Back to Journals » OncoTargets and Therapy » Volume 13
The Role of Methyltransferase NSD2 as a Potential Oncogene in Human Solid Tumors
Authors Chen R ![]() , Chen Y, Zhao W, Fang C, Zhou W, Yang X, Ji M
, Chen Y, Zhao W, Fang C, Zhou W, Yang X, Ji M
Received 25 April 2020
Accepted for publication 10 June 2020
Published 13 July 2020 Volume 2020:13 Pages 6837—6846
DOI https://doi.org/10.2147/OTT.S259873
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Rui Chen, Yan Chen, Weiqing Zhao, Cheng Fang, Wenjie Zhou, Xin Yang, Mei Ji
Department of Oncology, The Third Affiliated Hospital of Soochow University, The First People’s Hospital of Changzhou, Changzhou 213003, People’s Republic of China
Correspondence: Xin Yang; Mei Ji Email [email protected]; [email protected]
Abstract: Malignant solid tumors are the leading cause of death in humans, and epigenetic regulation plays a significant role in studying the mechanism of human solid tumors. Recently, histone lysine methylation has been demonstrated to be involved in the development of human solid tumors due to its epigenetic stability and some other advantages. The 90-kb protein methyltransferase nuclear receptor SET domain-containing 2 (NSD2) is a member of nuclear receptor SET domain-containing (NSD) protein lysine methyltransferase (KMT) family, which can cause epigenomic aberrations via altering the methylation states. Studies have shown that NSD2 is frequently over-expressed in multiple types of aggressive solid tumors, including breast cancer, renal cancer, prostate cancer, cervical cancer, and osteosarcoma, and such up-regulation has been linked to poor prognosis and recurrence. Further studies have identified that over-expression of NSD2 promotes cell proliferation, migration, invasion, and epithelial–mesenchymal transformation (EMT), suggesting its potential oncogenic role in solid tumors. Moreover, Gene Expression Profiling Interactive Analysis (GEPIA) was searched for validation of prognostic value of NSD2 in human solid tumors. However, the underlying specific mechanism remains unclear. In our present work, we summarized the latest advances in NSD2 expression and clinical applications in solid tumors, and our findings provided valuable insights into the targeted therapeutic regimens of solid tumors.
Keywords: NSD2, proliferation, migration, invasion, EMT, oncogene
Introduction
Malignant solid tumors are common neoplasms with high morbidity and mortality in humans. A number of studies have shown that epigenetic regulation plays a critical role in cancer initiation, development, and treatment, and the most active markers include DNA methylation or demethylation, histone modification, and chromatin remodeling.1,2 Histone lysine methylation, as one of the “histone codes”, has gradually been recognized as an emerging focus due to its epigenetic stability and some other advantages.3 The nuclear receptor-binding SET domain (NSD) family of histone H3 lysine 36 methyltransferases recognizes and catalyzes the methylation of histone. Recently, several studies have reported that NSD methyltransferases were amplified, mutated, or over-expressed in human cancers, implying their crucial role in oncogenesis and cancer development.4 Tri-methyltransferase SETD2 is mutated in a range of human cancers and acts as a potential tumor suppressor.5,6 NSD2, also known as MMSET (multiple myeloma SET domain) or WHSC1 (Wolf-Hirschhorn syndrome candidate 1), a member of the NSD protein family,7–9 has been reported as the responsible gene for the disease of Wolf-Hirschhorn syndrome. It is also closely related to accelerated disease progression and rapid relapse in 15–20% of multiple myeloma (MM) harboring the t (4;14) chromosomal translocation.4,10,11 In addition to these diseases, NSD2 over-expression is detected in several human solid tumors, such as breast cancer, lung cancer, renal cancer, and prostate cancer.12,13 NSD2 specifically catalyzes di-methylation of histone H3 lysine 36 (H3K36me2) in mammals.14–19 NSD2 has a series of biological functions, including DNA damage repair and epithelial–mesenchymal transformation (EMT) process, and it can co-act with enhancer of zeste homolog 2 (EZH2). Additionally, GEPIA based on The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) databases was further applied for validation of the expression of NSD2 and prognostic significance in seven different solid tumors. NSD2 may function as an oncogene, and it is expected to become a new therapeutic target for human solid tumors.14,20
Protein Structure of NSD2
NSD2 has the lowest molecular weight among all NSD protein family members,21 due to alternative splicing, it yields three main isoforms of protein, MMSET-I (containing 647 amino acids), MMSET-II (containing 1365 amino acids) and RE-IIBP (interleukin-5 response element II-binding protein) (containing 584 amino acids). MMSET-II and RE-IIBP possess a methyltransferase activity domain, AWS domain (associated with SET), SET domain (Su (var)3–9, Enhancer-of-zeste, Trithorax) and pro-SET domain (following the SET domain). Therefore, these types of protein enzyme can participate in the transfer of methyl groups during histone lysine methylation, while MMSET-I lacks enzymatic activity due to the absence of this important region.7 The PWWP (proline-tryptophan-tryptophan-proline) domain is the only known domain, which presents in all three isoforms of NSD2 protein and interacts with histone lysine to facilitate stability of NSD2 at chromatin. PWWP domain acts as the reader domain that preferentially combines with nucleosomes containing H3K36me2.22 In addition, MMSET-II is also composed of four PHD zinc finger domains and one HMG (high-mobility group) domain, whereas RE-IIBP is composed of two PHD zinc finger domains but lacks the HMG domain.
Biological Functions of NSD2
Multiple studies have identified that NSD2 is involved in a large number of cellular processes, many of which can cause tumorigenesis. It remains unclear how dysregulation of NSD2 affects these processes. Here, we reviewed the most crucial biological functions and their potential molecular mechanisms.
