")
Back to Journals » International Journal of Nanomedicine » Volume 12
The role of surface charge in the desolvation process of gelatin: implications in nanoparticle synthesis and modulation of drug release
Received 18 October 2016
Accepted for publication 3 December 2016
Published 24 January 2017 Volume 2017:12 Pages 795—808
DOI https://doi.org/10.2147/IJN.S124938
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Saad M Ahsan, Chintalagiri Mohan Rao
Centre for Cellular and Molecular Biology, Council of Scientific and Industrial Research, Hyderabad, Telangana, India
Abstract: The process of moving hydrophobic amino acids into the core of a protein by desolvation is important in protein folding. However, a rapid and forced desolvation can lead to precipitation of proteins. Desolvation of proteins under controlled conditions generates nanoparticles – homogeneous aggregates with a narrow size distribution. The protein nanoparticles, under physiological conditions, undergo surface erosion due to the action of proteases, releasing the entrapped drug/gene. The packing density of protein nanoparticles significantly influences the release kinetics. We have investigated the desolvation process of gelatin, exploring the role of pH and desolvating agent in nanoparticle synthesis. Our results show that the desolvation process, initiated by the addition of acetone, follows distinct pathways for gelatin incubated at different pH values and results in the generation of nanoparticles with varying matrix densities. The nanoparticles synthesized with varying matrix densities show variations in drug loading and protease-dependent extra- and intracellular drug release. These results will be useful in fine-tuning the synthesis of nanoparticles with desirable drug release profiles.
Keywords: protein desolvation, nanoparticle assembly, gelatin nanoparticle synthesis, protease susceptibility, intracellular drug release
Introduction
Proteins are excellent materials for nanoparticle-based drug/gene delivery applications.1 They are biocompatible, metabolizable and amenable to surface modifications and ligand attachments for targeted delivery.2–4 Desolvation is one of the common methods reported for protein nanoparticle synthesis, owing to its reproducibility and ease of preparation.5,6 The process of removing/replacing solvating water molecules, by a non-solvent, from the hydration shell of a macromolecule is called desolvation.7 Under physiological conditions, proteins tend to bury their nonpolar residues in the core to minimize solvent exposure along with increasing the surface exposure of polar residues.8 This process not only contributes in protein folding but also increases the solubility of proteins in water.7,9 When the critical balance between electrostatic and hydrophobic interactions is disturbed, proteins tend to undergo phase separation and precipitate.10 A rapid and forced desolvation of proteins thus leads to precipitation. However, desolvation under controlled experimental conditions generates homogeneous aggregates with a narrow size distribution which may be subsequently stabilized by chemical cross-linking.11–13
Several soluble and insoluble proteins such as elastin,14 albumin,15 gelatin,5 legumin,16 zein,17 soya18 and casein19 are being used to make protein nanoparticles. Gelatin and albumin have been extensively used for nanoparticle preparation by the desolvation method.20 Gelatin is a collagen derivative, and its properties are largely determined by the collagen source. A common characteristic feature of collagens, and hence gelatin, is the triple helical conformation,21 which is attributed to the conserved amino acid sequence repeat of Gly-X-Y, where glycine constitutes 33% of the sequence. The other major constituents are hydroxyproline, proline (combined 21%) and alanine (10%).22 The unique amino acid composition of gelatin is responsible for its unusual hydrogen bonding properties and hence a high water retention capacity.23,24
The preparation of nanoparticles by the desolvation method has been well studied and documented.25,26 The effect of various reaction parameters such as temperature and pH on nanoparticle size has been investigated.27 Protease-induced degradation of the nanoparticle matrix is considered the major mechanism of drug release, this could be either by bulk degradation or by surface erosion.26,28 Considering the surface area to volume ratio of nanoparticles, size is an important factor in determining the degradation rates by surface erosion. Yet another, rather less explored parameter important in determining the rate of drug release, is the matrix density. The desolvation process, under appropriate conditions, provides an opportunity to synthesize nanoparticles with varying matrix densities and hence a differential susceptibility toward proteases.
In the current study, we have investigated the mechanistic details of the desolvation process of gelatin, exploring the role of pH and desolvating agent in nanoparticle synthesis. Our results provide interesting insights into the mechanistic details of gelatin nanoparticle (GNP) assembly. We were able to produce nanoparticles with different matrix densities which show variations in their degradation kinetics, drug loading and protease-based extra- and intracellular drug release.
The results provided in the current study will be helpful in designing protein nanoparticles with the desired susceptibility toward proteases and hence tunable drug release profiles.
Materials and methods
Materials
Gelatin, from bovine skin, lime cured (type B), with bloom strength of 225, Nile Red (9-diethylamino-5H-benzo[α]phenoxazine-5-one) and fluorescein were obtained from Sigma-Aldrich (St Louis, MO, USA). Glutaraldehyde (25% aqueous solution) was obtained from Calbiochem. Acetone (high-performance liquid chromatography [HPLC] grade) was obtained from Spectrochem (Mumbai, India). Pronase was obtained from Roche. LysoTracker® Red DND-99 was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Vectashield antifade mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) was obtained from Vector Laboratories (Burlingame, CA, USA). All chemicals were of analytical reagent grade quality.
Zeta potential measurements
The zeta potential of gelatin in deionized water (10 mg mL−1) with pH ranging from 1.5 to 7.5 was recorded using a “Nanopartica” nanoparticle analyzer system (Horiba Scientific, Kyoto, Japan) at 25°C.
Probing hydrophobic surfaces of gelatin using Nile Red fluorescence
Nile Red (50 μM) was added to the sample of gelatin (0.5 mg mL−1), in water of pH ranging from 2.5 to 5.5. Fluorescence spectra were recorded from 550 to 800 nm using a Hitachi F-7000 fluorescence spectrophotometer (Hitachi High Technologies America Inc, Schaumburg, IL, USA) with the excitation wavelength set at 540 nm. The excitation and emission band passes were set at 5 nm.
Nanoparticle synthesis
The double desolvation method proposed by Coester et al5 was used for nanoparticle synthesis. Briefly, a 5% (w/v) gelatin solution was obtained by dissolving 2.5 g of gelatin type B (Bloom 225) in deionized water (50 mL) under gentle heating (50°C). The first desolvation step was carried out by a rapid addition of 50 mL of acetone to the gelatin solution. The supernatant containing low molecular weight gelatin fraction was discarded, and the precipitated high molecular weight gelatin fraction was resolubilized in deionized water and lyophilized. The lyophilized high molecular weight gelatin (0.1 g) was then dissolved in 10 mL of deionized water under gentle heating (50°C) to obtain a 1% gelatin solution. The solution was filtered with a 0.22 μm filter, and the pH was adjusted to the desired value by the addition of 0.1 N HCl or 0.1 N NaOH. Nanoparticle synthesis was then initiated by the dropwise addition of acetone under continuous stirring (500 rpm). After the addition of the desired amount of acetone, 20 μL of glutaraldehyde (25%) diluted in 1 mL of acetone was added to the reaction mixture for nanoparticle cross-linking. The solution was further stirred for 12 hours (h). The nanoparticles were subsequently purified by a three-step centrifugation (48,000× g for 10 min) and redispersion in acetone/water (30/70 mixture). The purified nanoparticles were stored as dispersion in deionized water at 4°C–8°C or lyophilized. The nanoparticles were analyzed for their size and polydispersity in a “Nanopartica” nanoparticle analyzer system (Horiba Scientific) equipped with a diode-pumped solid state (DPSS) laser of wavelength 532 nm and a temperature controller unit.
