Back to Journals » Journal of Inflammation Research » Volume 18
The Role of Mitochondrial Dysfunction and Inflammatory Response in the Pathogenesis of Sepsis-Induced Myocardial Injury: A Mechanistic Study
Authors Liu AB, Wang S ![]() , Shen Y, Ma L, Zhang JF
, Shen Y, Ma L, Zhang JF
Received 9 July 2025
Accepted for publication 19 October 2025
Published 30 October 2025 Volume 2025:18 Pages 15207—15235
DOI https://doi.org/10.2147/JIR.S552730
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Anh Ngo
An-Bu Liu,1– 3 Sheng Wang,4 Yue Shen,4 Lei Ma,1 Jun-Fei Zhang1,3
1Department of Emergency Medical, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 2State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia, Xinjiang Medical University, Wulumuqi, Xinjiang, People’s Republic of China; 3Ningxia Key Laboratory of Clinical and Pathogenic Microbiology, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 4School of Clinical Medicine, Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China
Correspondence: Jun-Fei Zhang, Email [email protected] Lei Ma, Email [email protected]
Abstract: Sepsis, a severe systemic infection triggered by the invasion of bacterial, viral, fungal, and other pathogens into human tissues, frequently results in substantial damage to the heart, which is one of the primary organs affected. This myocardial injury is strongly linked to poor patient outcomes in sepsis. Recent research has identified key factors such as mitochondrial dysfunction, metabolic disturbances, cell death, and dysregulated inflammatory responses as critical contributors to the pathogenesis of sepsis-induced myocardial injury (SIMI). These mechanisms not only enhance our understanding of SIMI but also offer potential therapeutic targets. The review aims to investigate the pathophysiological mechanisms driving myocardial injury in sepsis, particularly from the perspective of mitochondrial dysfunction. It will examine the complex interactions between inflammatory dysregulation, calcium homeostasis disruption, metabolic reprogramming, and mitochondrial dysfunction in the onset and progression of SIMI. By exploring therapeutic approaches focused on restoring mitochondrial function, this research aims to establish a theoretical framework for interventions targeting SIMI, thereby providing a robust foundation for the development of targeted therapies for SIMI.
Keywords: sepsis-induced myocardial injury, SIMI, inflammatory responses, mitochondrial dysfunction, RCD, metabolic reprogramming
Introduction
Sepsis is a severe infectious disease caused by the invasion of pathogens such as bacteria, viruses, and fungi into human tissues. In its most severe form, sepsis can progress to systemic inflammatory response syndrome (SIRS), multiple organ failure, and ultimately, death.1,2 Given its high mortality rate and the frequent onset of complex complications, sepsis represents a substantial global economic and social burden. In 2017, an estimated 48.9 million individuals worldwide were affected by sepsis, with 11 million sepsis-related deaths, accounting for 19.7% of all global fatalities.3 In the United States, over 1.7 million adults develop sepsis annually, resulting in nearly 350,000 deaths each year.4 A cross-sectional study conducted across 44 hospitals in China revealed a 90-day mortality rate of approximately 35.5% among patients hospitalized for sepsis.5
Sepsis typically begins with SIRS to infection, which can be partially managed by controlling the underlying infection. However, as the condition progresses, it frequently leads to multi-organ dysfunction or failure, involving critical organs such as the heart, brain, kidneys, and lungs. This multi-organ involvement is a central factor contributing to the high mortality observed in sepsis patients.6,7 Notably, sepsis can also result in additional complications such as acute respiratory distress syndrome (ARDS), renal failure, disseminated intravascular coagulation (DIC), and septic shock.8,9 These complications further exacerbate the severity of sepsis and pose significant challenges in its management. One of the key complications of sepsis is sepsis-induced myocardial injury (SIMI), which is associated with a markedly increased mortality rate, ranging from 30% to 70%, compared to non-cardiogenic septic patients.10,11 Traditional therapeutic strategies for SIMI include fluid resuscitation, infection management, vasopressor therapy, and organ support, yet their effectiveness remains constrained. In recent years, advanced treatment modalities such as extracorporeal membrane oxygenation (ECMO) and remote ischemic conditioning (RIC) have developed, offering heart-lung replacement therapy for sepsis patients. Despite their promise, these approaches are costly and add considerable financial strain on patients, and are typically restricted to intensive care units (ICUs) in large tertiary hospitals, limiting their widespread application. Furthermore, the multifactorial nature of SIMI complicates both its early diagnosis and effective treatment. Consequently, further research into the pathophysiological mechanisms underlying SIMI is essential to the development of targeted therapeutic strategies aimed at enhancing patient survival. Studies indicate that dysregulated systemic immune responses play a pivotal role in the development of myocardial injury in sepsis, although mitochondrial dysfunction, metabolic disturbances, myocardial edema, and cell death are also key contributors to its pathogenesis.12–14 For instance, exogenous fetuin-A can protect against sepsis-induced cardiac dysfunction in vivo via suppression of inflammation and oxidative damage.15 Additionally, interactions between these mechanisms may exist, necessitating a comprehensive approach to fully elucidate the condition.12–14 While much of the research has focused on these individual mechanisms in isolation, there is an increasing need to integrate and synthesize these findings to establish a robust scientific framework for targeted treatment strategies for SIMI. In this review, we will further explore the potential pathophysiological mechanisms of SIMI from the perspective of mitochondrial dysfunction, with particular emphasis on the role of dysregulated inflammatory responses, calcium homeostasis imbalance, and metabolic reprogramming in the pathogenesis of SIMI. Finally, we will provide a concise overview of therapeutic strategies targeting mitochondrial dysfunction and potential therapeutic targets for SIMI.
Mitochondrial Dysfunction Induces Dysregulation of the Inflammatory Response in SIMI
Mitochondria play a crucial role in regulating the inflammatory response, thereby contributing to the onset and progression of infectious diseases and tumors. First, mitochondria are believed to be evolutionary remnants of modern Gram-negative bacteria, specifically α-proteobacteria. Consequently, some mitochondrial components share similarities with bacterial molecules, suggesting that certain mitochondrial components could serve as ligands for pattern recognition receptors (PRRs).16 Secondly, both the inner mitochondrial membrane (IMM) and outer mitochondrial membrane (OMM) provide a double-membrane structure that controls the separation of mitochondrial damage-associated molecular patterns (mtDAMPs) and their homologous PRRs.17 Mitochondrial mtDAMPs are a series of molecules released into the cytoplasm or extracellular environment when the mitochondria are damaged or dysfunctional. These include mitochondrial DNA (mtDNA), mitochondrial peptides (such as N-formyl peptides), and adenosine triphosphate (ATP). These molecules possess damage-associated molecular pattern (DAMP) characteristics and can activate immune responses in host cells, leading to the induction of inflammation. Thirdly, and more extensively studied, mitochondria regulate various forms of programmed cell death, including apoptosis, pyroptosis, autophagy, and ferroptosis, which in turn trigger inflammatory responses and contribute to pathological states in the body.18 Sepsis is characterized by the acute release of multiple inflammatory mediators, including tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β. The excessive release of these mediators damages tissues and organs. The overactive inflammation triggered by damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) contributes to the disruption of endothelial barrier integrity and plays a key role in the pathogenesis of sepsis and sepsis-induced myocardial injury.19 Next, we will focus on the mechanisms by which mitochondrial dysfunction-induced dysregulation of inflammatory responses, through processes such as pyroptosis apoptosis, mitophagy, and ferroptosis, contributes to the development of septic cardiomyopathy.
Pyroptosis in SIMI
Pyroptosis is an emerging form of regulated cell death, characterized by progressive cell swelling culminating in membrane rupture, which results in the release of cellular contents and the subsequent activation of a potent inflammatory response.20 This process is primarily mediated by gasdermin proteins and plays an essential role in the innate immune response during both infection and inflammation. In the context of SIMI pathogenesis, mitochondrial membrane integrity is compromised, leading to the release of DAMPs that activate gasdermin-mediated immune responses.21 These reactive oxygen species(ROS) are subsequently released into the cytoplasm, where they interact with nucleotide- binding oligomerization domain, leucine- rich repeat and pyrin domain- containing 3(NLRP3), thereby activating the NLRP3 inflammasome.22 Furthermore, the excessive buildup of ROS activates Toll-like receptor (TLR)-mediated inflammatory pathways, exacerbating myocardial injury in the context of sepsis.23 Notably, Wang et al demonstrated that oxycodone mitigates pyroptosis through the modulation of the Nrf2/HO-1 signaling pathway, thereby alleviating inflammatory dysregulation and reducing lipopolysaccharide(LPS)-induced myocardial injury.24 In a similar vein, inhibition of the NLRP3 inflammasome/GSDMD signaling axis significantly dampens the inflammatory response and myocardial pyroptosis, yielding substantial therapeutic benefits in SIMI.25 In the subsequent sections, we will further explore the mechanistic underpinnings of pyroptosis in SIMI, with particular emphasis on its canonical pathways and critical molecular events.
The Classical Pathways of Pyroptosis in SIMI
In the pathogenesis of SIMI, pyroptosis primarily relies on the activation of inflammasomes, which trigger the activation of specific caspase family proteins. These caspases cleave gasdermin proteins, leading to the activation of gasdermin, which translocates to the membrane and forms pores, causing cell swelling, cytoplasmic leakage, and ultimately leading to cell membrane rupture and pyroptosis.26 Based on whether caspases are involved, pyroptosis is classified into two main pathways: the caspase-1-dependent and caspase-1-independent pyroptotic pathways. In the Caspase-1-dependent pyroptotic pathway, upon the invasion of various pathogens, inflammasomes such as NLRP3, NLRC4, AIM2, and Pyrin can recognize these signals and become activated. These inflammasomes subsequently interact with the adapter protein ASC and pro-caspase-1 to form a complex, activating caspase-1.27 On one hand, the activated caspase-1 cleaves Gasdermin D (GSDMD), exposing the N-terminal fragment of GSDMD, which interacts with phospholipids on the cell membrane, forming pores that release cellular contents and induce pyroptosis.20 On the other hand, activated caspase-1 also cleaves and activates the precursors of IL-1β and IL-18, which are then released extracellularly, thereby amplifying the inflammatory response. In the caspase-1-independent pyroptotic pathway, upon exposure to LPS, human caspases-4 and -5, as well as murine caspase-11, can directly bind to LPS and become activated. These caspases then cleave GSDMD, exposing its N-terminal fragment and initiating pyroptosis. Additionally, the activation of caspase-4/5/11 stimulates the activation of the Pannexin-1 channel, which facilitates the efflux of K+ ions, triggering the NLRP3 inflammasome. This in turn activates caspase-1, further promoting the caspase-1-dependent pyroptotic pathway. Thus, these two distinct pyroptotic pathways, whether caspase-1-dependent or caspase-1-independent, play crucial roles in mediating pyroptosis and driving the inflammatory processes that contribute to the myocardial injury observed in SIMI28 (Figure 1).
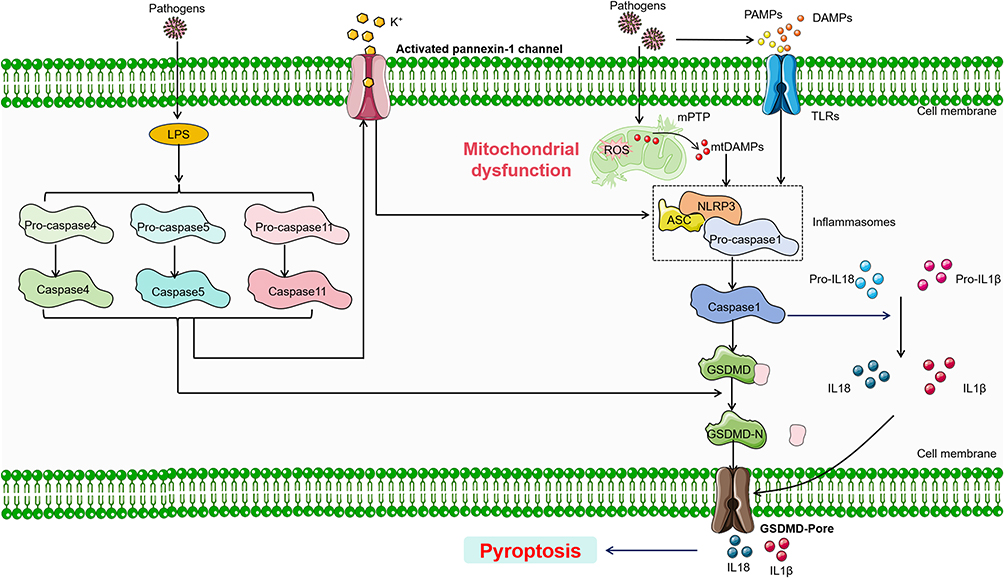 |
Figure 1 Main pathway of pyroptosis in SIMI. During the onset of SIMI, various stress stimuli lead to mitochondrial dysfunction, with mtDAMPs being released through the mPTP, thereby inducing pyroptosis. Concurrently, extracellular DAMPs and PAMPs can also trigger pyroptosis. Pyroptosis can be classified into two types based on its dependence on caspase-1. In caspase-1 dependent pyroptosis, the process is initiated by the assembly of inflammasomes. Conversely, in caspase-1 independent pyroptosis, it is triggered by the interaction between caspase-4, caspase-5, or caspase-11 (depending on the species) and LPS. Solid black arrows present for direct processes. Black colored dotted squared box present for complex. Abbreviations: ASC, apoptosis-associated speck like protein containing a CARD; NLRP3, nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain- containing 3; GSDMD, Gasdermin D. |
The Key Factors of Pyroptosis in SIMI
Activation of the NLRP3 inflammasome, formation of GSDMD pores, and secretion of pro-inflammatory cytokines are considered key events in the occurrence of pyroptosis in SIMI. The NLRP3 inflammasome is a large molecular protein complex, with an approximate molecular weight of 700,000 Da, consisting of NLRP3, the adaptor protein ASC, and the effector protein Caspase-1. The assembly of the NLRP3 inflammasome requires the interaction between the NLRP3 receptor, the apoptosis-associated speck-like protein (ASC) adapter, and pro-caspase-1. Under normal physiological conditions, NLRP3 is in an auto-inhibited state. In SIMI, mitochondrial dysfunction, as well as the release of PAMPs or DAMPs, leads to the deactivation of this auto-inhibition. The N-terminal pyrin domain (PYD) of NLRP3 recruits the ASC adaptor protein, which also contains a PYD domain, while the caspase recruitment domain (CARD) of ASC recruits pro-caspase-1, which contains a CARD domain, thereby facilitating the assembly of the inflammasome. ASC, composed of both PYD and CARD domains, is primarily located in the nucleus of human monocytes/macrophages, but rapidly translocates to the cytoplasm under stress, linking NLRP3 and pro-caspase-1 to promote the activation of the NLRP3 inflammasome. Caspase-1, also known as IL-1β-converting enzyme, is the effector protein of the NLRP3 inflammasome. It is activated through the auto-cleavage of the precursor molecule pro-caspase-1, resulting in the formation of active caspase-1. Its primary function is to facilitate the cleavage of pro-IL-1β and pro-IL-18 into their mature forms, IL-1β and IL-18. Clinical studies have shown that levels of NLRP3, GSDMD, IL-1β, and IL-18 are elevated in sepsis patients.29–31 In a SIMI model induced by intraperitoneal LPS injection in mice, targeting the NLRP3 inflammasome effectively inhibited pyroptosis and alleviated cardiac oxidative stress and inflammation.32
GSDMD belongs to the gasdermin (GSDM) family and comprises a cytotoxic N-terminal domain and a C-terminal inhibitory domain linked by a flexible linker. Upon cleavage of the C-terminal domain of GSDMD, the N-terminal fragment (GSDMD-N) is recruited to the cell membrane, where it interacts with lipids to form an intermediate structure known as the pre-pore. This pre-pore undergoes conformational rearrangement to form oligomeric arcs, which subsequently transition into gap-like structures. Finally, these gaps expand to form a ring-shaped oligomer, resulting in the formation of membrane pores. Electron microscopy reveals that the pore formed by GSDMD-N has an inner diameter of 10–15 nm, allowing the passage of certain pro-inflammatory cytokines such as IL-1β and IL-18, thus triggering pyroptosis.33,34 Yang et al observed that in a septic mouse model, GSDMD knockout (GSDMD−/−) mice exhibited alleviated organ damage compared to wild-type mice.35 Further mechanistic studies revealed that during sepsis, mitochondrial dysfunction leads to the release of ROS, and GSDMD-N directly interacts with oxidized cardiolipin, inducing cardiac dysfunction.36 Additionally, caspase-4/11 activates GSDMD-N pore formation, which in turn amplifies ROS production and upregulates the NLRP3 inflammasome, further enhancing GSDMD-N pore formation.36
Pro-inflammatory cytokines, including IL-1β and IL-18, play a central role in this process. In a cecal ligation and puncture (CLP)-induced sepsis mouse model, elevated levels of cardiac troponin I (cTnI), IL-1β, and TNF-α were observed, alongside mitochondrial dysfunction.37 Moreover, studies have demonstrated that Irisin can reduce the expression of IL-1β and decrease serum lactate dehydrogenase (LDH) and creatine kinase MB (CK-MB) levels, alleviating LPS-induced damage to H9C2 cells by upregulating mitochondrial ubiquitin ligase (MITOL) in a GSDMD-dependent manner.38
In summary, during sepsis, mitochondrial structural or functional dysfunction releases mitochondrial-derived DAMPs, which activate the NLRP3 inflammasome and promote GSDMD pore formation. This leads to the release of IL-1β and IL-18 into the bloodstream, inducing myocardial pyroptosis and widespread inflammatory responses, ultimately contributing to sepsis-induced myocardial injury. Therefore, targeting mitochondrial dysfunction and cell pyroptosis to mitigate excessive inflammatory responses represents a promising therapeutic strategy for SIMI.