NSD2 Promotes DNA Damage Repair
Previous studies have uncovered the role of NSD2 in human DNA damage repair.16,23,24 Pei et al have reported that p53-binding protein 1 (53BP1, an important mediator of DNA damage repair) and di-methylation of histone H4 lysine 20 (H4K20me2) are implicated in DNA damage repair. NSD2 is required for the methylation of H4K20 at DNA double-strand breaks (DSBs) and the recruitment of 53BP1 to sites of DNA damage. Down-regulation of NSD2 significantly decreases the methylation of H4K20, subsequently leading to the increased accumulation of 53BP1.16 Generally, H4K20me2 is established in the absence of DNA damage via mono-methylation of newly synthesized H4K20 mediated SETD8, and de-methylation of H4K20me1 mediated the SUV420H1 and SUV420H2 methyltransferases rather than de novo H4K20 methylation by NSD2.25,26 In addition, γH2AX-MDC1 signaling pathway is involved in the recruitment of NSD2 to DSBs.16 Zhang et al have reported that NSD2 can interact with phosphatase and tensin homolog deleted on chromosome 10 (PTEN) via its C-terminal domain and dramatically stimulate the di-methylation of PTEN in cells. The latter is recognized by the specific domain of 53BP1 to recruit PTEN into sites of DNA damage, which regulates the sensitivity of cells to DNA damage, and efficiently completes the DSB repair partly through dephosphorylation of γH2AX. Interestingly, only the MMSET-II has a high binding affinity to PTEN.23 Therefore, the γH2AX-MDC1-NSD2 axis participates in DNA damage repair and plays a major role.
NSD2 Regulates the Process of EMT
EMT is a crucial process in cancer development, in which the epithelial cell acquires characteristics of the mesenchymal cell during tumorigenesis, development, and progression.27–29 Recent studies have supported the role of NSD2 in the invasion and metastasis of tumor cells via EMT process. NSD2 regulates EMT-related protein, and down-regulation of NSD2, the expression level of E-cadherin protein increased while N-cadherin protein and vimentin protein decreased. The mechanism behind these processes has been linked to NSD-mediated twist family bHLH transcription factor 1 (TWIST1, a transcription factor that promotes cell invasion and metastasis in several cancers) and H3K36me2, and NSD2 can interact with TWIST1 and then lead to an increase in H3K36me2, thus promoting the EMT.30,31 A recent study identified that the over-expression of NSD2 occurs in 15% of patients with t (4;14)-positive MM and that Twist-1 participates in driving the expression of EMT-related genes and contributes to tumor migration.32
The EZH2-NSD2-Histone Methyltransferase (HMTase) Axis
Previous studies have proven that EZH2, which mediates the tri-methylation of H3K27 and is related to gene silencing, is an important oncogenic driver and may play a potential therapeutic role in human cancers.33–35 Recently, relevant studies have identified a tight relationship between NSD2 and EZH2. The EZH2-NSD2-HMTase axis is coordinated by a microRNA network, which is regulated via microRNA miR-31, miR-26a, and miR-203. EZH2 recognizes and mediates H3K27 to generate tri-methylation of histone H3 lysine K27 (H3K27me3), and the latter can suppress the expressions of the above-mentioned microRNAs, finally leading to the up-regulation of NSD2 at mRNA level.36,37 Another study has uncovered that NSD2 alters EZH2 binding and reprograms the myeloma epigenome via focal and global changes in the methylation of H3K27 and H3K36me2.38 Over-expression of NSD2 induced a global reduction of H3K27me3. However, specific genomic loci display increased recruitment of the EZH2 histone methyltransferase and become hypermethylated on this residue. Moreover, EZH2 has been reported to be coordinately expressed and functioning upstream of NSD2 (mediates di-methylation of H3K36 and is related to active transcription). Over-expression of NSD2 causes a global un-regulation in H3K36me2, redistributing this mark in a broad, increased level across the genome.38 NSD2 and EZH2 co-act and function as a master regulator of transcription activation, repression, and tumorigenesis, which may represent a crucial therapeutic target in human cancers.39
Other Functions
Other studies have also reported that NSD2 mediates Wnt pathway, NF-kB signaling for oncogenesis, proliferation, and survival.40,41 NSD2 boosts the proliferation of cancer cells via regulating the expressions of cancer targets, such as necrosis factor α (TNF-α), interleukin 6 (IL-6), and IL-8,41 or via reducing the p53 stability.42 NSD2 participates in regulating in cell cycle and facilitating normal DNA replication43,44 (Figure 1).
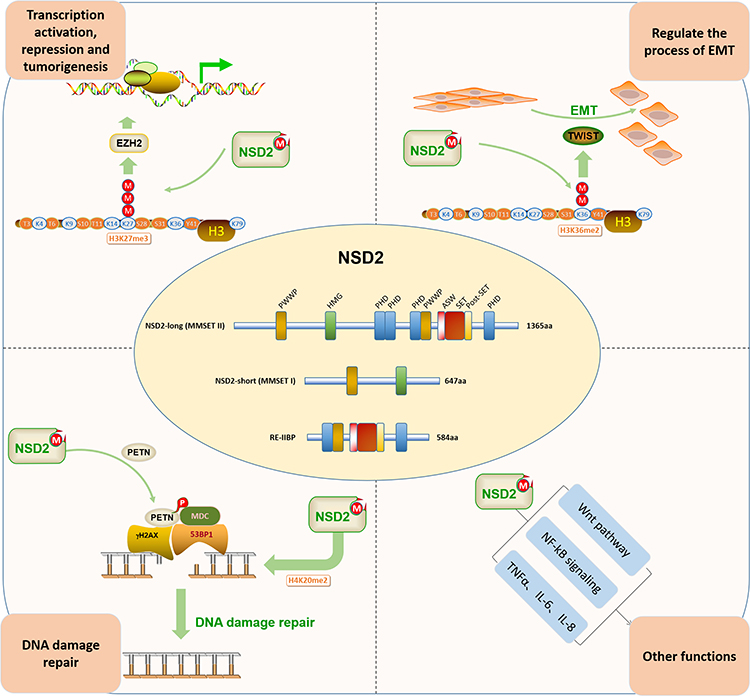 |
Figure 1 The biological functions of NSD2. DNA damage repair, epithelial–mesenchymal transformation (EMT) process, co-act with epithelial–mesenchymal transformation (EZH2) and other functions. |
Prognosis Analysis Using Bioinformatics Method
We clearly performed an exploration of the expression levels of NSD2 in seven different solid tumors based on GEPIA (http://gepia.cancer-pku.cn/index.html) (a significant and highly cited resource for gene expression analysis from the TCGA and the GTEx databases). Kaplan–Meier survival analysis with the Log-rank test was used to describe the correlation between NSD2 expression levels and overall survival (OS) and disease-free survival (DFS) in seven different solid tumors through the GEPIA databases, respectively, P<0.05 was considered to be statistically significant (Figures 2 and 3).
