")
Back to Journals » Risk Management and Healthcare Policy » Volume 14
The RB1 Mutation Spectrum and Genetic Management Consultation in Pediatric Patients with Retinoblastoma in Beijing, China
Received 31 May 2021
Accepted for publication 22 July 2021
Published 21 August 2021 Volume 2021:14 Pages 3453—3463
DOI https://doi.org/10.2147/RMHP.S322373
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jongwha Chang
Ying Xie,1,2 Xiao-Lin Xu,1 Wen-Bin Wei1
1Beijing Tongren Eye Center, Beijing Key Laboratory of Intraocular Tumor Diagnosis and Treatment, Beijing Ophthalmology and Visual Sciences Key Lab, Medical Artificial Intelligence Research and Verification Key Laboratory of the Ministry of Industry and Information Technology, Beijing Tongren Hospital, Capital Medical University, Beijing, 100730, People’s Republic of China; 2Department of Opthalmology, Shanxi Provincial People’s Hospital, Taiyuan, 030012, People’s Republic of China
Correspondence: Wen-Bin Wei
Beijing Tongren Eye Center, Beijing Key Laboratory of Intraocular Tumor Diagnosis and Treatment, Beijing Ophthalmology and Visual Sciences Key Lab, Medical Artificial Intelligence Research and Verification Key Laboratory of the Ministry of Industry and Information Technology, Beijing Tongren Hospital, Capital Medical University, No. 1 of Dong Jiao Min Lane, Dongcheng District, Beijing, 100730, People’s Republic of China
Tel +86 10 5826 9516
Fax +86 10 6512 5617
Email [email protected]
Objective: The present study screened the structural mutations of the retinoblastoma (RB1) gene using gene capture and a preliminary exploration of the correlation between the genotypes and phenotypes.
Methods: A total of 45 formalin-fixed paraffin-embedded (FFPE) tissue samples and 12 peripheral venous blood samples from patients with retinoblastoma (RB) confirmed by pathological examination at Beijing Tongren Hospital were collected between May 2019 and May 2021. DNA from the samples was extracted, sequenced, and analyzed to detect the mutations in the RB1 gene by designing the targeted capture probes for exons and the flanking sequences of the gene.
Results: Of the 45 FFPE tissue samples, 23 were from male patients and 22 were from female patients, all aged between 4 months and 10 years, with an average age of 2.5 ± 1.3 years. Two of these patients had bilateral RB and 43 had unilateral RB (23 in the right eye and 20 in the left eye). Of the 12 peripheral venous blood samples, 7 were from male patients and 5 were from female patients, all aged between 8 months and 4 years, with an average age of 1.3 ± 0.9 years. Two of these patients had bilateral RB and 10 had unilateral RB (8 in the right eye and 2 in the left eye). Three de novo pathogenic mutations were found in the FFPE tissues, along with one de novo potentially pathogenic mutation, while three de novo potentially pathogenic mutations were found in the blood samples.
Conclusion: Gene capture is a low-cost and efficient method for the gene sequencing of RB. A total of seven de novo mutations were identified through mutation testing of the pathogenic gene RB1 in 56 pediatric patients with RB. This complemented the mutation spectrum of the RB1 gene and helped to improve the molecular diagnosis of RB, thereby providing a basis for genetic counseling and prediction of the clinical phenotype, as well as for the genetic testing of the offspring of patients with RB.
Clinical Registration Number: ChiCTR-EPC-17013892.
Keywords: retinoblastoma, RB1 gene, gene capture
Introduction
Retinoblastoma (RB) is the most common primary intraocular malignancy in infants and children, accounting for 3% of all malignant tumors in pediatric patients, and the second most common malignant tumor in pediatrics overall.1,2 The global incidence of RB is 1 in 15,000–20,000 live births,3 and the incidence in China is 1 in 23,160.4 RB was first reported by the Dutch internist Pawius in 1597, and the first case in China was reported by Huade Bi in 1921.5,6 The increase in the incidence of RB is due to improved diagnosis and a gradual increase in recognition and awareness, which have greatly increased the detection rate, and the prevalence of early diagnosis and the rational treatment of patients, which have increased the cure rate. Due to the increased cure rate, there is also now a higher chance of survivors passing on the pathological gene to their offspring.
There are two types of RB—hereditary and non-hereditary—with hereditary RB accounting for approximately 45% of cases and non-hereditary RB accounting for approximately 55%.7 Hereditary RB is caused by germ-cell mutation and manifests as bilateral RB (accounting for 80% of cases) or unilateral multiple RB (accounting for 15% of cases), of which approximately 5% can be accompanied by intracranial tumors, called trilateral RB.8 Most patients with hereditary RB develop a second malignant neoplasm several years later.9 Non-hereditary RB is caused by somatic mutations and mostly manifests as unilateral RB.