Apoptosis in SIMI
Research has shown that LPS can disrupt the ultrastructure of mitochondria, leading to mitochondrial membrane potential (MMP) impairment. This disturbance increases the production of LDH, ROS, and malondialdehyde (MDA), while simultaneously decreasing the levels of superoxide dismutase (SOD).39 These changes trigger cell apoptosis and dysregulated inflammatory responses, ultimately contributing to SIMI.39 Apoptosis refers to a programmed, orderly cell death process that occurs under both physiological and pathological conditions to maintain homeostasis within the body. It is characterized by a series of morphological and biochemical alterations, including nuclear condensation, DNA fragmentation, cell membrane remodeling and blebbing, cellular shrinkage, and the formation of apoptotic bodies. The apoptotic cells are then engulfed by macrophages for removal.40 Apoptosis is a major feature of inflammatory system dysfunction, representing a hallmark of both human sepsis and experimental sepsis models in cells and animals.41 It has been observed that during SIMI, the accumulation of ROS leads to the oxidative modification of large molecular proteins and DNA structures, thereby disrupting mitochondrial integrity. This increases the permeability of the mitochondrial membrane, which in turn activates apoptotic pathways through the release of cytochrome c. This cascade of events results in myocardial cell apoptosis.42 This section will discuss the classic pathways and key molecules involved in apoptosis during the development and progression of SIMI.
The Classical Pathways of Apoptosis in SIMI
The process of apoptosis in SIMI primarily involves the extrinsic death receptor pathway, the intrinsic mitochondrial pathway, and the intrinsic endoplasmic reticulum (ER) pathway.43,44 The extrinsic death receptor pathway is mainly activated through the TNF receptor-associated death domain (TRADD) receptor. The activation of death receptors on the cell surface primarily involves the TNF superfamily and TNF-related apoptosis-inducing ligands, which specifically recognize and activate these receptors, leading to the activation of caspase-8. The intrinsic ER pathway is mainly triggered by various stress factors that cause calcium ions to be pumped out of the ER, which activates caspase-12 and, in turn, activates the effector caspase-3, initiating the apoptotic cascade. Notably, in SIMI, the pathway associated with mitochondrial dysfunction is the intrinsic mitochondrial pathway, which is primarily activated by stress-induced stimuli. When the body undergoes various stress conditions such as activation of oncogenes, DNA damage, hypoxia, or deprivation of growth factors, pro-apoptotic proteins from the Bcl-2 family, such as Bax, are upregulated. Bax is generally found in the cytoplasm, and when it receives apoptotic signals, it translocates to the mitochondrial membrane. At the mitochondrial surface, Bax forms pores across the mitochondrial membrane, leading to a decrease in membrane potential and increased membrane permeability.45 This results in the release of apoptotic factors such as cytochrome c and second mitochondria-derived activator of caspases (SMAC), which then induce apoptosis.45 Once cytochrome c is released into the cytoplasm, it interacts with Apaf-1, and with the assistance of ATP and dATP, forms the apoptosome. The apoptosome recruits and activates pro-caspase-9, forming the caspase-9 holoenzyme. Caspase-9 further activates the effector caspases-3 and −7, initiating the caspase cascade and cleaving over 100 substrates in the cell, such as α-tubulin, actin, PARP, and lamin, ultimately leading to cell apoptosis. Additionally, in sepsis, cytochrome c oxidase exerts a non-competitive inhibitory effect on cardiomyocytes, mainly by interrupting mitochondrial oxidative phosphorylation and inhibiting ATP production, which ultimately leads to SIMI.46 During SIMI, with changes in the mitochondrial membrane potential, other factors such as apoptosis-inducing factor(AIF) and endonuclease G (ENDOG) are also released into the cytoplasm.47,48 AIF and ENDOG are transported to the cell nucleus, where they induce chromatin condensation and DNA fragmentation, triggering apoptosis47,48 (Figure 2).
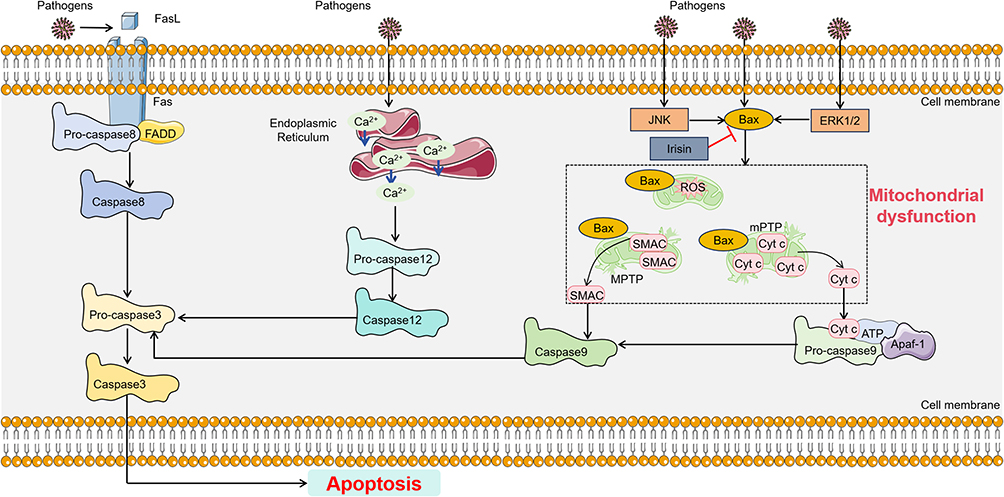 |
Figure 2 Main pathway of apoptosis in SIMI. The apoptotic pathway can be categorized into three distinct types: the extrinsic death receptor pathway, the intrinsic mitochondrial pathway, and the intrinsic endoplasmic reticulum (ER) pathway. The extrinsic apoptotic cascade is primarily activated through the specific activation of FADD receptors and caspase-8. The intrinsic ER pathway is primarily triggered by various stress factors that lead to the efflux of calcium ions from the ER, activating caspase-12, which in turn activates the effector caspase-3 and initiates the apoptosis cascade. The intrinsic mitochondrial apoptotic pathway is activated by the release of cytochrome c and SMAC, which leads to the upregulation of pro-apoptotic protein Bax and the induction of apoptosis. Additionally, the interaction between cytochrome c, Apaf-1, and caspase-9 can also trigger the extrinsic apoptotic cascade. Black arrows present for direct processes. Black colored dotted squared box present for mitochondrial dysfunction process. Abbreviations: FADD, Fas-associating protein with a novel death domain; SMAC, second mitochondria-derived activator of caspases; Apaf-1, apoptotic protease activating factor-1. |
The Key Factors of Apoptosis in SIMI
Caspases, or cysteine-aspartic acid proteases, are specialized proteases that possess a cysteine residue at their active site and cleave their substrates at the C-terminal side of aspartic acid residues. The caspase family plays a critical role in inhibiting cell survival pathways and specifically activating pro-apoptotic factors. Caspases are essential in myocardial cell apoptosis. In SIMI, the extrinsic apoptotic pathway is activated through the binding of ligands to death receptors, such as FAS, leading to the recruitment, dimerization, and activation of caspase-8 with the help of adapter proteins like FAS-associated death domain (FADD) and TRADD. Activated caspase-8 then cleaves and activates effector caspases (caspase-3, -6, and -7), directly initiating apoptosis, or it cleaves BID to activate the intrinsic apoptotic pathway and induce effective cell death. On the other hand, the intrinsic mitochondrial apoptosis pathway can be activated by various cytokines, leading to the release of cytochrome c from the mitochondria and the formation of the apoptosome composed of APAF1, cytochrome c, ATP, and caspase-9, which activates caspase-9. Activated caspase-9 subsequently cleaves and activates effector caspases, such as caspase-3, -6, and -7, which then execute apoptosis. Studies have shown that in sepsis, the molecular mechanisms of myocardial injury include apoptosis and increased caspase activity.49 Wang et al found that docosahexaenoic acid (DHA) enhances cell viability by reducing the release of LDH, expression levels of cleaved caspase-3, and caspase-3 activity.50 DHA also decreases the production of ROS and increases superoxide dismutase activity, thus alleviating LPS-induced myocardial cell apoptosis and mitochondrial damage. Similarly, the use of the broad-spectrum caspase inhibitor Z-Val-Ala-Asp (OMe)-FMK (SAD) to inhibit caspase activity significantly restored myocardial function in septic cardiomyopathy mouse models.51 Hayakawa et al demonstrated that using the multi-caspase inhibitor IDN-1965 in transgenic Gα(q) mouse models reduced caspase-3 activity, resulting in improved myocardial function and a marked reduction in mortality.52
The Bcl-2 family is a vital group of proteins involved in regulating mitochondrial apoptosis, primarily consisting of pro-apoptotic and anti-apoptotic proteins, which work in coordination during the apoptotic process. They mediate the signaling pathways through the mitochondrial pathway to determine whether a cell will undergo apoptosis. Among them, pro-apoptotic proteins can be further classified into those with BH1-3 domains and those with only the BH3 domain. Members of the pro-apoptotic group, such as Bak, and anti-apoptotic proteins, such as Bcl-2 and Bcl-xL, are primarily located on the mitochondrial membrane, while other members, such as Bid and Bad, are mainly present in the cytoplasm. In the pathogenesis of SIMI, the pro-apoptotic protein Bax plays a significant role. Under normal conditions, Bax exists in an inactive form in the cytoplasm. Two hypotheses explain the activation of Bax. One hypothesis suggests a direct activation model, where BH3-only proteins are divided into activators and sensitizers. In the absence of apoptotic signals, activators bind to anti-apoptotic proteins, inhibiting the activation of Bax. Upon receiving apoptotic signals, sensitizer proteins bind to the anti-apoptotic proteins, releasing the activators, which directly activate Bax. The other hypothesis proposes an indirect activation model, where BH3-only family members, upon receiving apoptotic signals, inhibit the activity of anti-apoptotic proteins, thereby indirectly activating Bax. Once activated, Bax relocates to the mitochondrial membrane, forming pores across the mitochondrial membrane, leading to a loss of membrane potential, increased membrane permeability, and the release of apoptotic factors. Studies have shown that during sepsis-induced myocardial cell apoptosis, the pro-apoptotic gene Bax mediates the opening of the mitochondrial permeability transition pore (mPTP) on the inner mitochondrial membrane, which inhibits the expression of the anti-apoptotic factor Bcl-2.53 The Bax/Bcl-2 ratio increases, and the myocardial cell apoptosis index progressively increases.53 Additionally, Bax can form homodimers under stress conditions, disrupting the protein hydrolysis cascade and causing the mitochondria to release a large amount of pro-apoptotic factors, ultimately leading to myocardial cell apoptosis. Research has indicated that mitochondrial damage induced by excessive ROS is the primary source of myocardial cell apoptosis. Dehydrocorydaline (Deh) has shown significant anti-inflammatory, anti-apoptotic, and antioxidant effects by inhibiting the TRAF6/NF-κB pathway, reducing levels of IL-1β, IL-6, TNFα, and IFNγ, lowering ROS levels, and upregulating Nrf2/HO-1, SOD, and GSH-PX expression.54 Zhang et al have found that by regulating the activity of extracellular signal-regulated kinase 1/2 (ERK1/2) and c-Jun N-terminal kinase (JNK), inhibiting the expression of Bax while upregulating the expression of Bcl-2, mitochondrial membrane potential is reduced, ATP generation and mtDNA expression are enhanced, thus alleviating oxidative stress and improving mitochondrial damage, providing therapeutic benefits for SIMI.55
Inhibiting the expression of pro-apoptotic proteins and their associated pathways, while promoting the expression of anti-apoptotic proteins and their respective pathways, represents a critical therapeutic approach for SIMI. For example, in SIMI mouse models, an increase in Bcl-2 protein expression, accompanied by a reduction in the expression of Bid, t-Bid, and caspase-9, effectively inhibits myocardial cell apoptosis.42 Li et al demonstrated that Irisin enhances Bcl-2 levels and decreases the expression of Bax and its downstream effector caspase-3 in septic myocardial cells, thereby mitigating sepsis-induced apoptosis and offering therapeutic potential for SIMI.56 Furthermore, Shao et al observed in SIMI mice that Gastrodin decreased the expression of Caspase-1, IL-1β, and Bax in myocardial cells while upregulating Bcl-2 expression, thereby reducing LPS-induced myocardial apoptosis and improving cell viability.57
In summary, during sepsis, the accumulation of ROS and mitochondrial dysfunction, induced by endotoxins and various stress stimuli, leads to an upregulation of pro-apoptotic proteins and a downregulation of anti-apoptotic proteins. Simultaneously, Bax is relocalized to the mitochondrial surface, forming pores in the mitochondrial membrane, which decreases membrane potential and increases membrane permeability. This facilitates the release of pro-apoptotic factors, thereby triggering cell apoptosis. These events contribute to the myocardial damage observed in sepsis. Collectively, these findings provide a robust theoretical foundation for the targeted treatment of myocardial cell apoptosis in SIMI.
Mitophagy in SIMI
Autophagy is a process that involves the sequestration of damaged proteins and organelles within double-membrane vesicles known as autophagosomes, which are subsequently delivered to lysosomes for degradation. It plays a key role in cellular homeostasis and is also implicated in mechanisms of cell death. In sepsis animal models induced by CLP or LPS, activation of autophagy and mitochondrial depletion have been observed in the heart.58–60 The role of autophagy in sepsis and organ damage remains a subject of controversy. In SIMI, autophagy activation has been shown to mitigate myocardial cell damage and aid in the restoration of cardiac function.61,62 Conversely, some studies have found that inhibition of autophagy can improve myocardial damage caused by sepsis.63 The prevailing view in the literature suggests that autophagy is suppressed in SIMI, and its activation holds promise for alleviating SIMI.64 Additionally, in sepsis animal models, a reduction in the co-localization of autophagosomes and lysosomes, coupled with impaired autophagic interaction and decreased degradation, has been observed, potentially leading to ATP depletion and cardiac dysfunction. Interestingly, stimulation with rapamycin has been shown to restore these processes.65
LPS stimulation not only induces myocardial autophagy but also triggers a more selective form of autophagy.58 Mitochondrial autophagy, which involves the removal of damaged mitochondria, may play a crucial role in mitigating cardiac and mitochondrial dysfunction. Mitochondrial autophagy is a selective form of autophagy that acts as a compensatory mechanism in response to the inflammatory response induced by sepsis. This process is triggered by severe stress, increased ROS expression, a higher number of damaged mitochondria, and the involvement of factors such as Bcl-2 family proteins, Parkin, and BNIP3.66 The primary mechanism underlying mitochondrial autophagy involves the activation of the PTEN induced putative kinase 1 (PINK1)/Parkin pathway, with PINK1 enhancing this process by increasing the dynamics of ATP levels.
