Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
The role of microglia in multiple sclerosis
Authors Luo C, Jian C, Liao Y, Huang Q, Wu Y, Liu X, Zou D ![]() , Wu Y
, Wu Y
Received 28 April 2017
Accepted for publication 22 May 2017
Published 26 June 2017 Volume 2017:13 Pages 1661—1667
DOI https://doi.org/10.2147/NDT.S140634
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Chun Luo,1 Chongdong Jian,1 Yuhan Liao,1 Qi Huang,1 Yuejuan Wu,1 Xixia Liu,1 Donghua Zou,2 Yuan Wu1
1Department of Neurology, The First Affiliated Hospital of Guangxi Medical University, 2Department of Neurology, The Fifth Affiliated Hospital of Guangxi Medical University and The First People’s Hospital of Nanning, Nanning, People’s Republic of China
Abstract: Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS). Microglia are the resident innate immune cells in the CNS; they play an important role in the processes of demyelination and remyelination in MS. Microglia can function as antigen-presenting cells and phagocytes. In the past, microglia were considered to be the same cell type as macrophages, and researchers have different opinions about the role of microglia in MS. This review focuses on the original classification of microglia and their role in the pathogenesis of MS. Moreover, we present a hypothetical model for the role of microglia in the pathogenesis of MS based on recent findings.
Keywords: microglia, multiple sclerosis, macrophage, myelin
Introduction
Multiple sclerosis (MS) is a chronic inflammatory and neurodegenerative disease of the central nervous system (CNS) characterized by focal lesions of inflammation, axonal loss, gliosis, and demyelination that affect the white and gray matter.1,2 In MS patients, destruction of myelin in the CNS is associated with activated macrophages or microglia,2–4 which are thought to be involved in the disease pathogenesis.5 Microglia are the resident innate immune cells of the CNS and play an important role in inflammatory and immune responses.6 Microglia express major histocompatibility (MHC) antigens I and II and secrete many proinflammatory and anti-inflammatory cytokines such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), and IL-10, as well as costimulatory molecules such as intercellular adhesion molecule-1 (ICAM-1), B7-1, and B7-2. Microglia also express Fc receptors (I–III) and complement receptors (CR1, CR2, and CR4).5,7 On one hand, microglia promote remyelination through the expression of anti-inflammatory molecules, phagocytosis of debris, and repair of tissues. On the other hand, microglia present antigens and secrete proinflammatory molecules that can damage the myelin sheath and/or oligodendrocytes.8 As antigen-presenting cells (APCs), microglia are involved in cross-talk with other immune cells, and such cross-talk leads to the activation of T cells during the course of demyelination and remyelination in MS.7,9
Origins of microglia
Activated microglia express many of the same cell surface markers as infiltrating monocyte-derived macrophages. It is also very difficult to distinguish these cell types because they are similar in morphology. In addition, they have many functional similarities, such as presentation of antigens and production of cytokines, oxidative radicals, chemokines, and NO. Because of the similarities in morphology and function, many researchers have referred to these cells collectively as macrophages/microglia. In addition, microglia and macrophages were originally thought to have originated from the same myeloid precursor and lineage in the bone marrow.10 However, recent reports showed that bone marrow-derived macrophages are very different from microglia. In fact, these two cell types are derived from distinct myeloid populations.11–13 On the basis of the serial block-face scanning electron microscopy, Yamasaki et al found that monocyte-derived macrophages are associated with nodes of Ranvier and initiated demyelination, whereas microglia clear debris in the experimental autoimmune encephalomyelitis (EAE) mouse model of MS.14 In the embryonic yolk sac, the erythromyeloid precursors migrate to the developing neural tube, and then differentiate into microglia.12,13 By contrast, macrophages are derived from the hematopoietic stem cells of the bone marrow, which differentiate into monocytes that circulate throughout the body, with the exception of the brain and spine parenchyma.