Although RB is a serious risk to the life of patients, the survival rate in developed countries can be as high as 95%, and more than 90% of pediatric patients with RB retain visual function in at least one eye.10 However, for many pediatric patients in China, the tumor is already large by the time RB is diagnosed, making it difficult—and even life threatening—to preserve the eye.
The RB1 gene was the first tumor-suppressor gene to be discovered in humans. Its discovery is recognized as an important milestone in the study of human oncology and the cell cycle. The gene is located at 13q14, with a total length of approximately 180 kb and 27 exons, encoding an RB protein (pRB) with 928 amino acid residues.11 The structure of pRB is divided into three domains, composed of an N-terminus, an R motif, an A/B pocket, and a C-terminus.12 Mutations in the RB1 gene deactivate the pRB, resulting in abnormal cell-cycle division and the transformation of benign retinal tumors into malignant RB.13
Current genetic testing methods involve extracting high-quality DNA from the blood and/or tumor tissue and using quantitative polymerase chain reaction (PCR), gene chips, or high-throughput sequencing to detect the presence of variants, such as rearrangements, deletions, and insertions in the genes; allele-specific PCR is also used for the known common gene-mutated loci, along with the sequencing of all exons and the intron shear site of the RB1 gene. Based on current understanding and technology, 5% of patients with RB are not found to have pathogenic gene mutations.14
In the present study, a new-generation sequencing platform based on sequence capture of the target region was used to sequence the RB1 gene in DNA extracted from the formalin-fixed paraffin-embedded (FFPE) tissues and peripheral venous blood of pediatric patients with RB. This platform can detect both the copy number and single-base mutations and has the advantages of high sensitivity, high specificity, high throughput, low costs, and rapid results. The findings from this statistical analysis of the RB1 gene contribute to current understanding of the high-frequency mutated loci of the RB1 gene in patients with RB in China, which will help with the early diagnosis of RB in pediatric patients and guide genetic consultation for families with RB.
Subjects and Methods
Study Subjects
DNA samples from the FFPE tissues of 45 pediatric patients with RB and from the peripheral venous blood of 12 pediatric patients with RB were studied between May 2019 and May 2021. One patient underwent simultaneous blood sample and FFPE tissue testing, so there were 27 female patients and 29 male patients enrolled in total.
The FFPE tissues were histopathologically diagnosed, and the clinical diagnosis of RB was made by clinical examination, radiological examination (ultrasound/computed tomography/magnetic resonance imaging), and RetCam imaging.
Methods
Collection of Blood Samples and Concentration Detection
A total of 2 mL of venous blood was collected from each patient, placed in a test tube, and anticoagulated with ethylenediaminetetraacetic acid. The DNA was extracted using a FlexiGene DNA Kit (Qiagen, Germany) and stored at –80°C for further detection. DNA content was determined using a NanoDrop 2000 ultra-micro spectrophotometer (Thermo Fisher Scientific, USA).
Collection of FFPE Tissues and Concentration Detection
Excess paraffin on each FFPE tissue was removed with a scalpel, and the sample was sliced to a thickness of 10 μm; the first 2–3 outside-exposed and air-exposed slices were discarded, and 1–2 internal slices were maintained for DNA extraction. The slices were immediately placed in 2 mL EP centrifuge tubes, and DNA was extracted using a FlexiGene DNA Kit (Qiagen, Germany) and stored at –80°C for further detection. DNA content was determined using a NanoDrop 2000 ultra-micro spectrophotometer (Thermo Fisher Scientific, USA).
Probe Design and Target-Gene Sequencing
Using the Online Mendelian Inheritance in Man database(OMIM), Human Gene Mutation Database (HGMD), and Leiden Open Variation Database 3.0 (LOVD) as references, target-capture probes targeting the exons and flanking sequences (± 10 bp) of the RB1 gene were designed using the SureDesign online design tool (Agilent, USA). The target-gene capture libraries were prepared using an Agilent SureSelect Target Enrichment System Kit (Agilent, USA) on the DNA samples from the 56 patients, after which the samples from the sequencing libraries were tested using an Agilent 2100 Bioanalyzer with DNA chips (Agilent, USA) and DNA 1000 Reagent (Agilent, USA). A NextSeq 500 sequencer (Illumina, USA) was then used to sequence the DNA samples according to the concentration and sequencing depth requirements of the captured library.
Data Analysis and Evaluation
The raw-data FASTQ files generated by the sequencing platform were analyzed using RTA software (Illumina, USA), CASAVA software v. 1.8.2 (Illumina, USA), and the genetic analysis toolkit bioinformatics software to assess the sequencing quality. The evaluation parameters included total number of reads, proportion of matches to human genome sequences, proportion located within the target sequence of the target gene, average sequencing depth, proportion of sequenced regions greater than 20× depth, and coverage uniformity analysis. After generating single-nucleotide variant (SNV) reports, variant annotation was performed using PolyPhen 2.2.2 software, Annotate Variation software, the HGMD database, the LOVD database, the Single Nucleotide Polymorphism Database (dbSNP), and the 1000 Genomes Project database.