In the process of mitophagy, cells initiate the clearance of damaged mitochondria through multiple pathways, which include both ubiquitin-dependent and -independent mechanisms. The ubiquitin-dependent pathway is regarded as the primary route for mitophagy. In this pathway, stress-induced stimuli result in the loss of mitochondrial membrane potential and the cleavage of OPA1. PINK1 is phosphorylated on the OMM, where it facilitates the recruitment of Parkin, an E3 ubiquitin ligase.67 PINK1-mediated phosphorylation of mitofusin 2 triggers the activation of Parkin, leading to the ubiquitination of various mitochondrial proteins, including mitofusins and voltage-dependent anion channels (VDAC).68 This process is followed by the recruitment of mitophagy receptors through Parkin’s ubiquitin-binding domain. The ubiquitin chains, generated by Parkin, are phosphorylated by PINK1, establishing a positive feedback loop that anchors Parkin to the mitochondrial membrane and further drives mitophagy.69 The rupture of the outer mitochondrial membrane subsequently releases IMM proteins, such as PHB2, which are recognized by autophagy receptors.70,71 These receptors, which possess an LC3-interaction region (LIR), activate autophagic machinery, facilitating the degradation and removal of dysfunctional mitochondria.70,71 Thus, PINK1 serves as a critical biological sensor for mitochondrial health, orchestrating the activation of Parkin to promote mitochondrial turnover (Figure 3).
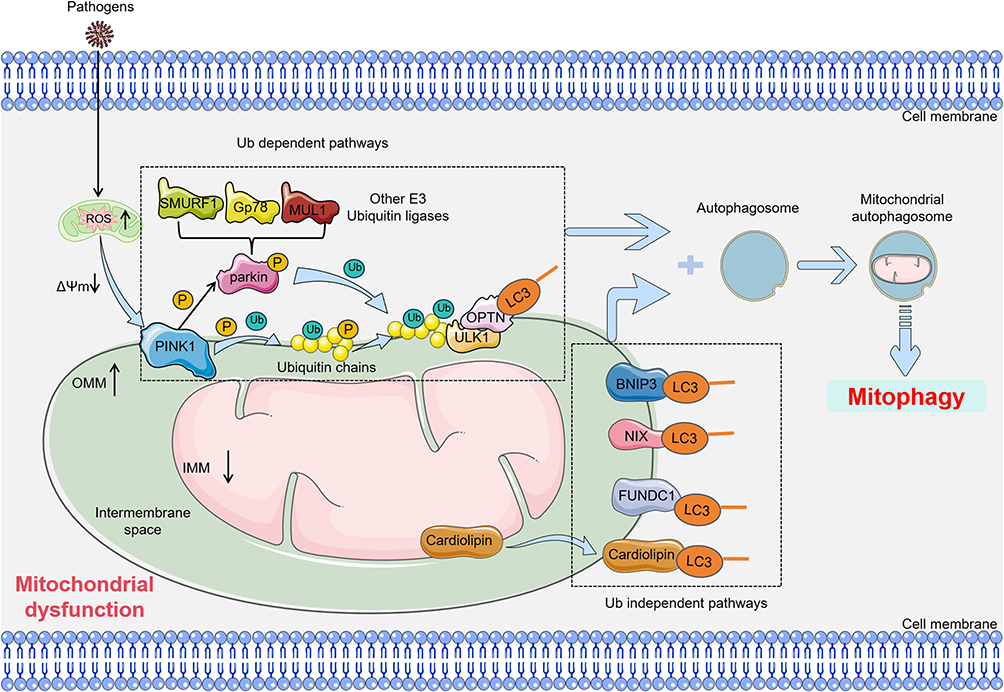 |
Figure 3 Main pathway of mitophagy in SIMI. Mitophagy primarily consists of two pathways: the ubiquitin-dependent pathway and the ubiquitin-independent pathway. In the ubiquitin-dependent pathway, stress-induced mitochondrial membrane potential depolarization leads to the accumulation of PINK1 on the outer mitochondrial membrane (OMM), where it activates the E3 ligase activity of Parkin. Subsequently, Parkin ubiquitinates outer mitochondrial membrane proteins, and the ubiquitin chains are further phosphorylated by PINK1. The phosphorylated ubiquitin-modified outer membrane proteins are then recognized by autophagy adaptors, such as OPTN, which initiate the mitophagy process. Additionally, PINK1 can directly recruit autophagy adaptors like OPTN to the mitochondria via ubiquitin phosphorylation, thereby promoting mitophagy. In the ubiquitin-independent pathway, proteins located in the outer mitochondrial membrane, such as BNIP3, FUNDC1, and Bcl2-L-13, as well as cardiolipin in the IMM, can bind to LC3 and subsequently initiate mitophagy. Blue and black colored arrows present for direct processes. Black colored dotted squared box present for ub dependent pathways or ub independent pathways. Abbreviations: OPTN, Optineurin; ULK1, UNC-51-like kinase 1; LC3, light chain 3; BNIP3, bcl2 interacting protein 3; NIX, NIP3-like protein X; FUNDC1, FUN14 domain-containing protein 1; SMURF1, Smad ubiquitin regulatory factor 1; MUL1, mitochondrial ubiquitin ligase 1; Gp78, glycoprotein 78. |
Moreover, studies have shown that melatonin treatment can enhance Parkin-dependent mitophagy by upregulating Beclin1 levels, thereby mitigating the pathological effects of cardiomyopathy and promoting the expression of PINK1 and Parkin.72 In addition to Parkin, mitochondrial ubiquitin ligase 1 (MUL1) also plays a role as a mitophagy receptor in the ubiquitin pathway. Research indicates that MUL1 can activate NF-κB and induce the ubiquitination and degradation of mitochondrial fusion proteins, even in the absence of Parkin.73
In contrast, the ubiquitin-independent pathway involves proteins located in the outer mitochondrial membrane, such as Bcl2 interacting protein 3(BNIP3), FUN14 domain-containing protein 1 (FUNDC1), and Bcl2-L-13, as well as cardiolipin in the IMM, which can directly interact with LC3 and bind to autophagosomes under conditions of extreme stress.74 For example, FUNDC1 facilitates Parkin-independent mitophagy in response to hypoxia by interacting with LC3, and inhibition of FUNDC1-mediated mitophagy increases mitochondrial apoptosis under pathological conditions.75 Both BNIP3 and NIP3-like protein X(NIX) integrate into the outer mitochondrial membrane through their glycine-zipper transmembrane structures and engage with LC3, thus indirectly promoting autophagy by disrupting the Bcl-2–Beclin1 interaction.76 Additionally, Bcl2-L-13 recruits LC3 to the mitochondrial surface via its LIR domain, driving the formation of mitophagosomes and promoting mitophagy.77 Cardiolipin, a direct mediator of mitophagy, translocates to the outer mitochondrial membrane upon membrane rupture and binds to the N-terminal region of LC3.78 Under conditions of severe stress, this interaction triggers the release of apoptotic factors, such as cytochrome c.78
The regulation of mitophagy has emerged as a promising therapeutic approach for SIMI. For example, in a murine model of sepsis induced by LPS, α-ketoglutarate (AKG) has been demonstrated to facilitate the clearance of damaged mitochondria through the enhancement of mitophagy and mitochondrial fission, ultimately alleviating the cardiac dysfunction associated with septic cardiomyopathy.79 In the context of SIMI, transmembrane Bax inhibitor motif containing 1 (TMBIM1) has been identified as a key mediator that interacts with Parkin, promoting its translocation from the cytosol to damaged mitochondria.80 This interaction activates mitophagy, enhancing the removal of dysfunctional mitochondria and mitigating myocardial injury.80 Additionally, in a rat model of sepsis induced by CLP, the traditional herbal medicine Po-Ge-Jiu-Xin decoction has been shown to activate the PINK1/Parkin pathway, thus facilitating mitophagy, preserving mitochondrial quality, and improving cardiac function in SIMI.81 Whereas, it is crucial to note that excessive activation of mitophagy, aimed at improving myocardial injury, should be approached with caution. Overstimulation of mitophagy may lead to residual dysfunctional mitochondria and insufficient ATP production, potentially exacerbating heart failure. For instance, 4-hydroxy-trans-2-nonenal has been reported to inhibit the expression of PINK1/Parkin by activating aldehyde dehydrogenase 2 (ALDH2), thereby preventing excessive mitophagy, reducing inflammation, and mitigating myocardial dysfunction in LPS-induced injury.82
Ferroptosis in SIMI
Ferroptosis is a form of iron-dependent, regulated cell death.83 Under normal conditions, an oxidative system composed of iron ions, the Fenton reaction, and ROS counteracts the antioxidant defense system, which includes the cystine/glutamate antiporter (system Xc−), glutathione peroxidase 4 (GPX4), and glutathione (GSH), as well as other recently identified pathways. This balance helps maintain cellular and organismal homeostasis. Research indicates that ferroptosis plays a critical role in the pathogenesis of various conditions such as atherosclerosis, myocardial ischemia-reperfusion injury, SIMI, drug-induced heart failure, arrhythmias, and diabetic cardiomyopathy.84,85
As an iron-dependent form of programmed cell death, ferroptosis typically occurs in environments characterized by metabolic dysregulation and oxidative stress.86 For instance, during the bacterial infection phase in severe pancreatitis, ferroptosis in intestinal epithelial cells can disrupt the intestinal barrier, facilitating the entry of harmful gut bacteria and toxins into the bloodstream, thereby exacerbating systemic inflammation and organ damage in sepsis.87 In the progression of sepsis, mitochondrial dysfunction in cardiomyocytes is considered a crucial factor in the release of inflammatory mediators and oxidative stress, representing a key mechanism of SIMI. Ferroptosis is closely associated with dysregulated inflammatory responses, and in LPS-induced myocardial injury models, morphological changes in mitochondria align with characteristic features of ferroptosis, suggesting a significant link between mitochondrial dysfunction and ferroptosis.88 Moreover, mitochondria, as the energy factories of cells, are essential for normal cellular metabolism, and mitochondrial dysfunction often serves as a catalyst for various diseases and widespread cell death.89,90 Thus, targeting ferroptosis may provide an effective strategy to maintain mitochondrial homeostasis in SIMI. By modulating ferroptosis, it may be possible to alleviate cellular damage and inflammatory responses in SIMI, thereby mitigating myocardial injury and improving immune function. The following sections will discuss the classical pathways of ferroptosis and the key molecules involved in its role during the development of SIMI.
The Classical Pathway of Ferroptosis in SIMI
Ferroptosis can be initiated through both exogenous (transport protein-dependent) and endogenous (enzyme-regulated) pathways. In the exogenous pathway, cellular stressors such as infection, inflammation, or oxidative stress can inhibit membrane transport proteins, particularly the cystine/glutamate antiporter (System Xc-). System Xc- is a vital component of the cellular antioxidant defense system, consisting of two subunits: solute carrier family 7 member 11(SLC7A11), responsible for the high-affinity transport of cystine and glutamate, and solute carrier family3 member 2 (SLC3A2), which functions as a chaperone. This system exchanges extracellular cystine (Cys2) for intracellular glutamate in a 1:1 ratio. Upon internalization, cystine is converted into GSH by the action of glutamate cysteine ligase (GCL) and glutathione synthetase (GSS). GSH plays a critical role as a reductive cofactor for GPX4, an enzyme involved in protecting cellular membranes from oxidative damage. Inhibition of System Xc- disrupts cystine uptake, limiting GSH synthesis and thereby impairing GPX4 activity.91,92 This results in a weakened cellular antioxidant defense, increasing susceptibility to ferroptosis91,92. Concurrently, transferrin (including serum transferrin or lactoferrin) facilitates iron uptake through the transferrin receptor (TFRC), and ferritin subunits (FTH1/FTL) contribute to iron accumulation via autophagic degradation, both facilitating ferroptosis.93,94
The endogenous pathway of ferroptosis is primarily regulated by the inhibition of antioxidant enzymes, such as GPX4. The accumulation of lipid peroxides, a hallmark of ferroptosis, is catalyzed by GPX4, which reduces lipid hydroperoxides (L-OOH) to their corresponding alcohols (L-OH).40,93 Direct inhibition of GPX4 activity, for instance by the ferroptosis inducer RSL3, suppresses its antioxidant function, leading to an increase in reactive oxygen species ROS and the accumulation of toxic lipid peroxides.95 Alternatively, indirect inhibition occurs through the depletion of selenium-cysteine, a critical amino acid in GPX4’s active site. This reduction can result from downregulation of the mevalonate pathway, which affects the maturation of selenium-cysteine tRNA, ultimately reducing GPX4 expression and activity, thereby triggering ferroptosis.96 In addition to GPX4 regulation, other enzymes are involved in ferroptosis. Long-chain acyl-CoA synthetase 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are crucial for the incorporation of polyunsaturated fatty acids (PUFAs) into phospholipids, generating PUFA-containing phospholipids (PUFA-PLs). These PUFA-PLs are prone to oxidation by free radicals, a process facilitated by lipoxygenases (ALOXs). The oxidation of PUFA-PLs leads to the disruption of the lipid bilayer, impairing cellular membrane integrity and further promoting ferroptosis97 (Figure 4).
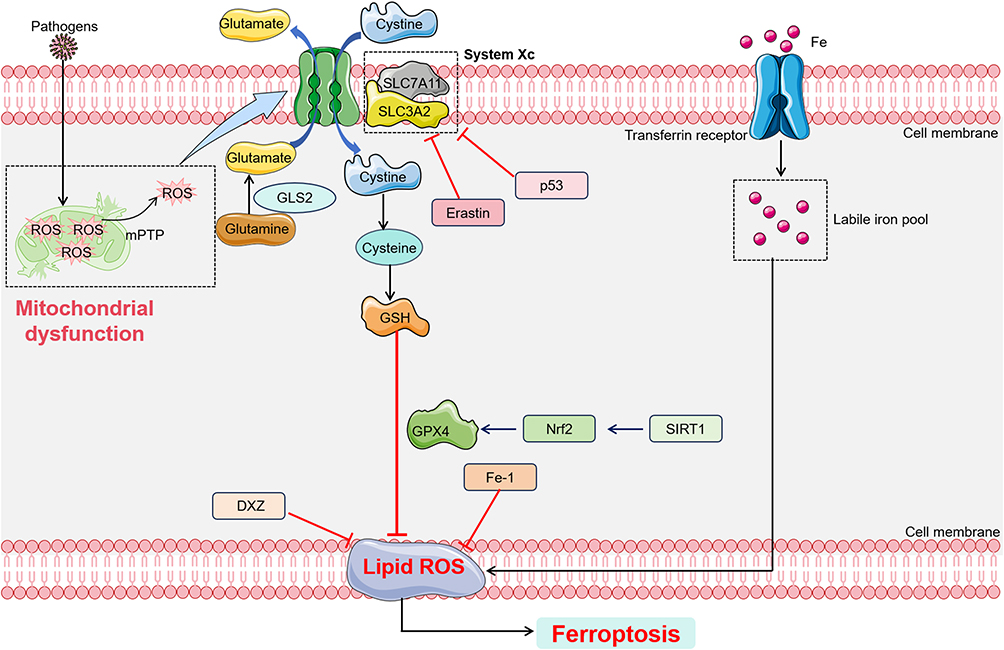 |
Figure 4 Main pathway of feroptosis in SIMI. Ferroptosis occurs through both exogenous (transport protein-dependent) and endogenous (enzyme-regulated) pathways. In the exogenous pathway, cysteine is transported into the cell via the System Xc- transporter, where it participates in the synthesis of GSH. Inhibition of this pathway impedes GSH synthesis, reduces the activity of the membrane lipid repair enzyme GPX4, and decreases the cell’s antioxidant capacity, thereby promoting ferroptosis. Additionally, iron enters the cell through transferrin, elevating intracellular iron levels and triggering ferroptosis. The endogenous/enzyme-regulated pathway is primarily mediated by intracellular antioxidant enzymes, such as GPX4. Black colored arrows present for direct processes. Black colored dotted squared box present for mitochondrial dysfunction process or labile iron pool. Abbreviations: GSH, glutathione; GPX4, glutathione peroxidase 4; GLS2, glutaminase2; DXZ, dexrazoxane; SIRT1, sirtuin1. |
The Key Factors of Ferroptosis in SIMI
GPX is a member of a protein superfamily, with its catalytic site containing cysteine as a redox-active residue, facilitating redox reactions.98,99 Among the eight members of human GPX, GPX4 is considered to play a critical role in ferroptosis. GPX4 plays a pivotal regulatory role during ferroptosis, and its unique physiological function is to convert lipid hydroperoxides into non-toxic lipid alcohols, thus inhibiting lipid peroxidation. When GPX4 synthesis is insufficient or its activity is inhibited, the accumulation of peroxides within cells leads to ferroptosis. GPX4 is often a key regulatory node in ferroptosis triggered by various inducers.100 Recent studies have also found the generation of ROS during sepsis, which triggers ferroptosis.101 In a mouse model of LPS-induced sepsis, LPS upregulates the expression of ferroptosis markers, such as prostaglandin-endoperoxide synthase 2 (PTGS2), malondialdehyde (MDA), and lipid ROS, leading to ferroptosis and mitochondrial damage, while iron chelators like Fe-1 and dexrazoxane (DXZ) alleviate this damage.102 Similarly, overexpression of GPX4 can attenuate palmitic acid-induced ferroptosis in myocardial cells, whereas low expression of GPX4 significantly reduces its protective effect against ferroptosis.103 GPX4 is crucial in regulating ferroptosis in the pathophysiology of septic myocardial injury. For instance, Oligo-peptide I-C-F-6 activates the Nrf2/HO-1/GPX4 pathway, alleviating myocardial oxidative damage and inhibiting ferroptosis in septic mice.104 Dapagliflozin, by inhibiting the translation of key proteins involved in ferroptosis such as GPX4, FTH1, and SLC7A11, suppresses ferroptosis and improves SIMI.105 Therefore, targeting GPX4 appears to be a promising strategy for improving SIMI.
