")
Back to Journals » Vascular Health and Risk Management » Volume 19
The Role of Palmitic Acid in the Co-Toxicity of Bacterial Metabolites to Endothelial Cells
Authors Choroszy M, Środa-Pomianek K, Wawrzyńska M, Chmielarz M, Bożemska E, Sobieszczańska B
Received 17 February 2023
Accepted for publication 18 May 2023
Published 4 July 2023 Volume 2023:19 Pages 399—409
DOI https://doi.org/10.2147/VHRM.S408897
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Harry Struijker-Boudier
Marcin Choroszy,1 Kamila Środa-Pomianek,2 Magdalena Wawrzyńska,3 Mateusz Chmielarz,1 Edyta Bożemska,1 Beata Sobieszczańska1
1Department of Microbiology, Wroclaw Medical University, Wroclaw, Poland; 2Department of Biophysics and Neuroscience, Wroclaw Medical University, Wroclaw, Poland; 3Department of Preclinical Studies, Faculty of Health Sciences, Wroclaw Medical University, Wroclaw, Poland
Correspondence: Marcin Choroszy, Chalubinskiego 4 Street, Wroclaw, 51-657, Poland, Tel +48-71-7840-065, Fax +48-71-784-0117, Email [email protected]
Introduction: Metabolic endotoxemia most often results from obesity and is accompanied by an increase in the permeability of the intestinal epithelial barrier, allowing co-absorption of bacterial metabolites and diet-derived fatty acids into the bloodstream. A high-fat diet (HFD) leading to obesity is a significant extrinsic factor in developing vascular atherosclerosis. In this study, we evaluated the effects of palmitic acid (PA) as a representative of long-chain saturated fatty acids (LCSFA) commonly present in HFDs, along with endotoxin (LPS; lipopolysaccharide) and uremic toxin indoxyl sulfate (IS), on human vascular endothelial cells (HUVECs).
Methods: HUVECs viability was measured based on tetrazolium salt metabolism, and cell morphology was assessed with fluorescein-phalloidin staining of cells’ actin cytoskeleton. The effects of simultaneous treatment of endothelial cells with PA, LPS, and IS on nitro-oxidative stress in vascular cells were evaluated quantitatively with fluorescent probes. The expression of vascular cell adhesion molecule VCAM-1, E-selectin, and occludin, an essential tight junction protein, in HUVECs treated with these metabolites was evaluated in Western blot.
Results: PA, combined with LPS and IS, did not influence HUVECs viability but induced stress on actin fibers and focal adhesion complexes. Moreover, PA combined with LPS significantly enhanced reactive oxygen species (ROS) production in HUVECs but decreased nitric oxide (NO) generation. PA also considerably increased the expression of VCAM-1 and E-selectin in HUVECs treated with LPS or IS but decreased occludin expression.
Conclusion: Palmitic acid enhances the toxic effect of metabolic endotoxemia on the vascular endothelium.
Keywords: metabolic endotoxemia, high-fat diet, palmitic acid, endotoxin, indoxyl sulfate
Introduction
Metabolic endotoxemia (ME) is defined as a 2–3-fold increase in plasma LPS levels that develop on a high saturated-fat diet.1,2 ME, in contrast with acute endotoxemia associated with a bacterial blood infection, e.g., bacteremia and sepsis, characterizes by the presence of low LPS levels in the blood and is associated with low-grade systemic inflammation, ultimately leading to the development of a range of diseases such as obesity, diabetes type 2, cardiometabolic diseases, non-alcoholic fatty liver disease, chronic kidney disease and many others.3,4 The gut microbiota is considered the primary source of endotoxin in the blood. HFD-induced ME is associated with alterations in the intestinal barrier, increasing the gut permeability and trafficking of endotoxin and other bacterial metabolites into circulation.2 Moreover, dietary fatty acids absorbed from the gut into circulation can modulate the immune system, contributing to inflammation.5 Animal and experimental studies demonstrated that dietary fats promote endotoxin translocation from the gut to circulation due to its absorption through chylomicrons formed within enterocytes.6,7 Hence, the effect of ME seems to be particularly adverse in obese individuals, in whom cumulative endotoxemia correlates with postprandial cumulative chylomicronemia, which increases with the amount of fat consumed.7 Although the level of endotoxemia occurs mainly after high-fat meals, frequent consumption of fats, especially LCSFA, over a long period eventually induces chronic inflammation in the vasculature, ultimately leading to cardiovascular diseases.8 PA is the most LCSFA, accounting for 20–30% of total fatty acids in the human body. The diet provides PA exogenously, but PA is also synthesized endogenously via de novo lipogenesis.9 It has been demonstrated that a diet rich in LCSFA, in particular PA, increases its concentration in the blood leading to obesity and inflammatory responses from endothelial cells, both risk factors in the development of cardiovascular diseases (CAD).10 PA exerts its lipotoxicity via up-regulating the expression of adhesion molecules, i.e. VCAM-1 in endothelial cells (ECs), and down-regulation of endothelial nitric oxide synthase (eNOS), resulting in decreased NO production.11–14 Our previous study demonstrated that even low LPS concentration (<10 ng/mL) induces increased thrombogenicity of ECs in vitro.15 Furthermore, the adverse effect of LPS on ECs was significantly potentiated by low levels of IS, indicating that even trace concentrations of some bacterial metabolites in the circulation may exert pathological consequences on vascular endothelium.15 Since ME, apart from the presence of endotoxin in the blood, is also associated with elevated LCSFA levels, of which PA is a significant component, in this study, we examined the simultaneous effects of PA, LPS, and IS on the vascular endothelium.