Results
General Characteristics
The general characteristics of the 56 patients are shown in Tables 1 and 2. Most of the probands were classified as group D or E (according to the International Classification of Intraocular Retinoblastoma) at the time of diagnosis, accounting for 96%, with two cases in the FFPE group being classified as group C, accounting for 4%.
 |
Table 1 General Clinical Profiles of RB Probands in This Study (FFPE) |
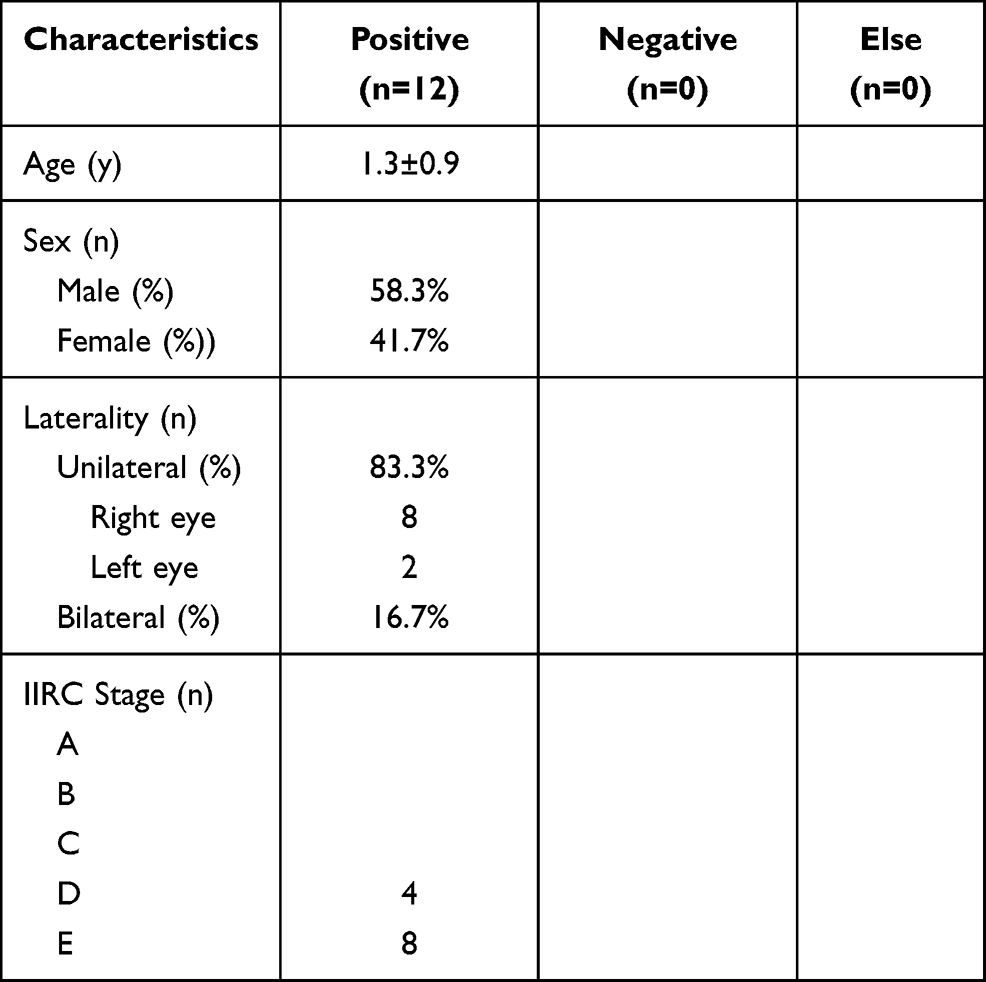 |
Table 2 General Clinical Profiles of RB Probands in This Study (Blood) |
Results of the Gene Detection in the FFPE Tissue Samples and Blood Samples
FFPE Tissue Samples
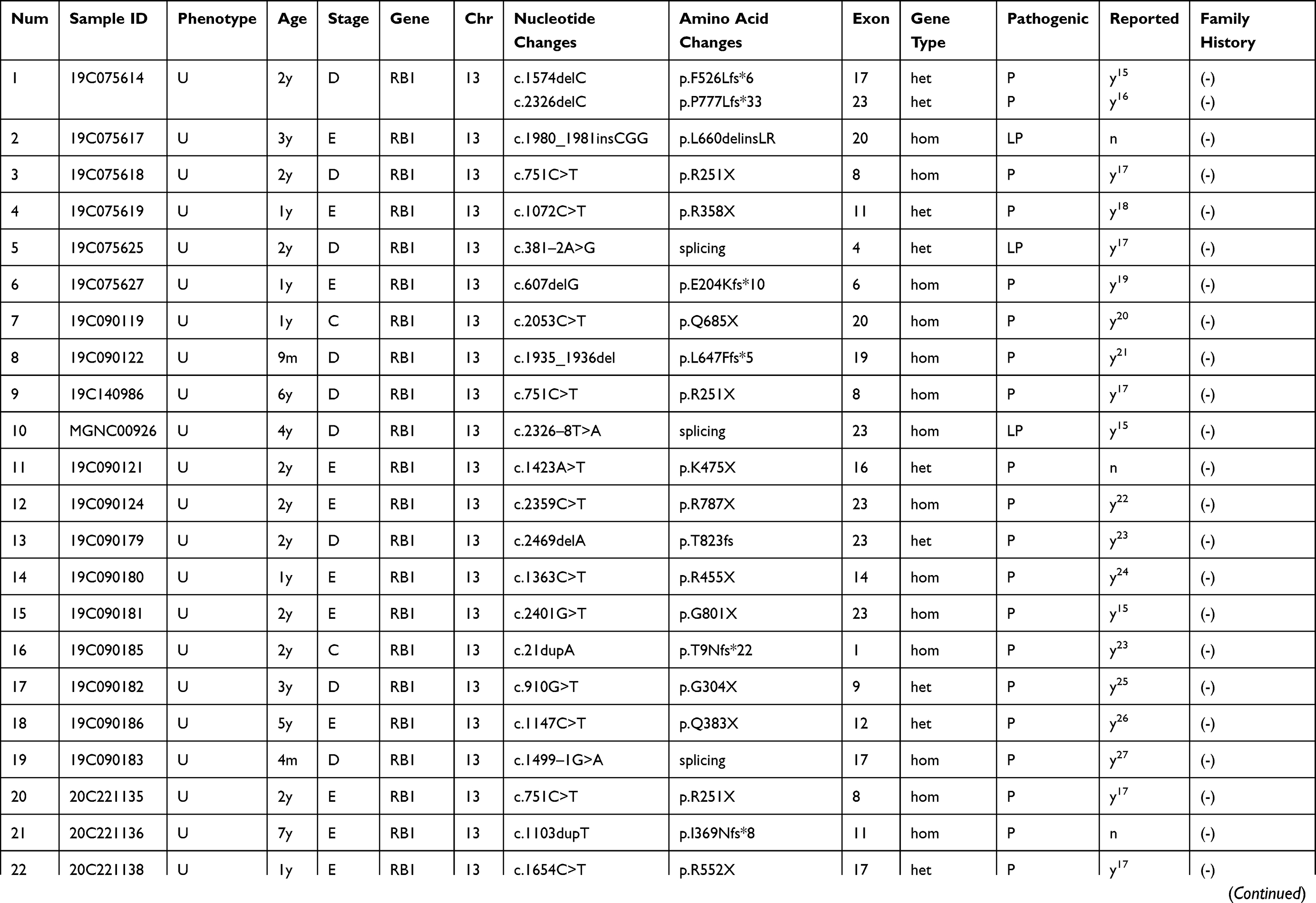
One case in the FFPE tissue samples had negative results, four cases were found to be non-RB1 mutations, and the remaining 40 cases showed RB1 mutations, with a positivity of 88.9%. The 44 cases are summarized in Table 3.
 |
 |
 |
Table 3 The Results of FFPE-DNA Gene Sequencing in 44 Cases Were Summarized |
Peripheral Blood Samples
All 12 peripheral blood specimens were found to have RB1 mutations, with a positivity of 100%. The mutations are summarized in Table 4. These results indicate that a high detection rate can be achieved using blood testing, the samples for which are easily available in clinic.
 |
Table 4 Summary of Blood-DNA Genetic Test Results |
Unreported Mutations
FFPE Tissue Samples
There were four cases of unreported RB1 mutations in the FFPE tissues, three of were pathogenic mutations and one of which was a potentially pathogenic mutation, as shown in Table 3.
Peripheral Blood Samples
Three types of potentially pathogenic mutations that had not been reported before were detected in the peripheral blood samples, as shown in Table 4.