Another key protein involved in ferroptosis is the cystine/glutamate antiporter SLC7A11, which primarily functions in the reverse transport of extracellular cystine into cells, contributing to the formation of GSH and antioxidant defense.106 Modulating the activity of SLC7A11 can target ferroptosis to improve sepsis-induced myocardial damage. METTL3-mediated m6A methylation of SLC7A11 enhances its mRNA methylation level, leading to mRNA decay and consequently promoting ferroptosis in SIMI.107 Erastin, a specific inducer of ferroptosis, reduces intracellular GSH levels by inhibiting System Xc-, thus triggering ferroptosis.40 In animal and cell models induced by LPS, Cao et al found that sodium hydrosulfide (NaHS) inhibits the phosphorylation of Beclin 1, an endogenous binding protein of SLC7A11, increases the expression levels of SLC7A11 and GPX4, and alleviates iron metabolism disorders and oxidative stress, thereby inhibiting ferroptosis and improving SIMI.108 Additionally, the traditional Chinese medicine YiQiFuMai injection targets the SLC7A11/GPX4 axis to suppress ferroptosis and improve SIMI.109 As a primary intracellular antioxidant and the source of GSH, SLC7A11 can inhibit lipid peroxidation. Similarly, in an LPS-induced sepsis mouse model, Cyclovirobuxine D upregulates the expression of SLC7A11, inhibits ferroptosis, and ameliorates sepsis-induced myocardial damage.110
In addition to GPX4 and SLC7A11, p53 plays an important role in ferroptosis during SIMI. Previous studies have confirmed that p53 specifically inhibits the System Xc- system by downregulating the expression of SLC7A11, thereby inducing ferroptosis.111 Moreover, mitochondrial dysfunction in a p53-dependent manner is observed in the pathogenesis of SIMI.112 Lin et al discovered that quercetin alleviates SIMI by upregulating GPX4 and ferritin levels and downregulating iron and prostaglandin-endoperoxide synthase 2 levels via the sirtuin 1 (SIRT1)/p53/SLC7A11 signaling pathway, thereby ameliorating ferroptosis.113 Other molecules also play important roles in ferroptosis. Heme oxygenase-1 (HO-1), a major source of intracellular iron, has been shown by Kwon et al to induce lipid peroxidation and consequently trigger ferroptosis.114
In summary, during infection, mitochondrial dysfunction, excessive ROS production, and dysregulated inflammatory responses inhibit key ferroptosis proteins, including GPX4 and SLC7A11, leading to iron overload and ferroptosis in myocardial cells, which contributes to SIMI. Thus, targeting ferroptosis and mitochondrial dysfunction represents a crucial strategy for improving SIMI.
Mitochondrial Dysfunction Induces Calcium Homeostasis Imbalance in SIMI
The precise regulation of intracellular calcium (Ca²⁺) levels is essential for cell survival, functional maintenance, and mitochondrial dynamics.115,116 Intracellular Ca²⁺ levels are primarily controlled through two major pathways: one is Ca²⁺ influx via ion channels across the plasma membrane, and the other is the release of Ca²⁺ from intracellular stores such as the ER. The ER and mitochondria are the principal intracellular Ca²⁺ storage reservoirs. Mitochondria play a particularly crucial role in calcium homeostasis, not only serving as the primary ATP production factories within the cell but also being intimately linked to mitochondrial dynamics, function, and metabolism. Mitochondria are also involved in the regulation of microdomains within the cell, which govern key cellular processes.117,118
Mitochondrial involvement in calcium signaling is regulated by two mechanisms.119 On the one hand, mitochondria possess a calcium buffering function, which allows them to handle intracellular Ca²⁺ levels ranging from 50 to 500 nM in various normal cell types.120,121 Through this buffering capacity, mitochondria dynamically manage transient changes in calcium concentrations and buffer calcium overload associated with pathological states.120 On the other hand, mitochondria are in close contact with the ER and plasma membrane calcium channels, forming microdomains that promote synchronized increases in mitochondrial Ca²⁺ concentrations and cytosolic Ca²⁺ signaling.122 Within the ER, inositol 1,4,5-trisphosphate (IP₃)-activated gated channels expose mitochondria to much higher Ca²⁺ concentrations than the cytosol, thereby increasing mitochondrial calcium levels.123 Moreover, extracellular Ca²⁺ entering through plasma membrane channels can also enter mitochondria, further linking cellular activation to mitochondrial function.120 The rapid diffusion of Ca²⁺ helps prevent calcium overload in organelles.
Furthermore, in cardiac mitochondria, calcium ion transport processes are facilitated by microdomains between the sarcoplasmic reticulum (SR) and mitochondria, which bring the calcium release and uptake sites closer together and are maintained by connecting proteins such as mitochondrial fusion proteins. This system ensures that mitochondrial calcium ions can synchronize with ATP production required for excitation-contraction coupling, thus regulating ATP generation.124 Additionally, calcium ion delivery to mitochondria is crucial for maintaining mitochondrial antioxidant capacity and declining ROS generation caused by increased ATP synthesis.125 Mitochondrial calcium overload is considered a major trigger for the opening of the mitochondrial permeability transition pore (mPTP), which, under oxidative stress, may lead to pathological organ damage.126 Therefore, dysfunction of the calcium transport system plays a significant role in cardiac diseases, such as heart failure.127
In cardiomyocytes, calcium homeostasis involves not only cytosolic Ca²⁺ but also mitochondrial Ca²⁺. Calcium channels on the IMM include the mitochondrial calcium uniporter (MCU), mitochondrial ryanodine receptors (mRyR), mPTP, mitochondrial sodium/calcium exchanger (mNCX), and hydrogen/calcium exchanger (mHCX).128,129 Among these, MCU operates through the mitochondrial membrane potential, dependent on matrix and extracellular calcium concentrations, and is closely linked to oxidative phosphorylation processes and the activity of dehydrogenases in the tricarboxylic acid (TCA) cycle, thus providing energy for ATP synthesis.130 The voltage-dependent anion channel (VDAC) is part of the mPTP, responsible for transporting calcium and metabolic products across the outer mitochondrial membrane.131 Notably, about 20% of the mitochondrial surface is adjacent to the ER, forming a high-calcium microenvironment known as mitochondrial-associated membranes (MAMs).132 During calcium-induced calcium release (CICR) in the autonomic nervous system, the ER releases large amounts of calcium through the RyR, which enters mitochondria via VDAC. Subsequently, the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) reabsorbs calcium to prevent excessive cytosolic calcium accumulation, maintaining calcium homeostasis. Dysfunction of these calcium channels, particularly inhibition of L-type calcium channels (LTCC), SERCA inhibition, increased RyR calcium leakage, and decreased myofilament calcium sensitivity, is closely associated with the occurrence and progression of SIMI.133 For instance, during SIMI, reduced sensitivity of L-type calcium channels leads to diminished RyR activity, impairing the influx of extracellular calcium into cardiomyocytes via L-type calcium channels, affecting calcium uptake and further hindering calcium binding to troponin, thereby disrupting myocardial contraction activation.134
In sepsis, due to calcium leakage from the SR and decreased calcium uptake by organelles, mitochondrial calcium concentration increases.135 Moreover, mitochondrial dysfunction activates NADPH oxidase 2 (NOX2), leading to abnormal calcium handling and damaging myocardial contraction. Targeting NOX2 and restoring calcium homeostasis is therefore considered a potential therapeutic strategy for improving SIMI. In murine models of sepsis induced by LPS and CLP, the NOX2 inhibitor apocynin alleviated oxidative stress in cardiomyocytes, maintained intracellular calcium homeostasis and mitochondrial function, and mitigated the contractile dysfunction induced by sepsis.136 Targeting mitochondrial calcium efflux or calcium uptake can also alleviate SIMI. Mitochondrial calcium efflux is regulated by the PINK1-PKA-NCLX axis. In CLP-induced sepsis mouse models, reduced expression of PINK1 in cardiomyocytes disrupts the PINK1-PKA-NCLX axis, leading to disordered mitochondrial calcium efflux, mitochondrial calcium overload, and inhibition of mitochondrial autophagy, preventing the clearance of damaged mitochondria, thereby contributing to SIMI.137 Jiang et al found that LPS increased the formation of MAMs and intracellular calcium levels, enhanced expression of Cav1.2 and RyR2, reduced mitochondrial membrane potential and intracellular ATP levels, promoted mitochondrial fragmentation, expression of mitophagy proteins, and ROS generation in H9c2 cells, while Bazedoxifene and Stattic reversed this phenomenon, alleviating SIMI.138 Similarly, after LPS administration, rodents exhibited increased mitochondrial Ca²⁺ content or mitochondrial Ca²⁺ overload, while preventing mitochondrial Ca²⁺ overload helped improve mitochondrial and myocardial dysfunction.136,139 Additionally, reduced mitochondrial Ca²⁺ uptake was associated with increased survival rates in septic rats.140 Therefore, the role of mitochondria in calcium homeostasis may serve as a promising target for management of SIMI.
Although existing research has demonstrated that disturbances in mitochondrial calcium efflux and uptake play a significant role in the exacerbation of SIMI, and that modulating this process holds potential for mitigating SIMI, the precise molecular mechanisms involved remain poorly understood. Therefore, extensive future research is required to comprehensively investigate and clarify these underlying mechanisms.
Mitochondrial Dysfunction Induces Metabolic Dysregulation in SIMI
Sepsis is a systemic inflammatory response triggered by infection, which leads to the dysfunction of multiple organs, particularly the heart. Mitochondria are crucial in cellular energy metabolism, predominantly producing ATP through oxidative phosphorylation (OXPHOS), which is essential for cell survival (Figure 5). However, during sepsis, alterations in cellular energy demands occur due to metabolic reprogramming, which subsequently affects the metabolic processes of immune cells and cardiomyocytes, ultimately leading to immune dysregulation and myocardial injury.
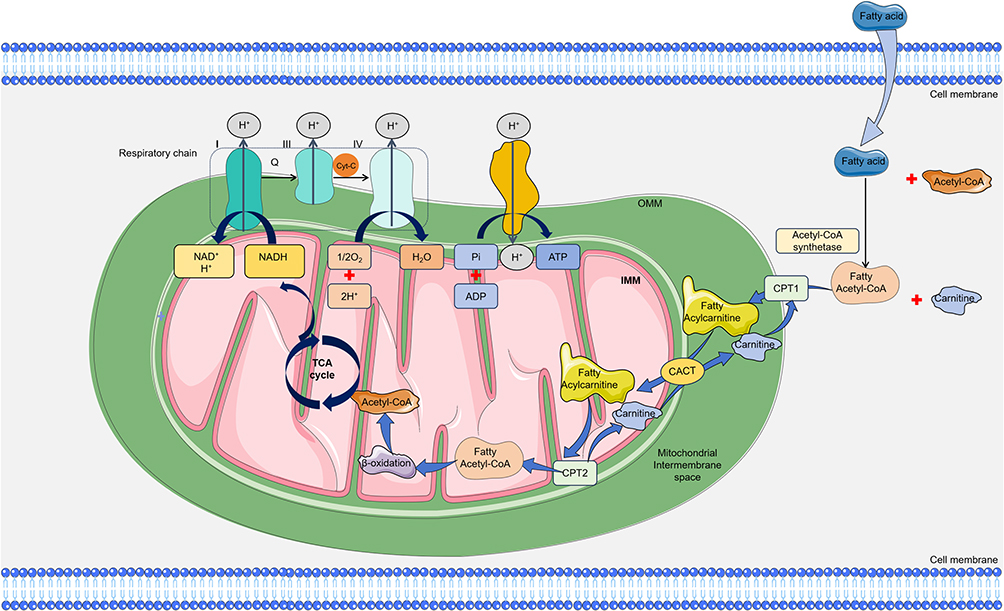 |
Figure 5 The Normal Metabolism in Myocardial Cells. Under normal conditions, myocardial cells uptake long-chain fatty acids via transport proteins, facilitating their oxidation within the mitochondria to produce acetyl-CoA and NADH. These metabolites are further involved in the tricarboxylic acid (TCA) cycle, subsequently entering the electron transport chain to generate ATP. Black or blue colored arrows present for direct processes. Black colored dotted squared box present for respiratory chain. Abbreviations: CPT1, carnitine palmitoyl transferase 1; CACT, carnitine-acylcarnitine translocase; CPT2, carnitine palmitoyl transferase 2; NADH, Nicotinamide adenine dinucleotide; TCA cycle, tricarboxylic acid cycle. |
Metabolic reprogramming refers to a shift in cellular metabolic pathways that occurs in response to various environmental challenges, enabling cells to adapt their energy production mechanisms to meet the stresses or challenges posed by external factors. In the case of sepsis, almost all types of cells undergo metabolic reprogramming. Notably, in immune cells and certain organ cells, glycolysis replaces oxidative phosphorylation as the primary energy source, a phenomenon referred to as the “Warburg effect”.141–143 This metabolic adaptation is critical for the function of immune cells during sepsis, and a similar reprogramming from oxidative phosphorylation to glycolysis has also been observed in cardiomyocytes144 (Figure 6).
 |
Figure 6 The cell shifts its metabolism from FAO-dependent oxidative phosphorylation (OXPHOS) to a predominantly glycolytic pathway in SIMI. During sepsis, cells undergo a metabolic shift from FAO-driven OXPHOS to glycolysis. Glycolysis generates ATP by producing pyruvate, which is subsequently converted to lactate, thereby fulfilling the energy demands of the immune response. Black or blue colored arrows present for direct processes. Black colored dotted squared box present for respiratory chain or aerobic glycolysis process. Abbreviations: GLUT, glucose transporter; HK2, hexokinase2; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; PKM2, Pyruvate kinase M2; LDH, lactate dehydrogenase; MCT, medium chain triglyceride. |
Research on metabolic reprogramming in SIMI has primarily focused on immune cells (such as macrophages) and cardiomyocytes.144,145 Under normal conditions, cells uptake long-chain fatty acids through transport proteins, promoting their oxidation in mitochondria to generate acetyl-CoA, FADH2, and NADH. These metabolites participate in the tricarboxylic acid (TCA) cycle and enter the electron transport chain to produce ATP. However, during sepsis, cells undergo a metabolic shift from fatty acid oxidation (FAO)-driven OXPHOS to glycolysis. Glycolysis generates ATP by producing pyruvate, which is then converted to lactate to meet the energy demands of the immune response. Studies suggest that moderate glycolysis plays a protective role in sepsis by activating immune responses and regulating inflammation, but excessive glycolysis can lead to immune suppression, further exacerbating myocardial injury induced by sepsis.