For studying the effect of pH on nanoparticle size, gelatin (10 mg mL−1) was incubated at the desired pH. The amount of acetone added to obtain the nanoparticles was, however, varied. All other parameters were kept constant.
For studying the effect of acetone concentration on nanoparticle size, gelatin type B (10 mg mL−1) was incubated at the desired pH, while all other parameters were kept constant. Desolvation was initiated by the addition of acetone. During the process of acetone addition, 1 mL of sample was withdrawn, treated with 2 μL of glutaraldehyde (25%) and kept on a rotator for 12 h. After stirring for 12 h, the nanoparticles were washed and analyzed for their size as described earlier.
The high matrix density-GNPs (HMD-GNPs) and the low matrix density-GNPs (LMD-GNPs) were synthesized by incubating gelatin type B (10 mg mL−1) at pH 4 and 3.25, respectively. The amount of acetone added to obtain nanoparticles was ~57% for HMD-GNPs and 73% for LMD-GNPs. All other parameters were kept constant, and the procedure described earlier was followed.
Loading of fluorescein into GNPs
GNPs were soaked in an aqueous fluorescein solution (fluorescein to GNP ratio, 1:10) at room temperature. After 24 h, the nanoparticles were purified by dialysis (molecular weight cutoff [MWCO], 8–12 K; Serva Electrophoresis GmbH, Heidelberg, Germany) against deionized water overnight to remove the unloaded free fluorescein. To determine the loading efficiency (LE) of fluorescein, the nanoparticles were digested with 10 μg mL−1 pronase at 37°C and the concentration of fluorescein in the solution was measured by monitoring the absorbance at 492 nm using a Perkin Elmer (Waltham, MA) spectrophotometer. A linear standard curve, obtained by fitting the absorbance and concentration of a series of fluorescein dilutions, was used to determine the concentration of fluorescein in the pronase digested nanoparticle samples. The LE was calculated by the formula:
|
|

Degradation kinetics of GNPs
GNPs (2.5 mg mL−1) were resuspended in phosphate-buffered saline (PBS), and pronase was added to a final concentration of 10 μg mL−1. Nanoparticle scattering was monitored at 365 nm with the temperature set at 37°C using a Hitachi F-7000 fluorescence spectrophotometer equipped with a temperature controller unit.
Fluorescein release rates from GNPs
Fluorescein-loaded GNPs were resuspended in PBS with varying concentrations of pronase (0 mg mL−1, 0.01 mg mL−1 and 0.1 mg mL−1) and incubated at 37°C. The GNPs were centrifuged at specific time intervals, and the supernatant was collected for fluorescein estimation. The pellet was resuspended in PBS with the desired amount of pronase and incubated further.
Intracellular stability of fluorescein-loaded GNPs
Mouse monocyte/macrophage cells (RAW 264.7) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics (5 μg mL−1 penicillin and 6 μg mL−1 streptomycin) at 37°C under 95% humidity and 5% CO2. Cells seeded on coverslips were incubated with 200 μg mL−1 of fluorescein-loaded GNPs for 4 h. The cells were washed and cultured in complete DMEM for different time intervals. At indicated time points (0 h, 8 h, 16 h and 24 h), cells were washed, stained with LysoTracker as per the manufacturer’s instructions and fixed with 4% formaldehyde. Cells were counterstained with DAPI, and images were acquired using a 63× objective lens on a Leica confocal microscope (TCS-SP8; Leica Microsystems, Wetzlar, Germany). Images were analyzed by Leica Application Suite AF software provided by the company.
Results and discussion
Nanoparticle synthesis by the desolvation method
The desolvation protocol developed by Marty et al6 and modified by Coester et al5 has been extensively used for the synthesis of protein-based nanoparticles. The process adopted in this study is described in Scheme 1, which includes adjusting the pH of the solution to impart a net charge on the protein molecules. Nanoparticle synthesis is then initiated by the addition of a desolvating agent. The size of the nanoparticles may be regulated by manipulating the reaction conditions. Previous studies have shown the size modulation by varying the amount and rate of addition of desolvating agent (acetone), pH, temperature, concentration of the protein, and the amount of the cross-linking agent.27,29,30 The size of the nanoparticles, which determines the surface area to volume ratio, is an important parameter in governing the degradation kinetics and hence the drug release profiles. In addition to nanoparticle size, the matrix density of the particles should also govern the drug release profiles. Two parameters that are most likely to play an important role in regulating the matrix densities of the particles are, 1) the pH (which determines the charge on gelatin), and 2) the amount of desolvating agent added.
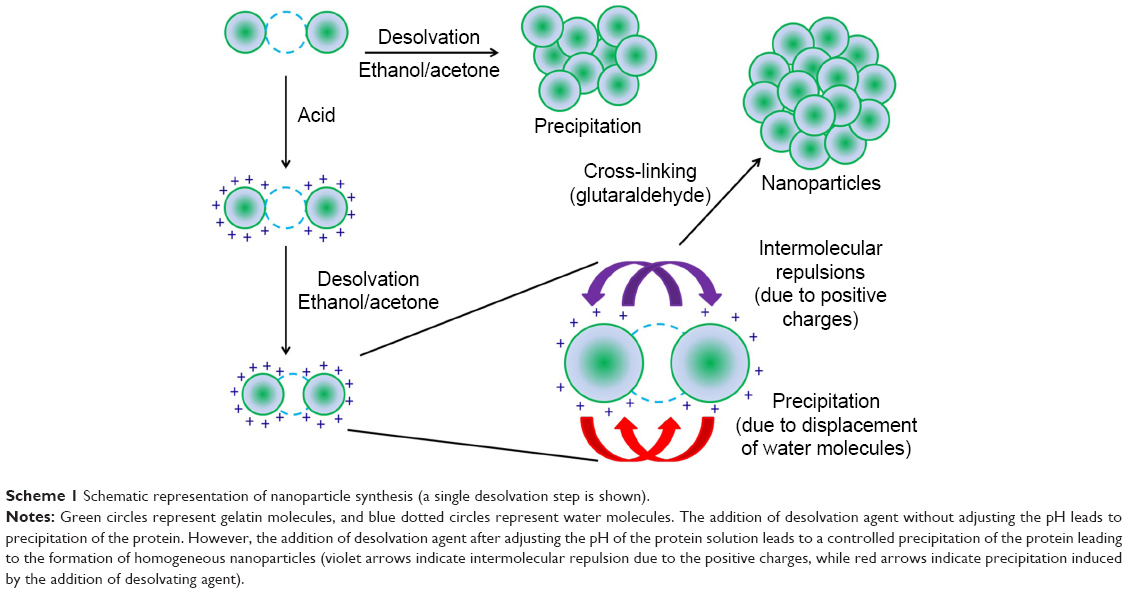 | Scheme 1 Schematic representation of nanoparticle synthesis (a single desolvation step is shown). |
Effect of pH on the surface charge of gelatin
The pH before the desolvation step, as mentioned earlier, imparts a net positive or negative charge on gelatin, thereby inducing intra- and intermolecular repulsions in gelatin. While a high surface charge leads to intermolecular repulsions and hence a stable suspension, a low surface charge may lead to precipitation.
