Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 15
The Relationship Between the Blood-Brain-Barrier and the Central Effects of Glucagon-Like Peptide-1 Receptor Agonists and Sodium-Glucose Cotransporter-2 Inhibitors
Authors Dong M ![]() , Wen S
, Wen S ![]() , Zhou L
, Zhou L
Received 21 May 2022
Accepted for publication 12 August 2022
Published 22 August 2022 Volume 2022:15 Pages 2583—2597
DOI https://doi.org/10.2147/DMSO.S375559
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ming-Hui Zou
Meiyuan Dong,1 Song Wen,1 Ligang Zhou1,2
1Department of Endocrinology, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai, 201399, People’s Republic of China; 2Shanghai Key Laboratory of Vascular Lesions Regulation and Remodeling, Shanghai Pudong Hospital, Shanghai, People’s Republic of China
Correspondence: Ligang Zhou, Tel +8613611927616, Email [email protected]
Abstract: Diabetes and obesity are growing problems worldwide and are associated with a range of acute and chronic complications, including acute myocardial infarction (AMI) and stroke. Novel anti-diabetic medications designed to treat T2DM, such as glucagon-like peptide-1 receptor agonists (GLP-1RAs) and sodium-glucose cotransporter-2 inhibitors (SGLT-2is), exert beneficial effects on metabolism and the cardiovascular system. However, the underlying mechanisms are poorly understood. GLP-1RAs induce anorexic effects by inhibiting the central regulation of food intake to reduce body weight. Central/peripheral administration of GLP-1RAs inhibits food intake, accompanied by an increase in c-Fos expression in neurons within the paraventricular nucleus (PVN), amygdala, the nucleus of the solitary tract (NTS), area postrema (AP), lateral parabrachial nucleus (LPB) and arcuate nucleus (ARC), induced by the activation of GLP-1 receptors in the central nervous system (CNS). Therefore, GLP-1RAs need to pass through the blood-brain barrier to exert their pharmacological effects. In addition, studies revealed that SGLT-2is could reduce the risk of chronic heart failure in people with type 2 diabetes. SGLT-2 is extensively expressed throughout the CNS, and c-Fos expression was also observed within 2 hours of administration of SGLT-2is in mice. Recent clinical studies reported that SGLT-2is improved hypertension and atrial fibrillation by modulating the “overstimulated” renin-angiotensin-aldosterone system (RAAS) and suppressing the sympathetic nervous system (SNS) by directly/indirectly acting on the rostral ventrolateral medulla. Despite extensive research into the central mechanism of GLP-1RAs and SGLT-2is, the penetration of the blood-brain barrier (BBB) remains controversial. This review discusses the interaction between GLP-1RAs and SGLT-2is and the BBB to induce pharmacological effects via the CNS.
Keywords: central nervous system, anorexic medications, sympathetic nervous system, chronic heart failure
Introduction
Type 2 diabetes mellitus (T2DM) and obesity are major public health problems affecting millions of people worldwide. T2DM is a metabolic disorder characterized by long-term hyperglycemia and is one of the leading causes of morbidity and mortality.1 Obesity is an important exo-promoting factor of T2DM. Accumulating evidence demonstrates that obesity is not only strongly related to the etiology and pathogenesis of T2DM but also to the development of diabetic complications.2 Hyperglycemia and glucose toxicity can induce oxidative stress, worsening the outcome of AMI (acute myocardial infarction) in patients with diabetes.3 In addition, both hypertension and diabetes are critical risk factors for stroke in patients with atherosclerotic cardiovascular impairment.4 Capability of entering the CNS is an effective way for drug development to improve metabolism, alleviate diabetes, and obesity. CNS is an important target for improving obesity and many obesity-improving drugs exert effect through the CNS.5 Therefore, whether it can cross the BBB determines the effect of the drug. GLP-1 is secreted during mealtime and reduces blood glucose levels by enhancing insulin secretion and inhibiting glucagon release. A growing number of GLP-1RAs have been produced, which can be classified into short-acting or long-acting compounds. The net effects of the former reduce postprandial blood glucose, while the latter affects both fasting and postprandial blood glucose.6 In rodents, the anorexigenic effect of GLP-1RA administration peripherally requires central GLP-1R, for it is not inhibited in mice with central specific GLP-1R deletion.7
Sodium-glucose cotransporter-2 inhibitors (SGLT-2is) are a novel class of oral drugs for the treatment of T2DM. These drugs block SGLT-2 in proximal tubules, through which 90% of glucose is reabsorbed, thus decreasing the reabsorption of glucose and lowering the blood glucose levels.8 In addition to the anti-diabetic effects, weight loss has also been observed in patients receiving SGLT-2i monotherapy.9–11 This drug class affects multiple systems, including the nervous, cardiovascular and endocrine systems, resulting in exceedingly complex metabolic effects beyond glycemic control.
Clinical studies have shown that both SGLT-2is and GLP-1RAs reduce body weight and lower the risk of hospitalization and mortality of cardiovascular disease such as AMI in patients with type 2 diabetes.12–14 These two agents possibly share similar CNS routes, but their modes of action might be very distinct.15,16 The blood-brain barrier (BBB) is a dynamic barrier that maintains the regular physiological state and metabolism of the central nervous system (CNS), but it also inhibits peripheral drugs from entering the CNS.17 Both the lipid-soluble GLP-1RAs and small-molecule SGLT-2is were considered to be able to cross the BBB in numerous preclinical trials.
Furthermore, increased levels of circulating free fatty acids, inflammatory cytokines (TNF-α, IL-1β, IL-6) and chemokines increase BBB permeability. These effects are compounded by the apoptosis of BBB microvascular endothelial cells caused by hyperglycemia and obesity.18–21 BBB impairment has also been reported in a variety of neurological disorders, including Parkinson’s disease and Alzheimer’s disease.22 Interestingly, oxidative stress is a common feature of all these cases. Elevated levels of reactive oxygen species (ROS) are observed during pathological processes, and ROS is considered to contribute to the increased permeability of the BBB. Mitochondria are the main source of ROS production in cells. The destruction of the mitochondrial electron transport chain (ETC) leads to an increase in ROS production, which triggers matrix metalloproteinases (MMP) induction, ultimately disrupting tight junctions and increasing BBB permeability.23 The integrity of the BBB is compromised in certain pathological states, and its temporary disruption allows drugs in the peripheral circulation to enter the CNS. Consequently, GLP-1RAs and SGLT-2is are able to cross the BBB. Terami et al revealed that diabetes-induced disruption of the BBB allowed SGLT-2is to pass through in a dose-dependent manner.24
This review introduces the basic structure and permeation mechanism of the BBB, discussing the relationship between the structural and/or chemical properties of SGLT-2is/GLP-1RAs’ and their permeability through the BBB.
The Physiology and Pathology of Blood–Brain Barrier (BBB) Related to Obesity and T2DM
Structure and Function of BBB
The blood–brain barrier (BBB) is composed of capillary endothelial cells linked by tight junctions, basal lamina, pericytes, and astrocytes. It acts as a regulatory interface between capillaries and nerve tissues in the brain and spinal cord that restricts the transfer of most drug substances from the bloodstream into the brain and serves as an important barrier to maintaining the brain’s homeostasis (Figure 1).17,25 However, some structures, such as circumventricular organs (CVOs), lack a complete BBB. CVOs are composed of malformed ependymal cells and many leaky capillaries that lack a full blood-brain barrier, allowing chemicals to enter the brain. CVOs include the pineal gland, the organum vasculosum of the lamina terminalis (OVLTs), the subfornical organ (SFO), the choroid plexus, the region postrema (AP), the pituitary gland’s posterior lobe, the subcommissural organ, and the median eminence (ME) of the mediobasal hypothalamus.26,27
 |
Figure 1 Structure of the BBB: capillary endothelial cell, tight junction, basal lamina, astrocyte, pericyte, interneuron. |
BBB Permeation Prediction Index
The semipermeable nature of BBB restricts the movement of large molecules, such as drugs or proteins >500 kDa. Compounds having logPoct≈2.0 have optimal brain penetration. The parameters used for predicting drug penetrability by modified Lipinski’s rules for CNS penetration include hydrogen bond donors (HBD) ≤3; hydrogen bond acceptors (HBA) ≤7; logPoct ≤ 5.0; 7.5 < pKa < 10.5; molecular weight ≤ 400Da.28
Transport Routes
Substances can cross the blood-brain barrier via paracellular transport and intercellular transport. Although most macromolecules in the blood are physically prevented from entering the brain due to the presence of the blood-brain barrier and tight junctions, specific and non-specific transcellular mechanisms transport various macromolecules and complexes across the blood-brain barrier. Figure 2 illustrates several primary routes across the BBB: positively charged macromolecules cross the BBB via adsorptive-mediated transcytosis (AMT); non-polar solutes and lipid-soluble drugs pass via passive diffusion; c) carrier-mediated transport (CMT) is responsible for the transport of a variety of essential polar molecules into the CNS; and receptor-mediated transcytosis (RMT), which requires specific ligands29 (Figure 2).