Moderate Glycolysis Activates the Immune System, Inducing Inflammatory Responses in SIMI
PRRs are pivotal in the innate immune response by detecting PAMPs and DAMPs. In the early phases of sepsis, the activation of PRRs initiates the immune response, triggering the onset of inflammation. During sepsis and SIRS, there is a metabolic shift in immune cells from oxidative phosphorylation to glycolysis. This metabolic reprogramming supports the rapid activation and proliferation of immune cells.146,147 Specifically, glycolysis leads to increased glucose uptake and enhances metabolic pathways, ensuring immune cells acquire sufficient energy to address inflammatory challenges.146,148 This reprogramming ensures a rapid supply of ATP, but the resulting metabolic intermediates also serve as essential nutrients for immune cell proliferation and biosynthesis. Studies have highlighted that the enhanced expression of GLUT1 facilitates glucose uptake, thereby promoting glycolysis and further supporting immune cell functions.149,150 Inhibition of GLUT1 expression significantly reduces glucose uptake and glycolytic flux, resulting in a marked decrease in the viability and functional capacity of effector T cells (Teff).149,151 The increased glucose uptake and glycolytic flux, alongside the high rate of ATP production via glycolysis, ensure that ATP derived from glycolysis meets the energy demands of activated immune cells, even in the presence of impaired OXPHOS. Metabolites such as glucose-6-phosphate (G-6-P), dihydroxyacetone phosphate (DHAP), and glyceraldehyde 3-phosphate (G-3-P) generated through glycolysis provide sufficient energy and biosynthetic precursors, thereby facilitating immune activation and enabling more efficient and rapid immune responses.
Consequently, limiting glycolysis may attenuate organ damage associated with the “cytokine storm” in the early stages of sepsis.152,153 Research has identified mTOR as a key regulator of cellular metabolism. Inhibition of mTOR through rapamycin or its derivatives activates downstream targets such as HIF-1α and GLUT1, thereby promoting glucose uptake and enhancing glycolysis.154,155 Furthermore, rapamycin suppresses LPS-induced pro-IL-1β synthesis and the subsequent ATP-driven release of IL-1β.156 HIF-1α, as a central regulator of immune cell metabolism during sepsis, drives the expression of GLUT1 and LDH, stimulating glycolytic metabolism in inflammatory cells, enhancing pro-inflammatory cytokine secretion, and triggering the cytokine storm in the early stages of sepsis. Thus, HIF-1α represents a promising therapeutic target for sepsis. SOCS1 serves as a negative regulator of the metabolic reprogramming during sepsis by inhibiting HIF-1α-induced hexokinase 1 (HK1) expression in macrophages.157 Additionally, lidocaine effectively reduces the secretion of LPS-induced inflammatory cytokines by inhibiting the HIF-1α-mediated glycolytic pathway, exerting anti-inflammatory effects.158 miR-223, by downregulating HIF-1α and modulating the glycolytic pathway, facilitates M2 macrophage polarization, thereby inducing an anti-inflammatory state.159
Macrophages play a critical role in the immune response during sepsis and SIMI, particularly in their polarization into pro-inflammatory (M1) and immune-suppressive (M2) phenotypes.160 M1 macrophages predominantly contribute to the inflammatory response through glycolysis and lipid biosynthesis. These cells are induced by LPS, TLR ligands, or interferon-γ (IFN-γ). Lipid biosynthesis is crucial for membrane remodeling and the production of inflammatory mediators in M1 macrophages.161 And it is transcriptionally regulated by sterol regulatory element-binding proteins (SREBPs). Notably, SREBP-1a is highly expressed in macrophages, regulating lipid biosynthesis and promoting M1 polarization.162 Fatty acid synthase (FAS), a key enzyme in fatty acid biosynthesis, is integral to the inflammatory responses in M1 macrophages. Wei et al have shown that FAS deficiency in macrophages prevents the recruitment of adipose tissue macrophages and inflammation in mice.163
Targeting glycolytic pathways can also promote M2 macrophage polarization, presenting a potential therapeutic strategy for sepsis.159 For example, methylsulfonylmethane promotes the expression of Arg1 via the lactate H3K18la pathway, inducing M2 macrophage polarization and offering protective effects against methicillin-resistant Staphylococcus aureus infections.164 Additionally, Sema7A induces metabolic reprogramming in macrophages through the activation of mTOR and AKT2 signaling pathways. This reprogramming is characterized by reduced fatty acid oxidation (FAO) and oxidative phosphorylation, enhanced glycolysis and the pentose phosphate pathway (PPP), and impaired Krebs cycle activity, ultimately favoring M2 macrophage polarization.165 Furthermore, exosomes derived from human bone marrow mesenchymal stem cells have been shown to downregulate glycolysis in sepsis-induced macrophages and promote M2 polarization.166 In summary, metabolic reprogramming during the early stages of sepsis enhances glycolysis, facilitating appropriate inflammatory responses that provide organ protection.
Excessive Glycolysis Can Induce Persistent Low-Grade Inflammation and Immune Paralysis in SIMI
Sepsis, particularly in its later stages, exhibits a complex shift in metabolic pathways that significantly affects the immune response. While glycolysis plays a crucial role in the early stages of sepsis by supporting immune cell activation, sustained glycolytic activity in the later phases can contribute to immune suppression. This immune dysregulation manifests as a reduced expression of genes encoding pro-inflammatory cytokines (such as TNF-α and IL-6) and chemokines that recruit T cells (such as CCL2), while the expression of anti-inflammatory cytokines, such as IL-4 and IL-10, is increased.167,168 Consequently, the host becomes less capable of mounting an effective response to secondary infections. This immune suppression is associated with a metabolic shift from glucose metabolism to fatty acid oxidation (FAO), where increased FAO facilitates the transition of immune cells from a pro-inflammatory to an anti-inflammatory phenotype. For instance, PPAR-γ, by promoting β-oxidation of fatty acids, suppresses the pro-inflammatory response of macrophages, thereby contributing to immune suppression.169 Sirtuins, key regulators of metabolism and energy sensing, further influence immune responses by modulating the transition between glycolysis and FAO. SIRT1 and SIRT6, through the SIRT1-RELB-SIRT6 axis, regulate metabolic reprogramming that supports the metabolic shift from glycolysis to FAO. Liu et al demonstrated that SIRT6 inhibits HIF-1α-dependent glycolysis, while SIRT1 enhances PGC-1-dependent fatty acid flux and oxidation.170 This transition from glucose to fatty acid metabolism represents a critical aspect of metabolic reprogramming associated with the early and late stages of sepsis and SIMI.170
In addition to its role in promoting an anti-inflammatory immune phenotype through FAO, lactate, produced during glycolysis, acts as an important signaling molecule in sepsis and organ injury.171,172 Lactate interacts with receptors such as GPR81 and GPR132, contributing to the pathophysiology of sepsis and organ damage.173–175 It has been shown that lactate inhibits TLR/NF-κB signaling and the production of pro-inflammatory cytokines by macrophages.176 Zhang et al found that elevated intracellular lactate levels inhibit RLR-mediated IFN activation in M1 macrophages.177 Liu et al demonstrated that increased lactate levels in macrophages promote an M2 phenotype through HIF-2α, inducing the expression of M2-associated genes such as vascular endothelial growth factor(VEGF), Mannose receptor C- type 1(MRC1), arginase 1(ARG1), and Retnla.178 Furthermore, lactate, when transported out of the cell via the Monocarboxylate Transporter 4(MCT4), can influence immune cell migration and cytokine production, inducing “migration cessation” signals mediated by lactate transporters SLC5A12 (in CD4+ T cells) and SLC16A1 (in CD8+ T cells).179 Thus, excessive lactate production exacerbates immune suppression and contributes to both sepsis progression and myocardial damage.
In summary, during sepsis and associated myocardial injury, metabolic reprogramming profoundly impacts the host’s immune response and organ function. While moderate metabolic reprogramming can effectively activate the immune system and protect organs from damage, excessive glycolysis may lead to immune paralysis and the onset of secondary infections. Therefore, targeting immune cell metabolism and limiting excessive glycolysis could represent promising therapeutic strategies in the treatment of sepsis.
Targeted Therapy of SIMI Focusing on Mitochondrial Dysfunction
The previous discussion comprehensively examined the role of mitochondrial dysfunction in Sepsis-induced Myocardial Injury (SIMI), delving into its contributions through various mechanistic pathways, including dysregulated inflammation and immune responses, disturbances in calcium homeostasis, and metabolic reprogramming. As the central organelle responsible for energy production and cellular homeostasis, mitochondria play a pivotal role in regulating these processes, and their dysfunction in the context of SIMI is emerging as a critical factor driving disease progression. Consequently, targeting mitochondrial dysfunction has been identified as a promising therapeutic strategy for mitigating SIMI, with several compounds demonstrating potential in reversing the deleterious effects of sepsis on myocardial tissue (Table 1).
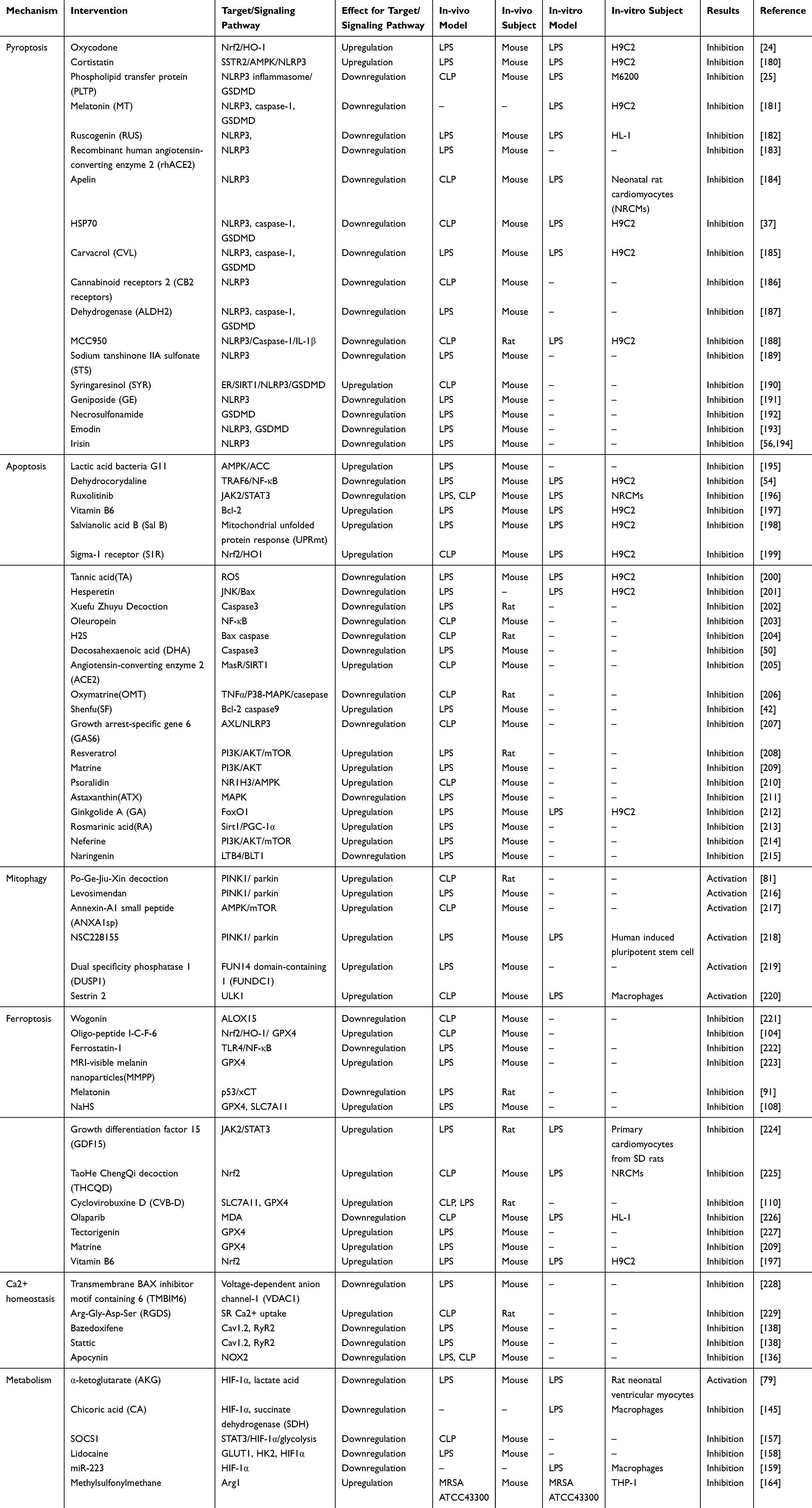 |
Table 1 Therapeutic Strategies Based on RCD, Calcium Homeostasis Imbalance and Metabolic Reprogramming Associated with Mitochondrial Dysfunction in SIMI |
For example, the selective estrogen receptor modulator Bazedoxifene and the Src kinase inhibitor Stattic have been shown to effectively counteract LPS-induced myocardial inflammation. These compounds not only improve mitochondrial membrane potential and restore intracellular ATP levels, but also reduce mitochondrial fragmentation, the expression of mitophagy-related proteins, and the production of ROS in H9c2 cells, thereby alleviating the cellular stress and dysfunction that characterize SIMI.138 In addition, the oligo-peptide I-C-F-6, through activation of the Nrf2/HO-1/GPX4 signaling pathway, has been demonstrated to significantly attenuate myocardial oxidative damage in sepsis mice, offering a potential therapeutic avenue by inhibiting ferroptosis, a form of regulated cell death linked to iron and ROS overload.104 Given the intricate interplay between dysregulated inflammatory responses, various forms of regulated cell death, calcium dysregulation, and metabolic shifts, these factors collectively contribute to the pathophysiology of sepsis and SIMI. However, targeting a single molecular pathway may prove insufficient in addressing the multifaceted nature of this condition. As such, multi-target therapeutic approaches have gained traction as a promising direction for future research. In this regard, therapeutic strategies that can simultaneously modulate mitochondrial function, restore calcium homeostasis, regulate inflammatory responses, and mitigate cellular metabolic shifts may hold the key to more effective interventions in SIMI. However, the complexity of sepsis-induced myocardial injury—characterized by a dynamic and interconnected network of factors such as inflammation, mitochondrial dysfunction, regulatory cell death, and metabolic alterations—calls for more comprehensive, multi-dimensional therapeutic approaches. Future research should prioritize the development of such multi-target strategies, combining novel molecular agents that target distinct but interconnected pathways involved in SIMI. This approach not only promises to provide a more holistic treatment framework but also has the potential to offer new theoretical insights into the prevention and management of SIMI. Ultimately, this comprehensive understanding could lead to the establishment of innovative therapeutic paradigms, enhancing the clinical outcomes of patients suffering from this devastating condition.
Conclusion
Mitochondrial dysfunction plays a central role in the pathogenesis of SIMI, directly influencing cardiomyocyte viability and cardiac function. As sepsis progresses, mitochondrial dysfunction initiates and amplifies a cascade of inflammatory responses, culminating in disturbances in calcium homeostasis, activation of regulated cell death pathways (including apoptosis, pyroptosis, mitophagy, and ferroptosis), and metabolic reprogramming. These processes contribute to the profound myocardial damage observed in SIMI, leading to significantly increased mortality rates in sepsis patients.
Initially, mitochondrial dysfunction in sepsis is marked by a reduction in mitochondrial membrane potential, impaired ATP production, and excessive ROS generation. This triggers the release of mtDAMPs, which activate abnormal inflammatory responses and further escalate cellular injury. Additionally, mitochondrial calcium overload exacerbates oxidative stress, creating a vicious cycle that accelerates myocardial injury. The reprogramming of cellular metabolism, shifting from oxidative phosphorylation to aerobic glycolysis in the early stages of sepsis, contributes to a low-grade inflammatory state and immune suppression in later stages, further worsening sepsis and systemic organ damage.
These findings underscore the multifactorial nature of SIMI and highlight the urgent need for more targeted therapeutic approaches that address both the mitochondrial dysfunction and the associated inflammatory, metabolic, and cellular pathways. While promising therapeutic strategies, including biomolecules and phytochemicals, have shown potential in preclinical studies, their clinical application remains limited. Future research should prioritize the identification of key signaling pathways involved in SIMI and the development of novel therapeutic targets. By improving our understanding of the complex interplay between mitochondrial dysfunction, inflammatory responses, and metabolic alterations, we can develop more effective strategies for treating SIMI, ultimately improving patient outcomes in sepsis.