As shown in Figure 1A and B, gelatin type A and type B show an increase in the zeta potential as the pH is lowered from the isoelectric point (pI) of gelatin. However, after reaching a peak value around pH 4 and 3.5 (for gelatin type A and type B, respectively), the zeta potential starts decreasing with further lowering of pH.
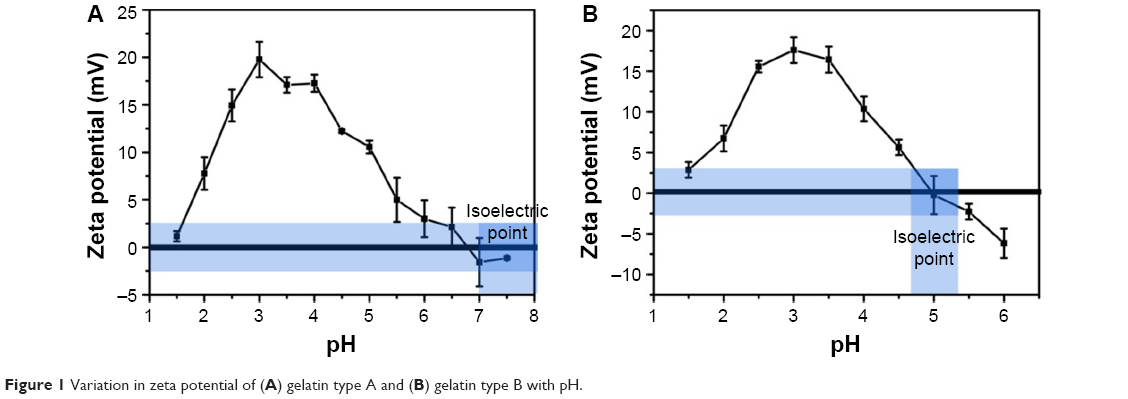 | Figure 1 Variation in zeta potential of (A) gelatin type A and (B) gelatin type B with pH. |
The pI of gelatin type A and type B varies in the range of 7–9 and 4.7–5.2, respectively. A decrease in pH below the pI thus leads to an increase in the zeta potential of gelatin. A further decrease in pH below 4 may lead to the protonation of aspartic acid and glutamic acid (pKa of 3.9 and 4.3, respectively), thereby accounting for the maximum zeta potential values observed at a pH of ~4 and 3.5 for type A and type B gelatin, respectively. However, a decrease in the zeta potential below this pH is unexpected and can be attributed to the counter ions (Cl−) added to the solution while adjusting the pH.31,32 As the pKa values of all the amino acid side chains are above pH 3.5, it can be assumed that at a pH below 3.5, all the side chains are maximally protonated and thus a further decrease in pH should, in principle, have no effect on the ionization state of the protein. As HCl is added for decreasing the pH of the gelatin solution, Cl− ions in the solution also increase. The introduction of anions may lead to ion pair formation and Debye–Huckel screening effect, thereby accounting for the decrease in the zeta potential of the protein.31
The possibility of an increase in surface hydrophobicity on gelatin molecules with decreasing pH was explored by incubating gelatin type B with a neutral dye, Nile Red.33 Nile Red binding remained low and constant with varying pH (Figure S1), thereby indicating no alteration in surface hydrophobicity.
Precipitation of gelatin by acetone: formation of nanoparticles
Acetone was utilized as a desolvating agent. The gradual addition of acetone leads to an increase in turbidity of the gelatin solution indicating the formation of nanoparticles. The amount of acetone required to precipitate gelatin varies and is dependent on the pH of the gelatin solution. Figure 2A shows the percentage of acetone required to initiate precipitation in gelatin type B solutions incubated at different pH values. Solutions incubated at pH values close to the pI of gelatin require lesser acetone (~55% acetone for gelatin at pH 4) for precipitation. However, as the pH is varied away from the pI of gelatin, the amount of acetone required to precipitate gelatin increases (~73% acetone for gelatin at pH 3.25).
 | Figure 2 (A) Acetone (%) required for the precipitation of gelatin solutions maintained at different pH values. (B) Effect of pH on nanoparticle size prepared from gelatin type A. (C) Effect of pH on nanoparticle size prepared from gelatin type B. |
The process of GNP synthesis involves a controlled precipitation, where the force of precipitation driven by the removal of water molecules is balanced by intermolecular charge repulsions induced by the altered pH. Apart from this, the increase in the water-holding capacity of gelatin at low pH may also lead to an increase in the acetone concentrations required.
The decrease in pH also has an effect on the water-holding capacity of gelatin molecules. In regular α-helices and β-sheets, the NH and CO groups of the peptide backbone are involved in intramolecular hydrogen bonding. However, the formation of triple helical structures in gelatin results in the availability of CO and NH groups (of the peptide backbone) which may also be involved in hydrogen bonding (Figure S2).34 A decrease in pH may lead to an increase in the interaction of the hydrophilic segments in gelatin with the solvating water molecules. A low pH thus makes the hydration network in gelatin more extensive and stronger.35,36 Moreover, the inter- and intra-chain interactions in gelatin arising either directly or through water-mediated hydrogen bonding (Figure S2) may increase its water retention capacity, thereby accounting for the increased amount of acetone required to initiate phase separation, precipitation and nanoparticle formation in gelatin.