 |
Figure 2 Mechanism of BBB transportation: a) Carrier-mediated influx, in which polar molecules are transported; b) Adsorptive-mediated transcytosis, in which positively charged macromolecules bind to receptors and are transported across the endothelial cell; c) Passive diffusion, in which most lipid-soluble molecules are transported; d) Receptor-mediated transcytosis, in which macromolecules bind to receptors and are transported to the CNS; e) Tight junction model (partially or completely open). Arrows indicate the direction of transportation. |
The Physiology of the Interactions Between GLP-1RAs and the BBB
Location of GLP-1R and Structure of GLP-1RAs
Jensen et al conducted a study in the CNS by isolating and purifying a novel specific monoclonal GLP-1R antibody and detecting the location of GLP-1R using an in situ hybridization technique.30,31 The results confirmed that GLP-1 receptors (GLP-1R) are widely distributed throughout the cerebral cortex, hypothalamus, hippocampus, thalamus, caudate nucleus, and globus pallidus. Numerous studies focusing on diverse brain illnesses such as Alzheimer’s disease (AD) and ischemic brain injury have corroborated the localization of GLP-1R in the CNS.32–34 In addition, a study reported GLP-1R expression on pericytes, which are multi-functional mural cells located at the center of the neurovascular unit (NVU).35 Other studies have mapped the action of GLP-1RAs, demonstrating that GABA neurons in the NTS express GLP-1 receptors and mediate the anorectic effects of liraglutide in rats.36 However, a study of single-cell RNA sequencing in the brain shows that the presence of GLP-1R on BBB endothelial cells is negligible.37
Exenatide,38 lixisenatide,39,40 beinaglutide,41 liraglutide,42 dulagutide43 and semaglutide44 are now commercially accessible as GLP-1 receptor agonists (Table 1). The GLP-1 receptor agonists can be classified into two groups based on their chemical structure. Exenatide is based on the exendin-4 structure and has a low amino acid sequence homology with human GLP-1, whereas natural GLP-1RAs, such as liraglutide, have a higher amino acid sequence homology of 95% with human GLP-1.45 According to the different molecular conformations (natural GLP-1 or long-acting GLP-1 analogs) and drug administration pathways, GLP-1 and its analogs transmit food reduction signals directly or indirectly to the central nervous system (CNS) through different signal pathways.
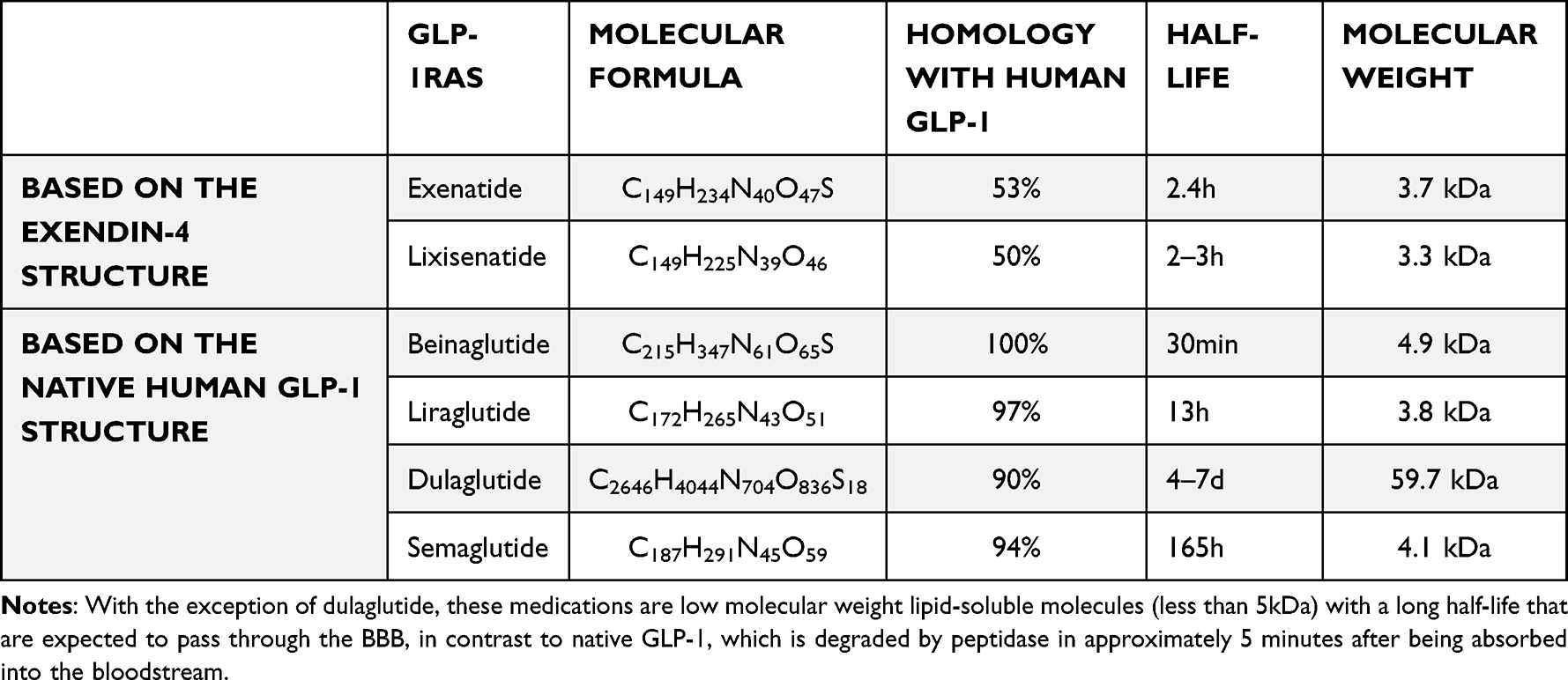 |
Table 1 The Physicochemical Properties of GLP-1 Receptor Agonists |
Native GLP-1, GLP-1RAs and BBB
Numerous studies have discovered the expression of native GLP-1 in CNS, implying that natural GLP-1 could cross the BBB from the peripheral circulation via passive transport due to their physiological, molecular biology, pharmacokinetic, and pharmacodynamic properties. Kastin et al observed and analyzed the influx rate following intravenous injection of 125I-[Ser8] GLP-1. The blood-brain flow of GLP-1 via passive diffusion may be determined by its physicochemical properties rather than lipophilicity, demonstrating the BBB’s permeability to GLP-1.46 Several preclinical and clinical studies on ischemia-induced brain injury, Alzheimer’s disease (AD), Parkinson’s disease (PD), and other neurodegenerative disorders have established that GLP-1RAs can cross the BBB and exert a significant neuroprotective effect.32–34 Furthermore, various studies have examined the anorexia-promoting properties of GLP-1RAs. The studies have demonstrated that GLP-1R is also expressed in nuclei controlling food intake, such as the arcuate nucleus (ARC) and paraventricular thalamic nucleus (PVN). Previous studies also demonstrated that GLP-1RAs might cross the BBB and promote bodyweight loss by acting on these nuclei.47–50 The effect of intravenous infusion of GLP-1 on food intake disappeared after vagotomy in humans, which suggested that the vagus afferent pathway is involved in the effect of GLP-1 on appetite.51 However, subdiaphragmatic vagotomy had no effect on GLP-1-induced acute activation of PKA in the brain, indicating that the vagus nerve is not involved in GLP-1RAs entering the brain.52 Interestingly, subphrenic vagus afferents can also attenuate the short-term appetite inhibitory effects of liraglutide and exendin-4, but their long-term effects on food intake do not depend entirely on vagus afferents.53 The above findings indicate that GLP-1RAs inhibit food intake and cause weight loss mainly by acting on different brain regions, which also supports the evidence of long-acting GLP-1RAs penetrating incomplete BBB in certain areas. GLP-1R–mediated transport occurs through circumventricular organs (CVOs) and maybe other parts of the hypothalamus via fenestrated capillaries and is partially dependent on tanycyte–mediated transport through certain localized compartments of the cerebrospinal fluid (CSF).54,55 Central administration of GLP-1RAs suppressed appetite, accompanied by increased expression of c-Fos in neurons within the PVN, amygdala, NTS, AP, LPB and ARC, which was consistent with the results of GLP-1R expression studies in the CNS.56 Decreased food intake and weight loss were observed with peripheral administration of GLP-1RAs, but inactivation of intestinal GLP-1 production did not interfere with food intake or weight control, suggesting that the GLP-1R-dependent signaling regulating appetite and body weight occurs in the CNS.50,57
GLP-1RAs Originating from the Structure of Exendin-4: Exendin-4, Exenatide and Lixisenatide
Exendin-4, a bioactive peptide produced from Heloderma suspectum venom, is a 39-amino-acid peptide.58 Exendin-4 improved cognitive behavioral tests in Alzheimer’s disease patients by protecting the brain against abnormal insulin signaling.59 Kastin and Akerstrom established in a landmark study that peripherally injected Exendin-4 passed readily through the BBB, facilitated by its lipophilicity, rather than becoming trapped in the endothelial cell.60 According to multiple time regression analyses, exendin-4 passes directly through the BBB. Exendin-4 has been shown to easily cross the BBB, which is critical for Parkinson’s disease treatment.61 In addition, Exendin-4 quickly enters the brain and brain microvascular endothelial cells via active ligands that bind to and activate GLP-1 receptors.52 Exenatide is based on exendin-4 structure.62 Numerous clinical trials established exenatide’s efficacy in individuals with Parkinson’s disease, demonstrating that it entered the CNS via the BBB and exerted neuroprotective and neurorestorative effects.63–65 Additionally, it was demonstrated that exendin-4 is also mediated by the adsorptive transcytosis across brain endothelial cells.66
On the other hand, Salameh’s research indicates that lixisenatide can penetrate the BBB via adsorptive transcytosis.66 Lixisenatide has been demonstrated to pass the blood-brain barrier and exert neuroprotective effects in Alzheimer’s disease.67 Hunter and Holscher reported increased cAMP levels in the CNS at all dosages after 30 minutes and 3 hours of lixisenatide injection, which indicated the activation of the GLP-1 receptor, and presented persuasive evidence that lixisenatide crossed the BBB.68
GLP-1RAs Originating from the Native Human GLP-1 Structure: Liraglutide, Dulaglutide and Semaglutide
Liraglutide is a GLP-1 agonist with neuroprotective properties in Alzheimer’s disease.69 It has demonstrated the ability to cross the BBB. The pharmacological concentration of GLP-1RA in the brain was proportional to the intraperitoneal injections, with 2.5, 25, or 250 nmol per kg of body weight protecting against impairment of recognition memory synaptic loss and degradation in the hippocampus of patients.68,70 Despite its varied ability to cross the BBB, liraglutide was more effective than exendin-4 in repairing the damage induced by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MTPT).71 While these findings suggest that GLP-1RAs can reach brain tissue by passive diffusion across the BBB, other research has shown that liraglutide could only enter the brain by binding to the central GLP-1 receptor.