Subgroups of microglia
Activated microglia can be divided into M1 and M2 subgroups.15 M1 microglia promote inflammation and oligodendrocyte damage, whereas M2 microglia regulate immune functions and promote repair in inflammatory diseases of the CNS.16 M1 microglia produce proinflammatory cytokines such as IL-1β and TNF-α and induce NO synthase activity. In vitro, adult human M1 microglia express CD86, CD16/32, CD40, CD74, and CCR7 on their surfaces. In contrast, M2 microglia secrete anti-inflammatory cytokines, such as IL-10, and express CD206 (mannose receptor), the anti-inflammatory cytokine CCL22, and Arg1 (arginase).17 Some researchers found that microglia can differentiate into many subgroups with various functions upon different types of stimulation. Activation was shown to be strongly affected by the environment, which resulted in different activation states characterized by the production of different kinds of mediators and expression of various cell surface markers.18–22 Erny et al found that the function of microglia changed with the host microbiota.23 In addition, Xue et al found that activated microglia can be classified into at least nine subgroups based on a transcriptomic signature, which demonstrates the complexity and diversity of microglia.24 Using the EAE mouse model of demyelination, Olah et al found that microglia have different morphologies during demyelination and remyelination.25 Miron et al also observed plasticity in microglia during different phases of EAE where M2 microglia regulated the responses of oligodendrocyte progenitor cells (OPCs).26 In addition, changes in the morphology of microglia were accompanied by changes in function; during the course of remyelination, microglia phagocytosed apoptotic cells and myelin debris.26 These studies have revealed the plasticity of microglia and the diversity of microglial function under different conditions.
Microglia in the pathogenesis of MS
Microglia have various immunologic and neurobiologic functions that are closely associated with chronic inflammatory diseases, such as MS. The hallmark of MS pathology is demyelinated plaques in the white and gray matter with preservation of axons.27 A systematic study of brain biopsies showed that subcortical and cortical demyelination was present in patients in the early stages of MS.28 In the cuprizone-induced demyelination EAE model, activated microglia were found in lesions of the CNS and were thought to contribute to the CNS inflammation in MS.29,30 Besides the physical presence of microglia at sites of demyelination, the role of microglia in the pathogenesis of MS is only beginning to be revealed.
Microglia and oxidative damage
Several studies have suggested that oxidative injury plays an important role in the pathogenesis of demyelinating diseases, such as MS and Alzheimer’s disease. Extensive loss and apoptosis of mature oligodendrocytes were found in MS lesions,31 and oxidized DNA and lipids are present in oligodendrocytes and the myelin of active MS lesions.32,33 OPCs are particularly vulnerable to oxidative stress because they have lower levels of antioxidant enzymes and antiapoptotic proteins and higher levels of proapoptotic proteins.34,35 The oxidative burst in activated microglia and macrophages is regarded as the major source of reactive oxygen species (ROS) in MS.36 The extent of lipid and DNA oxidation correlated significantly with inflammation and oxidative injury of oligodendrocytes and neurons, which was also associated with active demyelination and axonal and neuronal injury in MS.37 Furthermore, van Horssen et al found extensive oxidative injury and upregulated expression of oxidative molecules and antioxidant enzymes in MS lesions.38–40 Other recent data also showed that activated microglia produce ROS and NO radicals in MS lesions, which suggested their role in the demyelination and neurodegeneration process of MS.41–45 These findings indicate that activated microglia are an important source of oxidative damage: activated microglia promote oxidative stress in MS, which accounts for the features of MS pathological findings.46 Ablation of microglia protected the gray and white matter in EAE mice.47 Microglial Hv1 proton channels promoted cuprizone-induced demyelination through ROS production. Liu et al found that Hv1−/− mice had reduced microglial accumulation, increased OPC proliferation, ROS production, and partial protection from demyelination and motor deficits after cuprizone exposure.48
Microglia in remyelination
Remyelination is the process of myelin regeneration occurring simultaneously with or following demyelination, which is characterized by the appearance of myelin around the axon. Clearance of debris and proliferation of oligodendrocytes are key processes in remyelination, followed by recruitment and proliferation of OPCs, which differentiate into mature myelinating oligodendrocytes.49 However, we observed that remyelination failed in the majority of MS patients, who became progressively disabled. The extent of remyelination was partly related to the location of the lesion; remyelination was found more frequently in lesions of the cortical or subcortical white matter than in periventricular areas.50 Lampron et al found that CX3CR1 knockout mice had reduced myelin debris clearance and impaired remyelination due to the ineffective function of microglia/macrophages.51