We further elucidated the effect of zeta potential variations on GNP size. Nanoparticles were synthesized at different pH values for both gelatin type A and type B. Nanoparticles synthesized at pH values close to the pI (7–9 for gelatin type A and 4.7–5.2 for gelatin type B) were large and unstable with a tendency to aggregate. However, nanoparticles synthesized below pH 4 were small and stable with a low polydispersity index (PDI). As shown in Figure 2B and C, a trend of decreasing particle size with decreasing pH was observed until pH 4 for gelatin type A and pH 3.25 for gelatin type B. However, decreasing the pH below these values resulted in an increase in nanoparticle size. Nanoparticle formation was not possible at a pH below 3.5 for gelatin type A and pH below 3.0 for gelatin type B. Even a high concentration of desolvating agent failed to induce precipitation in gelatin at these low pH values. However, there are a few reports of nanoparticle synthesis at a pH as low as 2.5 for gelatin type A.5,26 This could be due to molecular crowding because of the significantly higher concentration of gelatin used. Moreover, an increase in gelatin concentration is known to increase its viscosity,37 which may promote nanoparticle formation at low pH and was also observed to increase the nanoparticle size (Figure S3). In the current study, gelatin type A and type B were used at a concentration of 1%, which failed to precipitate at pH values below 3.5 and 3, irrespective of the amount of acetone used.
Shutava et al25 have reported a similar trend in nanoparticle size variation with decreasing pH. While the decrease in the size of the nanoparticles as the pH is reduced below the pI of the protein is attributed to the effective precipitation of gelatin, the increase in particle size with a further decrease in pH (pH below 4 for gelatin type A and pH below 3.25 for gelatin type B) has been attributed to a decrease in solution viscosity. However, we suggest that apart from the viscosity, the surface charge on gelatin may also play an important role in determining the particle size. As shown in Figure 1, the zeta potential of gelatin type A and B reaches a maximum value around pH 4 and 3.5, respectively. Below these pH values, as the zeta potential starts decreasing, the particles size starts increasing, suggesting a decrease in repulsive forces and hence an increase in the precipitation tendency of gelatin. This could be the possible reason why nanoparticles formed at these extremely low pH values tend to have a larger size than the nanoparticles synthesized at pH 4 and 3.25 for gelatin type A and type B, respectively.
Mechanism of nanoparticle assembly at low and high pH
As mentioned earlier, the amount of acetone (desolvating agent) required for nanoparticle synthesis varies depending upon the pH of the solution. We, therefore, investigated the effect of acetone concentration on the particle size in an attempt to gain insights into the mechanism of nanoparticle assembly at different pH values. Figure 3A shows the size variation in gelatin (type B) nanoparticles, prepared at pH 4, as a function of acetone concentration. As evident from Figure 3A, precipitation is initiated at ~50%–55% of acetone and the nanoparticle size increases with increasing acetone concentration. Weber et al29 suggested that the desolvation process by the addition of acetone can be divided into two parts: in the initial part, nanoparticle size increases with an increase in acetone concentration while in the later part, the number of particles increase upon acetone addition. As shown in Figure 3A, the particle size gradually increases with acetone (until ~60%), then remains constant with no significant change. While the biphasic behavior of nanoparticle formation observed in the current study is consistent with the findings of Weber et al, we found that the second phase is characterized by an increase in the PDI of the nanoparticles which suggests, 1) addition of free gelatin to preformed nanoparticles, 2) a further removal of water from the preformed nanoparticles, and 3) interparticle aggregation due to low surface charges. We propose that the nanoparticle formation at pH 4 takes place via the formation of a dense core during the initial phase which may act as a nucleus upon which additional gelatin molecules are added by the further addition of acetone (Figure 4A). The whole process results in the formation of nanoparticles with a highly dense matrix as shown in Figure 4B.
 | Figure 3 Effect of acetone concentration on gelatin (type B) nanoparticle size prepared at pH 4 (A) and pH 3.25 (B). |