Dulaglutide is a GLP-1RA with a larger molecular weight, which may potentially cross a compromised BBB.72 However, there is no direct evidence proving that dulaglutide crosses the BBB.
In 2017, the US Food and Drug Administration (FDA) authorized semaglutide as the seventh clinically available GLP-1RA.44 The longer aliphatic chain in semaglutide, a long-acting formulation based on the structure of liraglutide, enhances hydrophobicity by altering the DPP-4 enzyme hydrolysis site with polyethylene glycol (PEG), allowing it to strongly bind to albumin. Semaglutide was detected within vascular endothelial cells and could not cross the BBB. According to the research by Gabery et al, semaglutide could bind to albumin and directly reach the brainstem, septal nucleus, and hypothalamus via CVOs, but showed no interaction with endothelial cells and could not cross the BBB. Interestingly, semaglutide seems to be able to pass through the tanycytes that line the ventricular wall or areas lacking a BBB.73
The Physiology of the Interaction Between SGLT-2 Inhibitors and the BBB
Location of SGLT-2 and Structure of SGLT-2 Inhibitors
SGLT-2 is mainly expressed in the apical membranes of kidney segments 1 and 2 of the proximal convoluted tubules.74 A previous study has demonstrated that SGLT-2 is present in the central nervous system, including the brain parenchymal and the BBB.75–77 SGLT-2 was shown to be expressed in ependymal cells and choroid plexus epithelial cells (CPE) in the brainstem, hypothalamus, periaqueductal grey (PAG), amygdala, and solitary nucleus of mice and humans.78
SGLT-2is are a novel class of diabetes medications and are well-known for their efficacy in the treatment of chronic heart failure in both diabetic and non-diabetic patients. RAAS is one of the most important arterial blood pressure regulators and plays a key role in cardiovascular and renal diseases. A study by Shin et al reported that dapagliflozin attenuated inflammatory and fibrotic markers.79 In addition to their anti-diabetic effects, SGLT-2is play a role in cardio-nephroprotection due to RAAS activation from their diuretic and sympathoinhibition effects.80 Phlorizin, the first SGLT-2i, is a naturally occurring chalcone compound discovered in apple bark in the 1850s by French researchers. It is a non-specific SGLT-1 and SGLT-2i that was not used as an anti-hyperglycemic medication due to gastrointestinal side effects until the chemical structure was modified in this decade. SGLT-2is are lipid soluble drugs with low molecular weight, as indicated by their chemical structures (Figure 3).
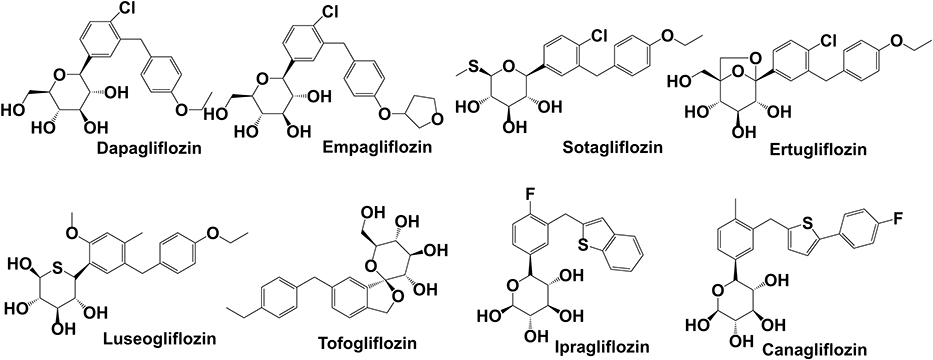 |
Figure 3 Chemical structure and molecule weight of SGLT-2 inhibitors. Dapagliflozin(2013) MF:C21H25ClO6 MW:408.88; Empagliflozin(2014) MF:C23H27ClO7, MW:450.91; Sotagliflozin(2019) MF:C21H25ClO5S MW:424.94; Ertugliflozin(2017) MF:C22H25ClO7 MW:436.88; Luseogliflozin(2014) MF:C23H30O6S MW:434.55; Tofogliflozin(2014) MF:C22H26O6 MW:386.44; Ipragliflozin(2014) MF:C21H21FO5S MW:404.45; Canagliflozin(2013) MF:C24H25FO5S MW:444.52. Abbreviations: MF, molecular formula; MW, molecular weight. |
SGLT-2 Inhibitors and BBB
Physicochemical properties, including molecular weight, pKa, LogPoct, HBA and HBD, are critical parameters related to the ability of drugs to cross the BBB. These properties can be obtained from the ChEMBL database, which is an open database that contains information on a large number of drug-like bioactive compounds: https://www.ebi.ac.uk/chembldb(Table 2).
