1992;120(4):510–515. | ||
Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–882. | ||
Otto GP, Sossdorf M, Claus RA, et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Critical Care. 2011;15(4):R183. | ||
van Vught LA, Klein Klouwenberg PMC, Spitoni C, et al. Incidence, risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. JAMA. 2016;315(14):1469. | ||
Souza ACP, Yuen PST, Star RA. Microparticles: markers and mediators of sepsis-induced microvascular dysfunction, immunosuppression, and AKI. Kidney Int. 2015;87(6):1100–1108. | ||
Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778(3):794–809. | ||
Bhattacharya J, Matthay MA. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol. 2013;75(1):593–615. | ||
Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121(13):2115–2122. | ||
Weksler BB, Subileau EA, Perrière N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. Faseb J. 2005;19(13):1872–1874. | ||
Anderson JM. Molecular structure of tight junctions and their role in epithelial transport. News Physiol Sci. 2001;16:126–130. | ||
Rodrigues SF, Granger DN. Blood cells and endothelial barrier function. Tissue Barriers. 2015;3(1–2):e978720. | ||
Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285(5430):1028–1033. | ||
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(24):4195–4200. | ||
Souza-Fernandes AB, Pelosi P, Rocco PRM. Bench-to-bedside review: the role of glycosaminoglycans in respiratory disease. Critical Care. 2006;10(6):237. | ||
Sukriti S, Tauseef M, Yazbeck P, Mehta D. Mechanisms regulating endothelial permeability. Pulm Circ. 2014;4(4):535–551. | ||
Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol Mech Dis. 2009;4(1):71–95. | ||
Lee KS, Jin SM, Kim HJ, Lee YC. Matrix metalloproteinase inhibitor regulates inflammatory cell migration by reducing ICAM-1 and VCAM-1 expression in a murine model of toluene diisocyanate-induced asthma. J Allergy Clin Immunol. 2003;111(6):1278–1284. | ||
Kuebler WM, Wittenberg C, Lee WL, et al. Thrombin stimulates albumin transcytosis in lung microvascular endothelial cells via activation of acid sphingomyelinase. Am J Physiol Lung Cell Mol Physiol. 2016;310(8):L720–L732. | ||
Wang L, Mehta S, Ahmed Y, Wallace S, Pape MC, Gill SE. Differential mechanisms of septic human pulmonary microvascular endothelial cell barrier dysfunction depending on the presence of neutrophils. Front Immunol. 2018;9:9. | ||
Rahimi N. Defenders and challengers of endothelial barrier function. Front Immunol. 2017;8:1847. | ||
Giannotta M, Trani M, Dejana E. VE-Cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26(5):441–454. | ||
Mehta D, Ravindran K, Kuebler WM. Novel regulators of endothelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2014;307(12):L924–L935. | ||
Dreymueller D, Uhlig S, Ludwig A. ADAM-family metalloproteinases in lung inflammation: potential therapeutic targets. Am J Physiol Lung Cell Mol Physiol. 2015;308(4):L325–L343. | ||
Flemming S, Burkard N, Renschler M, et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc Res. 2015;107(1):32–44. | ||
Lakhan SE, Kirchgessner A, Tepper D, Leonard A, Aidan L. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32. | ||
Nagase H, Woessner JF. Matrix metalloproteinases. J Biol Chem. 1999;274(31):21491–21494. | ||
Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115(19):3719–3727. | ||
Stetler-Stevenson WG. Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention. J Clin Invest. 1999;103(9):1237–1241. | ||
Dreymueller D, Pruessmeyer J, Groth E, Ludwig A. The role of ADAM-mediated shedding in vascular biology. Eur J Cell Biol. 2012;91(6–7):472–485. | ||
Dreymueller D, Martin C, Kogel T, et al. Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol Med. 2012;4(5):412–423. | ||
Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta – Mol Cell Res. 2010;1803(1):55–71. | ||
Masciantonio MG, Lee CKS, Arpino V, Mehta S, Gill SE. Chapter Three – the balance between metalloproteinases and TIMPs: critical regulator of microvascular endothelial cell function in health and disease. In: Khalil RABT. Matrix Metalloproteinases and Tissue Remodeling in Health and Disease: Cardiovascular Remodeling. Vol 147. Academic Press; 2017:101–131. | ||
Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011;12(11):233. | ||
Vaalamo M, Leivo T, Saarialho-Kere U. Differential expression of tissue inhibitors of metalloproteinases (TIMP-1, -2, -3, and -4) in normal and aberrant wound healing. Hum Pathol. 1999;30(7):795–802. | ||
Ashcroft GS, Herrick SE, Tarnuzzer RW, Horan MA, Schultz GS, Ferguson MWJ. Human ageing impairs injury-inducedin vivo expression of tissue inhibitor of matrix metalloproteinases (TIMP)-1 and -2 proteins and mRNA. J Pathol. 1997;183(2):169–176. | ||
Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal. 2008;1(27):re6. | ||
Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–573. | ||
Johnson JL. Metalloproteinases in atherosclerosis. Eur J Pharmacol. 2017;816(June):93–106. | ||
Kelwick R, Desanlis I, Wheeler GN, Edwards DR. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015;16(1):113. | ||
Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015;44–46:247–254. | ||
Selman M, Ruiz V, Cabrera S. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am J Physiol Cell Mol Physiol. 2000;279(3):L562–L574. | ||
Elkington PTG, Friedland JS. Matrix metalloproteinases in destructive pulmonary pathology. Thorax. 2006;61(3):259–266. | ||
Gill SE, Huizar I, Bench EM, et al. Tissue inhibitor of metalloproteinases 3 regulates resolution of inflammation following acute lung injury. Am J Pathol. 2010;176(1):64–73. | ||
Akahane T, Akahane M, Shah A, Connor CM, Thorgeirsson UP. TIMP-1 inhibits microvascular endothelial cell migration by MMP-dependent and MMP-independent mechanisms. Exp Cell Res. 2004;301(2):158–167. | ||
Chen P, Mcguire JK, Hackman RC, et al. Tissue inhibitor of metalloproteinase-1 moderates airway re-epithelialization by regulating matrilysin activity. Am J Pathol. 2008;172(5):1256–1270. | ||
Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13(9):649–665. | ||
Bernardo MM, Fridman R. TIMP-2 (tissue inhibitor of metalloproteinase-2) regulates MMP-2 (matrix metalloproteinase-2) activity in the extracellular environment after pro-MMP-2 activation by MT1 (membrane type 1)-MMP. Biochem J. 2003;374(3):739–745. | ||
Black RA. TIMP3 checks inflammation. Nat Genet. 2004;36(9):934–935. | ||
Mohammed FF, Smookler DS, Taylor SEM, et al. Abnormal TNF activity in Timp3 −/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet. 2004;36(9):969–977. | ||
Borrell-Pagès M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for the activation of the EGFR by TGF-α in tumors. Embo J. 2003;22(5):1114–1124. | ||
Hamilton LM, Torres-Lozano C, Puddicombe SM, et al. The role of the epidermal growth factor receptor in sustaining neutrophil inflammation in severe asthma. Clin Exp Allergy. 2003;33(2):233–240. | ||
Hashimoto G, Aoki T, Nakamura H, Tanzawa K, Okada Y. Inhibition of ADAMTS4 (aggrecanase-1) by tissue inhibitors of metalloproteinases (TIMP-1, 2, 3 and 4). FEBS Lett. 2001;494(3):192–195. | ||
Troeberg L, Fushimi K, Scilabra SD, et al. The C-terminal domains of ADAMTS-4 and ADAMTS-5 promote association with N-TIMP-3. Matrix Biol. 2009;28(8):463–469. | ||
Murphy AN, Unsworth EJ, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinases-2 inhibits bFGF-induced human microvascular endothelial cell proliferation. J Cell Physiol. 1993;157(2):351–358. | ||