Analysis of the Sequencing Results
RB1 mutations were present in 40 of the patients from whom FFPE tissue samples were taken, of which 33 were pathogenic mutations, including 20 single-nucleotide variant (SNV)/stopgain, eight deletion/frameshift mutations, three insertion/shift mutations, and two splicing/splicing mutations. These pathogenic mutations mainly manifested as stopgain and frameshift mutations. A further four cases were potentially pathogenic mutations, including three splicing/splicing and one insertion/non-frameshift, while three cases were mutations with uncertain significance, all of which were SNV/non-synonymous. After gene capture in combination with Sanger sequencing, exon sequencing identified the pathogenic mutations in the FFPE tissue samples as c.1423A>T p.K475X on exon 16, c.1103dupT p.I369Nfs*8 on exon 11, and c.618dupA p.Q207Tfs*5 on exon 7. The potentially pathogenic mutation identified was c.1980_1981insCGG p.L660delinsLR on exon 20. Unreported mutations in the RB1 gene were found in four patients (see Table 3). Three of these cases were pathogenic mutations and one were potentially pathogenic mutations.
All 12 peripheral blood samples exhibited RB1 mutations, of which three were pathogenic mutations, including one deletion/frameshift and two SNV/stopgain. A further six of the peripheral blood samples exhibited potentially pathogenic mutations, while three had mutations of uncertain significance, all of which were SNV/non-synonymous. The types of mutation were diverse and scattered across multiple exonic sequences of RB1 (see Figure 1). Testing of the DNA from the blood samples identified three potentially pathogenic mutations that had not been previously reported (see Table 4). These mutations included c.2252C>A p.S751Y (three cases), c.2260G>T p.V754L (two cases), and c.2213C>A p.T738K (one case) on exon 22. By searching the dbSNP and 1000 Genomes Project database to exclude the possibility of SNP, it was concluded that these mutations were pathogenic mutations for RB. No mutations were detected in the patients’ parents, suggesting that all these RB1 mutations were de novo mutations.
 |
Figure 1 The distribution of mutation types in the 27 exons and splice sites of the gene. The numbers in boxes refer to the exons, and the two grey oval regions represent functional domains A and B. The five symbols denote five mutation types; the full lines above and below the gene schematic refer to pathogenic and likely pathogenic mutations; dotted lines represent uncertain significance. |
In the present study, 22 variants (two bilateral and 20 unilateral) were located in the regions encoding two conserved domains of the pRb—A (residue 393–572) and B (residue 646–772)—of which nine were stopgain, five were frameshift, four were splicing, three were non-synonymous, and one was non-frameshift (see Figure 1). Of these, 72.7% (16/22) of cases were pathogenic, with two of these cases being de novo mutations, and 27.3% (6/22) of cases were potentially pathogenic, with four of these cases being de novo mutations.
Discussion
The RB1 gene, which has an anticancer effect as a tumor-suppressor gene, is the first human anticancer gene to have been isolated. Deletion or inactivation of the RB1 gene is an important mechanism for the occurrence of RB, which may occur as a result of the simultaneous deletion, mutation, or inactivation of one pair of RB alleles. The analysis of RB1 mutations and the identification of the genetic type of RB are important for the determination of a therapeutic plan and follow-up for patients with RB, as well as for the prevention of recurrence for their family members.
The RB1 gene is large, and its mutations are highly heterogeneous and can manifest not only at the chromosomal level, such as variations in chromosome structure and number, but also at the molecular level, such as changes in nucleic acids (substitutions, deletions, insertions, and duplication mutations), deletion of exons, loss of heterozygosity, and transcriptional inactivation due to hypermethylation of the CpG island in the promoter of the RB1 gene.32
The number of new cases of RB is increasing yearly, so it is necessary to establish a rapid and effective RB1 gene-testing platform so that the results of gene mutation can be identified quickly for early diagnosis and treatment. Gene capture allows high-throughput sequencing of multiple target regions of pathogenic genes, reducing sequencing costs, shortening the detection duration, and providing high detection rates.33 The technique also allows for the selection of different sequencing throughputs, with higher detection rates and better targeting at 1000× sequencing depth. The clinical selection of different throughputs for pediatric patients with different degrees of RB phenotype may improve the detection rate.
Among the 44 patients in the present study with positive FFPE results, 40 had RB1 gene mutations. Four of the cases were other types of mutations that had no correlation with the RB1 gene: the most common unrelated mutated genes were testis-specific protein Y-linked (TSPY [chr Y]), zinc finger protein 207 (ZNF207 [chr 17]), zinc finger protein 618 (ZNF618 [chr 9)]), otopetrin 3 (OTOP3 [chr 17]), and deleted in azoospermia (DAZ [chr Y]) (see Table 3). The MutRatios (number of mutant bases sequencing/total number of sequencing) of these genes were between 0.01 and 0.03, with a low value and no need reference. Their presence may have been caused by the DNA decomposition and fragmentation of FFPE tissue affecting the test results.