Acknowledgments
The authors thank Department of Emergency Medical, General Hospital of Ningxia Medical University, State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia, Xinjiang Medical University and Ningxia Key Laboratory of Clinical and Pathogenic Microbiology, General Hospital of Ningxia Medical University for supporting this work.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Natural Science Fund of Ningxia (2024AAC03606, 2024AAC03662), State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia Fund (SKL-HIDCA-2024-NX7) and the University level research project of Ningxia Medical University (XT2024033).
Disclosure
The authors declare that they have no competing interests.
References
1. Xu J-Q, Zhang W-Y, Fu -J-J, et al. Viral sepsis: diagnosis, clinical features, pathogenesis, and clinical considerations. Mil Med Res. 2024;11:78. doi:10.1186/s40779-024-00581-0
2. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:801–810. doi:10.1001/jama.2016.0287
3. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395:200–211. doi:10.1016/S0140-6736(19)32989-7
4. Dantes RB, Kaur H, Bouwkamp BA, et al. Sepsis program activities in acute care hospitals - national healthcare safety network, United States, 2022. MMWR Morb Mortal Wkly Rep. 2023;72:907–911. doi:10.15585/mmwr.mm7234a2
5. Xie J, Wang H, Kang Y, et al. The epidemiology of sepsis in Chinese ICUs: a national cross-sectional survey. Crit Care Med. 2020;48:e209–e218. doi:10.1097/CCM.0000000000004155
6. Levy RJ. Mitochondrial dysfunction, bioenergetic impairment, and metabolic down-regulation in sepsis. Shock. 2007;28:24–28. doi:10.1097/01.shk.0000235089.30550.2d
7. Court O, Kumar A, Parrillo JE, Kumar A. Clinical review: myocardial depression in sepsis and septic shock. Crit Care. 2002;6:500–508. doi:10.1186/cc1822
8. Chang Y, Yoo HJ, Kim SJ, et al. A targeted metabolomics approach for sepsis-induced ARDS and its subphenotypes. Crit Care. 2023;27:263. doi:10.1186/s13054-023-04552-0
9. Heberer G, Paumgartner G, Sauerbruch T, et al. A retrospective analysis of 3 year’s experience of an interdisciplinary approach to gallstone disease including shock-waves. Ann Surg. 1988;208:274–278. doi:10.1097/00000658-198809000-00003
10. Orde SR, Pulido JN, Masaki M, et al. Outcome prediction in sepsis: speckle tracking echocardiography based assessment of myocardial function. Crit Care. 2014;18:R149. doi:10.1186/cc13987
11. Vieillard-Baron A, Caille V, Charron C, et al. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med. 2008;36:1701–1706. doi:10.1097/CCM.0b013e318174db05
12. Fan Y, Guan B, Xu J, et al. Role of toll-like receptor-mediated pyroptosis in sepsis-induced cardiomyopathy. Biomed Pharmacother. 2023;167:115493. doi:10.1016/j.biopha.2023.115493
13. Fan D, Wu R. Mechanisms of the septic heart: from inflammatory response to myocardial edema. J Mol Cell Cardiol. 2024;195:73–82.
14. Lukić I, Mihić D, Varžić SC, et al. Septic cardiomyopathy. Rev Cardiovasc Med. 2024;25:23. doi:10.31083/j.rcm2501023
15. Sidheeque Hassan V, Hanifa M, Navik U, Bali A. Exogenous fetuin-A protects against sepsis-induced myocardial injury by inhibiting oxidative stress and inflammation in mice. Fundam Clin Pharmacol. 2023;37:607–617. doi:10.1111/fcp.12870
16. Roger AJ, Muñoz-Gómez SA, Kamikawa R. The origin and diversification of mitochondria. Curr Biol. 2017;27:R1177–R1192. doi:10.1016/j.cub.2017.09.015
17. Harapas CR, Idiiatullina E, Al-Azab M, et al. Organellar homeostasis and innate immune sensing. Nat Rev Immunol. 2022;22:535–549. doi:10.1038/s41577-022-00682-8
18. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi:10.1038/s41418-017-0012-4
19. Opal SM, van der Poll T. Endothelial barrier dysfunction in septic shock. J Intern Med. 2015;277:277–293. doi:10.1111/joim.12331
20. Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi:10.1038/nature18590
21. Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi:10.1038/ni.1960
22. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi:10.1038/nature09663
23. Ren G, Zhou Q, Lu M, Wang H. Rosuvastatin corrects oxidative stress and inflammation induced by LPS to attenuate cardiac injury by inhibiting the NLRP3/TLR4 pathway. Can J Physiol Pharmacol. 2021;99:964–973. doi:10.1139/cjpp-2020-0321
24. Wang Y, Feng W, Li S, et al. Oxycodone attenuates lipopolysaccharide-induced myocardial injury by inhibiting inflammation, oxidation and pyroptosis via Nrf2/HO-1 signalling pathway. Clin Exp Pharmacol Physiol. 2024;51:e13910. doi:10.1111/1440-1681.13910
25. Wang J, Hou J, Peng C. Phospholipid transfer protein ameliorates sepsis-induced cardiac dysfunction through NLRP3 inflammasome inhibition. Open Med. 2024;19:20240915. doi:10.1515/med-2024-0915
26. Jiang H, Gong T, Zhou R. The strategies of targeting the NLRP3 inflammasome to treat inflammatory diseases. Adv Immunol. 2020;145:55–93.
27. Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi:10.1038/nature15541
28. Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi:10.1038/nature18629
29. Dolinay T, Kim YS, Howrylak J, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 2012;185:1225–1234. doi:10.1164/rccm.201201-0003OC
30. Homsy E, Das S, Consiglio P, et al. Circulating gasdermin-D in critically ill patients. Crit Care Explor. 2019;1:e0039. doi:10.1097/CCE.0000000000000039
31. Huang W, Wang X, Xie F, Zhang H, Liu D. Serum NLRP3: a biomarker for identifying high-risk septic patients. Cytokine. 2022;149:155725. doi:10.1016/j.cyto.2021.155725
32. Zhu -X-X, Meng X-Y, Zhang A-Y, et al. Vaccarin alleviates septic cardiomyopathy by potentiating NLRP3 palmitoylation and inactivation. Phytomedicine. 2024;131:155771. doi:10.1016/j.phymed.2024.155771
33. Barnett KC, Ting JP-Y. Mitochondrial GSDMD pores DAMPen pyroptosis. Immunity. 2020;52:424–426. doi:10.1016/j.immuni.2020.02.012
34. Xiaodong L, Xuejun X. GSDMD-mediated pyroptosis in retinal vascular inflammatory diseases: a review. Int Ophthalmol. 2023;43:1405–1411. doi:10.1007/s10792-022-02506-z
35. Yang W, Wang Y, Huang Y, et al. Immune Response Gene-1 [IRG1]/itaconate protect against multi-organ injury via inhibiting gasdermin D-mediated pyroptosis and inflammatory response. Inflammopharmacology. 2024;32:419–432. doi:10.1007/s10787-023-01278-x
36. Tang Y, Wu J, Sun X, et al. Cardiolipin oxidized by ROS from complex II acts as a target of gasdermin D to drive mitochondrial pore and heart dysfunction in endotoxemia. Cell Rep. 2024;43:114237. doi:10.1016/j.celrep.2024.114237
37. Song C, Zhang Y, Pei Q, et al. HSP70 alleviates sepsis-induced cardiomyopathy by attenuating mitochondrial dysfunction-initiated NLRP3 inflammasome-mediated pyroptosis in cardiomyocytes. Burns Trauma. 2022;10:tkac043. doi:10.1093/burnst/tkac043
38. Xiong X, Lu L, Wang Z, et al. Irisin attenuates sepsis-induced cardiac dysfunction by attenuating inflammation-induced pyroptosis through a mitochondrial ubiquitin ligase-dependent mechanism. Biomed Pharmacother. 2022;152:113199. doi:10.1016/j.biopha.2022.113199
39. Huang J, Peng W, Zheng Y, et al. Upregulation of UCP2 expression protects against LPS-induced oxidative stress and apoptosis in cardiomyocytes. Oxid Med Cell Longev. 2019;2019:2758262. doi:10.1155/2019/2758262
40. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364. doi:10.1038/s41422-019-0164-5
41. Ward PA. Sepsis, apoptosis and complement. Biochem Pharmacol. 2008;76:1383–1388. doi:10.1016/j.bcp.2008.09.017
42. Xu P, Zhang W-Q, Xie J, et al. Shenfu injection prevents sepsis-induced myocardial injury by inhibiting mitochondrial apoptosis. J Ethnopharmacol. 2020;261:113068. doi:10.1016/j.jep.2020.113068
43. Levy RJ, Deutschman CS. Evaluating myocardial depression in sepsis. Shock. 2004;22:1–10. doi:10.1097/01.shk.0000129198.53836.15
44. Bratton SB, MacFarlane M, Cain K, Cohen GM. Protein complexes activate distinct caspase cascades in death receptor and stress-induced apoptosis. Exp Cell Res. 2000;256:27–33. doi:10.1006/excr.2000.4835
45. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi:10.1126/science.1099320
46. Pathan N, Franklin JL, Eleftherohorinou H, et al. Myocardial depressant effects of interleukin 6 in meningococcal sepsis are regulated by p38 mitogen-activated protein kinase. Crit Care Med. 2011;39:1692–1711. doi:10.1097/CCM.0b013e3182186d27
47. Zhang J, Yang Z, Liang Z, et al. Anti-Interleukin-16 neutralizing antibody treatment alleviates sepsis-induced cardiac injury and dysfunction via the nuclear factor erythroid-2 related factor 2 pathway in mice. Oxid Med Cell Longev. 2021;2021:6616422. doi:10.1155/2021/6616422
48. Wang W, Li J, Zhou Q. The biological function of cytoplasm-translocated ENDOG (endonuclease G). Autophagy. 2024;20:445–447. doi:10.1080/15548627.2023.2271750
49. Nevière R, Fauvel H, Chopin C, Formstecher P, Marchetti P. Caspase inhibition prevents cardiac dysfunction and heart apoptosis in a rat model of sepsis. Am J Respir Crit Care Med. 2001;163:218–225. doi:10.1164/ajrccm.163.1.2003109
50. Wang C, Han D, Feng X, Hu L, Wu J. Docosahexaenoic acid alleviates LPS-induced cytotoxicity in HL-1 cardiac cells via improving stress-induced mitochondrial fragmentation. Heliyon. 2023;9:e22465. doi:10.1016/j.heliyon.2023.e22465
51. Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141:2629–2634. doi:10.4049/jimmunol.141.8.2629
52. Hayakawa Y, Chandra M, Miao W, et al. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Gα(q) transgenic mice. Circulation. 2003;108:3036–3041. doi:10.1161/01.CIR.0000101920.72665.58
53. Zhang W, Yuan SL, Qiang JC, et al. Malvidin mitigates sepsis-induced cardiac injury by modulating the TLR4-iNOS-COX-2 inflammatory pathway and the Bax/Bcl-2/Cyto-C mitochondrial apoptosis pathway in a p38 MAPK-dependent manner. Biomed Environ Sci. 2024;37:221–227. doi:10.3967/bes2024.024
54. Li Y, Zhang L, Zhang P, Hao Z. Dehydrocorydaline protects against sepsis-induced myocardial injury through modulating the TRAF6/NF-κB pathway. Front Pharmacol. 2021;12:709604. doi:10.3389/fphar.2021.709604
55. Zhang L, Xiu L, Wang T, Zhao D. Effect of L-carnitine in ameliorating lipopolysaccharide-induced cardiomyocyte injury via MAPK signaling. Mol Biotechnol. 2024;66:79–89. doi:10.1007/s12033-023-00731-0
56. Li Q, Zhang M, Zhao Y, Dong M. Irisin protects against LPS-stressed cardiac damage through inhibiting inflammation, apoptosis, and pyroptosis. Shock. 2021;56:1009–1018. doi:10.1097/SHK.0000000000001775
57. Shao F, Zhou L, Zhang Y, et al. Gastrodin alleviates inflammatory injury of cardiomyocytes in septic shock mice via inhibiting NLRP3 expression. In Vitro Cell Dev Biol Anim. 2021;57:571–581. doi:10.1007/s11626-021-00593-3
58. Preau S, Delguste F, Yu Y, et al. Endotoxemia engages the RhoA kinase pathway to impair cardiac function by altering cytoskeleton, mitochondrial fission, and autophagy. Antioxid Redox Signal. 2016;24:529–542. doi:10.1089/ars.2015.6421
59. Vanasco V, Saez T, Magnani ND, et al. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radic Biol Med. 2014;77:1–9. doi:10.1016/j.freeradbiomed.2014.08.009
60. Drosatos K, Khan RS, Trent CM, et al. Peroxisome proliferator-activated receptor-γ activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail. 2013;6:550–562. doi:10.1161/CIRCHEARTFAILURE.112.000177
61. Jia J, Gong X, Zhao Y, et al. Autophagy enhancing contributes to the organ protective effect of alpha-lipoic acid in septic rats. Front Immunol. 2019;10:1491. doi:10.3389/fimmu.2019.01491
62. Zhao P, Kuai J, Gao J, et al. Delta opioid receptor agonist attenuates lipopolysaccharide-induced myocardial injury by regulating autophagy. Biochem Biophys Res Commun. 2017;492:140–146. doi:10.1016/j.bbrc.2017.06.029
63. Luo Y, Fan C, Yang M, et al. CD74 knockout protects against LPS-induced myocardial contractile dysfunction through AMPK-Skp2-SUV39H1-mediated demethylation of BCLB. Br J Pharmacol. 2020;177:1881–1897. doi:10.1111/bph.14959
64. Klionsky DJ, Petroni G, Amaravadi RK, et al. Autophagy in major human diseases. EMBO J. 2021;40:e108863. doi:10.15252/embj.2021108863
65. Yuan H, Perry CN, Huang C, et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009;296:H470–479. doi:10.1152/ajpheart.01051.2008
66. Wang J, Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm Sin B. 2020;10:1866–1879. doi:10.1016/j.apsb.2020.03.004
67. Green DR, Van Houten B. SnapShot: mitochondrial quality control. Cell. 2011;147:
68. Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi:10.1038/ncb2012
69. Ordureau A, Sarraf S, Duda D, et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. doi:10.1016/j.molcel.2014.09.007
70. Sedlackova L, Korolchuk VI. Mitochondrial quality control as a key determinant of cell survival. Biochim Biophys Acta Mol Cell Res. 2019;1866:575–587. doi:10.1016/j.bbamcr.2018.12.012
71. Wei Y, Chiang W-C, Sumpter R, Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–238.e10. doi:10.1016/j.cell.2016.11.042
72. Wang S, Zhao Z, Feng X, et al. Melatonin activates Parkin translocation and rescues the impaired mitophagy activity of diabetic cardiomyopathy through Mst1 inhibition. J Cell Mol Med. 2018;22:5132–5144. doi:10.1111/jcmm.13802
73. Yun J, Puri R, Yang H, et al. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife. 2014;3:e01958. doi:10.7554/eLife.01958
74. Cadete VJJ, Vasam G, Menzies KJ, Burelle Y. Mitochondrial quality control in the cardiac system: an integrative view. Biochim Biophys Acta Mol Basis Dis. 2019;1865:782–796. doi:10.1016/j.bbadis.2018.11.018
75. Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi:10.1038/ncb2422
76. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi:10.1038/cdd.2009.16
77. Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527. doi:10.1038/ncomms8527
78. Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. doi:10.1038/ncb2837
79. Wu W, Ma Q, Li B-T, et al. α‑ketoglutarate protects against septic cardiomyopathy by improving mitochondrial mitophagy and fission. Mol Med Rep. 2025;31:146. doi:10.3892/mmr.2025.13511
80. Huang W, Lou A, Wang J, et al. TMBIM1 ameliorates sepsis-induced cardiac dysfunction by promoting Parkin-mediated mitophagy. FASEB J. 2025;39:e70397. doi:10.1096/fj.202402599RR
81. Wang Z, Wang Y, Dong C, et al. Po-Ge-Jiu-Xin decoction alleviate sepsis-induced cardiomyopathy via regulating phosphatase and tensin homolog-induced putative kinase 1 /parkin-mediated mitophagy. J Ethnopharmacol. 2025;337:118952. doi:10.1016/j.jep.2024.118952
82. Ji W, Wan T, Zhang F, et al. Aldehyde dehydrogenase 2 protects against lipopolysaccharide-induced myocardial injury by suppressing mitophagy. Front Pharmacol. 2021;12:641058. doi:10.3389/fphar.2021.641058
83. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi:10.1016/S1535-6108(03)00050-3
84. Cheng X, Yu C, Yang X, Wang F, Min J. A panoramic view of ferroptosis in cardiovascular disease. Kidney Dis. 2023;9:173–186. doi:10.1159/000530046
85. Fang W, Xie S, Deng W. Ferroptosis mechanisms and regulations in cardiovascular diseases in the past, present, and future. Cell Biol Toxicol. 2024;40:17. doi:10.1007/s10565-024-09853-w
86. Zhu H, Santo A, Jia Z, Robert Li Y. GPx4 in bacterial infection and polymicrobial sepsis: involvement of ferroptosis and pyroptosis. React Oxyg Species. 2019;7:154–160.