Baker AH, Zaltsman AB, George SJ, Newby AC. Divergent effects of tissue inhibitor of metalloproteinase-1, -2, or -3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. J Clin Invest. 1998;101(6):1478–1487. | ||
Liu X-W, Bernardo MM, Fridman R, Kim H-RC, H-Rc K. Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells against intrinsic apoptotic cell death via the focal adhesion kinase/phosphatidylinositol 3-kinase and MAPK signaling pathway. Journal of Biological Chemistry. 2003;278(41):40364–40372. | ||
Savarin C, Bergmann CC, Hinton DR, Stohlman SA. MMP-independent role of TIMP-1 at the blood brain barrier during viral encephalomyelitis. ASN Neuro. 2013;5(5):AN20130033. | ||
Qi JH, Ebrahem Q, Moore N, et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9(4):407–415. | ||
Qi JH, Anand-Apte B. Tissue inhibitor of metalloproteinase-3 (TIMP3) promotes endothelial apoptosis via a caspase-independent mechanism. Apoptosis. 2015;20(4):523–534. | ||
Aird WC. Review article the role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. 2003;101(10):3765–3777. | ||
Yazdan-Ashoori P, Liaw P, Toltl L, et al. Elevated plasma matrix metalloproteinases and their tissue inhibitors in patients with severe sepsis. J Crit Care. 2011;26(6):556–565. | ||
Hästbacka J, Linko R, Tervahartiala T, et al. Serum MMP-8 and TIMP-1 in critically ill patients with acute respiratory failure: TIMP-1 is associated with increased 90-day mortality. Anesth Analg. 2014;118(4):790-8. | ||
Wang M, Zhang Q, Zhao X, Dong G, Li C. Diagnostic and prognostic value of neutrophil gelatinase-associated lipocalin, matrix metalloproteinase-9, and tissue inhibitor of matrix metalloproteinases-1 for sepsis in the Emergency Department: an observational study. Critical Care. 2014;18(6):634. | ||
Lorente L, Martín MM, Solé-Violán J, et al. Association of sepsis-related mortality with early increase of TIMP-1/MMP-9 ratio. PLoS One. 2014;9(4):e94318. | ||
Bojic S, Kotur-Stevuljevic J, Kalezic N, et al. Diagnostic value of matrix metalloproteinase-9 and tissue inhibitor of matrix metalloproteinase-1 in sepsis-associated acute kidney injury. Tohoku J Exp Med. 2015;237(2):103–109. | ||
Mühl D, Nagy B, Woth G, et al. Dynamic changes of matrix metalloproteinases and their tissue inhibitors in severe sepsis. J Crit Care. 2011;26(6):550–555. | ||
Bugno M, Witek B, Bereta J, Bereta M, Edwards DR, Kordula T. Reprogramming of TIMP-1 and TIMP-3 expression profiles in brain microvascular endothelial cells and astrocytes in response to proinflammatory cytokines. FEBS Lett. 1999;448(1):9–14. | ||
Shastry S, Tyagi SC. Homocysteine induces metalloproteinase and shedding of β-1 integrin in microvessel endothelial cells. J Cell Biochem. 2004;93(1):207–213. | ||
Arpino V, Mehta S, Wang L, et al. Tissue inhibitor of metalloproteinases 3-dependent microvascular endothelial cell barrier function is disrupted under septic conditions. American Journal of Physiology-Heart and Circulatory Physiology. 2016;310(11): H1455–H1467. | ||
Shen Q, Lee ES, Pitts RL, Mh W, Yuan SY. Tissue inhibitor of metalloproteinase-2 regulates matrix metalloproteinase-2–mediated endothelial barrier dysfunction and breast cancer cell transmigration through lung microvascular endothelial cells. Mol Cancer Res. 2010;8(7):939–951. | ||
Dollery CM, Mcewan JR, Wang M, Sang QA, Liu YE, Shi YE. TIMP-4 is regulated by vascular injury in rats. Circ Res. 1999;84(5):498–504. | ||
Schrimpf C, Xin C, Campanholle G, et al. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J Am Soc Nephrol. 2012;23(5):868–883. | ||
Schrimpf C, Koppen T, Duffield JS, et al. TIMP3 is regulated by pericytes upon shear stress detection leading to a modified endothelial cell response. Eur J Vasc Endovasc Surg. 2017;54(4):524–533. | ||