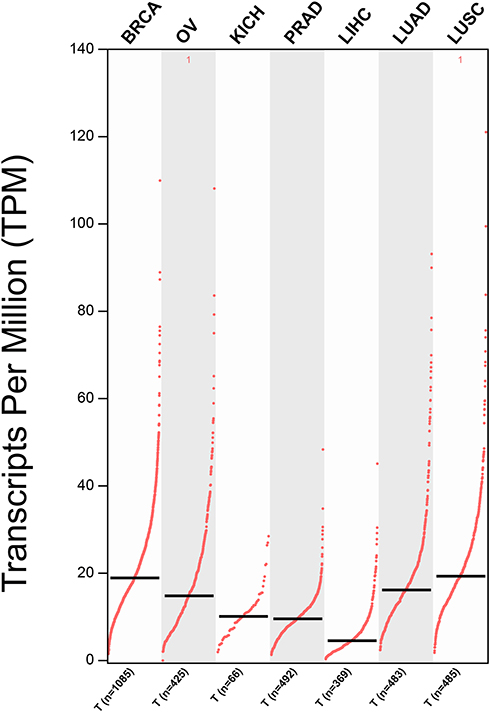 |
Figure 2 The expression levels of NSD2 were detected from the GEPIA public databases in seven different types of solid cancers. Abbreviations: BRCA, breast invasive carcinoma; OV, ovarian serous; KICH, kidney chromophobe; PRAD, prostate adenocarcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma. |
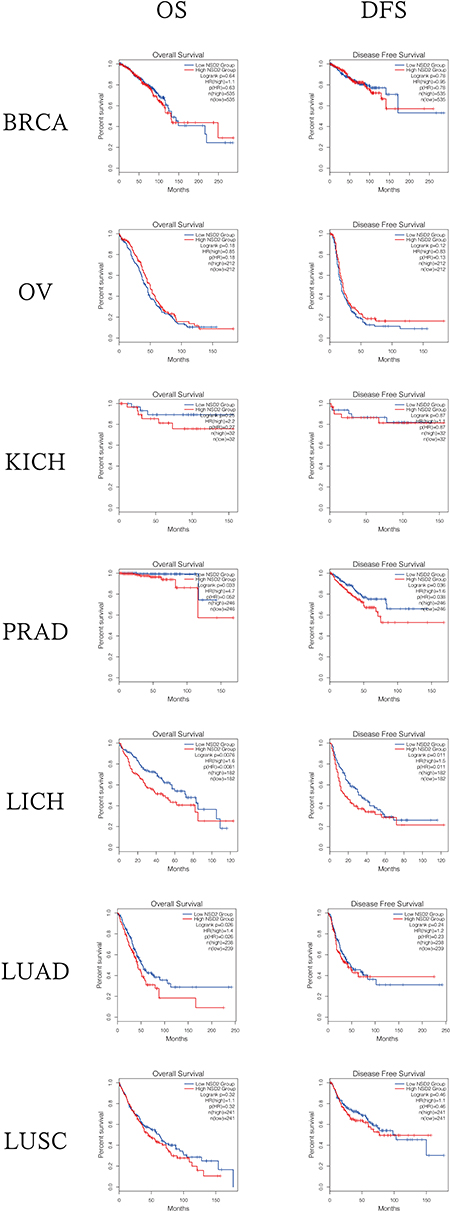 |
Figure 3 The overall survival (OS) analyses and disease-free survival (DFS) based on the Gene Expression Profiling Interactive Analysis (GEPIA) public databases. Abbreviations: BRCA, breast invasive carcinoma; OV, ovarian serous; KICH, kidney chromophobe; PRAD, prostate adenocarcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; HR, hazard ratio. |
The Clinical Applications of NSD2 and Solid Tumors
NSD2 has been clarified to play a significant role in blood cancers. The over-expression of NSD2 exhibit increased tumor dissemination, accelerated disease progression and rapid relapse in patients with MM.19,32 Studies have uncovered that mutation of NSD2 also occurs in acute lymphoblastic leukemia (ALL) and mutant NSD2 cell display reduced apoptosis and increasing proliferation, clonogenicity, migration, and adhesion.45,46 Similarly, the abnormal NSD2 expression exists in human solid tumors, we summarized the relevant researches about NSD2 expression level and its potential clinical applications.