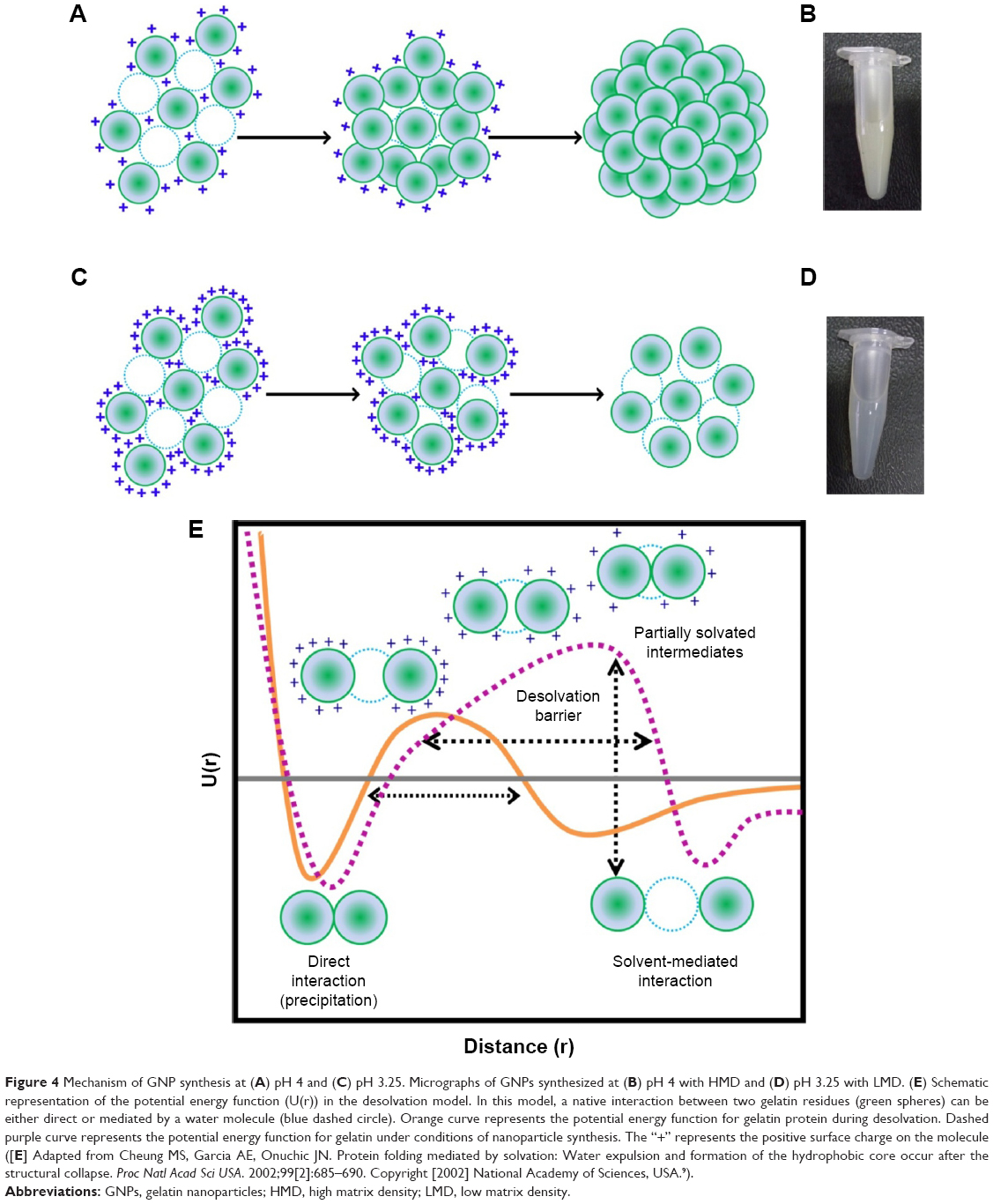 | Figure 4 Mechanism of GNP synthesis at (A) pH 4 and (C) pH 3.25. Micrographs of GNPs synthesized at (B) pH 4 with HMD and (D) pH 3.25 with LMD. (E) Schematic representation of the potential energy function (U(r)) in the desolvation model. In this model, a native interaction between two gelatin residues (green spheres) can be either direct or mediated by a water molecule (blue dashed circle). Orange curve represents the potential energy function for gelatin protein during desolvation. Dashed purple curve represents the potential energy function for gelatin under conditions of nanoparticle synthesis. The “+” represents the positive surface charge on the molecule ([E] Adapted from Cheung MS, Garcia AE, Onuchic JN. Protein folding mediated by solvation: Water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc Natl Acad Sci USA. 2002;99[2]:685–690. Copyright [2002] National Academy of Sciences, USA.9). |
In contrast to the nanoparticles synthesized at pH 4, those synthesized at pH 3.25 appear to follow a completely different mechanism of particle assembly. As shown in Figure 3B, we observed a decrease in particle size upon the addition of acetone. At pH 3.25, perhaps due to enhanced water retention capacity of gelatin and an incomplete extraction of water molecules by acetone, the nanoparticles formed were larger. However, with a further increase in acetone concentration, we observed a decrease in nanoparticle size. We propose that the initial particles formed, at low concentrations of acetone at pH 3.25, are large followed by a condensation of the particles with further increase in acetone concentration. This leads to a reduction in nanoparticle size along with the addition of new particles in the solution. Even the addition of excess acetone (beyond 78%) had no effect on the particle size and polydispersity. The particles synthesized at pH 3.25, thus, tend to have a less dense matrix as shown in Figure 4C and D.
From the current set of experiments, we conclude that the combined effect of pH and acetone determines the matrix density in addition to the particle size and number. The differences in the assembly of nanoparticles might be responsible for the differences in the matrix density observed.
The process of nanoparticle assembly can also be explained based on the desolvation barrier model proposed for protein folding by Cheung et al.9 They suggest that the process of protein folding via the desolvation model takes place through a partially hydrated, less stable state. The two minima (Figure 4E) in the potential energy curve represent either a stable water-mediated interaction or a stable direct interaction (between two different protein molecules or within the same protein molecule). In the context of GNP synthesis, the two minima may be a hydrated state and a precipitated state. Nanoparticles may occupy the region between the two minima, on the desolvation barrier. These nanoparticles are energetically in a less favorable state. Nanoparticles isolated at this level may either aggregate or dissolve. Glutaraldehyde cross-linking preserves these nanoparticles (Figure 4E).
Evaluating the protease susceptibility of the GNPs synthesized with different matrix densities
We synthesized nanoparticles with varying matrix densities with a rationale to develop nanoparticles with varying matrix densities. Figure 4B and D shows the pictures of the nanoparticles, in equal quantities (10 mg mL−1). The nanoparticles synthesized at pH 4 were found to be more turbid compared to the ones synthesized at pH 3.25. The hydrodynamic diameter of the nanoparticles synthesized at pH 3.25 and 4 was found to be 185.2±5.2 nm and 235.5±2.2 nm, respectively, with a PDI of <0.2.
We then studied the protease-induced degradation of GNPs synthesized at pH 3.25 with LMD-GNPs and at pH 4 with HMD-GNPs, by monitoring the light scatter at 465 nm in a fluorescence spectrophotometer. The degradation kinetics of nanoparticles with an LMD (Figure 5, black line) was faster as compared to the nanoparticles synthesized with a HMD (Figure 5, gray line).
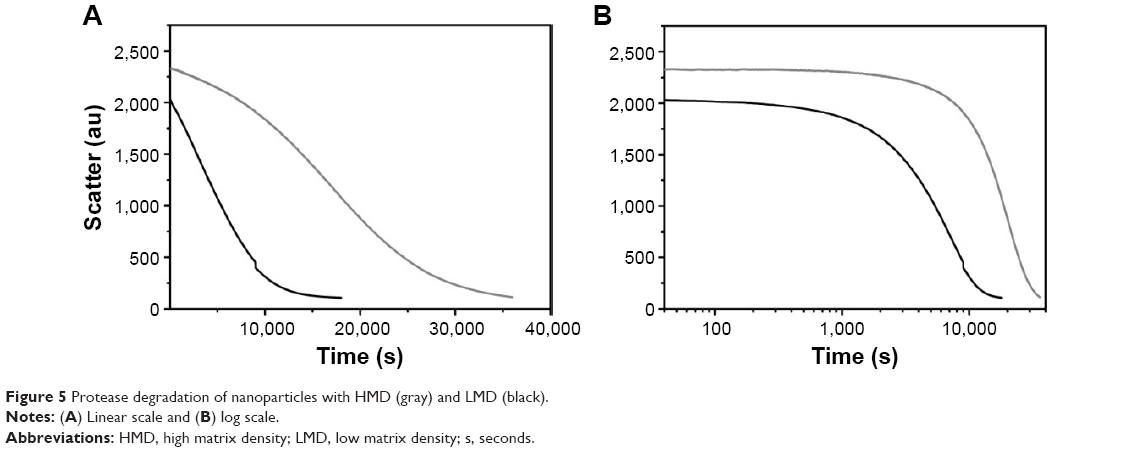 | Figure 5 Protease degradation of nanoparticles with HMD (gray) and LMD (black). |
Modulating the susceptibility of protein-based nanoparticles toward proteases can be an efficient tool for regulating nanoparticle degradation and hence drug release. The differences in degradation kinetics of the two nanoparticle systems described in the current study can thus be utilized to regulate drug release. Such a system can be specifically advantageous in conditions such as aggressive cancers38 and keratitis,39 where the protease concentration in the microenvironment is dependent on the severity of the pathological situation. By carefully utilizing such a system, the drug release can be modulated more efficiently.
Evaluation of fluorescein loading and release from nanoparticles synthesized with varying matrix densities
To evaluate the LE of nanoparticles, fluorescein-loaded LMD-GNPs and HMD-GNPs were weighed and resuspended in PBS. The particles were subsequently digested with pronase, and the amount of fluorescein released was estimated by monitoring the absorbance at 492 nm and comparison to fluorescein standards. The LE for LMD-GNPs and HMD-GNPs was found to be 52.2%±2.3% and 46.3%±3.5%, respectively. The slight difference in the LEs of the two types of GNPs is possibly due to the high porosity of the LMD-GNPs and hence more free space to accommodate fluorescein.