Materials and Methods
Chemicals Used in the Study
HUVEC cell line (cat. No.: C0035C), Human Large Vessel Endothelial Cell Basal Medium Cat. No.: M200500), Low Serum Growth Supplement (LSGS; Cat. No.: S00310), and M199 were purchased in ThermoFisher Scientific, UK. Palmitic acid, LPS of E. coli, fatty-acid-free bovine serum albumin (BSA), IS, (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT), phalloidin-conjugated with fluorescein isothiocyanate (FITC), and rat tail type I collagen were all purchased in Sigma Aldrich-Merck, Germany. The 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCF-DA), diacetate-(4-amino-5-methylamino-2′,7′-difluorescein diacetate) (DAF-FM), rabbit anti-human E-selectin (CD62E; cat. No.: PA5-85877) and anti-human VCAM-1 (CD106; cat. No.: MA5-31965), and mouse anti-human occludin (Cat. No.: 33–1500), a horseradish peroxidase-conjugated secondary anti-rabbit and anti-mouse antibodies, bovine serum (FBS), phosphate-buffered saline (PBS; pH 7.4), and penicillin-streptomycin (PS) 100 × concentrated solution for cell culture were all purchased in ThermoFisher Scientific, UK.
Cell Culture
HUVEC cells were routinely cultured in Human Large Vessel Endothelial Cell Basal Medium supplemented with LSGS and used at early-passage numbers from 4 to 6. For all experiments, HUVEC cells were cultured overnight in a cell-culture plate coated with 10 µg/mL rat tail type I collagen in M199 medium supplemented in 10% FBS and 1% PS without any growth factors that could influence the results. Cells were maintained at 37°C in a humid atmosphere with 5% CO2. The culture medium was changed every second day.
HUVEC Stimulation
PA was dissolved to the concentration of 1 mM in 150 mM NaCl and 0.17 mM BSA to obtain a 1 mM stock solution (final molar ratio 6:1), which was then diluted in a cell culture medium to 200 µM working solution. A 150 mM NaCl/BSA solution dissolved in a cell culture medium served as vehicle control for HUVEC cells treated with PA alone or PA combined with LPS and IS. PA and vehicle control stock solutions were stored at −20°C. LPS was dissolved in cell-grade water to obtain a stock solution of 1 mg/mL, which was further diluted in a cell culture medium to appropriate concentrations. IS was diluted in PBS to a stock solution of 100 mM following dissolving in a cell culture medium to appropriate concentrations.
HUVEC Cell’s Viability and Morphology
The viability of HUVECs seeded into 96-well plates (5000 cells per well) and incubated overnight to 80% confluency and then treated for 20 h with PA alone at a concentration of 200 µM or simultaneously treated with LPS (5 ng/mL and 50 ng/mL) and/or IS (20 µM and 200 µM) was assessed with 5 mg/mL of MTT. After incubation, the cell culture medium in wells was discarded and replaced with a cell culture medium containing MTT at a final concentration of 0.5 mg/mL. Following 3 h incubation, colored formazan crystals were dissolved in DMSO, and the reaction was read at 570 nm in a spectrophotometer UV/VIS 340. The morphology of HUVECs growing on round glass slides in a 24-well culture plate and stimulated with PA, LPS, and IS, as described above, was assessed with phalloidin-FITC. HUVEC cells were washed three times with pre-warmed PBS and fixed with 4% buffered formalin for 10 min at room temperature. After four washes with PBS, cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min and washed thrice with PBS. Then, cells were stained in the darkness for 40 min with phalloidin-FITC at 10 µg/mL in PBS. After two washes with PBS, DAPI (10 µg/mL) in PBS was added for 5 min to stain the nuclei of the cells. After drying, the slides were analyzed under a fluorescence microscope.
Determination of NO and Intracellular ROS Production
ROS and NO production in HUVEC cells treated for 4h with PA, LPS, and IS were assessed with an H2DCF-DA and DAF-FM diacetate fluorescence probes. H2DCF-DA probe measures intracellular ROS production, including singlet oxygen, superoxide, hydroxyl radicals, and various peroxide and hydroperoxides. DAF-FM detects low concentrations of intracellular NO. In ROS detection assays, the cells treated with 50 µM H2O2 and untreated cells served as positive and negative controls, respectively. After treatment, cells were washed three times with Hank’s Balanced Solution (HBSS), and 10 µM H2DCF-DA or DAF-FM in HBSS was added to cells for 30 min incubation at 37°C in 5% CO2 following two washes in HBSS. The fluorescent product was quantified with a spectrophotometer Tecan Infinite M200 plate reader at 488/525 nm for ROS and 495/515 nm for NO quantification. Blank wells were subtracted from the treated wells to obtain normalized well fluorescence intensity.
Western Blotting Analysis
The expression of VCAM-1 and E-selectin and the tight-junction protein occludin was assessed via blotting. HUVECs stimulated for 20 h with PA, LPS, and IS, as described above, were harvested and lysed in a radioimmunoprecipitation (RIPA) buffer. Protease and Phosphatase Inhibitor cocktail was used directly to the cell lysis buffer (100x, added 10 µL/mL). Protein concentration was determined using the Bradford assay. Equal amounts of proteins (30 μg per lane) were loaded onto the gels. The isolates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis for 1 h at 120 V, and the separated fractions were transferred by wet blotting to a polyvinylidene difluoride membrane. The membrane was incubated in Tris-buffered saline with 0.1% Tween 20 detergent and BSA (TBST-BSA) buffer (20 mmol/L Tris, 137 mmol/L NaCl, pH 7.6, 0.1% Tween, 25 mg/mL BSA) at 4°C for 1.5 h. Subsequently, the membrane was washed with TBST (20 mmol/L Tris, 137 mmol/L NaCl, pH 7.6, 0.1% Tween) at 4°C for 10 min. The incubations with anti-VCAM-1 (diluted 1:500), E-selectin (diluted 1:500), and occludin monoclonal antibodies at 1 µg/mL were performed in TBST-BSA at 4°C for 1 h. The membrane was rewashed with TBST and incubated with the horse-radish peroxidase (HRP)-conjugated secondary antibody at 4°C for 30 min. The membrane was finally exposed to 3 amino-9-ethyl carbazole (AEC), the substrate for HRP. The reaction was carried out in the presence of 30% H2O2 and terminated by washing with water. The relative level of protein normalized to the control derived from nontreated cells was determined by detecting the optical density of the bands on the electropherograms using the Image J program.