NSD2 in Breast Cancers
The latest global cancer statistics has shown that breast cancer is a malignancy in women with high incidence and mortality worldwide,47,48 and the diagnosis and treatment of breast cancers, especially triple-negative breast cancers (TNBC), remain a challenge all over the world. Wang et al have shown that NSD2 is over-expressed in both breast tissues and cells. Kaplan–Meier analysis has shown that such abnormal expression of NSD2 is significantly correlated with earlier disease-related death, which is consistent with the analysis result from a series of published tumor datasets that over-expression of NSD2 is associated with adverse survival of patients with TNBC (P=0.027). Further research has shown that NSD2 participates in regulating TNBC cell survival and invasion via mediating the ADAM9-EFGR-AKT signaling pathway. Additionally, the up-regulation of NSD2 activates the EGFR-AKT signaling pathway and facilitates the TNBC cell resistance to EGFR inhibitors.49 In another study, Zhao et al have detected the NSD2 expression in breast cancers by real-time PCR and Western blotting analysis. Their results have indicated that NSD2 is strongly over-expressed in cancer cells and clinical specimens and that a higher expression of NSD2 is correlated with the poor prognosis and clinicopathological characteristics. The silencing of NSD2 leads to markedly decreased cell proliferation and metastasis via inhibiting the Wnt/β-catenin pathway.50 Endocrine therapy, such as tamoxifen, is commonly used for the prevention and treatment of patients with estrogen receptor alpha (ERα)-positive postmenopausal breast cancers. Wang et al have shown that NSD2 is over-expressed in tamoxifen-resistant breast cancers and associated with disease recurrence and poor prognosis. According to Wang et al, NSD2 was recruited and di-methylated H3K36 at the promoters of key glucose metabolic enzyme genes. The over-expression of NSD2 coordinately up-regulated glucose-6-phosphate dehydrogenases (G6PD), hexokinase 2 (HK2), and TP-53-mediated glycolysis regulatory phosphatase TIGAR. Therefore, NSD2-mediated tamoxifen-resistant cells and tumor exhibited a heightened activity of pentose phosphate pathway (PPP), a higher level of nicotinamide adenine dinucleotide phosphate (NADPH) and a reduced level of ROS, yet leaving the glycolysis process intact. As a result, the whole glucose metabolism process was affected.51 Currently, another study in 2020 has reported that DZNep, an indirect small-molecule inhibitor, induces NSD2 protein degradation and inhibits NSD2 target genes expression, such as G6PD, HK2, TIGAR, and GLUT1.52 Another study has identified that NSD2 positively regulates the expression of ERα via BET protein BRD3/4.53
NSD2 in Cancers of the Urinary System
Han et al have shown that NSD2 mRNA is over-expressed in renal cancer based on GEO database, and immunohistochemical results have shown that the expression of NSD2 at the protein level is higher in clear cell renal cancer, while such up-regulation is not associated with pathological grading. The depletion of NSD2 represses cell migration and invasion. Meanwhile, the expression of E-cadherin is increased, while the expressions of N-cadherin and vimentin are decreased. Overall, down-regulation of NSD2 can inhibit the metastasis of renal cancer cells through suppressing EMT in renal cancer.54 Similarly, NSD2 is over-expressed in the prostate cancer cells, and depletion of NSD2 significantly inhibits cell proliferation, migration, and invasion, while up-regulation of NSD can facilitate cell migration, invasion, and EMT. Another study has found that NSD2 may promote the EMT process and invasion through activating TWIST1.30 Aytes et al have shown that NSD2 is strongly up-regulated in lethal human prostate cancer, and silencing of NSD2 can repress the metastasis in vivo.55 Yang et al have reported that the up-regulation of NSD2 is correlated with the expressions of IL-6 and TNF-α in the prostate, emphasizing that NSD2 plays a crucial role in the proliferation and survival of cancer cells.41 Li et al have identified that increased activity of AKT due to phosphatase and tensin homolog (PTEN) loss directly phosphorylates NSD2 at S172, preventing the degradation of NSD2. Up-regulated of NSD2 expression transcriptionally increases expression of RICTOR (a key component of mTOR complex 2, mTORC2), to further enhance the activity of AKT. Moreover, NSD2 positively regulates Rac1 transcription to increase cell motility. Therefore, it is believed that there exists a pivotal link between NDS2, AKT, RICTOR, and Rac1, which can co-drive the prostate metastasis.56
NSD2 in Cancers of Female Reproductive System
Previous studies have reported that NSD2 is over-expressed in the cervical cancer cells and tissues and associated with unfavorable prognosis.57,58 Wu et al have displayed that a high level of NSD2 is associated with differentiation and stage, depletion of NSD2 inhibits cell proliferation, migration, and invasion, and this is affected by endothelial nitric oxide synthase (eNOS) signaling pathway.57 In agreement with Wu, Yin et al have shown that up-regulation of NSD2 significantly promotes the proliferation, migration, and invasion of cancer cells, and the underlying mechanism may be attributed to the activation of the AKT/metalloproteinase-2 (MMP-2) signaling pathway upon over-expression of NSD2.58 Zhu et al have confirmed that the growth of cancer cells is reduced after depletion of NSD2 in vivo and in vitro. Meanwhile, cancer metastasis is inhibited through the regulation of TGF-1 (transforming growth factor-1)/TGF-RI/SMAD signaling pathway.59 In endometrial cancers, NSD2 is over-expressed in cancer tissue compared with normal tissue, and patients with a higher NSD2 expression have a poorer OS and DFS compared with the rest.60 Taken together, NSD2 may act as a novel biomarker in female reproductive cancers.