The phagocytosis of myelin debris by microglia and macrophages plays an important role in the initiation of remyelination.52 The reaction of macrophages to demyelination affects the local microenvironment and subsequently impacts the efficiency of remyelination.53 Due to a block in the differentiation of OPCs, remyelination usually fails in chronic MS.54 Kuhlmann et al found that microglia produced TNF-α, IGF-1, and FGF-2, which were very important for the proliferation of oligodendrocytes.55 IL-4 injection increased oligodendrocyte proliferation in the spinal cord of EAE mice, which suggested the potential involvement of microglia/macrophages in remyelination.56 Miron et al found that oligodendrocyte differentiation in cerebellar slices was improved in vitro by culture in M2 microglia-conditioned medium and decreased in vivo after depletion of M2 microglia; blocking of activin A secreted by M2 microglia suppressed oligodendrocyte differentiation during remyelination.57 The functions of microglia/macrophages in demyelination and remyelination are summarized in Table 1.
 | Table 1 Summary of the function of microglia/macrophage in demyelination and remyelination |
Microglia interactions with T cells
T cells play an important role in the pathogenesis of MS. Although the etiology of inflammation is still unclear, it has been found that T cells, specifically Th1 and Th17 cells, have an important link to MS pathology.59,60 In MS patients and animal models, infiltrating T cells and activated microglia are found in the CNS lesions. Microglia play a key role in recruitment of adaptive immune cells to the CNS.61 Activated microglia act as APCs through the expression of class I and II MHCs simultaneously with costimulatory molecules,62 through which T cells can recognize activated microglia. The microglia can interact with CD28 or cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) on T cells. The costimulatory molecules B7-1 and B7-2 expressed by microglia bind CD28 to positively stimulate the T cell to proliferate, differentiate, and secrete cytokines. Conversely, binding to CTLA4 causes T-cell anergy or apoptosis.63 The costimulatory molecule CD40 on microglia is also a key factor in T-cell activation. A study of mouse bone marrow chimeras found that CD40 knockout significantly abrogated T-cell activation in the CNS. Different kinds of T cells have different effects on microglia through cell-to-cell contact. Myelin basic protein (MBP)-primed Th1 cells (isolated from MBP-immunized mice) induced the expression of the proinflammatory molecules NO, IL-1, and TNF-α in mouse primary microglia and astroglia.64 MBP-primed Th2 cells, however, induced the production of brain-derived neurotrophic factor and neurotrophin-3.64 Ebner et al found that microglia, as inducible APCs, induced proliferation of Tregs and, therefore, promoted the balance between beneficial and inflammatory T-cell responses in EAE.65 Bsibsi et al found that IFN-γ, released by infiltrating T cells, along with heat shock protein (HSP)B5 changed the protective response of microglia into a robust proinflammatory response as shown by the release of TNF-α, IL-6, IL-12, IL-1β, and ROS.66 Oligodendrocytes are a pivotal local source of HSPB5 in MS-affected brains.66 The interaction between T cells and microglia likely affects many aspects of MS pathobiology. Under different conditions, T-cell interaction with microglia may generate different outcomes.
Hypothetical model of the pathogenesis of MS
In the quest to understand the pathogenesis of MS, many studies have focused on the T cell and have provided much insight. In particular, Th1 and Th17 cells have been considered central players in MS pathology. It has been widely accepted that the disease process is initiated by the adaptive immune response, then infiltration of activated T cells into the CNS with associated upregulation of proinflammatory mediators activate local microglia/macrophages, leading to inflammation and demyelination.67,68 This hypothesis considers T-cell-dependent and microglia/macrophage-mediated inflammation as central to this autoimmune disease.69,70 However, some researchers have also found that oligodendrocyte apoptosis in MS lesions or damage is not associated with T cells or peripheral macrophages.71–74 Insufficiency or the absence of lymphocyte recruitment after the initial relapsing phase of the disease indicates that other factors drive pathology in late-stage disease. There is increasing evidence of plasma cell and innate immunity involvement in progressive disease.75 These findings challenge the old theory.