Statistical Analysis
The statistical significance of differences between means of results was determined by independent t-test and analysis of variance (one-way ANOVA with post hoc Tukey’s honestly significant difference) with p ≤ 0.05 considered statistically significant.
Results
Palmitic Acid Does Not Affect Endothelial Cells Viability
PA at 200 µM, like LPS and IS at both concentrations, did not significantly affect HUVECs viability. Although PA, combined with IS 20 µM and 200 µM, reduced HUVECs viability by 14.4% and 12%, respectively, compared to the vehicle control, the decrease did not reach statistical significance (Figure 1A). At concentrations used in the study, all metabolites examined individually did not significantly affect HUVECs morphology. However, HUVECs treated with PA in combination with IS and LPS, used at high and low concentrations, increased F-actin stress fibers and focal adhesion complexes (Figure 1B).
 |
Figure 1 (A) The viability of HUVEC cells stimulated for 20 h with PA alone or in combination with LPS and IS relative to the negative control (untreated cells) or vehicle control (VC; PA solvent). The results are the mean ± SD of three repeats performed in eight wells each. (B) Representative images of HUVECs morphology after treatment with PA alone and combined with LPS and IS. The actin cytoskeleton of endothelial cells was stained with phalloidin-FITC and the cell’s nuclei with DAPI. Fluorescence microscope, magnification 400x. |
Palmitic Acid Induces Oxidative Stress and Decreases NO Production in Endothelial Cells
PA significantly increased ROS production in HUVECs by over 170% (p = 0.0001) compared to vehicle control (Figure 2A). Similarly, LPS and IS at both concentrations significantly enhanced ROS production by over 150% (p < 0.05) compared to untreated control cells. Simultaneous treatment of HUVECs with PA, LPS, and IS at low and high concentrations further increased ROS by 27.6% (p = 0.01) and 53.2% (p = 0.0009), respectively, confirming the co-toxicity of LPS and IS, and the role of PA in enhancing LPS- and IS-induced ROS generation in ECs.
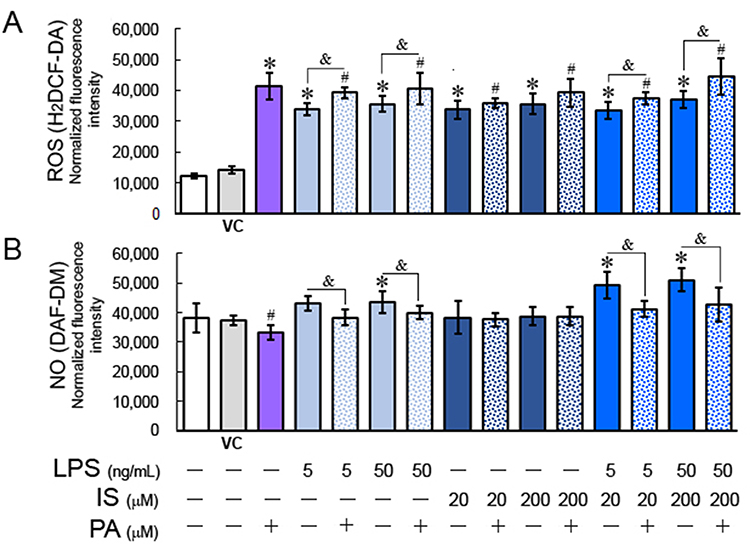 |
Figure 2 ROS (A) and NO (B) generation in HUVECs treated with PA, LPS, and IS for 4 h. Statistically significant differences vs control *p < 0.05; vs vehicle control (VC) #p < 0.05; with vs without PA &p < 0.05. |
NO levels in HUVECs treated with PA decreased by 10.6% (p = 0.03) (Figure 2B). Similarly, PA combined with IS at both concentrations reduced NO levels, although IS alone had no significant effect on NO production in ECs (Figure 2B). In contrast, LPS at both concentrations increased NO levels by 13% (p > 0.05) and 14.6% (p = 0.04) compared to the negative control. A combination of LPS with IS at both concentrations further increased NO levels but, when combined with PA, reduced it by 19.2% (p = 0.013) compared to the vehicle control, indicating that PA reduces NO generation in ECs.