The results of the present study confirm the diversity of the types of mutation of the RB1 gene. The reported and unreported mutation types identified in the results included deletion, insertion, SNV, and splicing mutation, and the loci were distributed in multiple exons, with exons 22, 8, 23, 17, 20, 10, and 14 being the most common ones. Previous literature has reported that RB1 mutations are mostly nonsense mutations, insertion or deletion mutations, missense mutations, large-fragment deletions, and other mutation types.34,35 Valverde et al36 conducted a meta-analysis of 932 reported RB1 mutations in Europe and the USA, identifying three characteristics of the RB1 mutation profile: (1) the mutations are most frequently nonsense mutations, with the incidence of shift mutations, splice-site mutations, and missense mutations caused by small deletions/insertions decreasing in descending order. pRB is frequently inactivated by nonsense and deletion mutations; (2) although RB1 mutations are scattered throughout the gene sequence, there are 16 hotspot mutations with high recurrence frequency and seven exons with high mutation frequency (exons 9, 10, 14, 17, 18, 20, and 23); (3) the prevalence of RB1 nonsense mutations and splice mutations differs significantly between patients of different races, countries, and regions.
In a study by Sagi et al in Israel37 and another by Zou et al38 in Shanghai, China, the most common type of RB1 mutation found was nonsense mutation, followed by splice and shift mutations, with no statistically significant differences between different types of mutation in terms of average age at diagnosis and the side affected by RB. The results of the present study identified the same high-frequency mutation types and mutation loci.
Patients with bilateral RB carry germ-cell mutations, and the literature indicates that 25% of patients have a family history of the mutation—ie, the gene is inherited from parents with RB—while the other 75% are scattered cases and the result of germ-cell mutations during the early embryonic stage.39 In the present study, germ-cell mutations were found in four patients with bilateral RB, none of whose parents were found to have RBl mutations, which is consistent with the scattered genotype.
In the present study, one patient was selected for simultaneous sequencing of FFPE tissue and peripheral blood DNA, the results of which are shown in Table 5. The sequencing results showed that the FFPE tissue sample exhibited more pathogenic mutation loci than the blood sample, but the MutRatio of the two different mutations was 0.01, which could be ignored. Thus, the common mutation was c.958C>T p.R320X, indicating concordance between the FFPE tissue and blood sample. This suggests that, when the genetic testing of children is performed clinically, venous blood has the same detection rate as pathological tissue sections. However, it is easier to obtain venous blood samples and implement the testing in a clinical setting.
 |
Table 5 FFPE-Blood-DNA RB1 Gene Test Results |
RB is the most common intraocular malignant tumor in infants and children, and many cases in China have severely affected vision or blindness at the time of diagnosis. The condition can even be life threatening to children. Early detection and early treatment can save a patient’s vision, preserve the eye structure, and prolong the patient’s life. There is currently an urgent need to improve the coverage of genetic testing and counseling for patients with RB in China, and RB1 genetic testing can help achieve individualized, comprehensive treatment and genetic guidance for patients with RB. The application of genetic testing in the clinical diagnosis and treatment of RB also needs to be promoted. The China Retinoblastoma Diagnosis and Treatment Guidelines (2019) state that the first signs of RB are leukocoria, strabismus, and other less common signs. After appearance of these signs, patients should be examined by an ophthalmologist and, in case of a possible tumor, should be referred to a specialist for urgent implementation of treatment. After initiation of treatment, genetic analysis should be performed: probands should be tested for the RB1 gene, and patients with chromosome 13q14 deletion should be examined at an ophthalmology department within 48–72 hours; different control measures should be conducted for individuals with known family mutations in the RB1 gene.14
Once RB is diagnosed, its treatment depends on its laterality and the stage of the tumor. Genetic diagnosis is important in predicting the risk of the disease in the siblings or children of patients with RB, thereby reducing the incidence of children being born with RB, improving the overall quality of the population, reducing the psychological and financial burden on families of patients with RB, and saving medical resources.40 In the present study, it was found that patients with unilateral RB might carry germline mutations that can be passed on to their offspring, so the application of genetic diagnosis should also be emphasized in patients with unilateral RB, genetic counseling should be actively provided to patients and their families, and early diagnosis and treatment should be provided to high-risk families.
In this study, there were 52 patients with unilateral RB and four with bilateral RB. No standardized selection was made in quantity. Due to the influence of the COVID-19 pandemic, there was a significant decrease in the number of children seeking medical treatment. Most of the patients seeking medical treatment had unilateral RB, and most of those undergoing eye extraction also had unilateral RB. A further study needs to select more patients with bilateral RB, which will be advantageous to the analysis of germ-cell mutation types. In the present study, the patients with unilateral RB all tested positive for RB1 mutation, which may be due to the severe condition of the children who visited the hospital, all of whom were grade D or E. Therefore, the number of blood samples collected should be increased in the next study in order to improve the objectivity of the results.