87. Ma D, Jiang P, Jiang Y, Li H, Zhang D. Effects of lipid peroxidation-mediated ferroptosis on severe acute pancreatitis-induced intestinal barrier injury and bacterial translocation. Oxid Med Cell Longev. 2021;2021:1–12. doi:10.1155/2021/6644576
88. Conrad M, Proneth B. Broken hearts: iron overload, ferroptosis and cardiomyopathy. Cell Res. 2019;29:263–264. doi:10.1038/s41422-019-0150-y
89. Liu B-H, Xu C-Z, Liu Y, et al. Mitochondrial quality control in human health and disease. Mil Med Res. 2024;11:32. doi:10.1186/s40779-024-00536-5
90. Long X, Liu M, Nan Y, et al. Revitalizing ancient mitochondria with nano-strategies: mitochondria-remedying nanodrugs concentrate on disease control. Adv Mater. 2024;36:e2308239. doi:10.1002/adma.202308239
91. Jing X, Chen Z, Zhang M, et al. Melatonin mitigates the lipopolysaccharide-induced myocardial injury in rats by blocking the p53/xCT pathway-mediated ferroptosis. Naunyn Schmiedebergs Arch Pharmacol. 2025;398:1653–1663. doi:10.1007/s00210-024-03367-2
92. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–296. doi:10.1038/s41571-020-00462-0
93. Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi:10.1126/science.1164680
94. Dixon SJ, Lemberg K, Lamprecht M, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi:10.1016/j.cell.2012.03.042
95. Lv Y, Chen D, Tian X, et al. Protectin conjugates in tissue regeneration 1 alleviates sepsis-induced acute lung injury by inhibiting ferroptosis. J Transl Med. 2023;21:293. doi:10.1186/s12967-023-04111-9
96. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21:648–657. doi:10.1111/jcmm.13008
97. Zhang LJ, Salekeen R, Soto-Palma C, et al. Identification of lipid senolytics targeting senescent cells through ferroptosis induction. bioRxiv. 2024;2024:
98. Xie Y, Kang R, Klionsky DJ, Tang D. GPX4 in cell death, autophagy, and disease. Autophagy. 2023;19:2621–2638. doi:10.1080/15548627.2023.2218764
99. Flohé L, Toppo S, Orian L. The glutathione peroxidase family: discoveries and mechanism. Free Radic Biol Med. 2022;187:113–122. doi:10.1016/j.freeradbiomed.2022.05.003
100. Li F-J, Long H-Z, Zhou Z-W, et al. System Xc -/GSH/GPX4 axis: an important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol. 2022;13:910292. doi:10.3389/fphar.2022.910292
101. Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41:274–286. doi:10.1016/j.tibs.2015.11.012
102. Li N, Wang W, Zhou H, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–318. doi:10.1016/j.freeradbiomed.2020.08.009
103. Wang N, Ma H, Li J, et al. HSF1 functions as a key defender against palmitic acid-induced ferroptosis in cardiomyocytes. J Mol Cell Cardiol. 2021;150:65–76. doi:10.1016/j.yjmcc.2020.10.010
104. Li H, Li G, Gao Y, et al. Oligo-peptide I-C-F-6 mitigates polymicrobial sepsis-induced cardiac dysfunction in mice. Eur J Pharmacol. 2025;996:177545. doi:10.1016/j.ejphar.2025.177545
105. Hu K, Jiang P, Hu J, et al. Dapagliflozin attenuates LPS-induced myocardial injury by reducing ferroptosis. J Bioenerg Biomembr. 2024;56:361–371. doi:10.1007/s10863-024-10020-3
106. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599–620. doi:10.1007/s13238-020-00789-5
107. Shen H, Xie K, Tian Y, Wang X. N6-methyladenosine writer METTL3 accelerates the sepsis-induced myocardial injury by regulating m6A-dependent ferroptosis. Apoptosis. 2023;28:514–524. doi:10.1007/s10495-022-01808-y
108. Cao G, Zeng Y, Zhao Y, et al. H2S regulation of ferroptosis attenuates sepsis‑induced cardiomyopathy. Mol Med Rep. 2022;26:335. doi:10.3892/mmr.2022.12851
109. Guo L, Li P, Wang Y, et al. Yiqifumai injection ameliorated sepsis-induced cardiomyopathy by inhibition of ferroptosis via Xct/Gpx4 axis. Shock. 2024;61:638–645. doi:10.1097/SHK.0000000000002257
110. Wang J, Guan P, Chen Y, et al. Cyclovirobuxine D pretreatment ameliorates septic heart injury through mitigation of ferroptosis. Exp Ther Med. 2023;26:407. doi:10.3892/etm.2023.12106
111. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi:10.1038/nature14344
112. Mukherjee R, Tetri LH, Li S-J, et al. Drp1/p53 interaction mediates p53 mitochondrial localization and dysfunction in septic cardiomyopathy. J Mol Cell Cardiol. 2023;177:28–37. doi:10.1016/j.yjmcc.2023.01.008
113. Lin X, Zhao X, Chen Q, et al. Quercetin ameliorates ferroptosis of rat cardiomyocytes via activation of the SIRT1/p53/SLC7A11 signaling pathway to alleviate sepsis‑induced cardiomyopathy. Int J Mol Med. 2023;52:116. doi:10.3892/ijmm.2023.5319
114. Yang W, Wang Y, Zhang C, et al. Maresin1 protect against ferroptosis-induced liver injury through ROS inhibition and Nrf2/HO-1/GPX4 activation. Front Pharmacol. 2022;13:865689. doi:10.3389/fphar.2022.865689
115. Yang S, Huang X-Y. Ca2+ influx through L-type Ca2+ channels controls the trailing tail contraction in growth factor-induced fibroblast cell migration. J Biol Chem. 2005;280:27130–27137. doi:10.1074/jbc.M501625200
116. Tsai F-C, Seki A, Yang HW, et al. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat Cell Biol. 2014;16:133–144. doi:10.1038/ncb2906
117. Xiong J, Camello PJ, Verkhratsky A, Toescu EC. Mitochondrial polarisation status and [Ca2+]i signalling in rat cerebellar granule neurones aged in vitro. Neurobiol Aging. 2004;25:349–359. doi:10.1016/S0197-4580(03)00123-4
118. Tang S, Wang X, Shen Q, et al. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem Biophys Res Commun. 2015;458:186–193. doi:10.1016/j.bbrc.2015.01.092
119. Deak AT, Blass S, Khan MJ, et al. IP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. J Cell Sci. 2014;127:2944–2955. doi:10.1242/jcs.149807
120. Hartmann J, Verkhratsky A. Relations between intracellular Ca2+ stores and store-operated Ca2+ entry in primary cultured human glioblastoma cells. J Physiol. 1998;513(Pt 2):411–424. doi:10.1111/j.1469-7793.1998.411bb.x
121. Imbert N, Cognard C, Duport G, Guillou C, Raymond G. Abnormal calcium homeostasis in Duchenne muscular dystrophy myotubes contracting in vitro. Cell Calcium. 1995;18:177–186. doi:10.1016/0143-4160(95)90062-4
122. Ahuja K, Motiani RK. Calcium acts as a critical determinant of mitochondria-nuclear networking driven retrograde signaling. Cell Calcium. 2025;127:103017. doi:10.1016/j.ceca.2025.103017
123. Gomez-Suaga P, Paillusson S, Stoica R, et al. The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr Biol. 2017;27:371–385. doi:10.1016/j.cub.2016.12.038
124. Kohlhaas M, Maack C. Calcium release microdomains and mitochondria. Cardiovasc Res. 2013;98:259–268. doi:10.1093/cvr/cvt032
125. Kohlhaas M, Liu T, Knopp A, et al. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi:10.1161/CIRCULATIONAHA.109.914911
126. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu -S-S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–833. doi:10.1152/ajpcell.00139.2004
127. Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A. 2015;112:11389–11394. doi:10.1073/pnas.1513047112
128. Hurst S, Hoek J, Sheu -S-S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J Bioenerg Biomembr. 2017;49:27–47. doi:10.1007/s10863-016-9672-x
129. Kim B, Takeuchi A, Koga O, Hikida M, Matsuoka S. Mitochondria Na(+)-Ca (2+) exchange in cardiomyocytes and lymphocytes. Adv Exp Med Biol. 2013;961:193–201.
130. Luongo TS, Lambert JP, Yuan A, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34. doi:10.1016/j.celrep.2015.06.017
131. Szabadkai G, Bianchi K, Várnai P, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi:10.1083/jcb.200608073
132. Giorgi C, Missiroli S, Patergnani S, et al. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal. 2015;22:995–1019. doi:10.1089/ars.2014.6223
133. Hobai IA, Edgecomb J, LaBarge K, Colucci WS. Dysregulation of intracellular calcium transporters in animal models of sepsis-induced cardiomyopathy. Shock. 2015;43:3–15. doi:10.1097/SHK.0000000000000261
134. Zaky A, Deem S, Bendjelid K, Treggiari MM. Characterization of cardiac dysfunction in sepsis: an ongoing challenge. Shock. 2014;41:12–24. doi:10.1097/SHK.0000000000000065
135. Stanzani G, Duchen MR, Singer M. The role of mitochondria in sepsis-induced cardiomyopathy. Biochim Biophys Acta Mol Basis Dis. 2019;1865:759–773. doi:10.1016/j.bbadis.2018.10.011
136. Joseph LC, Kokkinaki D, Valenti M-C, et al. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight. 2017;2:e94248. doi:10.1172/jci.insight.94248
137. Zhou Q, Xie M, Zhu J, et al. PINK1 contained in huMSC-derived exosomes prevents cardiomyocyte mitochondrial calcium overload in sepsis via recovery of mitochondrial Ca2+ efflux. Stem Cell Res Ther. 2021;12:269. doi:10.1186/s13287-021-02325-6
138. Jiang T, Peng D, Shi W, et al. IL-6/STAT3 signaling promotes cardiac dysfunction by upregulating FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes formation in sepsis mice. Front Cardiovasc Med. 2021;8:790612. doi:10.3389/fcvm.2021.790612
139. Hassoun SM, Marechal X, Montaigne D, et al. Prevention of endotoxin-induced sarcoplasmic reticulum calcium leak improves mitochondrial and myocardial dysfunction. Crit Care Med. 2008;36:2590–2596. doi:10.1097/CCM.0b013e3181844276
140. Pinto BB, Dyson A, Umbrello M, et al. Improved survival in a long-term rat model of sepsis is associated with reduced mitochondrial calcium uptake despite increased energetic demand. Crit Care Med. 2017;45:e840–e848. doi:10.1097/CCM.0000000000002448
141. Everts B, Amiel E, Huang SC-C, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15:323–332. doi:10.1038/ni.2833
142. He Y, Wang N, Shen Y, Zheng Z, Xu X. Inhibition of high glucose-induced apoptosis by uncoupling protein 2 in human umbilical vein endothelial cells. Int J Mol Med. 2014;33:1275–1281. doi:10.3892/ijmm.2014.1676
143. Soto‐Heredero G, Gómez De Las Heras MM, Gabandé‐Rodríguez E, Oller J, Mittelbrunn M. Glycolysis – a key player in the inflammatory response. FEBS J. 2020;287:3350–3369. doi:10.1111/febs.15327
144. Schilling J, Lai L, Sambandam N, et al. Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor γ coactivator-1 signaling. Circ Heart Fail. 2011;4:474–482. doi:10.1161/CIRCHEARTFAILURE.110.959833
145. Sun H-J, Zheng G-L, Wang Z-C, et al. Chicoric acid ameliorates sepsis-induced cardiomyopathy via regulating macrophage metabolism reprogramming. Phytomedicine. 2024;123:155175. doi:10.1016/j.phymed.2023.155175
146. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi:10.1111/j.1600-065X.2012.01146.x
147. Donnelly RP, Finlay DK. Glucose, glycolysis and lymphocyte responses. Mol Immunol. 2015;68:513–519. doi:10.1016/j.molimm.2015.07.034
148. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi:10.1146/annurev-cellbio-092910-154237
149. Macintyre AN, Gerriets V, Nichols A, et al. The glucose transporter glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20:61–72. doi:10.1016/j.cmet.2014.05.004
150. Freemerman AJ, Johnson AR, Sacks GN, et al. Metabolic Reprogramming of Macrophages. J Biol Chem. 2014;289:7884–7896. doi:10.1074/jbc.M113.522037
151. Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi:10.4049/jimmunol.1003613
152. Gauthier T, Chen W. Modulation of macrophage immunometabolism: a new approach to fight infections. Front Immunol. 2022;13:780839. doi:10.3389/fimmu.2022.780839
153. Pålsson-McDermott EM, O’Neill LAJ. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020;30:300–314. doi:10.1038/s41422-020-0291-z
154. Cheng S-C, Quintin J, Cramer RA, et al. mTOR- and HIF-1α–mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi:10.1126/science.1250684
155. Kasprzak A. Insulin-Like Growth Factor 1 (IGF-1) signaling in glucose metabolism in colorectal cancer. IJMS. 2021;22:6434. doi:10.3390/ijms22126434
156. Torp M-K, Yang K, Ranheim T, et al. Mammalian Target of Rapamycin (mTOR) and the proteasome attenuates IL-1β expression in primary mouse cardiac fibroblasts. Front Immunol. 2019;10:1285. doi:10.3389/fimmu.2019.01285
157. Piñeros Alvarez AR, Glosson-Byers N, Brandt S, et al. SOCS1 is a negative regulator of metabolic reprogramming during sepsis. JCI Insight. 2017;2:e92530. doi:10.1172/jci.insight.92530
158. Lin S, Jin P, Shao C, et al. Lidocaine attenuates lipopolysaccharide-induced inflammatory responses and protects against endotoxemia in mice by suppressing HIF1α-induced glycolysis. Int Immunopharmacol. 2020;80:106150. doi:10.1016/j.intimp.2019.106150
159. Dang CP, Leelahavanichkul A. Over-expression of miR-223 induces M2 macrophage through glycolysis alteration and attenuates LPS-induced sepsis mouse model, the cell-based therapy in sepsis. PLoS One. 2020;15:e0236038. doi:10.1371/journal.pone.0236038
160. Awasthi D, Nagarkoti S, Sadaf S, et al. Glycolysis dependent lactate formation in neutrophils: a metabolic link between NOX-dependent and independent NETosis. Biochim Biophys Acta Mol Basis Dis. 2019;1865:165542. doi:10.1016/j.bbadis.2019.165542
161. Batista-Gonzalez A, Vidal R, Criollo A, Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol. 2019;10:2993. doi:10.3389/fimmu.2019.02993
162. Im -S-S, Yousef L, Blaschitz C, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011;13:540–549. doi:10.1016/j.cmet.2011.04.001
163. Wei X, Song H, Yin L, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. 2016;539:294–298. doi:10.1038/nature20117
164. Ma W, Ao S, Zhou J, et al. Methylsulfonylmethane protects against lethal dose MRSA-induced sepsis through promoting M2 macrophage polarization. Mol Immunol. 2022;146:69–77. doi:10.1016/j.molimm.2022.04.001
165. Körner A, Bernard A, Fitzgerald JC, et al. Sema7A is crucial for resolution of severe inflammation. Proc Natl Acad Sci U S A. 2021;118:e2017527118. doi:10.1073/pnas.2017527118