Palmitic Acid Alters the Expression of Adhesive Molecules and Occludin in Endothelial Cells
The PA at 200 µM insignificantly increased VCAM-1 expression in HUVECs compared to the vehicle control. However, PA combined with low and ten-times higher LPS concentrations increased VCAM-1 by 3.8-fold (p = 0.01) and 7.3-fold (p = 0.0002), respectively, compared to the vehicle control and 2.8-fold (p = 0.03) and 4.9-fold (p = 0.001), respectively, compared to HUVECs stimulated with LPS without PA. PA combined with IS increased VCAM-1 expression by 3.7-fold (p = 0.01) but only when IS was used at 200 µM (Figure 3A and B).
 |
Figure 3 Expression of adhesive molecules and occludin in HUVEC cells treated with LPS, IS, and PA. (A) Western-bott results; (B–D) The band signal strength of VCAM-1, E-selectin, and occludin. The band signal strength of controls (untreated and vehicle-treated cells) was set to 1, and the values were normalized to β-actin levels. Statistically significant differences vs control *p < 0.05; vs VC #p < 0.05; with vs without PA &p < 0.05. |
LPS and IS individually at both concentrations increased E-selectin levels in HUVECs. However, their combination increased E-selectin levels by ca. ten times more, confirming their co-stimulatory effect on ECs. PA individually increased E-selectin level insignificantly by 0.9-fold but decreased it in HUVECs treated with IS at both concentrations, compared to cells treated with IS alone. A similar inhibitory effect on E-selectin expression was observed when PA was combined with LPS at 5 ng/mL. On the contrary, when PA was combined with LPS at 50 ng/mL, the level of E-selectin increased by 8.4-fold (p = 0.0001), almost twice the level induced by LPS alone (Figure 3A and C). The combination of PA, LPS, and IS at low and high concentrations also significantly decreased E-selectin levels in HUVEC.
In contrast to VCAM-1 and E-selectin, all metabolites tested individually or in combination reduced occludin expression in HUVECs (Figure 3A and D). Interestingly, PA did not further reduce occludin expression in cells treated with LPS and IS and their combination at high concentrations. However, PA combined with LPS and IS at low concentrations significantly (p = 0.002) reduced occludin expression. These results indicated that metabolic endotoxemia has a prominent effect on adherence molecules and tight-junction proteins in ECs.
Discussion
A diet rich in saturated fatty acids is one of the most important factors influencing the incidence of atherosclerosis. The diet produces gut dysbiosis with associated metabolic endotoxemia and exerts a direct toxic effect on vascular ECs.2 HFD is associated with endothelium dysfunction due to diminished NO bioavailability, increased ROS generation, and inflammation.16,17 The present study examined the simultaneous toxicity of PA combined with LPS and IS on ECs.
ROS are reactive intermediates of molecular oxygen produced physiologically in ECs as byproducts of cellular metabolism and acts as important second messengers in many intracellular signaling pathways in innate and adaptive immune responses.18,19 However, excessive ROS generation, often associated with the dysfunction of mechanisms and enzymes preventing excessive ROS production, leads to vascular senescence, atherosclerosis, plaque instability, and endothelial dysfunction in the vascular system.20 Several factors may induce oxidative stress in ECs, including HFD.21 The role of PA in causing oxidative stress in ECs has been well documented in numerous studies.16,22–24 Chen et al14 demonstrated that PA-induced ROS generation in HUVECs has been associated with autophagy activation via the calcium 2+/protein kinase C-α/NADPH oxidase 4 (Ca2+/PKCα/NOX4) pathway. Moreover, it has been shown that PA’s lipotoxicity to ECs is compounded by endotoxemia.25–37 LPS increases the generation of ROS, primarily superoxide anion (O2−), hydrogen peroxide (H2O2), and peroxynitrite, in ECs by enhancing the activity of NADPH oxidase 4 (Nox4), which is highly expressed in endothelial cells.16,28–31 According to the study of Park et al,29 COOH-terminal regions of Nox4 becomes directly coupled with TLR4 in response to LPS, and the complex of Nox4 with TLR4 induces ROS generation.
The results of the present study confirmed that PA significantly enhanced LPS-induced ROS generation in HUVECs. However, the impact of PA and IS combination on ROS generation in ECs was not studied so far. IS mediates oxidative stress in ECs by disrupting the balance between pro– and anti-oxidant mechanisms.32 Dou et al32 demonstrated that IS significantly increased ROS production in HUVECs by increasing NADPH oxidase activity and decreasing glutathione (GSH), the essential cellular ROS scavenger. Similarly, Yu et al33 have shown that IS reduced GSH levels in ECs. Although the present study demonstrated that IS alone at low and high concentrations significantly increased ROS production, confirming the role of this uremic toxin on endothelial oxidative stress, its combination with PA did not further affect ROS generation in ECs. Nevertheless, our results confirmed that HFD rich in SFA enhances the detrimental effect of ME-associated low-grade endotoxemia.