Conclusion
Gene capture is a low-cost and efficient method for the gene sequencing of RB. A total of seven de novo mutations were identified through mutation testing of the pathogenic gene RB1 in 56 pediatric patients with RB. This complemented the mutation spectrum of the RB1 gene and helped to improve the molecular diagnosis of RB, thereby providing a basis for genetic counseling and prediction of the clinical phenotype, as well as for the genetic testing of the offspring of patients with RB.
Data Sharing Statement
The datasets used and/or analysed during the current study available from the corresponding author ([email protected]) on reasonable request.
Ethics Approval and Consent to Participate
This study was conducted in accordance with the declaration of Helsinki.This study was conducted with approval from the Ethics Committee of Beijing Tongren Eye Center. Ethical Approval number TRECKY2018-056.A written informed consent was obtained from legal guardians of all participants.
Funding
Supported by The Capital Health Research and Development of Special (2020-1-2052); Science & Technology Project of Beijing Municipal Science & Technology Commission (Z201100005520045, Z181100001818003); the Beijing Municipal Administration of Hospitals’ Ascent Plan (DFL20150201).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Knudson AG
2. Pandey AN. Retinoblastoma: anoverview. Saudi J Ophthalmol. 2014;28(4):310–315. doi:10.1016/j.sjopt.2013.11.001
3. Kivela T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009;93(9):1129–1131. doi:10.1136/bjo.2008.150292
4. Abramson DH, Frank CM. Second nonocular tumors in survivors of bilateral retinoblastoma: apossible age effect on radiation-related risk. Ophthalmology. 1998;105(4):573–580. doi:10.1016/S0161-6420(98)94006-4
5. Wu ZY. Orbital Pathology of Modern Eye Tumors. Beijing: People’s Military Medical Press; 2002:251–272
6. Rehurek J. History of retinoblastoma. Cesk Slov Oftalmol. 1997;53(6):351–355.
7. Naumova A, Sapienza C. The genetica of retinoblastoma, revisited. Am J Hum Genet. 1994;54(2):264–273.
8. de Jong MC, Kors WA, de Graaf P, Castelijns JA, Kivelä T, Moll AC. Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol. 2014;15(10):1157–1167. doi:10.1016/S1470-2045(14)70336-5
9. Temming P, Arendt M, Viehmann A, et al. How eye-preserving therapy affects long-term overall survival in heritable retinoblastoma survivors. J Clin Oncol. 2016;34(26):3183–3188. doi:10.1200/JCO.2015.65.4012
10. Alberti WE, Sagerman RH. Diagnosis and management of retinoblastoma. Cancer Control. 2004;11(5):317–327. doi:10.1177/107327480401100506
11. Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323(6089):643–646. doi:10.1038/323643a0
12. Hensey CE, Hong F, Durfee T, Qian YW, Lee EY, Lee WH. Identification of discrete structural domains in the retinoblastoma protein. N-terminal domain is required for its oligomerization. J Biol Chem. 1994;269(2):1380–1387. PMID:8288605. doi:10.1016/S0021-9258(17)42269-1
13. Dimaras H, Khetan V, Halliday W, et al. Loss of RB1 induces non-proliferative retinoma: increasinggenomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17(10):1363–1372. PMID:18211953. doi:10.1093/hmg/ddn024
14. Xu X, Zhao MW. Chinese guidelines for the diagnosis and treatment of retinoblastoma (2019). Chin J Ophthalmol. 2019;55(10):726–738. doi:10.1159/000509424
15. Woods MO, Williams P, Careen A, et al. A new variant database for mismatch repair genes associated with lynch syndrome. Hum Mutat. 2007;28(7):669–673. doi:10.1002/humu.20502
16. Lohmann DR, Brandt B, Höpping W, Passarge E, Horsthemke B. Spectrum of small length germline mutations in the RB1 gene. Hum Mol Genet. 1994;3(12):2187–2193. doi:10.1093/hmg/3.12.2187
17. Cowell JK, Smith T, Bia B. Frequent constitutional C to T mutations in CGA-arginine codons in the RB1 gene produce premature stop codons in patients with bilateral (hereditary) retinoblastoma. Eur J Hum Genet. 1994;2(4):281–290. doi:10.1159/000472372
18. Blanquet V, Turleau C, Gross-Morand MS, Sénamaud-Beaufort C, Doz F, Besmond C. Spectrum of germline mutations in the RB1 gene: a study of 232 patients with hereditary and non hereditary retinoblastoma. Hum Mol Genet. 1995;4(3):383–388. doi:10.1093/hmg/4.3.383
19. Houdayer C, Dehainault C, Mattler C, et al. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum Mutat. 2008;29(7):975–982. doi:10.1002/humu.20765
20. Nájera C, Sánchez F, Mateu E, Prieto F, Beneyto M. Diagnóstico temprano del retinoblastoma: importancia de la búsqueda de mutaciones en el gen RB1 [Early diagnosis of retinoblastoma: usefulness of searching for RB1 gene mutations]. Med Clin (Barc). 2001;116(10):365–372. doi:10.1016/S0025-7753(01)71832-5
21. Szijan I, Lohmann DR, Parma DL, Brandt B, Horsthemke B. Identification of RB1 germline mutations in Argentinian families with sporadic bilateral retinoblastoma. J Med Genet. 1995;32(6):475–479. doi:10.1136/jmg.32.6.475
22. Yandell DW, Campbell TA, Dayton SH, et al. Oncogenic point mutations in the human retinoblastoma gene: their application to genetic counseling. N Engl J Med. 1989;321(25):1689–1695. doi:10.1056/NEJM198912213212501
23. Price EA, Price K, Kolkiewicz K, et al. Spectrum of RB1 mutations identified in 403 retinoblastoma patients. J Med Genet. 2014;51(3):208–214. doi:10.1136/jmedgenet-2013-101821
24. Lohmann DR, Brandt B, Höpping W, Passarge E, Horsthemke B. The spectrum of RB1 germ-line mutations in hereditary retinoblastoma. Am J Hum Genet. 1996;58(5):940–949.