The rate of fluorescein release from GNPs synthesized with varying matrix densities was evaluated. The nanoparticle solution was supplemented with varying concentrations of pronase to induce particle degradation. As shown in Figure 6, the fluorescein release from the GNPs synthesized with different matrix densities followed a different release profile. In the absence of pronase (Figure 6A), there was an initial burst (first 30 minutes) release of fluorescein from both the particles. However, the release from LMD-GNPs was more (44.6%±1.3%) as compared to that from HMD-GNPs (31.7%±1.0%). The higher burst release from the LMD-GNPs could be attributed to the high porosity of these particles and the effect of a mild sonication to disperse these particles prior to the experiment.
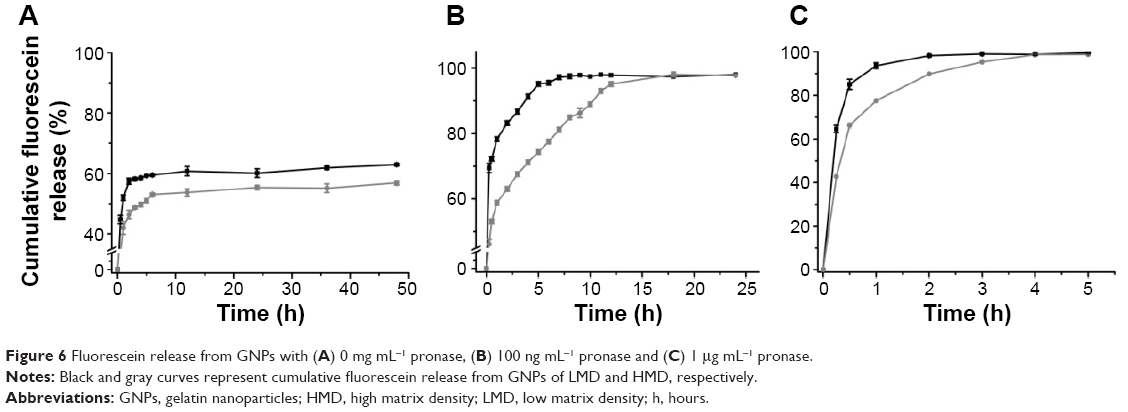 | Figure 6 Fluorescein release from GNPs with (A) 0 mg mL−1 pronase, (B) 100 ng mL−1 pronase and (C) 1 μg mL−1 pronase. |
In the presence of low concentrations of pronase (100 ng mL−1; Figure 6B), the release profile of fluorescein showed a clear difference between LMD-GNPs and HMD-GNPs. While LMD-GNPs showed an initial burst (first 30 minutes) of 72.3%±0.7% release, the burst release in the case of HMD-GNPs was found to be 53.1%±1.8%. However, the release of fluorescein from HMD-GNP was found to be slow and sustained over a period of 18 h (98.0%±2.3%) compared to a 95.5%±4.3% fluorescein release from LMD-GNPs in the first 6 h.
However, in the presence of a higher amount of pronase (1 μg mL−1), the fluorescein release profiles from both the particles were similar with a slight retardation of fluorescein release in the case of HMD-GNPs (Figure 6C).
GNP clearance by mouse macrophage (RAW 264.7) cell line
To evaluate the intracellular degradation and fluorescein release from GNPs, RAW 264.7 cells were treated with fluorescein-loaded LMD-GNPs and HMD-GNPs. The cells were then visualized under a confocal microscope at indicated time points. As shown in Figure 7, both LMD-GNPs and HMD-GNPs (green fluorescence) were rapidly taken up by the cells and localized with the lysosomes (red fluorescence) at the 0 h time point. At later time points, viz 8 h and 16 h, the nanoparticle-associated fluorescence was found to be similar in the LMD-GNPs- and HMD-GNP-treated cells. However, at the 24 h time point, cells treated with LMD-GNPs showed more diffused fluorescence compared to the HMD-GNP-treated cells, where particle-associated fluorescence was still visible (Figure 7, white arrows). The diffused fluorescence in the case of LMD-GNP-treated cells is indicative of nanoparticle degradation and fluorescein release. It could be thus concluded that the intracellular degradation of LMD-GNPs was significantly faster compared to HMD-GNPs.
 | Figure 7 Clearance of GNPs in RAW 264.7 cell line. |
Conclusion
Gelatin due to its extensive hydration network holds 5–10 times its weight of water. The extraction of these water molecules by a desolvating agent, under extreme pH values, forms the basis of the desolvation process utilized in GNP synthesis. We found that the pH at which gelatin is incubated determines the mechanism by which nanoparticles are formed. By altering the pH, hence the mechanism, it is possible to synthesize nanoparticles with different matrix densities. These nanoparticles when subjected to protease-based degradation show varying degradation susceptibility. They also show minor variations in the drug-loading efficiencies. However, they show significant differences in intra- and extracellular drug release rates. We believe that the current study should prove useful in designing GNPs with a variable response to proteases and aid in providing a better regulation over drug release rates.
Acknowledgments
This work was supported by the Department of Science and Technology (DST), Government of India funded project, Nanotechnology for biomedical applications. SMA acknowledges the Council for Scientific and Industrial Research (CSIR) for financial support. MRC acknowledges the funding from the Sir JC Bose National Fellowship of the Department of Science and Technology (DST), India.
Disclosure
The authors report no conflicts of interest in this work.