ROS and oxidative stress affect numerous signaling pathways in ECs, including NO availability.16 In ECs, NO can be generated by two different isoforms of nitric oxide synthase (NOS), i.e., inducible (iNOS) and eNOS. NO produced constitutively by eNOS is a physiologic vasodilator that controls blood pressure and has a vasoprotective and anti-thrombotic effect. Moreover, NO decreases endothelium permeability, the expression of chemoattractant cytokine MCP-1, and several surface molecules, preventing leukocyte adherence. In contrast, iNOS expression increases in ECs in response to LPS and generates large amounts of NO, contributing to the pathophysiology of cardiovascular diseases.28,34 Additionally, NO produced by iNOS by negative feedback inhibits the expression and activation of eNOS.35 In the study, we examined the effect of LPS and IS alone and in combination with PA on NO generation in HUVECs using a fluorescent probe. The results demonstrated that IS at low and high concentrations used in the study had no significant effect on NO production. In contrast, PA lowered NO synthesis in HUVECs, in contrast to LPS, which increased NO production. In concordance with our results, Dou et al32 demonstrated that IS at range concentrations from 100 to 1000 µM did not affect eNOS mRNA levels, and so NO generation in HUVECs. Several other studies have indicated that IS reduced NO signaling in HUVECs by inhibiting eNOS activation.36–38 Similarly, PA impairs NO production in ECs by decreasing eNOS activity via downregulating the insulin receptor substrate-phosphatidylinositol kinase (IRS-1/Akt)/eNOS pathway and the activation of inhibitor ĸB kinase (IKKβ), a critical cellular mediator of the response to inflammatory stimuli.39 On the contrary, LPS increases NO production in ECs via increased iNOS expression.40 These opposing effects of PA and LPS on NO production can lead to ECs dysfunction through the production of peroxynitrite (ONOO-), a marker of the oxidative stress responsible for protein nitration and ECs dysfunction. An increased iNOS expression in endothelial cells leads to an increased NO and oxygen radical, i.e., O2•− production, resulting in severe oxidative, nitro-oxidative, and nitrosative stress.19,41
VCAM-1 is an inducible adhesion molecule predominantly expressed in endothelial cells upon proinflammatory cytokines, ROS, oxidized low-density lipoprotein, high glucose concentration, toll-like receptor agonists like lipid A of LPS molecule, and shear stress.42 VCAM-1 ligation during inflammation activates calcium flux and Rac1, an intracellular transducer regulating multiple signaling pathways in ECs that control cytoskeleton organization, transcription, and cell proliferation. This process induces the activation of NADPH2 oxidase (Nox2), leading to ROS generation and activation of matrix metalloproteinases and protein kinase Cα (PKCα), which in turn increases the serine phosphorylation of the protein tyrosine phosphatase 1B (PTP1B) on the endoplasmic reticulum, activating PTP1B, required for VCAM-1-dependent leukocyte transendothelial migration.42,43 The recognition of LPS via TLR4 in ECs results in the induction of endothelial adhesion molecules, such as VCAM-1, intracellular adhesion molecule (ICAM-1), and E-selectin, via several downstream signaling pathways.44 Although VCAM-1 is considered a significant regulator of leukocyte adhesion and transendothelial migration, the interactions between ECs and leukocytes are mediated partly through E-selectin.
E-selectin is not constitutively expressed on ECs but can be rapidly induced by cytokines and LPS.44,45 LCSFAs, by activating proinflammatory signaling pathways via NF-κB, induce the expression of leukocyte adhesion molecules VCAM-1, ICAM-1, and E-selectin in ECs.46 Fratantonio et al46 demonstrated that PA at 100 µM significantly increased E-selectin and VCAM-1 expression in HUVECs, and the effect was attenuated by NFĸB inhibitor. Similar results are presented in the study of Pillon et al,12 where PA at 200 µM increased E-selectin and VCAM-1 expression. In our study, only weak expression of these molecules was observed after the stimulation of HUVECs with PA at 200 µM. However, when PA combined with LPS was used to stimulate HUVECs, the expression of VCAM-1 increased significantly, indicating the amplifying effect of PA on VCAM-1 expression. The amplifying effect was also observed for E-selectin when ECs were stimulated with PA and high, but not low, LPS concentration. LPS at both concentrations enhanced both E-selectin and VCAM-1 expression, although the increase was not significant in the case of VCAM-1. Contrary to LPS, IS had an insignificant impact on VCAM-1 and E-selectin expression. However, the combination of LPS and IS at both concentrations significantly increased the expression of these molecules, and PA markedly potentiated their effect on VCAM-1 but not on E-selectin expression.
The effect of IS on the expression of adhesion molecules in ECs has not been intensively studied, and the results are the opposite. Ito et al47 demonstrated that IS at high concentration (2 mM) had no effect on VCAM-1 and E-selectin expression in HUVECs, but enhanced their expression upon treatment of ECs with tumor necrosis factor-α (TNFα). In turn, Lee et al48 have shown that treatment of HUVECs with IS at 100 µM induced a two-fold increase in VCAM-1 but did not affect E-selectin expression. In this study, IS, combined with LPS and PA, significantly increased VCAM-1 expression but decreased the E-selectin level. These results suggest that IS, which exerts its toxic effect on ECs mainly via inducing inflammation and proinflammatory cytokine release,49 in combination with ME and HFD, may amplify the ECs dysfunction caused by both these metabolites.
Occludin is one of the most representative tight junction proteins (TJs) crucial for tight junction stability and the endothelial barrier function, involved in forming ECs polarity, cell migration, and proliferation.50 The abnormal expression and modification of occludin are accompanied by damage to the endothelial structure of blood vessels in tissues and organs, resulting in increased vascular permeability, inflammatory cell infiltration, and apoptosis.51 In this study, PA significantly decreased occludin expression in HUVECs. These results concord with those presented by Gorzelak-Pabiś et al52 who demonstrated that even a single high-fat meal could destabilize vascular endothelium barrier function and PA at 800 µM significantly decreased significantly occludin mRNA in HUVECs. According to Wang et al,53 palmitate at a concentration as low as 20 µM can stimulate Nlrp3 inflammasomes in ECs, which results in caspase-1 activation, and IL-1β production and, in consequence, reduces the expression of endothelial tight junction proteins that are essential for maintaining the endothelial barrier function and vascular integrity. Moreover, our study demonstrated that LPS combined with IS at high concentrations significantly reduced occludin expression in HUVECs, and PA has no further effect on occludin levels. However, simultaneous treatment of ECs with PA, LPS, and IS at low concentrations reduced occludin expression more than LPS and IS did individually or in combination, indicating that even low IS and LPS concentrations in LCSFA-rich blood reduce tight junction proteins and increase endothelial permeability.