Fujimoto M, Takagi Y, Aoki T, et al. Tissue inhibitor of metalloproteinases protect blood—brain barrier disruption in focal cerebral ischemia. J Cereb Blood Flow Metab. 2008;28(10):1674–1685. | ||
Chen F, Ohashi N, Li W, Eckman C, Nguyen JH. Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology. 2009;50(6):1914–1923. | ||
Menge T, Zhao Y, Zhao J. mesenchymal stem cells regulate blood brain barrier integrity in traumatic brain injury through production of the soluble factor TIMP3. Sci Transl Med. 2012;4(161):161ra150–161161. | ||
Koskivirta I, Rahkonen O, Mäyränpää M, et al. Tissue inhibitor of metalloproteinases 4 (TIMP4) is involved in inflammatory processes of human cardiovascular pathology. Histochem Cell Biol. 2006;126(3):335–342. | ||
Rorive S, Lopez XM, Maris C, et al. TIMP-4 and CD63: new prognostic biomarkers in human astrocytomas. Mod Pathol. 2010;23(10):1418–1428. | ||
Bauer H, Zweimueller-Mayer J, Steinbacher P, Lametschwandtner A, Bauer HC. The dual role of zonula occludens (ZO) proteins. J Biomed Biotechnol. 2010;2010(4):1–11. | ||
Ichikawa-Tomikawa N, Sugimoto K, Satohisa S, Nishiura K, Chiba H. Possible involvement of tight junctions, extracellular matrix and nuclear receptors in epithelial differentiation. J Biomed Biotechnol. 2011;2011(2):1–10. | ||
Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. Journal of Neuroscience. 2012;32(9):3044–3057. | ||
Sidibé A, Mannic T, Arboleas M, et al. Soluble VE-cadherin in rheumatoid arthritis patients correlates with disease activity: evidence for tumor necrosis factor α-induced VE-cadherin cleavage. Arthritis Rheum. 2012;64(1):77–87. | ||
Schulz B, Pruessmeyer J, Maretzky T, et al. ADAM10 regulates endothelial permeability and T-cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res. 2008;102(10):1192–1201. | ||
Lominadze D, Roberts AM, Tyagi N, Moshal KS, Tyagi SC. Homocysteine causes cerebrovascular leakage in mice. Am J Physiol Heart Circ Physiol. 2006;290(3):H1206–H1213. | ||
Passi A, Negrini D, Albertini R, de Luca G, Miserocchi G. Involvement of lung interstitial proteoglycans in development of hydraulic- and elastase-induced edema. Am J Physiol Lung Cell Mol Physiol. 1998;275(3):L631–L635. | ||
Partridge CA, Jeffrey JJ, Malik AB. A 96-kDa gelatinase induced by TNF-alpha contributes to increased microvascular endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 1993;265(5): L438–L447. | ||
Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. | ||
Bruegmann E, Gruemmer R, Neulen J, Motejlek K. Regulation of soluble vascular endothelial growth factor receptor 1 secretion from human endothelial cells by tissue inhibitor of metalloproteinase 1. Mol Hum Reprod. 2009;15(11):749–756. | ||
Ebrahem Q, Qi JH, Sugimoto M, et al. Increased neovascularization in mice lacking tissue inhibitor of metalloproteinases-3. Invest Ophthalmol Vis Sci. 2011;52(9):6117–6123. | ||
Lee S-J, Tsang PS, M Diaz T, Wei B-Yang, Stetler-Stevenson WG. TIMP-2 modulates VEGFR-2 phosphorylation and enhances phosphodiesterase activity in endothelial cells. Lab Invest. 2010;90(3):374–382. | ||
Singh R, Mason J, Lidington E, et al. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc Res. 2005;67(1):39–49. | ||
Ponnuchamy B, Khalil RA. Role of ADAMs in endothelial cell permeability: cadherin shedding and leukocyte rolling. Circ Res. 2008;102(10):1139–1142. | ||
Sithu SD, English WR, Olson P, et al. Membrane-type 1-matrix metalloproteinase regulates intracellular adhesion molecule-1 (ICAM-1)-mediated monocyte transmigration. J Biol Chem. 2007;282(34):25010–25019. | ||
Garton KJ, Gough PJ, Philalay J, et al. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17). J Biol Chem. 2003;278(39):37459–37464. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.