References
Maham A, Tang Z, Wu H, Wang J, Lin Y. Protein-based nanomedicine platforms for drug delivery. Small. 2009;5(15):1706–1721. | ||
Weber C, Reiss S, Langer K. Preparation of surface modified protein nanoparticles by introduction of sulfhydryl groups. Int J Pharm. 2000;211(1–2):67–78. | ||
Balthasar S, Michaelis K, Dinauer N, von BH, Kreuter J, Langer K. Preparation and characterisation of antibody modified gelatin nanoparticles as drug carrier system for uptake in lymphocytes. Biomaterials. 2005;26(15):2723–2732. | ||
Coester C, Kreuter J, von BH, Langer K. Preparation of avidin-labelled gelatin nanoparticles as carriers for biotinylated peptide nucleic acid (PNA). Int J Pharm. 2000;196(2):147–149. | ||
Coester CJ, Langer K, van BH, Kreuter J. Gelatin nanoparticles by two step desolvation – a new preparation method, surface modifications and cell uptake. J Microencapsul. 2000;17(2):187–193. | ||
Marty JJ, Oppenheim RC, Speiser P. Nanoparticles: a new colloidal drug delivery system. Pharm Acta Helv. 1978;53:17–23. | ||
Rank JA, Baker D. A desolvation barrier to hydrophobic cluster formation may contribute to the rate-limiting step in protein folding. Protein Sci. 1997;6:347–354. | ||
Aksel T, Majumdar A, Barrick D. The contribution of entropy, enthalpy, and hydrophobic desolvation to cooperativity in repeat-protein folding. Structure. 2011;19(3):349–360. | ||
Cheung MS, Garcia AE, Onuchic JN. Protein folding mediated by solvation: Water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc Natl Acad Sci USA. 2002;99(2):685–690. | ||
Dumetz AC, Chockla AM, Kaler EW, Lenhoff AM. Protein phase behavior in aqueous solutions: crystallization, liquid-liquid phase separation, gels, and aggregates. Biophys J. 2008;94(2):570–583. | ||
Mohanty B, Bohidar HB. Microscopic structure of gelatin coacervates. Int J Biol Macromol. 2005;36(1–2):39–46. | ||
Wartlick H, Spankuch-Schmitt B, Strebhardt K, Kreuter J, Langer K. Tumour cell delivery of antisense oligonuclceotides by human serum albumin nanoparticles. J Control Release. 2004;96(3):483–495. | ||
Kommareddy S, Amiji MM. Protein nanospheres for gene delivery. CSH Protoc. 2008;2008:db.to30. | ||
Megeed Z, Cappello J, Ghandehari H. Genetically engineered silk-elastinlike protein polymers for controlled drug delivery. Adv Drug Deliv Rev. 2002;54:1075–1091. | ||
Langer K, Anhorn MG, Steinhauser I, et al. Human serum albumin (HSA) nanoparticles: reproducibility of preparation process and kinetics of enzymatic degradation. Int J Pharm. 2008;347(1–2):109–117. | ||
Irache JM, Bergougnoux L, Ezpeleta I, Gueguen J, Orecchioni A-M. Optimization and in vitro stability of legumin nanoparticles obtained by a coacervation method. Int J Pharm. 1995;126:103–109. | ||
Podaralla S, Perumal O. Preparation of zein nanoparticles by pH controlled nanoprecipitation. J Biomed Nanotechnol. 2010;6(4):312–317. | ||
Teng Z, Luo Y, Wang Q. Nanoparticles synthesized from soy protein: preparation, characterization, and application for nutraceutical encapsulation. J Agric Food Chem. 2012;60(10):2712–2720. | ||
Elzoghby AO, Helmy MW, Samy WM, Elgindy NA. Novel ionically crosslinked casein nanoparticles for flutamide delivery: formulation, characterization, and in vivo pharmacokinetics. Int J Nanomedicine. 2013;8:1721–1732. | ||
Herrera Estrada LP, Champion JA. Protein nanoparticles for therapeutic protein delivery. Biomater Sci. 2015;3(6):787–799. | ||