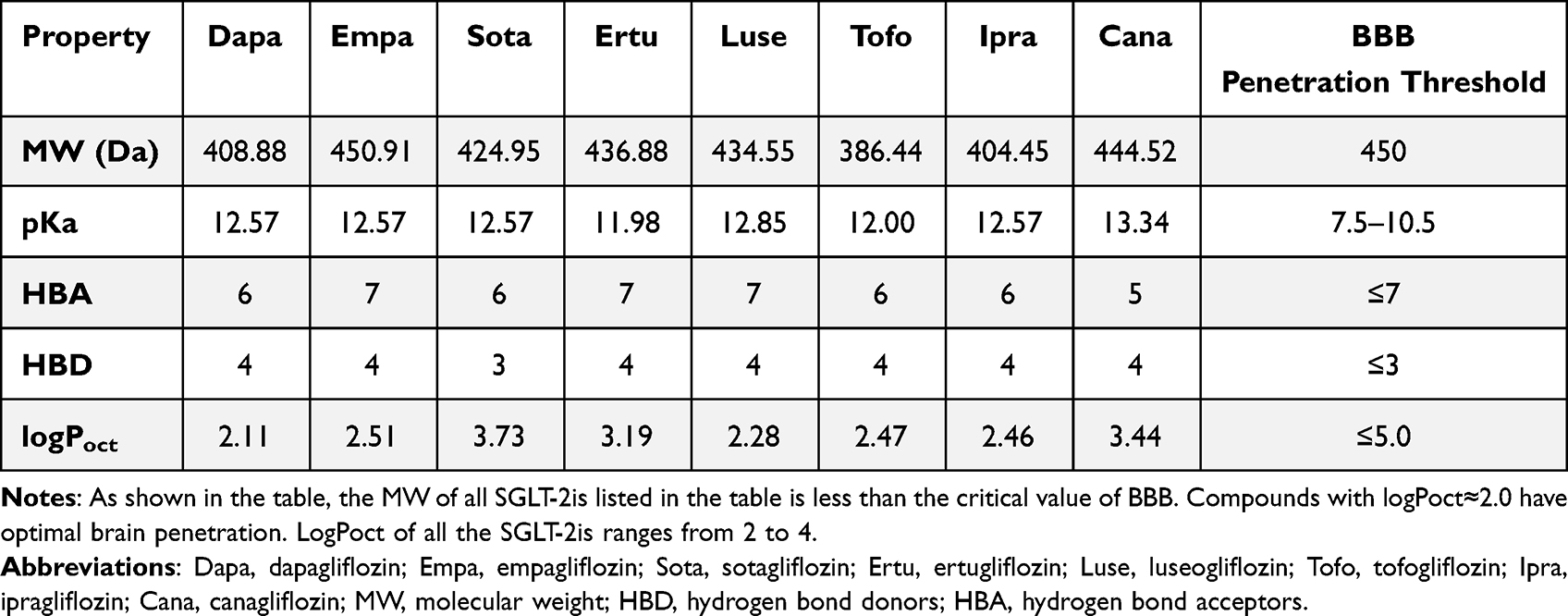 |
Table 2 Physicochemical properties related to penetration capacity of the BBB of SGLT-2is. |
Notably, given that SGLT-2 is expressed in the brain, which is consistent located with enhanced cFos expression induced by SGLT-2is, we hypothesize that SGLT-2is may cross the BBB via receptor-mediated transcytosis.76,81 In a recent study, we discovered that the SGLT-2i dapagliflozin might cross the BBB and interact with SGLT-2 in the brain.81 In addition to its hypoglycemic effect, the neuroprotective effect of SGLT-2is may be another evidence that it can cross the BBB. Dapagliflozin may enter the brain through impaired BBB, irrespective of its lipid-soluble properties, as evidenced by dramatically increased BBB permeability and disruption of tight junction between endothelial cells in Parkinson’s disease.82 Empagliflozin improved cognitive performance in diabetic rats by lowering brain oxidative stress. In this context, the increase in cognitive function associated with empagliflozin and canagliflozin may also support the notion that these medicines are capable of penetrating the damaged BBB.83 Inflammation in the CNS of patients with diabetes or other chronic diseases may increase the permeability of the BBB, allowing SGLT-2is to pass through. El-Sahar et al examined dapagliflozin’s neuroprotective effect in rats with Huntington’s disease and discovered that it was capable of modulating aberrant neurotransmission. Other abnormal conditions in the rat striatum, such as apoptosis, glycolysis, and autophagy, may allow SGLT-2is to cross the BBB.84 On the other hand, some research shows that the neuroprotective effect of SGLT-2is is most likely caused by the increased native GLP-1 concentration and decreased corticosterone concentration following SGLT-2is administration.85
Dapagliflozin was the first SGLT-2i to be licensed as a commercially accessible drug by the US Food and Drug Administration in 2000. Our previous study found that dapagliflozin may act on the rostral ventrolateral medulla (RVLM) and affect the sympathetic outflow of sympathetic preganglionic neurons to the intermediolateral nucleus of the spinal cord (IML), thereby promoting parasympathetic activity. Dapagliflozin also inhibits SGLT-2 in mice and regulates central autonomic activity by stimulating neurons in the central nervous system and controls cardiovascular functions. SGLT-2 was localized to specific regions involved in autonomic control. Expression of c-Fos was significantly higher in major critical nuclei in the aforementioned regions in the mice group treated with dapagliflozin.81 Shaikh et al reported that dapagliflozin is a potent dual inhibitor of SGLT-2 and acetylcholinesterase, which could be used as the basis for future dual therapy for patients with diabetes and diabetes-related neurological disorders.86
In 2014, the drug empagliflozin (EMPA) was approved for the treatment of T2DM. Extensive investigation revealed that EMPA had been shown to have a neuroprotective impact on illnesses like Alzheimer’s disease and Parkinson’s disease. In a recent study, EMPA was shown to traverse the damaged BBB and to improve ultrastructural remodeling of the neurovascular unit (NVU) and neuroglia in female diabetic mice.87 Entesar et al also confirmed that EMPA exerted neuroprotective effects in hyperglycemic rats.88 Increased expression of cerebral caspase-3 was observed in ischemia/reperfusion(I/R)-injured rats administered with oral EMPA, whereas apoptotic cells decreased, indicating that EMPA may help protect neurons. The researchers speculated that EMPA could cross through a compromised BBB and exert neuroprotective benefits. The above findings suggest that EMPA can enter areas with compromised BBB integrity (Figure 3).
Sotagliflozin, often known as a “dual SGLT-1/SGLT-2 inhibitor”, has the highest affinity for SGLT1 receptors but is not yet commonly used in diabetic patients as it is the newest SGLT-2is.89 In theory, sotagliflozin may have a greater neuroprotective effect than other SGLT-2is due to increased SGLT1 expression in the brain.90 Sotagliflozin should be able to penetrate the BBB via passive diffusion based on its lipid-soluble molecular structure. However, no current investigations have examined this concept.
Ertugliflozin was licensed for clinical usage in 2017 after it was demonstrated to decrease the risk of heart failure hospitalization (HHF).91 However, minimal research has been conducted on its BBB permeability. Based on physicochemical and polypharmacological data, ertugliflozin is likely unable to cross the BBB.92
Luseogliflozin is a selective SGLT-2 inhibitor that received marketing approval on March 24, 2014, for the treatment of T2DM.93 Many studies have shown that luseogliflozon could reverse cerebrovascular dysfunction and cognitive impairments in diabetic animals and elderly diabetics.94,95
The FDA authorized tofogliflozin for diabetic therapy in 2014. Takeda et al discovered that tofogliflozin could enhance food intake. They hypothesized that delivering tofogliflozin intracerebroventricularly (ICV) could stimulate food intake by acting directly on the CNS, whereas intraperitoneal administration showed no such effects.96 Tofogliflozin administered via ICV enhanced food intake, indicating the presence of SGLT-2 in the brain. However, the BBB permeability of tofogliflozin remains unknown (Figure 4). Additionally, c-fos (a neuronal activation indicator) was significantly enhanced in the arcuate nucleus (ARC) within 2–3 hours after tofogliflozin treatment, suggesting that tofogliflozin could permeate these regions and bind to receptors on the ARC.
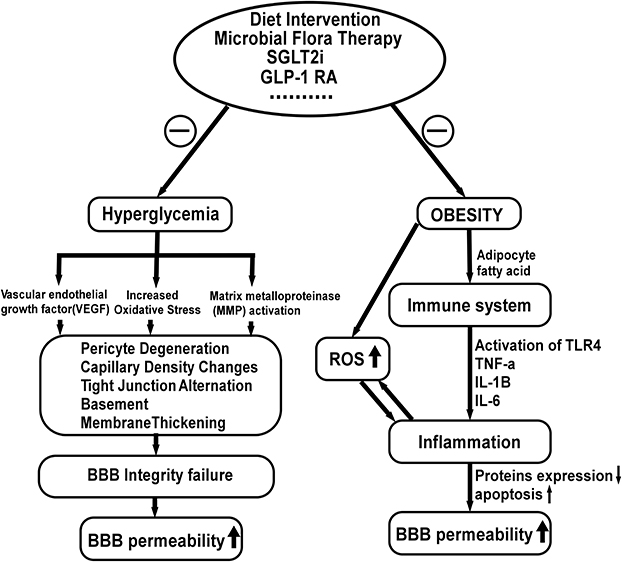 |
Figure 4 Factors influencing the blood-brain barrier permeability and possible treatment strategy. Increased oxidative stress, decreased VEGF levels and increased matrix metalloproteinase (MMP) activity result in basement membrane expansion, pericyte disintegration, increased paracellular diffusion, and decreased tight junction protein expression. In addition, fatty acids induce inflammatory responses, which reduce protein expression in BBB microvessels and increase BBB apoptosis, resulting in increased BBB permeability. Evidence shows that modification of the gut microbiota with prebiotics or probiotics and high-fiber diets improve systemic inflammation, restoring BBB integrity. In addition, treatment with GLP-1RAs and SGLT-2is inhibit the activation of inflammatory factors and attenuate oxidative stress via blood glucose and fatty acids reduction. |
Ipraglifozin has a five-membered ring structure with low selectivity for SGLT-2 and was first approved in Japan in 2014. Destruction of mitochondrial ETC leads to increased ROS production, which triggers matrix metalloproteinases (MMP) induction, ultimately leading to compromised BBB integrity. Studies have shown that ipragliflozin can reduce body fat mass and alleviate mitochondrial dysfunction.97,98 Therefore, it has the effect of both anti-obesity, and anti-oxidant to improve BBB permeability. However, there are no adequate studies revealing the mechanism of ipragliflozin and its ability to cross the BBB.