IS increases endothelial permeability via upregulating the aryl hydrocarbon receptor AHR/Src kinase pathway and VE-cadherin phosphorylation.54 However, the impact of IS on occludin expression in ECs has not been studied so far. On the contrary, LPS-induced increase in endothelial vascular permeability is associated with elevated expression of proinflammatory cytokines, e.g., TNFα, to disrupt the microtubule arrangement.55 Moreover, LPS and TRL4 ligation triggers the RhoA pathway, increasing the abundance of the F-actin microfilaments to increase transendothelial permeability and ECs stiffness.56,57 This effect we also observed in the present study, where LPS, especially in combination with PA and IS at high concentration, induced stress in actin filaments and focal adhesion complexes in HUVECs. Okamoto et al58 demonstrated that human aortic endothelial cells and HUVECs, in response to LPS-induced inflammation, enhanced the formation of focal adhesion and stress fibers, leading to cellular stiffening. According to Kasa et al,59 LPS-induced cytoskeletal rearrangements and ECs contractile response lead to disruption of intercellular contacts and endothelium permeability increase. In turn, IS induces cytoskeleton remodeling, cell contraction, and intercellular gaps formation via superoxide ani-on-phosphorylation of myosin light chain kinase (MLCK) and myosin light chains (MLC) as well as activation of extracellular-signal-regulated protein kinase (ERK1/ERK2).60 Hence, LPS, in combination with IS, can enhance ECs’ stiffness and increase endothelial permeability. By activating the proinflammatory TLR4 signaling pathways, PA can enhance LPS-induced inflammatory response in ECs and increase the effect of cytoskeleton remodeling.61
Overall, our study showed that LCSFAs, represented in the study by PA, exacerbate the toxic effects of ME on ECs. PA at 200 µM significantly potentiated LPS- and LPS/IS-induced VCAM-1 expression, ROS generation, and decreased occludin levels. Additionally, increased HUVECs’ stiffness and endothelial permeability demonstrated in the study strongly suggest that combining PA with LPS and uremic toxin at clinically irrelevant concentrations promotes atherosclerosis. However, further in vivo studies are needed to confirm the co-toxicity of PA with LPS and IS at such low concentrations.
Conclusion
Palmitic acid in vitro enhances the toxic effect of ME on the vascular endothelium.
Funding
The study was prepared under the project financed from the funds granted by the Ministry of Education and Science in Poland, in the “Regional Initiative of Excellence” program for the years 2019–2022, project number 016/RID/2018/19 (individual project number RID Z501.20.009).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi:10.2337/db06-1491
2. Mohammad S, Thiemermann C. Role of metabolic endotoxemia in systemic inflammation and potential interventions. Front Immunol. 2021;11:1–16. doi:10.3389/fimmu.2020.594150
3. Brown BI. Nutritional management of metabolic endotoxemia: a clinical review. Altern Ther Health Med. 2017;23(4):42–54.
4. André P, Laugerette F, Féart C. Metabolic endotoxemia: a potential underlying mechanism of the relationship between dietary fat intake and risk for cognitive impairments in humans? Nutrients. 2019;11(8):1887. doi:10.3390/nu11081887
5. Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50(1):90–97. doi:10.1194/jlr.M800156-JLR200
6. Neves AL, Coelho J, Couto L, Leite-Moreira A, Roncon-Albuquerque R. Metabolic endotoxemia: a molecular link between obesity and cardiovascular risk. J Mol Endocrinol. 2013;51(2):R51–R64. doi:10.1530/JME-13-0079
7. Vors C, Pineau G, Drai J, et al. Postprandial endotoxemia linked with chylomicrons and lipopolysaccharides handling in obese versus lean men: a lipid dose-effect trial. J Clin Endocrinol Metab. 2015;100(9):3427–3435. doi:10.1210/jc.2015-2518
8. Fasano A. Gut permeability, obesity, and metabolic disorders: who is the chicken and who is the egg? Am J Clin Nutr. 2017;105(1):3–4. doi:10.3945/ajcn.116.148338
9. Carta G, Murru E, Banni S, Manca C. Palmitic acid: physiological role, metabolism and nutritional implications. Front Physiol. 2017;8:1–14. doi:10.3389/fphys.2017.00902
10. Korbecki J, Bajdak-Rusinek K. The effect of palmitic acid on inflammatory response in macrophages: an overview of molecular mechanisms. Inflamm Res. 2019;68(11):915–932. doi:10.1007/s00011-019-01273-5
11. Sanadgol N, Mostafaie A, Mansouri K, Bahrami G. Effect of palmitic acid and linoleic acid on expression of ICAM-1 and VCAM-1 in human bone marrow endothelial cells (HBMECs). Arch Med Sci. 2012;8(2):192–198. doi:10.5114/aoms.2012.28544
12. Pillon NJ, Azizi PM, Li YE, et al. Palmitate-induced inflammatory pathways in human adipose microvascular endothelial cells promote monocyte adhesion and impair insulin transcytosis. Am J Physiol Endocrinol Metab. 2015;309(1):E35–E44. doi:10.1152/ajpendo.00611.2014