25. Aggarwala V, Ganguly A, Voight BF. De novo mutational profile in RB1 clarified using a mutation rate modeling algorithm. BMC Genomics. 2017;18(1):155. doi:10.1186/s12864-017-3522-z
26. Richter S, Vandezande K, Chen N, et al. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003;72(2):253–269. doi:10.1086/345651
27. Blanquet V, Gross MS, Turleau C, Sénamaud-Beaufort C, Doz F, Besmond C. Three novel germline mutations in exons 8 and 18 of the retinoblastoma gene. Hum Mol Genet. 1994;3(7):1185–1186. doi:10.1093/hmg/3.7.1185
28. Zhang Q, Minoda K, Zeng R, et al. Exon-by-exon screening for RB germline mutations using Heteroduplex-SSCP analysis. Yan Ke Xue Bao. 1997;13(1):5–11.
29. Onadim Z, Hogg A, Baird PN, Cowell JK. Oncogenic point mutations in exon 20 of the RB1 gene in families showing incomplete penetrance and mild expression of the retinoblastoma phenotype. Proc Natl Acad Sci U S A. 1992;89(13):6177–6181. doi:10.1073/pnas.89.13.6177
30. Tomar S, Sethi R, Sundar G, Quah TC, Quah BL, Lai PS. Mutation spectrum of RB1 mutations in retinoblastoma cases from Singapore with implications for genetic management and counselling. PLoS One. 2017;12(6):e0178776. doi:10.1371/journal.pone.0178776
31. Lohmann D, Horsthemke B, Gillessen-Kaesbach G, Stefani FH, Höfler H. Detection of small RB1 gene deletions in retinoblastoma by multiplex PCR and high-resolution gel electrophoresis. Hum Genet. 1992;89(1):49–53. doi:10.1007/BF00207041
32. de Andrade AF, da Hora Barbosa R, Vargas FR, et al. A molecular study of first and second RBl mutational hits in retinoblastoma patients. Cancer Genet Cytogenet. 2006;167(1):43–46. doi:10.1016/j.cancergencyto.2005.08.017
33. Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–276. doi:10.1038/nature08250
34. Ayari-Jeridi H, Moran K, Chebbi A, et al. Mutation spectrum of RB1 gene in unilateral retinoblastoma cases from Tunisia and correlations with clinical features. PLoS One. 2015;10(1):e0116615. doi:10.1371/journal.pone.0116615
35. Abidi O, Knari S, Sefri H, et al. Mutational analysis of the RB1 gene in Moroccan patients with retinoblastoma. Mol Vis. 2011;17:354l–3547.
36. Valverde JR, Alonso J, Palacios I, Pestaña A. RB1 gene mutation up-date, a meta-analysis based on 932 reported mutations available in a searchable database. BMC Genet. 2005;6(1):53. doi:10.1186/1471-2156-6-53
37. Sagi M, Frenkel A, Eilat A, et al. Genetic screening in patients with Retinoblastoma in Israel. Fam Cancer. 2015;14(3):471–480. PMID:25754945. doi:10.1007/s10689-015-9794-z
38. Zou Y, Li J, Hua P, Liang T, Ji X, Zhao P. Spectrum of germline mutations in RB1 in Chinese patients with retinoblastoma: application of targeted next-generation sequencing. Mol Vis. 2021;27:1–16.
39. Rodriguez-Galindo C, Orbach DB, VanderVeen D. Retinoblastoma. Pediatr Clin North Am. 2015;62(1):201–223. doi:10.1016/j.pcl.2014.09.014
40. Chen CZ, Yu XX. Attention should be paid to the genetic diagnosis of retinoblastoma. Chin J Exp Opthalmol. 2013;31(7):617–620.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.