In 2013, the FDA approved canagliflozin as an anti-diabetic medicine. Canagliflozin (CAN) has been demonstrated to bind to both AChE and SGLT-2, inhibiting both enzymes simultaneously.99 Its effects on neurotransmitters may also be linked to the activation of its receptors. Therefore, canagliflozin may potentially exert direct effects on CNS SGLT-2. Although a study found that canagliflozin had a cognitive protective effect in high-fat diet mice by decreasing obesity-related neuroinflammation,100 there is no conclusive evidence that it can cross the BBB. Due to its lipophilicity and low molecular weight, CAN could partially cross the blood-brain barrier via passive diffusion.101
The Role of Impaired BBB in Diabetes and Obesity with GLP-1RAs and SGLT-2is
DM and obesity influence BBB integrity through a number of mechanisms (Figure 4). In our previous studies, hyperglycemia and inflammation were shown to lead to pathological capillary changes in the retina.102 The current findings in the diabetic brain suggest a striking similarity, indicating a common pathophysiological mechanism between the retina and the brain microvessels. Magnetic resonance (MR) brain imaging with intravenous gadolinium (Gd) diethylenetriamine pentaacetic acid revealed increased BBB permeability in diabetic patients (Gd-DTPA). IgG has been demonstrated to leak into the brain interstitium of DMHC pigs via the permeable BBB.18,19 According to recent research, chronic hyperglycemic exposure resulted in compromised BBB integrity, characterized by altered tight junctions, thickened basement membrane, capillary density changes, and pericyte deterioration. In addition, pericyte loss caused by the Krebs cycle processing of excessive glucose resulted in increased oxidative stress. Furthermore, advanced glycation end products activate vascular endothelial growth factor (VEGF), which increases matrix metalloproteinase synthesis and alters tight junction proteins. In diabetic patients and rodents, elevated plasma matrix metalloproteinase (MMP) activity lowered BBB tight junction protein synthesis and increased BBB permeability.103–105 Microvascular diseases may lead to BBB decomposition, allowing serum-derived components to flow into the brain parenchyma.106 Hyperglycemia also induces the production of reactive oxygen species107 and increases pro-inflammatory cytokines and chemokines in many cells.108,109
Obesity is considered a low-grade chronic inflammation and has already been associated with functional alterations in the BBB (Figure 4). Dietary components such as fatty acids stimulate lipopolysaccharide (LPS) receptors and toll-like receptor 4(TLR4), triggering an inflammatory cascade that releases inflammatory mediators(TNF-α, IL-1β, IL-6). Studies have shown a decrease in energy consumption in BBB microvessels and a suppression of expression of 47 types of BBB proteins in diet-induced obesity (DIO) mice. Additionally, the activities of enzymes, transporters, and cytoskeleton proteins on BBB microvessels are preferentially suppressed in these DIO animals.110,111 Moreover, a study indicated that a high-energy (HE) diet is related to increased BBB permeability as elevations in peripheral proinflammatory cytokines compromise BBB function in specific areas of the brain.112
Decreased BBB permeability and upregulated expression of tight junction proteins were observed in germ-free adult mice.113 Intestinal flora disorders may compromise the BBB integrity due to the decreased expression of tight junction proteins.114 Disorders of the gut-microbial flora may cause systemic inflammation by destroying the intestinal epithelial barrier and introducing toxic by-products in the blood circulation.115 In humans, changes in the composition of gut-microbial flora are associated with obesity and T2DM.116,117 Furthermore, increasing evidence indicates that modifying the microbiota with prebiotics or probiotics benefits the host, while high-fiber diets also induce faster resolution of inflammatory responses.118,119
Temporary disruption of BBB has been widely studied as a prevalent approach for delivering drugs into the CNS from the circulatory system.120 BBB malfunction is a hallmark of a variety of neurological disorders, including Alzheimer’s disease, Parkinson’s disease, cerebral ischemia, stroke, hyperlipidemia, and diabetes.121 GLP-1RAs and SGLT-2is could cross the temporarily disrupted BBB in the brains of experimental animals and individuals with neurological disorders, diabetes, and obesity via a variety of transmembrane routes. GLP-1RAs and SGLT-2is both have the ability to penetrate damaged basement membranes and tight junctions (Figure 5).
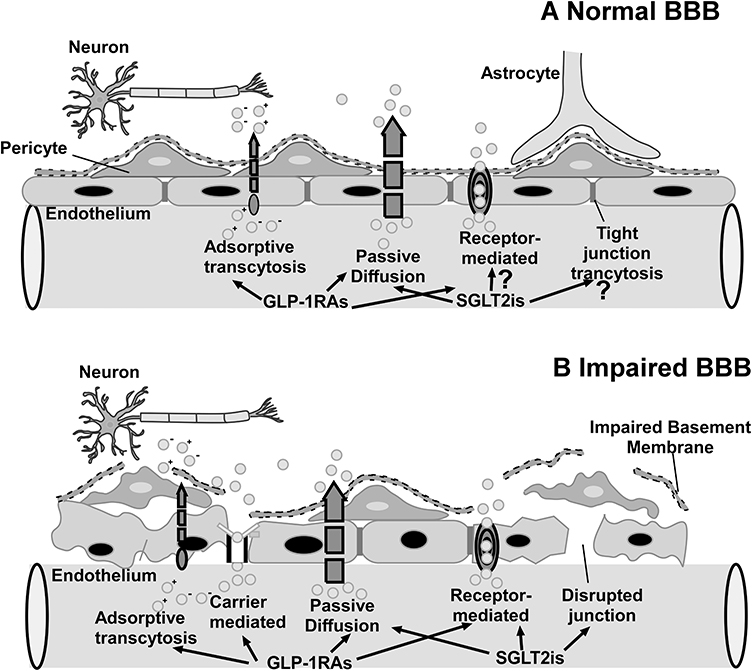 |
Figure 5 (A) Normal BBB: GLP-1RAs can cross the normal BBB by receptor-mediated transport and passive diffusion, but whether SGLT-2is may cross the compromised BBB via passive diffusion is still being investigated. (B) Impaired BBB. |
Limitations
Although providing a first glimpse at the capacity of GLP-1RAs and SGLT-2is to traverse the BBB, the current review has not disclosed the specific signaling mechanisms, particularly for SGLT-2is. In addition, it is yet unknown how the physical and chemical features of SGLT-2is regulate their transportation in the CNS. The interaction of GLP-1RAs and SGLT-2is with the blood-brain barrier (BBB) requires additional research and direct evidence at the molecular or cellular level.
Conclusion
In conclusion, the preclinical and clinical studies demonstrated that GLP-1RAs and SGLT-2is act directly or indirectly on their receptors by crossing the BBB. Thus, they can not only lower glucose levels but also be applied in the treatment of CNS diseases. Central administration of GLP-1RAs or SGLT-2is could increase the expression of c-Fos in neurons within the PVN, amygdala, NTS, AP, and ARC,56,96 which was consistent with the expression of GLP-1R/SGLT-2 in the whole CNS.30,31,75–77 This may be considered evidence that these drugs could pass through the BBB and activate the neurons in these nuclei. Some of the GLP-1RAs have been proven to cross through the BBB via passive diffusion.34,46,48,68 In addition, GLP-1RAs could cross the BBB via the GLP-1 receptor-mediated uptake mechanism.52 Liraglutide has access to specific areas of the brain associated with appetite regulation and was measured in selected CVOs and specific hypothalamic regions, of which the signal was GLP-1R-dependent.54 Adsorption transcytosis could be another mechanism involved in exendin-4 and lixisenatide crossing the BBB.66 Semaglutide may be unable to cross the regular BBB directly but could bind to serum albumin and pass through the tanycytes lining the ventricle wall.73 In summary, peripheral administration of GLP-1RAs either acts directly on the hypothalamus and hindbrain, which lack BBB, or directly crosses the BBB into CNS, which are then projected to other key feeding areas of the brain (Figure 6). Notably, SGLT-2is are medications of small molecular weight, with lower LogPoct than the permeation prediction index of BBB (listed in Table 2). SGLT-2is activated c-Fos expression in the CNS, and the relationship between the chemical structure/properties of SGLT-2is and BBB could be interpreted as evidence for penetrating the BBB.81 Due to the increased permeability of the BBB in hyperglycemia and obesity, SGLT-2is and GLP-1RAs are likely to pass through the compromised BBB.18 As such, the prospects of comprehensively elucidating the mechanism of SGLT-2is and GLP-1RAs interacting with BBB are promising, through which corresponding strategies for the prevention and treatment of DM, AD, PD and CVD can be developed.