NSD2 in Other Solid Tumors
García-Carpizo has reported that NSD2 promotes the proliferation of a subset of lung cancer cells through supporting RAS-driven transcription.61 Studies have found that NSD2 is over-expressed in osteosarcoma cells and is associated with adverse prognosis and poor 5-year OS. Moreover, up-regulation of NSD2 promotes the proliferation and invasion of cancer cells, and this may be attributed to the induction of EMT process.62 Another study has reported that down-regulation of NSD2 significantly decreases the apoptosis of osteosarcoma cells. On the contrary, the effect of cisplatin on osteosarcoma cells is markedly enhanced, thereby inhibiting the properties associated with cancer stem cells, and all of these may originate from the regulation of apoptotic regulatory proteins SOX2 and BCL2 via the AKT and ERK pathways.63 Zhou et al have investigated the expression of NSD2 in hepatocellular carcinoma from 103 patients, and the results have shown that NSD2 is over-expressed in hepatocellular carcinoma and correlated with Edmondson stage and vascular invasion. In addition, a higher expression of NSD2 has been linked to a shorter OS and DFS, indicating that NSD2 acts as an independent prognostic factor in patients with hepatocellular carcinoma.64 Saloura et al have shown that the expression of NSD2 is higher in head and neck squamous cell carcinoma compared with normal tissues and that such up-regulation is linked to a poor grade. Depletion of NSD2 inhibits the viability of cancer cells as well as the methylation level of H3K36. Furthermore, NSD2 affects cell growth, apoptosis, and progression by mediating a cell cycle regulator NIMA-related-kinase-7 (NEK7).65,66 NSD2 is also over-expressed in human skin squamous cell carcinoma, and NSD2 and microRNA-154 negatively regulate the proliferation of cancer cells via mediating the p53 signaling pathway.67 NSD2 is significantly over-expressed in neuroblastoma (NSD2 positivity 75%, n=164), and such up-regulation is correlated with a poor survival (P=0.012) than those with lower or no expression. Interestingly, Hudlebusch et al have found that the expression of NSD2 is strongly decreased after chemotherapy at the protein level, which is consistent with the result that NSD2 is markedly down-regulated in neuroblastoma cells induced by retinoic acid in vitro68 (Table 1).
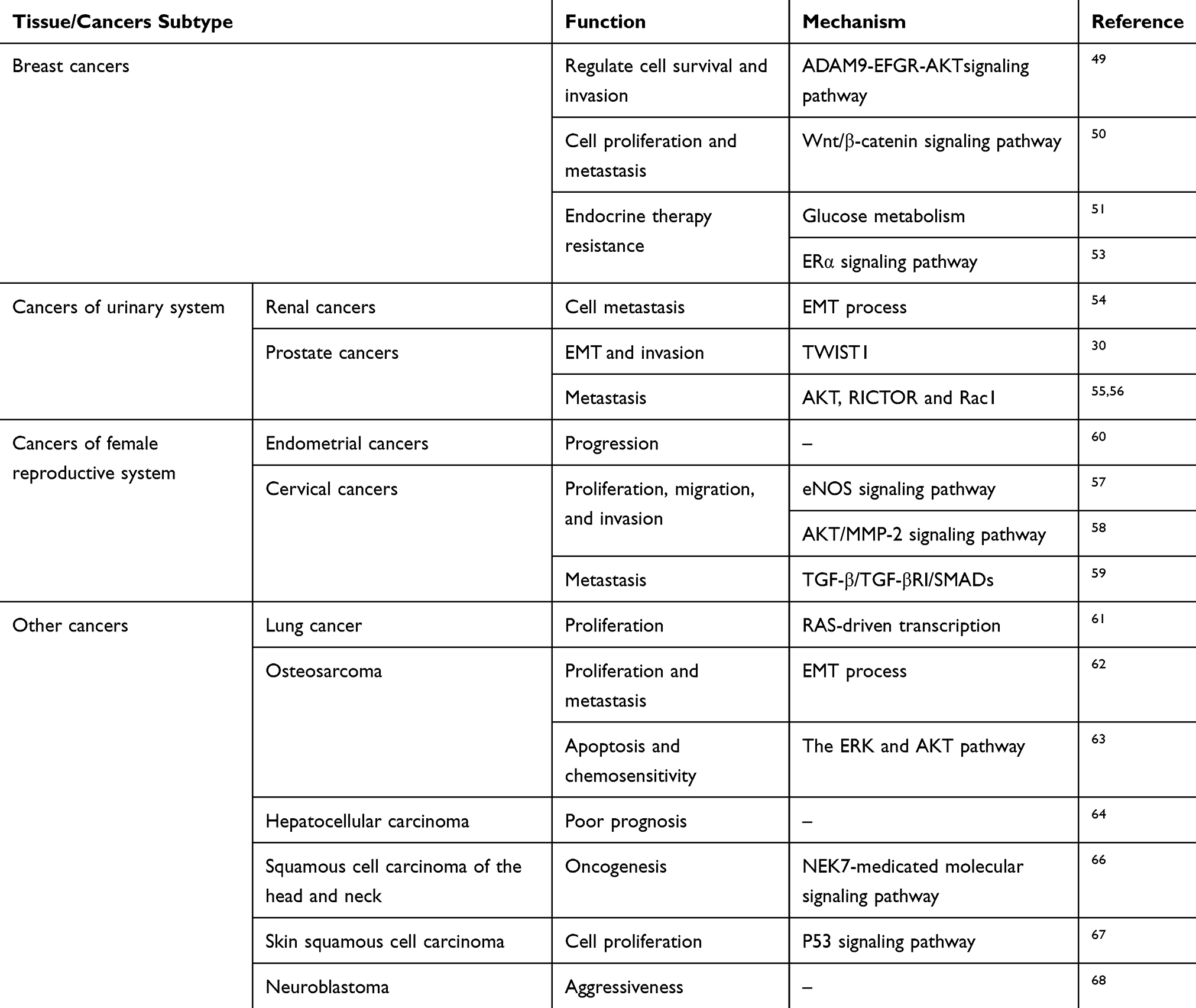 |
Table 1 Overview of NSD2 in a Selection of Several Human Solid Tumors Based on This Work |
NSD2 Small-Molecule Inhibitors
NSD2 is a potential therapeutic target for malignant solid tumors, it is therefore important to explore and develop the selective, potent, and bioactive small-molecule inhibitors. Coussens et al established a platform for the study and development of NSD2 inhibitors, and optimized and accomplished multiple assays. Five conformed NSD2 inhibitors were selected for follow-up studies, and the results showed that all five inhibitors successfully bind the catalytic SET domain and one displayed apparent activity.14 In addition, studies have reported that MCTP39, Sinefungin, and an N-Alkyl Sinefungin derivative, LEM-14, as NSD2 inhibitors.69,70
Conclusions and Perspectives
Malignant cancers have become the major cause of death due to the worsening of environmental pollution, hereditation, genetic abnormality, higher pressure, and some other factors, becoming a leading health challenge in the world. A great deal of attention has been paid to the modification of histone lysine, one of “histone codes”, because of its epigenetic stability, and activation of certain chromatin-related diseases, especially in H3K36. NSD2 is a histone lysine methyltransferase, which can catalyze histone lysine in the mono-methylated state into histone lysine in the di-methylated state through the transfer of methyl group. A large number of studies have shown that NSD2 is over-expressed in a variety of solid tumors, and such abnormal expression is related to the occurrence and development of cancers, often indicating poor prognosis. Depletion of NSD2 significantly inhibits cell proliferation, migration, invasion, and EMT process, while over-expression of NSD2 shows the opposite effect, suggesting that NSD2 functions as an oncogene in solid tumors. However, the specific mechanism underlying such process remains largely unexplored.