Wetzel R, Buder E, Hermel H, Hüttner A. Conformations of different gelatins in solutions and in films an analysis of circular dichroism (CD) measurements. Colloid Polym Sci. 1987;265:1036–1045. | ||
Djabourov M, Lechaire JP, Gaill F. Structure and rheology of gelatin and collagen gels. Biorheology. 1993;30:191–205. | ||
Brodsky B, Persikov AV. Molecular structure of the collagen triple helix. Adv Protein Chem. 2005;70:301–339. | ||
Lloyd DJ, Pleass WB. The absorption of water by gelatin. Biochem J. 1927;21:1352–1367. | ||
Shutava TG, Balkundi SS, Vangala P, et al. Layer-by-layer-coated gelatin nanoparticles as a vehicle for delivery of natural polyphenols. ACS Nano. 2009;3(7):1877–1885. | ||
Wong C, Stylianopoulos T, Cui J, et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci USA. 2011;108(6):2426–2431. | ||
Farrugia CA, Groves MJ. Gelatin behaviour in dilute aqueous solution: designing a nanoparticulate formulation. J Pharm Pharmacol. 1999;51(6):643–649. | ||
Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric systems for controlled drug release. Chem Rev. 1999;99:3181–3198. | ||
Weber C, Coester C, Kreuter J, Langer K. Desolvation process and surface characterisation of protein nanoparticles. Int J Pharm. 2000;194(1):91–102. | ||
Azarmi S, Huang Y, Chen H, et al. Optimization of a two-step desolvation method for preparing gelatin nanoparticles and cell uptake studies in 143B osteosarcoma cancer cells. J Pharm Pharm Sci. 2006;9(1):124–132. | ||
Goto Y, Takahashi N, Fink AL. Mechanism of acid-induced folding of proteins. Biochemistry. 1990;29(14):3480–3488. | ||
Goto Y, Calciano LJ, Fink AL. Acid-induced folding of proteins. Proc Natl Acad Sci USA. 1990;87(2):573–577. | ||
Sackett DL, Wolff J. Nile red as a polarity-sensitive fluorescent probe of hydrophobic protein surfaces. Anal Biochem. 1987;167(2):228–234. | ||
Brodsky B, Ramshaw JA. The collagen triple-helix structure. Matrix Biol. 1997;15(8–9):545–554. | ||
Koli JM, Basu S, Kannuchamy N, Gudipati V. Effect of pH and ionic strength on functional properties of fish gelatin in comparison to mammalian gelatin. Fish Technol. 2013;50:126–132. | ||
Nurul AG, Sarbon NM. Effects of pH on functional, rheological and structural properties of eel (Monopterus sp.) skin gelatin compared to bovine gelatin. Int Food Res J. 2015;22:572–583. | ||
Cumper CWN, Alexander AE. The viscosity and rigidity of gelatin in concentrated aqueous systems. I. Viscosity. Aust J Sci Res. 1952;5:146–152. | ||
Kohrmann A, Kammerer U, Kapp M, Dietl J, Anacker J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: new findings and review of the literature. BMC Cancer. 2009;9:188. | ||
Gopinathan U, Ramakrishna T, Willcox M, et al. Enzymatic, clinical and histologic evaluation of corneal tissues in experimental fungal keratitis in rabbits. Exp Eye Res. 2001;72(4):433–442. |
Supplementary materials
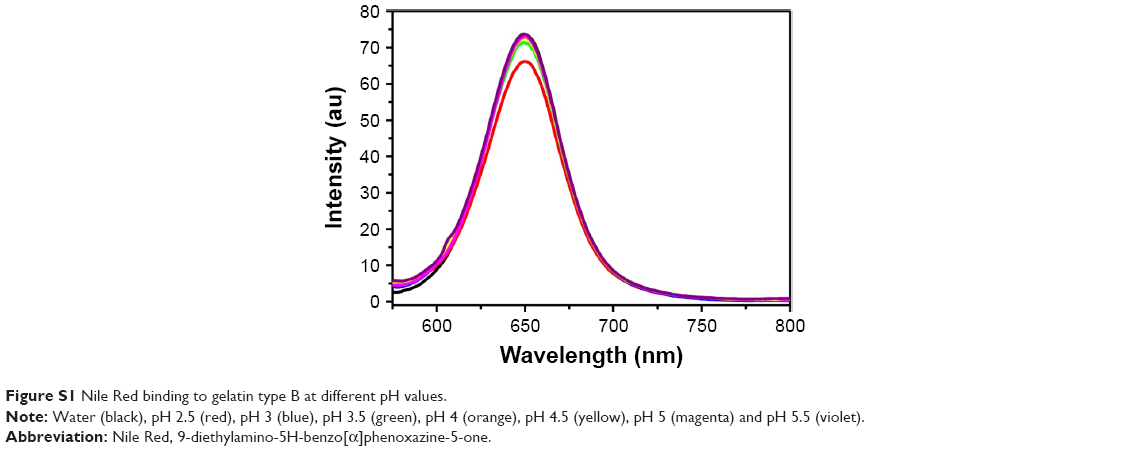 | Figure S1 Nile Red binding to gelatin type B at different pH values. |
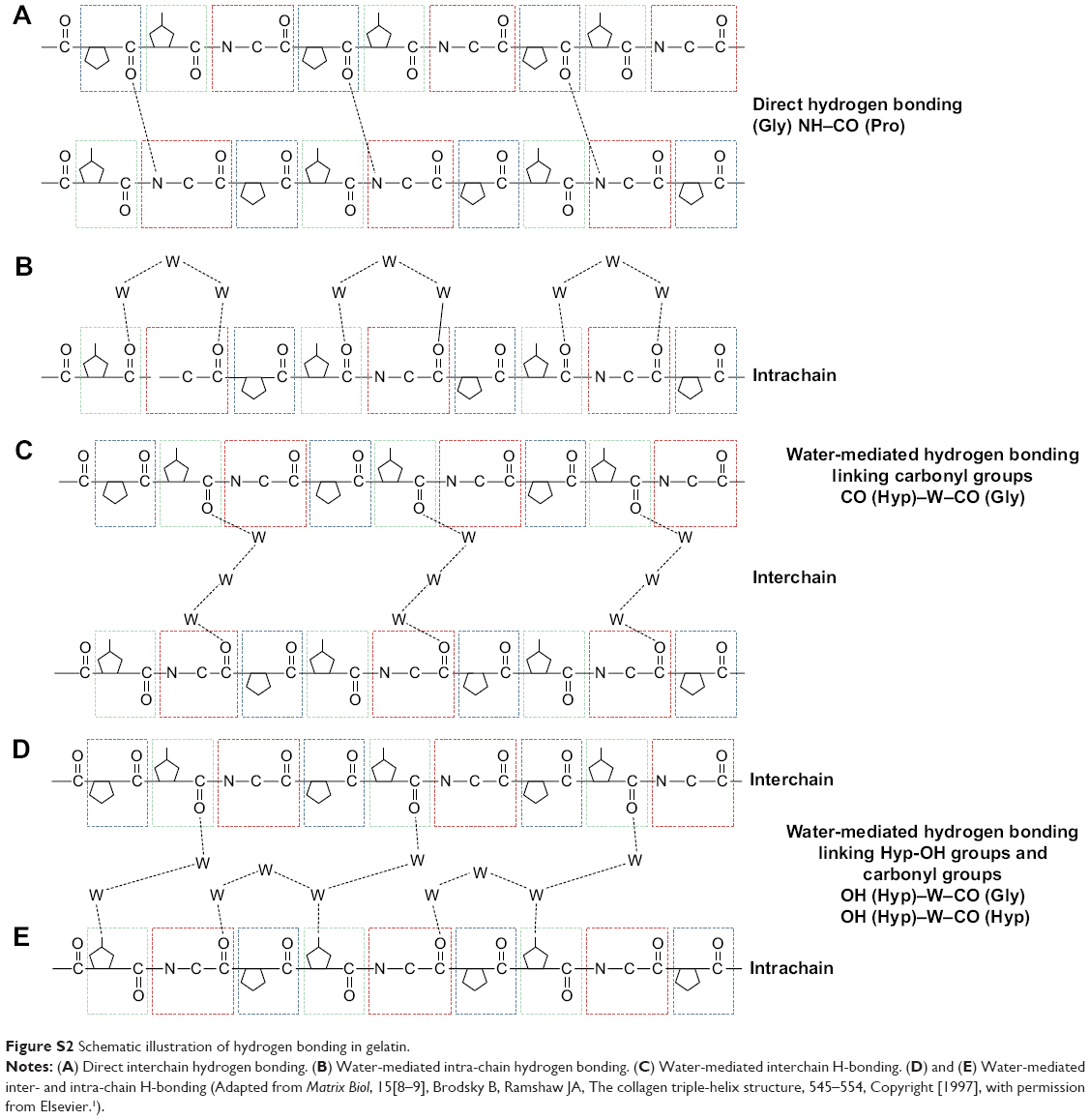 | Figure S2 Schematic illustration of hydrogen bonding in gelatin. |
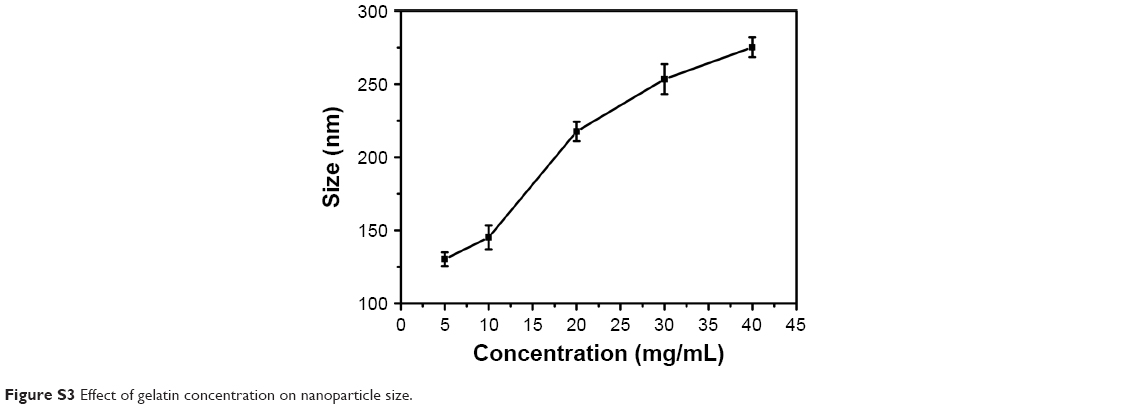 | Figure S3 Effect of gelatin concentration on nanoparticle size. |
Reference
Brodsky B, Ramshaw JA. The collagen triple-helix structure. Matrix Biol. 1997;15(8–9):545–554. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.