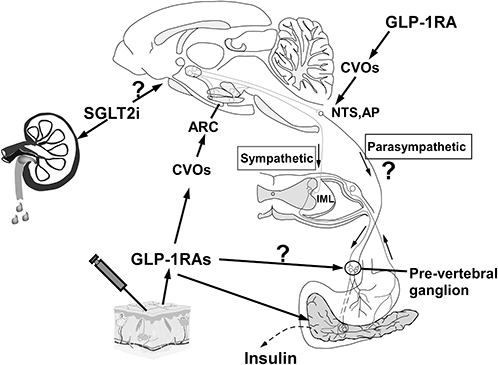 |
Figure 6 The arcuate nucleus (ARC) in the hypothalamus, the area postrema (AP) and the Nucleus of Solitary (NTS) in the brainstem are 3 examples of CVOs that are not protected by an effective blood-brain barrier. Once GLP-1RAs enter the bloodstream by subcutaneous injection, they can activate ARC hypothalamic POMC/CART neurons, which subsequently stimulate sympathetic efferent output, controlling insulin secretion. It is unclear if GLP-1-RAs stimulate the pre-vertebral ganglia directly. GLP-1RAs also activate GLP-1 receptors on the surface of pancreatic cells, resulting in the conversion of adenosine to cyclic adenosine phosphate (cAMP) in islet cells through conjugation of active G protein with adenosine cyclase. As cAMP levels rise, the pancreatic beta cell membrane depolarizes, which promotes insulin secretion. Further work is required to determine whether SGLT-2is may cross the BBB and exert similar effects on the brain. |
Funding
This work was supported by the Project of Key Medical Discipline of Pudong Hospital of Fudan University (Zdxk2020-11), Project of Key Medical Specialty and Treatment Center of Pudong Hospital of Fudan University (Zdzk2020-24), Integrative Medicine special fund of Shanghai Municipal Health Planning Committee (ZHYY- ZXYJHZX-2-201712), Special Department Fund of the Pudong New Area Health Planning Commission (PWZzk2017-03), Outstanding Leaders Training Program of Pudong Health Bureau of Shanghai (PWR12014-06), Pudong New Area Clinical Plateau Discipline Project (PWYgy-2021-03), the Natural Science Foundation of China (21675034), National Natural Science Foundation of China (81370932), Shanghai Natural Science Foundation (19ZR1447500) and Education Funding in Wenzhou Medical University (JG2021197).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14:88–98. doi:10.1038/nrendo.2017.151
2. Bluher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. 2019;15:288–298. doi:10.1038/s41574-019-0176-8
3. Filippo CD, Cuzzocrea S, Rossi F, Marfella R, D’Amico M. Oxidative stress as the leading cause of acute myocardial infarction in diabetics. Cardiovasc Drug Rev. 2006;24:77–87. doi:10.1111/j.1527-3466.2006.00077.x
4. Alloubani A, Saleh A, Abdelhafiz I. Hypertension and diabetes mellitus as a predictive risk factors for stroke. Diabetes Metab Syndr. 2018;12:577–584. doi:10.1016/j.dsx.2018.03.009
5. Gong M, Wen S, Nguyen T, et al. Converging relationships of obesity and hyperuricemia with special reference to metabolic disorders and plausible therapeutic implications. Diabetes Metab Syndr Obes. 2020;13:943–962. doi:10.2147/DMSO.S232377
6. Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8:728–742. doi:10.1038/nrendo.2012.140
7. Kanoski SE, Fortin SM, Arnold M, Grill HJ, Hayes MR. Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology. 2011;152(8):3103–3112. doi:10.1210/en.2011-0174
8. Gallo LA, Wright EM, Vallon V. Probing SGLT2 as a therapeutic target for diabetes: basic physiology and consequences. Diab Vasc Dis Res. 2015;12:78–89. doi:10.1177/1479164114561992
9. Brown RE, Gupta N, Aronson R. Effect of dapagliflozin on glycemic control, weight, and blood pressure in patients with type 2 diabetes attending a specialist endocrinology practice in canada: a retrospective cohort analysis. Diabetes Technol Ther. 2017;19:685–691. doi:10.1089/dia.2017.0134
10. Cai X, Yang W, Gao X, et al. The association between the dosage of SGLT2 inhibitor and weight reduction in type 2 diabetes patients: a meta-analysis. Obesity. 2018;26:70–80. doi:10.1002/oby.22066
11. Rosenstock J, Frias J, Pall D, et al. Effect of ertugliflozin on glucose control, body weight, blood pressure and bone density in type 2 diabetes mellitus inadequately controlled on metformin monotherapy (VERTIS MET). Diabetes Obes Metab. 2018;20:520–529. doi:10.1111/dom.13103
12. Khat DZ, Husain M. Molecular mechanisms underlying the cardiovascular benefits of SGLT2i and GLP-1RA. Curr Diab Rep. 2018;18:45. doi:10.1007/s11892-018-1011-7
13. Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–1844. doi:10.1056/NEJMoa1607141
14. Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393:31–39. doi:10.1016/S0140-6736(18)32590-X
15. Wen S, Nguyen T, Gong M, et al. An overview of similarities and differences in metabolic actions and effects of central nervous system between glucagon-like peptide-1 receptor agonists (GLP-1RAs) and sodium glucose co-transporter-2 inhibitors (SGLT-2is). Diabetes Metab Syndr Obes. 2021;14:2955–2972. doi:10.2147/DMSO.S312527
16. Zhou L, Sutton GM, Rochford JJ, et al. Serotonin 2C receptor agonists improve type 2 diabetes via melanocortin-4 receptor signaling pathways. Cell Metab. 2007;6:398–405. doi:10.1016/j.cmet.2007.10.008
17. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi:10.1016/j.nbd.2009.07.030
18. Acharya NK, Levin EC, Clifford PM, et al. Diabetes and hypercholesterolemia increase blood-brain barrier permeability and brain amyloid deposition: beneficial effects of the LpPLA2 inhibitor darapladib. J Alzheimers Dis. 2013;35:179–198. doi:10.3233/JAD-122254
19. Starr JM, Wardlaw J, Ferguson K, MacLullich A, Deary IJ, Marshall I. Increased blood–brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2003;74:70–76. doi:10.1136/jnnp.74.1.70
20. Mauro C, De Rosa V, Marelli-Berg F, Solito E. Metabolic syndrome and the immunological affair with the blood-brain barrier. Front Immunol. 2014;5:677. doi:10.3389/fimmu.2014.00677
21. Miller AA, Spencer SJ. Obesity and neuroinflammation: a pathway to cognitive impairment. Brain Behav Immun. 2014;42:10–21. doi:10.1016/j.bbi.2014.04.001
22. Pan Y, Nicolazzo JA. Impact of aging, Alzheimer’s disease and Parkinson’s disease on the blood-brain barrier transport of therapeutics. Adv Drug Deliv Rev. 2018;135:62–74. doi:10.1016/j.addr.2018.04.009
23. Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43:348–364. doi:10.1080/10715760902751902
24. Terami N, Ogawa D, Tachibana H, et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS One. 2014;9:e100777. doi:10.1371/journal.pone.0100777
25. Abbott NJ. Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol. 2005;25:5–23. doi:10.1007/s10571-004-1374-y
26. Alexander JJ. Blood-brain barrier (BBB) and the complement landscape. Mol Immunol. 2018;102:26–31. doi:10.1016/j.molimm.2018.06.267
27. Norsted E, Gomuc B, Meister B. Protein components of the blood-brain barrier (BBB) in the mediobasal hypothalamus. J Chem Neuroanat. 2008;36:107–121. doi:10.1016/j.jchemneu.2008.06.002
28. Bagchi S, Chhibber T, Lahooti B, et al. In-vitro blood-brain barrier models for drug screening and permeation studies: an overview. Drug Des Devel Ther. 2019;13:3591–3605. doi:10.2147/DDDT.S218708
29. Gawdi R, Emmady PD, Jindal C. Physiology, blood brain barrier. J Thorac Dis. 2021;13(10):5617–5626. doi:10.21037/jtd-21-1018
30. Jensen CB, Pyke C, Rasch MG, et al. Characterization of the glucagon like peptide-1 receptor in male mouse brain using a novel antibody and in situ hybridization. Endocrinology. 2018;159(2):665–675. doi:10.1210/en.2017-00812
31. Farr OM, Sofopoulos M, Tsoukas MA, et al. GLP-1 receptors exist in the parietal cortex, hypothalamus and medulla of human brains and the GLP-1 analogue liraglutide alters brain activity related to highly desirable food cues in individuals with diabetes: a crossover, randomised, placebo-controlled trial. Diabetologia. 2016;59:954–965. doi:10.1007/s00125-016-3874-y
32. Holscher C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology. 2018;136:251–259. doi:10.1016/j.neuropharm.2018.01.040
33. Lv M, Xue G, Cheng H, et al. The GLP-1/GIP dual-receptor agonist DA5-CH inhibits the NF-kappaB inflammatory pathway in the MPTP mouse model of Parkinson’s disease more effectively than the GLP-1 single-receptor agonist NLY01. Brain Behav. 2021;11:e2231. doi:10.1002/brb3.2231
34. McGovern SF, Hunter K, Holscher C. Effects of the glucagon-like polypeptide-1 analogue (Val8)GLP-1 on learning, progenitor cell proliferation and neurogenesis in the C57B/16 mouse brain. Brain Res. 2012;1473:204–213. doi:10.1016/j.brainres.2012.07.029
35. Bailey J, Barrett A, Coucha M, Abdelsaid MA. Abstract P729: glp-1 receptor nitration contributes to brain pericytes dysfunction in diabetes. Stroke. 2021;52:AP729–AP729. doi:10.1161/str.52.suppl_1.P729