In recent years, the contribution of innate immune cells to the pathogenesis of MS has been appreciated.76 Microglia comprise 10%–20% of glial cells and are the most common immune cells in the CNS.77,78 Microglia are considered resident macrophages of the CNS that express all known toll-like receptors (1–9).79 They are involved in phagocytosis, antigen presentation, and production of cytokines.60 Therefore, we postulate that the immune reaction initiates in the CNS and immune cells (including T cells/B cell) infiltrate into the CNS as a result. An unknown causative agent sets off a local inflammatory reaction, after which activated microglia are stimulated by the microenvironment to differentiate into many subgroups that then serve as APCs, phagocytes, and immune effector cells to activate T cells. Activated T cells can then cross the blood–brain barrier and result in a peripheral immune response. Subsequently, activated T cells, B cells, and macrophages migrate from the periphery into the CNS, which exacerbates the inflammation or leads to relapse. This hypothesis could explain some phenomena in MS patients. First, the APCs (microglia and macrophages) originate in the CNS, are recognized by CNS-resident cells, and remain localized within the CNS, which could explain why MS patients experience no peripheral complications, such as autoimmune nephritis and arthritis. Second, the inflammatory cascade occurs entirely within the CNS, which could explain the insufficiency or the absence of lymphocyte recruitment into the CNS after the initial relapsing phase of MS (Figure 1).71
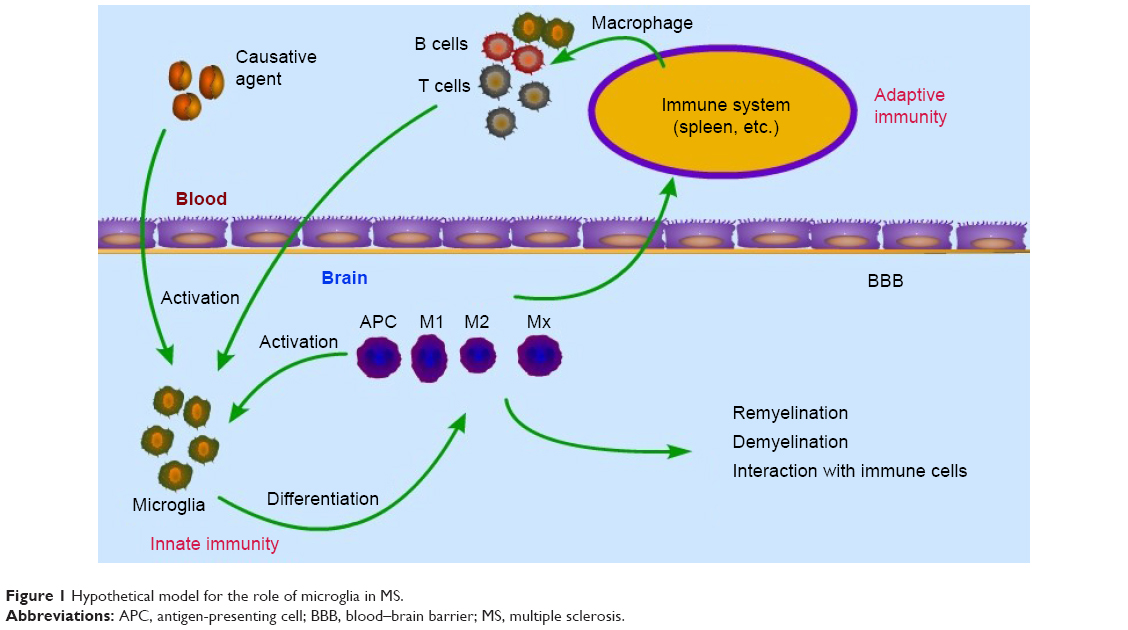 | Figure 1 Hypothetical model for the role of microglia in MS. |
Conclusion
There are likely more subgroups of microglia than presently known; therefore, determining the phenotypes and functions of each subgroup will be crucial for understanding the role of microglia in MS. We suggest the importance of defining macrophages80 according to their stimulation under experimental conditions and phenotype based on a combination of cell surface markers; this also could be an informative approach for microglia. A better understanding of the diversity of roles of microglia during inflammation and regeneration in the CNS will contribute to establishing new therapeutic strategies for MS patients.