In the present review, all of these findings show that NSD2 is abnormally expressed in multifarious solid tumors, including thoracic cancers, urinary and female reproductive cancers, osteosarcoma, hepatocellular carcinoma, head and neck squamous cell carcinoma, skin cancer, and neuroblastoma, suggesting that NSD2 potentially plays a key role in cell growth, proliferation, migration, invasion, and EMT process. Collectively, NSD2 may play a fundamental role in determining the recurrence and prognosis of various cancers, and can be used as a potential therapeutic target in human solid tumors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Costa-Pinheiro P, Montezuma D, Henrique R, Jerónimo C. Diagnostic and prognostic epigenetic biomarkers in cancer. Epigenomics. 2015;7(6):1003–1015. doi:10.2217/epi.15.56
2. Verma M. The role of epigenomics in the study of cancer biomarkers and in the development of diagnostic tools. Adv Exp Med Biol. 2015;867:59–80.
3. Fang D, Gan H, Lee JH, et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. 2016;352(6291):1344–1348. doi:10.1126/science.aae0065
4. Vougiouklakis T, Hamamoto R, Nakamura Y, Saloura V. The NSD family of protein methyltransferases in human cancer. Epigenomics. 2015;7(5):863–874. doi:10.2217/epi.15.32
5. Chen R, Zhao WQ, Fang C, Yang X, Ji M. Histone methyltransferase SETD2: a potential tumor suppressor in solid cancers. J Cancer. 2020;11(11):3349–3356. doi:10.7150/jca.38391
6. Hu M, Liu X, Lai J, et al. SETD2, an epigenetic tumor suppressor: a focused review on GI tumor. Front Biosci Landmark. 2020;25(4):781–797. doi:10.2741/4834
7. Bennett RL, Swaroop A, Troche C, Licht JD. The role of nuclear receptor-binding set domain family histone lysine methyltransferases in cancer. Cold Spring Harb Perspect Med. 2017;7(6):
8. Kuo AJ, Cheung P, Chen K, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44(4):609–620. doi:10.1016/j.molcel.2011.08.042
9. Poulin MB, Schneck JL, Matico RE, et al. Transition state for the NSD2-catalyzed methylation of histone H3 lysine 36. Proc Natl Acad Sci U S A. 2016;113(5):1197–1201. doi:10.1073/pnas.1521036113
10. Lhoumaud P, Badri S, Rodriguez-Hernaez J, et al. NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat Commun. 2019;10(1):4843. doi:10.1038/s41467-019-12811-4
11. Xie Z, Bi C, Chooi JY, Chan ZL, Mustafa N, Chng WJ. MMSET regulates expression of IRF4 in t(4;14) myeloma and its silencing potentiates the effect of bortezomib. Leukemia. 2015;29(12):2347–2354. doi:10.1038/leu.2015.169
12. Hudlebusch HR, Santoni-Rugiu E, Simon R, et al. The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin Cancer Res. 2011;17(9):2919–2933. doi:10.1158/1078-0432.CCR-10-1302
13. Kassambara A, Klein B, Moreaux J. MMSET is overexpressed in cancers: link with tumor aggressiveness. Biochem Biophys Res Commun. 2009;379(4):840–845. doi:10.1016/j.bbrc.2008.12.093
14. Coussens NP, Kales SC, Henderson MJ, et al. High-throughput screening with nucleosome substrate identifies small-molecule inhibitors of the human histone lysine methyltransferase NSD2. J Biol Chem. 2018;293(35):13750–13765. doi:10.1074/jbc.RA118.004274
15. Huang X, LeDuc RD, Fornelli L, et al. Defining the NSD2 interactome: PARP1 PARylation reduces NSD2 histone methyltransferase activity and impedes chromatin binding. J Biol Chem. 2019;294(33):12459–12471. doi:10.1074/jbc.RA118.006159
16. Pei H, Zhang L, Luo K, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470(7332):124–128. doi:10.1038/nature09658
17. Poulin MB, Schneck JL, Matico RE, et al. Nucleosome binding alters the substrate bonding environment of histone h3 lysine 36 methyltransferase NSD2. J Am Chem Soc. 2016;138(21):6699–6702. doi:10.1021/jacs.6b01612
18. Martinez-Garcia E, Popovic R, Min DJ, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117(1):211–220. doi:10.1182/blood-2010-07-298349
19. Huang Z, Wu H, Chuai S, et al. NSD2 is recruited through its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer Res. 2013;73(20):6277–6288. doi:10.1158/0008-5472.CAN-13-1000
20. Bobby R, Peciak K, Milbradt AG. Backbone resonance assignments for the SET domain of the human methyltransferase NSD2. Biomol NMR Assign. 2016;10(2):307–310. doi:10.1007/s12104-016-9689-4
21. Nimura K, Ura K, Shiratori H, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to wolf-hirschhorn syndrome. Nature. 2009;460(7252):287–291. doi:10.1038/nature08086
22. Sankaran SM, Wilkinson AW, Elias JE, Gozani O. A PWWP domain of histone-lysine n-methyltransferase nsd2 binds to dimethylated lys-36 of histone h3 and regulates nsd2 function at chromatin. J Biol Chem. 2016;291(16):8465–8474. doi:10.1074/jbc.M116.720748