36. Fortin SM, Lipsky RK, Lhamo R, et al. GABA neurons in the nucleus tractus solitarius express GLP-1 receptors and mediate anorectic effects of liraglutide in rats. Sci Transl Med. 2020;12. doi:10.1126/scitranslmed.aay8071
37. Vanlandewijck M, He L, Mae MA, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554:475–480. doi:10.1038/nature25739
38. Bhavsar S, Mudaliar S, Cherrington A. Evolution of Exenatide as a Diabetes Therapeutic. Curr Diabetes Rev. 2013;9:161–193. doi:10.2174/1573399811309020007
39. Werner U, Haschke G, Herling AW, Kramer W. Pharmacological profile of lixisenatide: a new GLP-1 receptor agonist for the treatment of type 2 diabetes. Regul Pept. 2010;164:58–64. doi:10.1016/j.regpep.2010.05.008
40. Tang C, Li Q, Deng X, et al. Discovery of lixisenatide analogues as long-acting hypoglycemic agents using novel peptide half-life extension technology based on mycophenolic acid. RSC Adv. 2020;10:12089–12104. doi:10.1039/D0RA01002B
41. Zhang YL, Zhou C, Li XF, et al. Beinaglutide showed significant weight-loss benefit and effective glycaemic control for the treatment of type 2 diabetes in a real-world setting: a 3-month, multicentre, observational, retrospective, open-label study. Obes Sci Pract. 2019;5:366–375. doi:10.1002/osp4.342
42. Buse JB, Rosenstock J, Sesti G, et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet. 2009;374:39–47. doi:10.1016/S0140-6736(09)60659-0
43. Geiser JS, Heathman MA, Cui X, et al. Clinical pharmacokinetics of dulaglutide in patients with type 2 diabetes: analyses of data from clinical trials. Clin Pharmacokinet. 2016;55:625–634. doi:10.1007/s40262-015-0338-3
44. Kalra S, Sahay R. A review on semaglutide: an oral glucagon-like peptide 1 receptor agonist in management of type 2 diabetes mellitus. Diabetes Ther. 2020;11:1965–1982. doi:10.1007/s13300-020-00894-y
45. Jacobsen LV, Flint A, Olsen AK, Ingwersen SH. Liraglutide in type 2 diabetes mellitus: clinical pharmacokinetics and pharmacodynamics. Clin Pharmacokinet. 2016;55:657–672. doi:10.1007/s40262-015-0343-6
46. Kastin AJ, Akerstrom V, Pan W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci. 2002;18:7–14. doi:10.1385/JMN:18:1-2:07
47. Zhou L, Williams T, Lachey JL, et al. Serotonergic pathways converge upon central melanocortin systems to regulate energy balance. Peptides. 2005;26(10):1728–1732. doi:10.1016/j.peptides.2004.12.028
48. Zhou L, Yueh CY, Lam DD, et al. Glucokinase inhibitor glucosamine stimulates feeding and activates hypothalamic neuropeptide Y and orexin neurons. Behav Brain Res. 2011;222:274–278. doi:10.1016/j.bbr.2011.03.043
49. Heisler LK, Jobst EE, Sutton GM, et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron. 2006;51:239–249. doi:10.1016/j.neuron.2006.06.004
50. Brierley DI, Holt MK, Singh A, et al. Central and peripheral GLP-1 systems independently suppress eating. Nature Metabol. 2021;3:258–273. doi:10.1038/s42255-021-00344-4
51. Plamboeck A, Veedfald S, Deacon CF, et al. The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am J Physiol Gastrointest Liver Physiol. 2013;304:G1117–G1127. doi:10.1152/ajpgi.00035.2013
52. Fu Z, Gong L, Liu J, et al. Brain endothelial cells regulate glucagon-like peptide 1 entry into the brain via a receptor-mediated process. Front Physiol. 2020;11:555. doi:10.3389/fphys.2020.00555
53. Labouesse MA, Stadlbauer U, Weber E, et al. Vagal afferents mediate early satiation and prevent flavour avoidance learning in response to intraperitoneally infused exendin-4. J Neuroendocrinol. 2012;24(12):1505–1516. doi:10.1111/j.1365-2826.2012.02364.x
54. Secher A, Jelsing J, Baquero AF, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest. 2014;124:4473–4488. doi:10.1172/JCI75276
55. Salinas CBG, Lu TT, Gabery S, et al. Integrated brain atlas for unbiased mapping of nervous system effects following liraglutide treatment. Sci Rep. 2018;8:10310. doi:10.1038/s41598-018-28496-6
56. Larsen PJ, Tang-Christensen M, Jessop DS. Central administration of glucagon-like peptide-1 activates hypothalamic neuroendocrine neurons in the rat. Endocrinology. 1997;138:4445–4455. doi:10.1210/endo.138.10.5270
57. McLean BA, Wong CK, Campbell JE, et al. Revisiting the complexity of GLP-1 action from sites of synthesis to receptor activation. Endocr Rev. 2021;42(2):101–132. doi:10.1210/endrev/bnaa032
58. Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from Guinea pig pancreas. J Biol Chem. 1992;267:7402–7405. doi:10.1016/S0021-9258(18)42531-8
59. Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J Clin Invest. 2012;122:1339–1353. doi:10.1172/JCI57256
60. Kastin A, Akerstrom V. Entry of exendin-4 into brain is rapid but may be limited at high doses. Int J Obes. 2003;27(3):313–318. doi:10.1038/sj.ijo.0802206
61. Harkavyi A, Abuirmeileh A, Lever R, et al. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J Neuroinflammation. 2008;5:19. doi:10.1186/1742-2094-5-19
62. Mishra SH, Bhavaraju S, Schmidt DR, Carrick KL. Facilitated structure verification of the biopharmaceutical peptide exenatide by 2D heteronuclear NMR maps. J Pharm Biomed Anal. 2021;203:114136. doi:10.1016/j.jpba.2021.114136
63. Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664–1675. doi:10.1016/S0140-6736(17)31585-4
64. Aviles-Olmos I, Dickson J, Kefalopoulou Z, et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J Parkinsons Dis. 2014;4:337–344. doi:10.3233/JPD-140364
65. Aviles-Olmos I, Dickson J, Kefalopoulou Z, et al. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest. 2013;123:2730–2736. doi:10.1172/JCI68295
66. Salameh TS, Rhea EM, Talbot K, Banks WA. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem Pharmacol. 2020;180:114187. doi:10.1016/j.bcp.2020.114187
67. McClean PL, Holscher C. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology. 2014;86:241–258. doi:10.1016/j.neuropharm.2014.07.015
68. Hunter K, Holscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 2012;13:33. doi:10.1186/1471-2202-13-33
69. McClean PL, Holscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. 2014;76(Pt A):57–67. doi:10.1016/j.neuropharm.2013.08.005
70. McClean PL, Parthsarathy V, Faivre E, Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31:6587–6594. doi:10.1523/JNEUROSCI.0529-11.2011
71. Liu W, Jalewa J, Sharma M, et al. Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience. 2015;303:42–50. doi:10.1016/j.neuroscience.2015.06.054
72. Glaesner W, Vick AM, Millican R, et al. Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an Fc fusion protein. Diabetes Metab Res Rev. 2010;26:287–296. doi:10.1002/dmrr.1080
73. Gabery S, Salinas CG, Paulsen SJ, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020;5. doi:10.1172/jci.insight.133429
74. Chen J, Williams S, Ho S, et al. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010;1:57–92. doi:10.1007/s13300-010-0006-4
75. Yu AS, Hirayama BA, Timbol G, et al. Functional expression of SGLTs in rat brain. Am J Physiol Cell Physiol. 2010;299:C1277–C1284. doi:10.1152/ajpcell.00296.2010
76. Yu AS, Hirayama BA, Timbol G, et al. Regional distribution of SGLT activity in rat brain in vivo. Am J Physiol Cell Physiol. 2013;304:C240–C247. doi:10.1152/ajpcell.00317.2012
77. Shah K, Desilva S, Abbruscato T. The role of glucose transporters in brain disease: diabetes and Alzheimer’s Disease. Int J Mol Sci. 2012;13:12629–12655. doi:10.3390/ijms131012629
78. Chiba Y, Sugiyama Y, Nishi N, et al. Sodium/glucose cotransporter 2 is expressed in choroid plexus epithelial cells and ependymal cells in human and mouse brains. Neuropathology. 2020;40(5):482–491. doi:10.1111/neup.12665
79. Shin SJ, Chung S, Kim SJ, et al. Effect of sodium-glucose co-transporter 2 inhibitor, dapagliflozin, on renal renin-angiotensin system in an animal model of type 2 diabetes. PLoS One. 2016;11:e0165703. doi:10.1371/journal.pone.0165703
80. Puglisi S, Rossini A, Poli R, et al. Effects of SGLT2 inhibitors and GLP-1 receptor agonists on renin-angiotensin-aldosterone system. Front Endocrinol. 2021;12:738848. doi:10.3389/fendo.2021.738848
81. Nguyen T, Wen S, Gong M, et al. Dapagliflozin activates neurons in the central nervous system and regulates cardiovascular activity by inhibiting SGLT-2 in mice. Diabetes Metab Syndr Obes. 2020;13:2781–2799. doi:10.2147/DMSO.S258593
82. Gray MT, Woulfe JM. Striatal blood-brain barrier permeability in Parkinson’s disease. J Cereb Blood Flow Metab. 2015;35:747–750. doi:10.1038/jcbfm.2015.32