Acknowledgments
This research was supported by funding from the Innovation Project of Guangxi Graduate Education, the Guangxi Natural Science Foundation (Grant No 2016GXNSFCA380012), the basic Ability Enhancement Program for Young and Middle-age Teachers of Guangxi (Grant No 2017KY056), and the Clinical Key Disciplines Construction Projects of Guangxi Zhuang Autonomous Region.
Disclosure
The authors report no conflicts of interest in this work.
References
De Groot CJ, Bergers E, Kamphorst W, et al. Post-mortem MRI-guided sampling of multiple sclerosis brain lesions: increased yield of active demyelinating and (p)reactive lesions. Brain. 2001;124(Pt 8):1635–1645. | ||
Lassmann H, Brück W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007;17(2):210–218. | ||
Babinski J. Recherches sur l’anatomie pathologique de la sclerose en plaque et étude comparative des diverses variétés de la scleroses de la moelle. Arch Physiol (Paris). 1885;5(6):186–207. | ||
Prineas JW, Kwon EE, Cho ES, et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol. 2001;50(5):646–657. | ||
Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mole Med (Berl). 1997;75(3):165–173. | ||
Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81(3):374–389. | ||
Frohman EM, Racke MK, Raine CS. Multiple sclerosis – the plaque and its pathogenesis. N Engl J Med. 2006;354(9):942–955. | ||
Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300–312. | ||
Voss EV, Škuljec J, Gudi V, et al. Characterization of microglia during de-and remyelination: can they create a repair promoting environment? Neurobiol Dis. 2012;45(1):519–528. | ||
Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: a review. Glia. 1988;1(5):301–307. | ||
Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. | ||
Schulz C, Perdiguero EG, Chorro L, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. | ||
Kierdorf K, Erny D, Goldmann T, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16(3):273–280. | ||
Yamasaki R, Lu H, Butovsky O, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211(8):1533–1549. | ||
Walker DG, Lue L. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimer’s ResTher. 2015;7(1):56. | ||
Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJ. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin- induced demyelination. Glia. 2001;35(3):204–212. | ||
Peferoen LA, Vogel DY, Ummenthum K, et al. Activation status of human microglia is dependent on lesion formation stage and remyelination in multiple sclerosis. J Neuropathol Exp Neurol. 2015;74(1):48–63. | ||
Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39(1):3–18. | ||
Durafourt BA, Moore CS, Zammit DA, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60(5):717–727. | ||
Giunti D, Parodi B, Cordano C, Uccelli A, Kerlero De Rosbo N. Can we switch microglia’s phenotype to foster neuroprotection? Focus on multiple sclerosis. Immunology. 2014;141(3):328–339. | ||
Melief J, Koning N, Schuurman KG, et al. Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia. 2012;60(10):1506–1517. | ||
Melief J, Schuurman KG, Garde MD, et al. Microglia in normal appearing white matter of multiple sclerosis are alerted but immunosuppressed. Glia. 2013;61(11):1848–1861. | ||
Erny D, de Angelis ALH, Jaitin D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965–977. | ||
Xue J, Schmidt SV, Sander J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–288. | ||
Olah M, Amor S, Brouwer N, et al. Identification of a microglia phenotype supportive of remyelination. Glia. 2012;60(2):306–321. | ||
Miron VE, Boyd A, Zhao J, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16(9):1211–1218. | ||
Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55(4):458–468. | ||