23. Zhang J, Lee YR, Dang F, et al. PTEN methylation by NSD2 controls cellular sensitivity to DNA damage. Cancer Discov. 2019;9(9):1306–1323. doi:10.1158/2159-8290.CD-18-0083
24. Shah MY, Martinez-Garcia E, Phillip JM, et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene. 2016;35(45):5905–5915. doi:10.1038/onc.2016.116
25. Hsiao KY, Mizzen CA. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J Mol Cell Biol. 2013;5(3):157–165. doi:10.1093/jmcb/mjs066
26. Wu H, Siarheyeva A, Zeng H, et al. Crystal structures of the human histone H4K20 methyltransferases SUV420H1 and SUV420H2. FEBS Lett. 2013;587(23):3859–3868. doi:10.1016/j.febslet.2013.10.020
27. Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi:10.1016/j.cell.2004.06.006
28. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5(9):744–749. doi:10.1038/nrc1694
29. Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119(6):1438–1449. doi:10.1172/JCI38019
30. Ezponda T, Popovic R, Shah MY, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene. 2013;32(23):2882–2890. doi:10.1038/onc.2012.297
31. Kwok WK, Ling MT, Lee TW, et al. Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. 2005;65(12):5153–5162. doi:10.1158/0008-5472.CAN-04-3785
32. Cheong CM, Mrozik KM, Hewett DR, et al. Twist-1 is upregulated by NSD2 and contributes to tumour dissemination and an epithelial-mesenchymal transition-like gene expression signature in t(4;14)-positive multiple myeloma. Cancer Lett. 2020;475:99–108. doi:10.1016/j.canlet.2020.01.040
33. Wu K, Jiang Y, Zhou W, et al. Long noncoding RNA RC3H2 facilitates cell proliferation and invasion by targeting MicroRNA-101-3p/EZH2 axis in OSCC. Mol Ther Nucleic Acids. 2020;20:97–110. doi:10.1016/j.omtn.2020.02.006
34. Jin B, Zhang P, Zou H, et al. Verification of EZH2 as a druggable target in metastatic uveal melanoma. Mol Cancer. 2020;19(1):52. doi:10.1186/s12943-020-01173-x
35. Min DJ, Ezponda T, Kim MK, et al. MMSET stimulates myeloma cell growth through microRNA-mediated modulation of c-MYC. Leukemia. 2013;27(3):686–694. doi:10.1038/leu.2012.269
36. Sun S, Huang DW, Huo LT, Li PL. The role of NSD2 and EZH2 in the pathogenesis of endometrial carcinoma. J Biol Regul Homeost Agents. 2019;33(4):1233–1239.
37. Kojima M, Sone K, Oda K, et al. The histone methyltransferase WHSC1 is regulated by EZH2 and is important for ovarian clear cell carcinoma cell proliferation. BMC Cancer. 2019;19(1):455. doi:10.1186/s12885-019-5638-9
38. Popovic R, Martinez-Garcia E, Giannopoulou EG, et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014;10(9):e1004566. doi:10.1371/journal.pgen.1004566
39. Asangani IA, Ateeq B, Cao Q, et al. Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol Cell. 2013;49(1):80–93. doi:10.1016/j.molcel.2012.10.008
40. Toyokawa G, Cho HS, Masuda K, et al. Histone lysine methyltransferase wolf-hirschhorn syndrome candidate 1 is involved in human carcinogenesis through regulation of the Wnt pathway. Neoplasia. 2011;13(10):887–898. doi:10.1593/neo.11048
41. Yang P, Guo L, Duan ZJ, et al. Histone methyltransferase NSD2/MMSET mediates constitutive NF-κB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol Cell Biol. 2012;32(15):3121–3131. doi:10.1128/MCB.00204-12
42. Park JW, Chae YC, Kim JY, Oh H, Seo SB. Methylation of Aurora kinase A by MMSET reduces p53 stability and regulates cell proliferation and apoptosis. Oncogene. 2018;37(48):6212–6224. doi:10.1038/s41388-018-0393-y
43. Evans DL, Zhang H, Ham H, et al. MMSET is dynamically regulated during cell-cycle progression and promotes normal DNA replication. Cell Cycle. 2016;15(1):95–105. doi:10.1080/15384101.2015.1121323
44. Brito JL, Walker B, Jenner M, et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica. 2009;94(1):78–86. doi:10.3324/haematol.13426
45. Jaffe JD, Wang Y, Chan HM, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 2013;45(11):1386–1391. doi:10.1038/ng.2777
46. Swaroop A, Oyer JA, Will CM, et al. An activating mutation of the NSD2 histone methyltransferase drives oncogenic reprogramming in acute lymphocytic leukemia. Oncogene. 2019;38(5):671–686. doi:10.1038/s41388-018-0474-y
47. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
48. DeSantis CE, Ma J, Goding Sauer A, Newman LA, Jemal A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J Clin. 2017;67(6):439–448. doi:10.3322/caac.21412