13. Gu YY, Tan XH, Song WP, et al. Icariside II attenuates palmitic acid-induced endothelial dysfunction through SRPK1-Akt-eNOS signaling pathway. Front Pharmacol. 2022;13:1–12. doi:10.3389/fphar.2022.920601
14. Chen P, Liu H, Xiang H, et al. Palmitic acid-induced autophagy increases reactive oxygen species via the Ca2+/PKCα/NOX4 pathway and impairs endothelial function in human umbilical vein endothelial cells. Exp Ther Med. 2019:2425–2432. doi:10.3892/etm.2019.7269
15. Choroszy M, Litwinowicz K, Łaczmanski Ł, Roleder T, Sobieszczanska B. Co-toxicity of bacterial metabolites, to vascular endothelial cells in coronary arterial disease accompanied by gut dysbiosis. Nutrients. 2022;14:424. doi:10.3390/nu14030424
16. Togo M, Konari N, Tsukamoto M, et al. Effects of a high-fat diet on superoxide anion generation and membrane fluidity in liver mitochondria in rats. J Int Soc Sports Nutr. 2018;15(1):1–8. doi:10.1186/s12970-018-0217-z
17. Dow CA, Stauffer BL, Greiner JJ, DeSouza CA. Influence of habitual high dietary fat intake on endothelium-dependent vasodilation. Appl Physiol Nutr Metab. 2015;40(7):711–715. doi:10.1139/apnm-2015-0006
18. Sena CM, Leandro A, Azul L, Seiça R, Perry G. Vascular oxidative stress: impact and therapeutic approaches. Front Physiol. 2018;9:1–11. doi:10.3389/fphys.2018.01668
19. Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1–19. doi:10.1016/J.VPH.2017.05.005
20. Sakurada R, Odagiri K, Hakamata A, Kamiya C, Wei J, Watanabe H. Calcium release from endoplasmic reticulum involves calmodulin-mediated NADPH oxidase-derived reactive oxygen species production in endothelial cells. Int J Mol Sci. 2019;20(7):1644. doi:10.3390/ijms20071644
21. Scioli MG, Storti G, D’amico F, et al. Oxidative stress and new pathogenetic mechanisms in endothelial dysfunction: potential diagnostic biomarkers and therapeutic targets. J Clin Med. 2020;9(6):1–39. doi:10.3390/jcm9061995
22. Zhu Q, Dong Q, Wang X, et al. Palmitic acid, a critical metabolite, aggravates cellular senescence through reactive oxygen species generation in Kawasaki disease. Front Pharmacol. 2022;13:1–14. doi:10.3389/fphar.2022.809157
23. Kim JE, Song SE, Kim YW, et al. Adiponectin inhibits palmitate-induced apoptosis through suppression of reactive oxygen species in endothelial cells: involvement of cAMP/protein kinase A and AMP-activated protein kinase. J Endocrinol. 2010;207(1):35–44. doi:10.1677/JOE-10-0093
24. Zhou X, Yang J, Zhou M, et al. Resveratrol attenuates endothelial oxidative injury by inducing autophagy via the activation of transcription factor EB. Nutr Metab. 2019;16(1):1–12. doi:10.1186/s12986-019-0371-6
25. Li W, Yang X, Zheng T, et al. TNF-α stimulates endothelial palmitic acid transcytosis and promotes insulin resistance. Sci Rep. 2017;7:1–18. doi:10.1038/srep44659
26. Lu Z, Li Y, Ru JH, Lopes-Virella MF, Lyons TJ, Huang Y. Interaction of palmitate and LPS regulates cytokine expression and apoptosis through sphingolipids in human retinal microvascular endothelial cells. Exp Eye Res. 2019;178:61–71. doi:10.1016/j.exer.2018.09.016
27. Park WJ, Han JS. Gryllus bimaculatus extract protects against lipopolysaccharide and palmitate-induced production of proinflammatory cytokines and inflammasome formation. Mol Med Rep. 2021;23(3):1–10. doi:10.3892/mmr.2021.11845
28. Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86(1):9–22. doi:10.1038/labinvest.3700366
29. Park HS, Chun JN, Jung HY, Choi C, Bae YS. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc Res. 2006;72(3):447–455. doi:10.1016/j.cardiores.2006.09.012
30. Montezano AC, Touyz RM. Reactive oxygen species and endothelial function - role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin Pharmacol Toxicol. 2012;110(1):87–94. doi:10.1111/j.1742-7843.2011.00785.x
31. Duarte S, Arango D, Parihar A, Hamel P, Yasmeen R, Doseff AI. Apigenin protects endothelial cells from lipopolysaccharide (LPS)-induced inflammation by decreasing caspase-3 activation and modulating mitochondrial function. Int J Mol Sci. 2013;14(9):17664–17679. doi:10.3390/ijms140917664
32. Dou L, Jourde-Chiche N, Faure V, et al. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost. 2007;5(6):1302–1308. doi:10.1111/j.1538-7836.2007.02540.x
33. Yu M, Kim YJ, Kang D-H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via induction of oxidative stress. Clin J Am Soc Nephrol. 2011;6(1):30–39. doi:10.2215/CJN.05340610
34. Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):829–837. doi:10.1093/eurheartj/ehr304
35. Connelly L, Madhani M, Hobbs AJ. Resistance to endotoxic shock in endothelial nitric-oxide synthase (eNOS) knock-out mice: a pro-inflammatory role for eNOS-derived no in vivo. J Biol Chem. 2005;280(11):10040–10046. doi:10.1074/jbc.M411991200
36. Matsumoto T, Takayanagi K, Kojima M, Taguchi K, Kobayashi T. Acute exposure to indoxyl sulfate impairs endothelium-dependent vasorelaxation in rat aorta. Int J Mol Sci. 2019;20(2). doi:10.3390/ijms20020338
37. Tumur Z, Niwa T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am J Nephrol. 2009;29(6):551–557. doi:10.1159/000191468