Lucchinetti CF, Popescu BF, Bunyan RF, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365(23):2188–2197. | ||
Sun SW, Liang HF, Trinkaus K, Cross AH, Armstrong RC, Song SK. Noninvasive detection of cuprizone induced axonal damage and demyelination in the mouse corpus callosum. Magn Reson Med. 2006;55(2):302–308. | ||
Gudi V, Gingele S, Skripuletz T, Stangel M. Glial response during cuprizone-induced de-and remyelination in the CNS: lessons learned. Front Cell Neurosci. 2014;8:73. | ||
Wolswijk G. Oligodendrocyte survival, loss and birth in lesions of chronic-stage multiple sclerosis. Brain. 2000;123(1):105–115. | ||
Smith KJ, Kapoor R, Felts PA. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9(1):69–92. | ||
Bizzozero OA, DeJesus G, Callahan K, Pastuszyn A. Elevated protein carbonylation in the brain white matter and gray matter of patients with multiple sclerosis. J Neurosci Res. 2005;81(5):687–695. | ||
Butts BD, Houde C, Mehmet H. Maturation-dependent sensitivity of oligodendrocyte lineage cells to apoptosis: implications for normal development and disease. Cell Death Differ. 2008;15(7):1178–1186. | ||
Blomgren K, Hagberg H. Free radicals, mitochondria, and hypoxia–ischemia in the developing brain. Free Radic Biol Med. 2006;40(3):388–397. | ||
Miller E, Wachowicz B, Majsterek I. Advances in antioxidative therapy of multiple sclerosis. Curr Med Chem. 2013;20(37):4720–4730. | ||
Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134(Pt 7):1914–1924. | ||
van Horssen J, Drexhage JA, Flor T, Gerritsen W, van der Valk P, de Vries HE. Nrf2 and DJ1 are consistently upregulated in inflammatory multiple sclerosis lesions. Free Radic Biol Med. 2010;49(8):1283–1289. | ||
van Horssen J, Schreibelt G, Bö L, et al. NAD(P)H: quinone oxidoreductase 1 expression in multiple sclerosis lesions. Free Radic Biol Med. 2006;41(2):311–317. | ||
Van Horssen J, Schreibelt G, Drexhage J, et al. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic Biol Med. 2008;45(12):1729–1737. | ||
Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated myeloperoxidase activity in white matter in multiple sclerosis. Neurosci Lett. 2008;444(2):195–198. | ||
Liu JS, Zhao M, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol. 2001;158(6):2057–2066. | ||
Fischer MT, Sharma R, Lim JL, et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. 2012;135(3):886–899. | ||
Fischer MT, Wimmer I, Höftberger R, et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain. 2013;136(Pt 6):1799–1815. | ||
Zeis T, Probst A, Steck AJ, Stadelmann C, Brück W, Schaeren Wiemers N. Molecular changes in white matter adjacent to an active demyelinating lesion in early multiple sclerosis. Brain Pathol. 2009;19(3):459–466. | ||
Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134(Pt 7):1914–1924. | ||
Heppner FL, Greter M, Marino D, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11(2):146–152. | ||
Liu J, Tian D, Murugan M, et al. Microglial Hv1 proton channel promotes cuprizone – induced demyelination through oxidative damage. J Neurochem. 2015;135(2):347–356. | ||
Domingues HS, Portugal CC, Socodato R, Relvas JB. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol. 2016;4:71. | ||
Albert M, Antel J, Brück W, Stadelmann C. Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol. 2007;17(2):129–138. | ||
Lampron A, Larochelle A, Laflamme N, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. 2015;212(4):481–495. | ||
Neumann H, Kotter MR, Franklin R. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132(Pt 2):288–295. | ||
Kotter MR, Zhao C, van Rooijen N, Franklin RJ. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol Dis. 2005;18(1):166–175. | ||
Kuhlmann T, Miron V, Cuo Q, Wegner C, Antel J, Brück W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131(Pt 7):1749–1758. | ||