49. Wang JJ, Zou JX, Wang H, et al. Histone methyltransferase NSD2 mediates the survival and invasion of triple-negative breast cancer cells via stimulating ADAM9-EGFR-AKT signaling. Acta Pharmacol Sin. 2019;40(8):1067–1075. doi:10.1038/s41401-018-0199-z
50. Zhao X, Xie T, Zhao W, Cai W, Su X. Downregulation of MMSET impairs breast cancer proliferation and metastasis through inhibiting Wnt/β-catenin signaling. Onco Targets Ther. 2019;12:1965–1977. doi:10.2147/OTT.S196430
51. Wang J, Duan Z, Nugent Z, et al. Reprogramming metabolism by histone methyltransferase NSD2 drives endocrine resistance via coordinated activation of pentose phosphate pathway enzymes. Cancer Lett. 2016;378(2):69–79. doi:10.1016/j.canlet.2016.05.004
52. Wang Q, Zheng J, Zou JX, et al. S-adenosylhomocysteine (AdoHcy)-dependent methyltransferase inhibitor DZNep overcomes breast cancer tamoxifen resistance via induction of NSD2 degradation and suppression of NSD2-driven redox homeostasis. Chem Biol Interact. 2020;317:108965. doi:10.1016/j.cbi.2020.108965
53. Feng Q, Zhang Z, Shea MJ, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014;24(7):809–819. doi:10.1038/cr.2014.71
54. Han X, Piao L, Yuan X, Wang L, Liu Z, He X. Knockdown of NSD2 suppresses renal cell carcinoma metastasis by inhibiting epithelial-mesenchymal transition. Int J Med Sci. 2019;16(10):1404–1411. doi:10.7150/ijms.36128
55. Aytes A, Giacobbe A, Mitrofanova A, et al. NSD2 is a conserved driver of metastatic prostate cancer progression. Nat Commun. 2018;9(1):5201. doi:10.1038/s41467-018-07511-4
56. Li N, Xue W, Yuan H, et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J Clin Invest. 2017;127(4):1284–1302. doi:10.1172/JCI91144
57. Wu J, Luo M, Duan Z, et al. WHSC1 acts as a prognostic indicator and functions as an oncogene in cervical cancer. Onco Targets Ther. 2019;12:4683–4690. doi:10.2147/OTT.S204701
58. Yin Z, Sun Y, Ge S, Sun J. Epigenetic activation of WHSC1 functions as an oncogene and is associated with poor prognosis in cervical cancer. Oncol Rep. 2017;37(4):2286–2294. doi:10.3892/or.2017.5463
59. Zhu L, Yu CL, Zheng Y. NSD2 inhibition suppresses metastasis in cervical cancer by promoting TGF-β/TGF-βRI/SMADs signaling. Biochem Biophys Res Commun. 2019;519(3):489–496. doi:10.1016/j.bbrc.2019.08.020
60. Xiao M, Yang S, Chen J, et al. Overexpression of MMSET in endometrial cancer: a clinicopathologic study. J Surg Oncol. 2013;107(4):428–432. doi:10.1002/jso.23234
61. García-Carpizo V, Sarmentero J, Han B, et al. NSD2 contributes to oncogenic RAS-driven transcription in lung cancer cells through long-range epigenetic activation. Sci Rep. 2016;6(1):32952. doi:10.1038/srep32952
62. Lu MH, Fan MF, Yu XD. NSD2 promotes osteosarcoma cell proliferation and metastasis by inhibiting E-cadherin expression. Eur Rev Med Pharmacol Sci. 2017;21(5):928–936.
63. He C, Liu C, Wang L, Sun Y, Jiang Y, Hao Y. Histone methyltransferase NSD2 regulates apoptosis and chemosensitivity in osteosarcoma. Cell Death Dis. 2019;10(2):65. doi:10.1038/s41419-019-1300-3
64. Zhou P, Wu LL, Wu KM, et al. Overexpression of MMSET is correlation with poor prognosis in hepatocellular carcinoma. Pathol Oncol Res. 2013;19(2):303–309. doi:10.1007/s12253-012-9583-z
65. Saloura V, Vougiouklakis T, Sievers C, et al. The role of protein methyltransferases as potential novel therapeutic targets in squamous cell carcinoma of the head and neck. Oral Oncol. 2018;81:100–108. doi:10.1016/j.oraloncology.2018.04.014
66. Saloura V, Cho HS, Kiyotani K, et al. WHSC1 promotes oncogenesis through regulation of NIMA-related kinase-7 in squamous cell carcinoma of the head and neck. Mol Cancer Res. 2015;13(2):293–304. doi:10.1158/1541-7786.MCR-14-0292-T
67. Chen H-Q, Gao D. Inhibitory effect of microRNA-154 targeting WHSC1 on cell proliferation of human skin squamous cell carcinoma through mediating the P53 signaling pathway. Int J Biochem Cell Biol. 2018;100:22–29. doi:10.1016/j.biocel.2018.04.021
68. Hudlebusch HR, Skotte J, Santoni-Rugiu E, et al. MMSET is highly expressed and associated with aggressiveness in neuroblastoma. Cancer Res. 2011;71(12):4226–4235. doi:10.1158/0008-5472.CAN-10-3810
69. Tisi D, Chiarparin E, Tamanini E, et al. Structure of the epigenetic oncogene MMSET and inhibition by N-alkyl sinefungin derivatives. ACS Chem Biol. 2016;11(11):3093–3105. doi:10.1021/acschembio.6b00308
70. Shen Y, Morishita M, Lee D, et al. Identification of LEM-14 inhibitor of the oncoprotein NSD2. Biochem Biophys Res Commun. 2019;508(1):102–108. doi:10.1016/j.bbrc.2018.11.037
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.