38. Kharait S, Haddad DJ, Springer ML. Nitric oxide counters the inhibitory effects of uremic toxin indoxyl sulfate on endothelial cells by governing ERK MAP kinase and myosin light chain activation. Biochem Biophys Res Commun. 2011;409(4):758–763. doi:10.1016/j.bbrc.2011.05.084
39. Kim F, Tysseling KA, Rice J, et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler Thromb Vasc Biol. 2005;25(5):989–994. doi:10.1161/01.ATV.0000160549.60980.a8
40. Suzuki K, Susaki EA, Nagaoka I. Lipopolysaccharides and cellular senescence: involvement in atherosclerosis. Int J Mol Sci. 2022;23(19):11148. doi:10.3390/ijms231911148
41. Kar S, Kavdia M. Endothelial NO and O2− production rates differentially regulate oxidative, nitroxidative, and nitrosative stress in the microcirculation. Free Radic Biol Med. 2013;63:161–174. doi:10.1016/j.freeradbiomed.2013.04.024
42. Kong DH, Kim YK, Kim MR, Jang JH, Lee S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int J Mol Sci. 2018;19(4):13–17. doi:10.3390/ijms19041057
43. Wittchen ES. Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front Biosci. 2009;14(7):2522–2545. doi:10.2741/3395
44. Dayang EZ, Plantinga J, Ter Ellen B, Van Meurs M, Molema G, Moser J. Identification of LPS-activated endothelial subpopulations with distinct inflammatory phenotypes and regulatory signaling mechanisms. Front Immunol. 2019;10:1–12. doi:10.3389/fimmu.2019.01169
45. Wong D, Dorovini-Zis K. Regulation by cytokines and lipopolysaccharide of E-selectin expression by human brain microvessel endothelial cells in primary culture. J Neuropathol Exp Neurol. 1996;55(2):225–235. doi:10.1097/00005072-199602000-00011
46. Fratantonio D, Speciale A, Ferrari D, Cristani M, Saija A, Cimino F. Palmitate-induced endothelial dysfunction is attenuated by cyanidin-3-O-glucoside through modulation of Nrf2/Bach1 and NF-κB pathways. Toxicol Lett. 2015;239(3):152–160. doi:10.1016/j.toxlet.2015.09.020
47. Ito S, Osaka M, Higuchi Y, Nishijima F, Ishii H, Yoshida M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J Biol Chem. 2010;285(50):38869–38875. doi:10.1074/JBC.M110.166686
48. Lee CT, Lee YT, Ng HY, et al. Lack of modulatory effect of simvastatin on indoxyl sulfate-induced activation of cultured endothelial cells. Life Sci. 2012;90(1–2):47–53. doi:10.1016/j.lfs.2011.10.014
49. Matsumoto T, Kojima M, Takayanagi K, Taguchi K, Kobayashi T. Role of s-equol, indoxyl sulfate, and trimethylamine n-oxide on vascular function. Am J Hypertens. 2020;33(9):793–803. doi:10.1093/ajh/hpaa053
50. Cong X, Kong W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell Signal. 2020;66:109485. doi:10.1016/j.cellsig.2019.109485
51. Higashi Y, Sukhanov S, Shai SY, et al. Endothelial deficiency of insulin-like growth factor-1 receptor reduces endothelial barrier function and promotes atherosclerosis in apoe-deficient mice. Am J Physiol Heart Circ Physiol. 2020;318(5):H730–H743. doi:10.1152/AJPHEART.00064.2020
52. Gorzelak-Pabiś P, Wozniak E, Wojdan K, Chalubinski M, Broncel M. Single triglyceride-rich meal destabilizes barrier functions and initiates inflammatory processes of endothelial cells. J Interferon Cytokine Res. 2020;40(1):43–53. doi:10.1089/jir.2018.0173
53. Wang L, Chen Y, Li X, Zhang Y, Gulbins E, Zhang Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget. 2016;7(45):73229–73241. doi:10.18632/oncotarget.12302
54. Assefa EG, Yan Q, Gezahegn SB, et al. Role of resveratrol on indoxyl sulfate-induced endothelial hyperpermeability via aryl hydrocarbon receptor (AHR)/Src-dependent pathway. Oxid Med Cell Longev. 2019;2019:1–15. doi:10.1155/2019/5847040
55. Petrache I, Birukova A, Ramirez SI, Garcia JGN, Verin AD. The role of the microtubules in tumor necrosis factor-α-induced endothelial cell permeability. Am J Respir Cell Mol Biol. 2003;28(5):574–581. doi:10.1165/rcmb.2002-0075OC
56. Prasain N, Stevens T. The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res. 2009;77(1):53–63. doi:10.1016/j.mvr.2008.09.012
57. Kubra K-T, Barabutis N. Brefeldin A and kifunensine modulate LPS-induced lung endothelial hyperpermeability in human and bovine cells. Am J Physiol Cell Physiol. 2021;321(2):C214–C220. doi:10.1152/ajpcell.00142.2021
58. Okamoto T, Kawamoto E, Usuda H, et al. Recombinant human soluble thrombomodulin suppresses monocyte adhesion by reducing lipopolysaccharide-induced endothelial cellular stiffening. Cells. 2020;9(8):1–16. doi:10.3390/cells9081811
59. Kása A, Csortos C, Verin AD. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers. 2015;3(1):1–2. doi:10.4161/21688370.2014.974448
60. Sen PY, Lin YT, Chen Y, Hung KY, Wang SM. Effects of indoxyl sulfate on adherens junctions of endothelial cells and the underlying signaling mechanism. J Cell Biochem. 2012;113(3):1034–1043. doi:10.1002/jcb.23435
61. Kotlyarov S, Kotlyarova A. Involvement of fatty acids and their metabolites in the development of inflammation in atherosclerosis. Int J Mol Sci. 2022;23(3):1308. doi:10.3390/ijms23031308
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.