Voß EV, Škuljec J, Gudi V, et al. Characterization of microglia during de- and remyelination: can they create a repair promoting environment? Neurobiol Dis. 2012;45(1):519–528. | ||
Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase +/IL-1β+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Rα after traumatic spinal cord injury. J Neurosci. 2014;34(26):8904–8917. | ||
Miron VE, Boyd A, Zhao J, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nature Neurosci. 2013;16(9):1211–1218. | ||
Di Penta, Chiba A, Alloza I, et al. A trifluoromethyl analogue of celecoxib exerts beneficial effects in neuroinflammation. Plos One. 2013;8(12):e83119. | ||
Jadidi Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol. 2011;74(1):1–13. | ||
Benveniste EN. Cytokines: influence on glial cell gene expression and function. Neuroimmunoendocrinology. 1997;69:31–75. | ||
Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Ann Rev Immunol. 2009;27:119–145. | ||
Almolda B, González B, Castellano B. Activated microglial cells acquire an immature dendritic cell phenotype and may terminate the immune response in an acute model of EAE. J Neuroimmunol. 2010;223(1–2):39–54. | ||
Bull ME, Vahlenkamp TW, Dow JL, et al. Spontaneous T cell apoptosis in feline immunodeficiency virus (FIV)-infected cats is inhibited by IL2 and anti-B7.1 antibodies. Vet Immunol Immunopathol. 2004;99(1–2):25–37. | ||
Roy A, Liu X, Pahan K. Myelin basic protein-primed T cells induce neurotrophins in glial cells via α5β3 integrin. J Biol Chem. 2007;282(44):32222–32232. | ||
Ebner F, Brandt C, Thiele P, et al. Microglial activation milieu controls regulatory T cell responses. J Immunol. 2013;191(11):5594–5602. | ||
Bsibsi M, Peferoen LA, Holtman IR, et al. Demyelination during multiple sclerosis is associated with combined activation of microglia/macrophages by IFN-γ and alpha B-crystallin. Acta Neuropathol. 2014;128(2):215–229. | ||
Adams C, Poston RN, Buk SJ. Pathology, histochemistry and immunocytochemistry of lesions in acute multiple sclerosis. J Neurol Sci. 1989;92(2):291–306. | ||
Hickey WF. The pathology of multiple sclerosis: a historical perspective. J Neuroimmunol. 1999;98(1):37–44. | ||
Prineas JW, Graham JS. Multiple sclerosis: capping of surface immunoglobulin G on macrophages engaged in myelin breakdown. Ann Neurol. 1981;10(2):149–158. | ||
Sriram S, Rodriguez M. Indictment of the microglia as the villain in multiple sclerosis. Neurology. 1997;48(2):464–470. | ||
Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55(4):458–468. | ||
Barnett MH, Henderson AP, Prineas JW. The macrophage in MS: just a scavenger after all? Pathology and pathogenesis of the acute MS lesion. Mult Scler J. 2006;12(2):121–132. | ||
Gay FW. Early cellular events in multiple sclerosis: intimations of an extrinsic myelinolytic antigen. Clin Neurol Neurosurg. 2006;108(3):234–240. | ||
Henderson AP, Barnett MH, Parratt JD, Prineas JW. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol. 2009;66(6):739–753. | ||
Weiner HL. A shift from adaptive to innate immunity: a potential mechanism of disease progression in multiple sclerosis. J Neurol. 2008;255(Suppl 1):3–11. | ||
Gandhi R, Laroni A, Weiner HL. Role of the innate immune system in the pathogenesis of multiple sclerosis. J Neuroimmunol. 2010;221(1):7–14. | ||
Bauer MK, Lieb K, Schulze Osthoff K, et al. Expression and regulation of cyclooxygenase-2 in rat microglia. Eur J Biochem. 1997;243(3):726–731. | ||
Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40(2):133–139. | ||
Aravalli RN, Peterson PK, Lokensgard JR. Toll-like receptors in defense and damage of the central nervous system. J Neuroimmune Pharmacol. 2007;2(4):297–312. | ||
Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.