166. Deng H, Zhu L, Zhang Y, et al. Differential lung protective capacity of exosomes derived from human adipose tissue, bone marrow, and umbilical cord mesenchymal stem cells in sepsis-induced acute lung injury. Oxid Med Cell Longev. 2022;2022:7837837. doi:10.1155/2022/7837837
167. Zhang Q, Hu Y, Zhang J, Deng C. iTRAQ‑based proteomic analysis of endotoxin tolerance induced by lipopolysaccharide. Mol Med Rep. 2019. doi:10.3892/mmr.2019.10264
168. Morris M, Li L. Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch Immunol Ther Exp. 2012;60:13–18. doi:10.1007/s00005-011-0155-9
169. Koutroulis I, Batabyal R, McNamara B, et al. Sepsis immunometabolism: from defining sepsis to understanding how energy production affects immune response. Crit Care Explor. 2019;1:e0061. doi:10.1097/CCE.0000000000000061
170. Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem. 2012;287:25758–25769. doi:10.1074/jbc.M112.362343
171. Manoharan I, Prasad PD, Thangaraju M, Manicassamy S. Lactate-dependent regulation of immune responses by dendritic cells and macrophages. Front Immunol. 2021;12:691134. doi:10.3389/fimmu.2021.691134
172. Brooks GA. The tortuous path of lactate shuttle discovery: from cinders and boards to the lab and ICU. J Sport Health Sci. 2020;9:446–460. doi:10.1016/j.jshs.2020.02.006
173. Chen P, Zuo H, Xiong H, et al. Gpr132 sensing of lactate mediates tumor–macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci U S A. 2017;114:580–585. doi:10.1073/pnas.1614035114
174. Errea A, Cayet D, Marchetti P, et al. Lactate inhibits the pro-inflammatory response and metabolic reprogramming in murine macrophages in a GPR81-independent manner. PLoS One. 2016;11:e0163694. doi:10.1371/journal.pone.0163694
175. Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate reduces liver and pancreatic injury in toll-like receptor– and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology. 2014;146:1763–1774. doi:10.1053/j.gastro.2014.03.014
176. Zhou H-C, Yu WW, Yan XY, et al. Lactate-driven macrophage polarization in the inflammatory microenvironment alleviates intestinal inflammation. Front Immunol. 2022;13:1013686. doi:10.3389/fimmu.2022.1013686
177. Zhang W, Wang G, Xu Z-G, et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell. 2019;178:176–189.e15. doi:10.1016/j.cell.2019.05.003
178. Liu N, Luo J, Kuang D, et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α-mediated tumor progression. J Clin Invest. 2019;129:631–646. doi:10.1172/JCI123027
179. Haas R, Smith J, Rocher-Ros V, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 2015;13:e1002202. doi:10.1371/journal.pbio.1002202
180. Duan F, Li L, Liu S, et al. Cortistatin protects against septic cardiomyopathy by inhibiting cardiomyocyte pyroptosis through the SSTR2-AMPK-NLRP3 pathway. Int Immunopharmacol. 2024;134:112186. doi:10.1016/j.intimp.2024.112186
181. Rahim I, Sayed RK, Fernández-Ortiz M, et al. Melatonin alleviates sepsis-induced heart injury through activating the Nrf2 pathway and inhibiting the NLRP3 inflammasome. Naunyn Schmiedebergs Arch Pharmacol. 2021;394:261–277. doi:10.1007/s00210-020-01972-5
182. Wang R-Y, Wang M-G, Tang H-Z, et al. The protective effects of ruscogenin against lipopolysaccharide-induced myocardial injury in septic mice. J Cardiovasc Pharmacol. 2024;84:175–187. doi:10.1097/FJC.0000000000001563
183. Wu C, Chen Y, Zhou P, Hu Z. Recombinant human angiotensin-converting enzyme 2 plays a protective role in mice with sepsis-induced cardiac dysfunction through multiple signaling pathways dependent on converting angiotensin II to angiotensin 1-7. Ann Transl Med. 2023;11:13. doi:10.21037/atm-22-6016
184. Cao Z, Li W, Shao Z, et al. Apelin ameliorates sepsis-induced myocardial dysfunction via inhibition of NLRP3-mediated pyroptosis of cardiomyocytes. Heliyon. 2024;10:e24568. doi:10.1016/j.heliyon.2024.e24568
185. Joshi S, Kundu S, Priya VV, et al. Anti-inflammatory activity of carvacrol protects the heart from lipopolysaccharide-induced cardiac dysfunction by inhibiting pyroptosis via NLRP3/Caspase1/Gasdermin D signaling axis. Life Sci. 2023;324:121743. doi:10.1016/j.lfs.2023.121743
186. Zhang J, Zhu Y, Chen S, et al. Activation of cannabinoid receptors 2 alleviates myocardial damage in cecal ligation and puncture-induced sepsis by inhibiting pyroptosis. Immunol Lett. 2023;264:17–24. doi:10.1016/j.imlet.2023.10.007
187. Zhang Y, Lv Y, Zhang Q, et al. ALDH2 attenuates myocardial pyroptosis through breaking down Mitochondrion-NLRP3 inflammasome pathway in septic shock. Front Pharmacol. 2023;14:1125866. doi:10.3389/fphar.2023.1125866
188. Li S, Guo Z, Zhang ZY. Protective effects of NLRP3 inhibitor MCC950 on sepsis-induced myocardial dysfunction. J Biol Regul Homeost Agents. 2021;35:141–150. doi:10.23812/20-662-A
189. Chen P, An Q, Huang Y, Zhang M, Mao S. Prevention of endotoxin-induced cardiomyopathy using sodium tanshinone IIA sulfonate: involvement of augmented autophagy and NLRP3 inflammasome suppression. Eur J Pharmacol. 2021;909:174438. doi:10.1016/j.ejphar.2021.174438
190. Wei A, Li D, Lu Y, et al. Syringaresinol attenuates sepsis-induced cardiac dysfunction by inhibiting inflammation and pyroptosis in mice. Eur J Pharmacol. 2021;913:174644. doi:10.1016/j.ejphar.2021.174644
191. Song P, Shen DF, Meng YY, et al. Geniposide protects against sepsis-induced myocardial dysfunction through AMPKα-dependent pathway. Free Radic Biol Med. 2020;152:186–196. doi:10.1016/j.freeradbiomed.2020.02.011
192. Rathkey JK, Zhao J, Liu Z, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3:eaat2738. doi:10.1126/sciimmunol.aat2738
193. Dai S, Ye B, Chen L, et al. Emodin alleviates LPS-induced myocardial injury through inhibition of NLRP3 inflammasome activation. Phytother Res. 2021;35:5203–5213. doi:10.1002/ptr.7191
194. Wang J, Zhu Q, Wang Y, et al. Irisin protects against sepsis-associated encephalopathy by suppressing ferroptosis via activation of the Nrf2/GPX4 signal axis. Free Radic Biol Med. 2022;187:171–184. doi:10.1016/j.freeradbiomed.2022.05.023
195. Zhang J, Tian Y, Yan H, et al. Protective effect and mechanism of plant-derived Lactobacillus G11 on septic myocardial injury. Int Immunopharmacol. 2025;148:114021. doi:10.1016/j.intimp.2025.114021
196. Yang B, Li T, Wang Z, et al. Ruxolitinib-based senomorphic therapy mitigates cardiomyocyte senescence in septic cardiomyopathy by inhibiting the JAK2/STAT3 signaling pathway. Int J Biol Sci. 2024;20:4314–4340. doi:10.7150/ijbs.96489
197. Shan M, Yu X, Li Y, Fu C, Zhang C. Vitamin B6 alleviates lipopolysaccharide-induced myocardial injury by ferroptosis and apoptosis regulation. Front Pharmacol. 2021;12:766820. doi:10.3389/fphar.2021.766820
198. Chen R, Zheng A, Wang Y, et al. Salvianolic acid B improves mitochondrial dysfunction of septic cardiomyopathy via enhancing ATF5-mediated mitochondrial unfolded protein response. Toxicol Appl Pharmacol. 2024;491:117072. doi:10.1016/j.taap.2024.117072
199. Li Z, Zhou J, Cui S, et al. Activation of sigma-1 receptor ameliorates sepsis-induced myocardial injury by mediating the Nrf2/HO1 signaling pathway to attenuate mitochondrial oxidative stress. Int Immunopharmacol. 2024;127:111382. doi:10.1016/j.intimp.2023.111382
200. Yang Y-P, Zhao J-Q, Gao H-B, et al. Tannic acid alleviates lipopolysaccharide‑induced H9C2 cell apoptosis by suppressing reactive oxygen species‑mediated endoplasmic reticulum stress. Mol Med Rep. 2021;24:535. doi:10.3892/mmr.2021.12174
201. Yang Z, LIU Y, DENG W, et al. Hesperetin attenuates mitochondria-dependent apoptosis in lipopolysaccharide-induced H9C2 cardiomyocytes. Mol Med Rep. 2014;9:1941–1946. doi:10.3892/mmr.2014.2002
202. Meng F, Lai H, Luo Z, et al. Effect of Xuefu Zhuyu decoction pretreatment on myocardium in sepsis rats. Evid Based Complement Alternat Med. 2018;2018:2939307. doi:10.1155/2018/2939307
203. Xing C, Xu L, Yao Y. Beneficial role of oleuropein in sepsis-induced myocardial injury. Possible Involvement of GSK-3β/NF-kB pathway. Acta Cir Bras. 2021;36:e360107. doi:10.1590/acb360107
204. Liu J, Li J, Tian P, et al. H2S attenuates sepsis-induced cardiac dysfunction via a PI3K/Akt-dependent mechanism. Exp Ther Med. 2019;17:4064–4072. doi:10.3892/etm.2019.7440
205. Wan -T-T, Li Y, Li JX, et al. ACE2 activation alleviates sepsis-induced cardiomyopathy by promoting MasR-Sirt1-mediated mitochondrial biogenesis. Arch Biochem Biophys. 2024;752:109855.
206. Zhang M, WANG X, BAI B, et al. Oxymatrine protects against sepsis-induced myocardial injury via inhibition of the TNF-α/p38-MAPK/caspase-3 signaling pathway. Mol Med Rep. 2016;14:551–559. doi:10.3892/mmr.2016.5250
207. Ji T, Liu Q, Yu L, et al. GAS6 attenuates sepsis-induced cardiac dysfunction through NLRP3 inflammasome-dependent mechanism. Free Radic Biol Med. 2024;210:195–211. doi:10.1016/j.freeradbiomed.2023.11.007
208. Shang X, Lin K, Yu R, et al. Resveratrol protects the myocardium in sepsis by activating the Phosphatidylinositol 3-Kinases (PI3K)/AKT/Mammalian Target of Rapamycin (mTOR) pathway and inhibiting the Nuclear Factor-κB (NF-κB) signaling pathway. Med Sci Monit. 2019;25:9290–9298. doi:10.12659/MSM.918369
209. Xiao Y, Yu Y, Hu L, et al. Matrine alleviates sepsis-induced myocardial injury by inhibiting ferroptosis and apoptosis. Inflammation. 2023;46:1684–1696. doi:10.1007/s10753-023-01833-2
210. Yang Y, Lei W, Qian L, et al. Activation of NR1H3 signaling pathways by psoralidin attenuates septic myocardial injury. Free Radic Biol Med. 2023;204:8–19. doi:10.1016/j.freeradbiomed.2023.04.006
211. Xie W-J, Hou G, Wang L, Wang -S-S, Xiong -X-X. Astaxanthin suppresses lipopolysaccharide‑induced myocardial injury by regulating MAPK and PI3K/AKT/mTOR/GSK3β signaling. Mol Med Rep. 2020;22:3338–3346. doi:10.3892/mmr.2020.11443
212. Wang L, Zhao Y, Su Z, et al. Ginkgolide A targets forkhead box O1 to protect against lipopolysaccharide-induced septic cardiomyopathy. Phytother Res. 2023;37:3309–3322. doi:10.1002/ptr.7802
213. Peng K, Yang F, Qiu C, Yang Y, Lan C. Rosmarinic acid protects against lipopolysaccharide-induced cardiac dysfunction via activating Sirt1/PGC-1α pathway to alleviate mitochondrial impairment. Clin Exp Pharmacol Physiol. 2023;50:218–227. doi:10.1111/1440-1681.13734
214. Qi Z, Wang R, Liao R, Xue S, Wang Y. Neferine ameliorates sepsis-induced myocardial dysfunction through anti-apoptotic and antioxidative effects by regulating the PI3K/AKT/mTOR signaling pathway. Front Pharmacol. 2021;12:706251. doi:10.3389/fphar.2021.706251
215. Ye G, Wang M, Liu D, et al. Mechanism of naringenin blocking the protection of LTB4/BLT1 receptor against septic cardiac dysfunction. Ann Clin Lab Sci. 2020;50:769–774.
216. Shi J, Chen Y, Zhi H, An H, Hu Z. Levosimendan protects from sepsis-inducing cardiac dysfunction by suppressing inflammation, oxidative stress and regulating cardiac mitophagy via the PINK-1-Parkin pathway in mice. Ann Transl Med. 2022;10:212. doi:10.21037/atm-22-483
217. Ma W, Huang Z, Miao Y, et al. ANXA1sp modulates the protective effect of Sirt3-induced mitophagy against sepsis-induced myocardial injury in mice. Acta Physiol. 2024;240:e14184. doi:10.1111/apha.14184
218. Jiang Y, Li Y, Zhang Y, et al. NSC228155 alleviates septic cardiomyopathy via protecting mitochondria and inhibiting inflammation. Int Immunopharmacol. 2023;116:109847. doi:10.1016/j.intimp.2023.109847
219. Tan Y, Zhang Y, He J, et al. Dual specificity phosphatase 1 attenuates inflammation-induced cardiomyopathy by improving mitophagy and mitochondrial metabolism. Mol Metab. 2022;64:101567. doi:10.1016/j.molmet.2022.101567
220. Kim M-J, Bae SH, Ryu J-C, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 2016;12:1272–1291. doi:10.1080/15548627.2016.1183081
221. Ye H, Wu L, Liu Y-M, et al. Wogonin attenuates septic cardiomyopathy by suppressing ALOX15-mediated ferroptosis. Acta Pharmacol Sin. 2025;46:2407–2422. doi:10.1038/s41401-025-01547-1
222. Xiao Z, Kong B, Fang J, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered. 2021;12:9367–9376. doi:10.1080/21655979.2021.2001913
223. Liu C, Zou Q, Tang H, et al. Melanin nanoparticles alleviate sepsis-induced myocardial injury by suppressing ferroptosis and inflammation. Bioact Mater. 2023;24:313–321. doi:10.1016/j.bioactmat.2022.12.026
224. Li X, Sun H, Zhang L, et al. GDF15 attenuates sepsis-induced myocardial dysfunction by inhibiting cardiomyocytes ferroptosis via the SOCS1/GPX4 signaling pathway. Eur J Pharmacol. 2024;982:176894. doi:10.1016/j.ejphar.2024.176894
225. Lu S-M, Yang B, Tan Z-B, et al. TaoHe ChengQi decoction ameliorates sepsis-induced cardiac dysfunction through anti-ferroptosis via the Nrf2 pathway. Phytomedicine. 2024;129:155597. doi:10.1016/j.phymed.2024.155597
226. Liu R, Li F, Hao S, et al. Low-dose Olaparib improves septic cardiac function by reducing ferroptosis via accelerated mitophagy flux. Pharmacol Res. 2024;200:107056. doi:10.1016/j.phrs.2023.107056
227. Fang X, Fu W, Zou B, Zhang F. Tectorigenin relieved sepsis-induced myocardial ferroptosis by inhibiting the expression of Smad3. Toxicol Res. 2023;12:520–526. doi:10.1093/toxres/tfad038
228. Zhou H, Dai Z, Li J, et al. TMBIM6 prevents VDAC1 multimerization and improves mitochondrial quality control to reduce sepsis-related myocardial injury. Metabolism. 2023;140:155383. doi:10.1016/j.metabol.2022.155383
229. Ji Y, Zhao M, Qi Y, et al. Effects of Arg-Gly-Asp-Ser on Ca2+ transport of myocardial sarcoplasmic reticulum in rat septic shock. Zhongguo Yao Li Xue Bao. 1996;17:129–132.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.