83. Bowen Lin NK, Hasegawa Y, Sueta D, et al. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol. 2014;13:1–5.
84. El-Sahar AE, Rastanawi AA, El-Yamany MF, Saad MA. Dapagliflozin improves behavioral dysfunction of Huntington’s disease in rats via inhibiting apoptosis-related glycolysis. Life Sci. 2020;257:118076. doi:10.1016/j.lfs.2020.118076
85. Millar P, Pathak N, Parthsarathy V, et al. Metabolic and neuroprotective effects of dapagliflozin and liraglutide in diabetic mice. J Endocrinol. 2017;234:255–267. doi:10.1530/JOE-17-0263
86. Shaikh S, Rizvi SMD, Shakil S, et al. Forxiga (dapagliflozin): plausible role in the treatment of diabetes-associated neurological disorders. Biotechnol Appl Biochem. 2016;63:145–150. doi:10.1002/bab.1319
87. Hayden MR, Grant DG, Aroor AR, DeMarco VG. Empagliflozin ameliorates type 2 diabetes-induced ultrastructural remodeling of the neurovascular unit and neuroglia in the female db/db mouse. Brain Sci. 2019;9(3):57. doi:10.3390/brainsci9030057
88. Amin EF, Rifaai RA, Abdel-Latif RG. Empagliflozin attenuates transient cerebral ischemia/reperfusion injury in hyperglycemic rats via repressing oxidative-inflammatory-apoptotic pathway. Fundam Clin Pharmacol. 2020;34:548–558. doi:10.1111/fcp.12548
89. Bhatt DL, Szarek M, Steg PG, et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. 2021;384:117–128. doi:10.1056/NEJMoa2030183
90. Pawlos A, Broncel M, Wozniak E, Gorzelak-Pabis P. Neuroprotective effect of SGLT2 inhibitors. Molecules. 2021;26(23):7213. doi:10.3390/molecules26237213
91. Cosentino F, Cannon CP, Cherney DZI, et al. Efficacy of ertugliflozin on heart failure-related events in patients with type 2 diabetes mellitus and established atherosclerotic cardiovascular disease: results of the VERTIS CV trial. Circulation. 2020;142:2205–2215. doi:10.1161/CIRCULATIONAHA.120.050255
92. Matharu K, Chana K, Ferro CJ, Jones AM. Polypharmacology of clinical sodium glucose co-transport protein 2 inhibitors and relationship to suspected adverse drug reactions. Pharmacol Res Perspect. 2021;9:e00867. doi:10.1002/prp2.867
93. Markham A, Elkinson S. Luseogliflozin: first global approval. Drugs. 2014;74:945–950. doi:10.1007/s40265-014-0230-8
94. Wang S, Fan F. Oral antihyperglycemic therapy with a SGLT2 inhibitor reverses cognitive impairments in elderly diabetics. Hypertension. 2019;74:A051–A051. doi:10.1161/hyp.74.suppl_1.051
95. Wang S, Jiao F, Border JJ, et al. Luseogliflozin, a sodium-glucose cotransporter-2 inhibitor, reverses cerebrovascular dysfunction and cognitive impairments in 18-mo-old diabetic animals. Am J Physiol Heart Circ Physiol. 2022;322:H246–H259. doi:10.1152/ajpheart.00438.2021
96. Takeda K, Ono H, Ishikawa K, et al. Central administration of sodium-glucose cotransporter-2 inhibitors increases food intake involving adenosine monophosphate-activated protein kinase phosphorylation in the lateral hypothalamus in healthy rats. BMJ Open Diabetes Res. 2021;9(1):e002104. doi:10.1136/bmjdrc-2020-002104
97. Lin KJ, Wang TJ, Chen SD, et al. Two birds one stone: the neuroprotective effect of antidiabetic agents on parkinson disease-focus on sodium-glucose cotransporter 2 (SGLT2) inhibitors. Antioxidants. 2021;10:1935. doi:10.3390/antiox10121935
98. Yokono M, Takasu T, Hayashizaki Y, et al. SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol. 2014;727:66–74. doi:10.1016/j.ejphar.2014.01.040
99. Rizvi SM, Shakil S, Biswas D, et al. Invokana (Canagliflozin) as a dual inhibitor of acetylcholinesterase and sodium glucose co-transporter 2: advancement in Alzheimer’s disease- diabetes type 2 linkage via an enzoinformatics study. CNS Neurol Disord Drug Targets. 2014;13(3):447–451. doi:10.2174/18715273113126660160
100. Naznin F, Sakoda H, Okada T, et al. Canagliflozin, a sodium glucose cotransporter 2 inhibitor, attenuates obesity-induced inflammation in the nodose ganglion, hypothalamus, and skeletal muscle of mice. Eur J Pharmacol. 2017;794:37–44. doi:10.1016/j.ejphar.2016.11.028
101. Arafa NM, Marie MA, AlAzimi SA. Effect of canagliflozin and metformin on cortical neurotransmitters in a diabetic rat model. Chem Biol Interact. 2016;258:79–88. doi:10.1016/j.cbi.2016.08.016
102. Sha W, Wen S, Chen L, et al. The role of SGLT2 inhibitor on the treatment of diabetic retinopathy. J Diabetes Res. 2020;2020:8867875. doi:10.1155/2020/8867875
103. Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50:202–211. doi:10.1007/s00125-006-0485-z
104. Bogush M, Heldt NA, Persidsky Y. Blood brain barrier injury in diabetes: unrecognized effects on brain and cognition. J Neuroimmune Pharmacol. 2017;12:593–601. doi:10.1007/s11481-017-9752-7
105. Banks WA. The blood-brain barrier interface in diabetes mellitus: dysfunctions, mechanisms and approaches to treatment. Curr Pharm Des. 2020;26:1438–1447. doi:10.2174/1381612826666200325110014
106. Serlin Y, Levy J, Shalev H. Vascular pathology and blood-brain barrier disruption in cognitive and psychiatric complications of type 2 diabetes mellitus. Cardiovasc Psychiatry Neurol. 2011;2011:609202. doi:10.1155/2011/609202
107. Thackeray JT, Hupe HC, Wang Y, et al. Myocardial Inflammation Predicts Remodeling and Neuroinflammation After Myocardial Infarction. J Am Coll Cardiol. 2018;71:263–275. doi:10.1016/j.jacc.2017.11.024
108. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018;9:119. doi:10.1038/s41419-017-0135-z
109. Stentz FB, Umpierrez GE, Cuervo R, Kitabchi AE. Proinflammatory cytokines, markers of cardiovascular risks, oxidative stress, and lipid peroxidation in patients with hyperglycemic crises. Diabetes. 2004;53:2079–2086. doi:10.2337/diabetes.53.8.2079
110. Shu CJ, Benoist C, Mathis D. The immune system’s involvement in obesity-driven type 2 diabetes. Semin Immunol. 2012;24:436–442. doi:10.1016/j.smim.2012.12.001
111. Ouyang S, Hsuchou H, Kastin AJ, et al. Diet-induced obesity suppresses expression of many proteins at the blood-brain barrier. J Cereb Blood Flow Metab. 2014;34:43–51. doi:10.1038/jcbfm.2013.166
112. Davidson TL, Monnot A, Neal AU, et al. The effects of a high-energy diet on hippocampal-dependent discrimination performance and blood-brain barrier integrity differ for diet-induced obese and diet-resistant rats. Physiol Behav. 2012;107:26–33. doi:10.1016/j.physbeh.2012.05.015
113. Braniste V, Al-Asmakh M, Kowal C, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. 2014;6:263ra158–263ra158. doi:10.1126/scitranslmed.3009759
114. Frohlich EE, Farzi A, Mayerhofer R, et al. Cognitive impairment by antibiotic-induced gut dysbiosis: analysis of gut microbiota-brain communication. Brain Behav Immun. 2016;56:140–155. doi:10.1016/j.bbi.2016.02.020
115. O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7:688–693. doi:10.1038/sj.embor.7400731
116. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi:10.1038/4441022a
117. Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut. 2014;63:1513–1521. doi:10.1136/gutjnl-2014-306928
118. Vieira AT, Galvao I, Macia LM, et al. Dietary fiber and the short-chain fatty acid acetate promote resolution of neutrophilic inflammation in a model of gout in mice. J Leukoc Biol. 2017;101:275–284. doi:10.1189/jlb.3A1015-453RRR
119. Liu X, Cao S, Zhang X. Modulation of gut microbiota-brain axis by probiotics, prebiotics, and diet. J Agric Food Chem. 2015;63:7885–7895. doi:10.1021/acs.jafc.5b02404
120. Toccaceli G, Barbagallo G, Peschillo S. Low-intensity focused ultrasound for the treatment of brain diseases: safety and feasibility. Theranostics. 2019;9:537–539. doi:10.7150/thno.31765
121. Xie J, Shen Z, Anraku Y, Kataoka K, Chen X. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials. 2019;224:119491. doi:10.1016/j.biomaterials.2019.119491
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.