Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
The Prognostic Value of DNA Methylation, Post-Translational Modifications and Correlated with Immune Infiltrates in Gynecologic Cancers
Authors Shang C ![]() , Li Y, Wu Z, Han Q, Zhu Y, He T, Guo H
, Li Y, Wu Z, Han Q, Zhu Y, He T, Guo H ![]()
Received 24 November 2020
Accepted for publication 26 December 2020
Published 12 January 2021 Volume 2021:14 Pages 39—53
DOI https://doi.org/10.2147/PGPM.S293399
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Chunliang Shang,1 Yuan Li,1 Zhangxin Wu,1 Qin Han,1 Yuan Zhu,2 Tianhui He,1 Hongyan Guo1,3
1Department of Obstetrics and Gynecology, Peking University Third Hospital, Beijing 100191, People’s Republic of China; 2Department of Obstetrics and Gynecology, The First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, People’s Republic of China; 3National Clinical Research Center for Obstetrics and Gynecology (Peking University Third Hospital), Beijing 100191, People’s Republic of China
Correspondence: Hongyan Guo
Department of Obstetrics and Gynecology, Peking University Third Hospital, No. 49 Huayuan North Road, Haidian District, Beijing 100191, People’s Republic of China
Tel +86-17301281996
Fax +86 1082267842
Email [email protected]
Background: To depict the prognostic landscape of gynecological cancers from the perspective of DNA methylation, alternative splicing (AS) and polyadenylation (APA) events and investigate their correlation with immune infiltrates.
Methods: Methylation and RNA-seq data and corresponding clinical information regarding gynecologic cancers were used to explore the relationships between changes in DNA methylation, AS and APA events and gynecologic cancer prognosis. QRT-PCR and multiple bioinformatics tools were employed to construct a gene interaction network and explore immune infiltrates.
Results: Only the mRNA levels of CIRBP and INPP5K were simultaneously significantly decreased in gynecologic cancers and negatively associated with overall survival, which verified by qrt-PCR. We also identified that CIRBP or INPP5K DNA methylation, AS and APA events are prognostic indicators of gynecologic cancers. The activation of T cells might be the main signaling pathway by which these genes modulate cancer progression. CIRBP/INPP5K expression is positively associated with immune infiltration and is a major risk factor of survival, especially among uterine corpus endometrial carcinoma (UCEC) patients.
Conclusion: According to these findings, the DNA methylation, AS and APA events of CIRBP and INPP5K may serve as important prognostic biomarkers and targets in gynecological cancers by modulating T cell infiltration.
Keywords: DNA methylation, alternative splicing, alternative polyadenylation, activation of T cells, immune infiltration
Introduction
Cancers of the female reproductive system remain an important cause of cancer morbidity and mortality. Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) continues to be the second leading cause of cancer death in women aged 20 to 39 years,1 with an overall 5-year survival rate in China of 45.4%.2 Uterine corpus endometrial carcinoma (UCEC) is the second most common cancer of the female genital system in China and increasing –as in Western countries—from 2013 to 2015 from 17.9 to 21.8 thousand, respectively, with an overall 5-year survival rate of 55.1%.2 Ovarian serous cystadenocarcinoma (OV) is the 3rd most common cancer of the female genital system and is primarily diagnosed at an advanced stage with an average 5-year survival rate of 38.9%.2 Traditionally these three cancers are treated with surgery, radiation, and chemotherapy. However, approximately 70% of patients experience disease relapse after receiving combination therapy.3 Antiangiogenic therapies, immunotherapy (eg, programmed cell death-1 (PD1)) and polyADP ribose polymerase (PARP) inhibitors are now often added.4 There is a need of exploiting new genomic and epigenomic information to integrate novel targeted therapies in the management of these cancers.
Using The Cancer Genome Atlas (TCGA) database and bioinformatic analysis, we discovered that cold-inducible RNA-binding protein (CIRBP) and inositol polyphosphate-5-phosphatase K (INPP5K) are novel prognostic markers and that the high expression of these factors is favorable in CESC, UCEC and OV. CIRBP was originally identified as a cold shock protein in the mammalian testis that is induced by ultraviolet radiation, cold, and hypoxia.5,6 CIRBP specifically acts as a translational activator to affect posttranscriptional regulation by binding the 3′-untranslated regions (3′-UTRs) of many target mRNAs.7 CIRBP has also been reported to be involved in the progression and inflammation of distinct tumors. INPP5K, which is also known as skeletal muscle and kidney-enriched inositol 5-phosphatase (SKIP), belongs to the phosphoinositide 5-phosphatase family located in a part of the endoplasmic reticulum.8 INPP5K reportedly participates in the regulation of tumor cell adhesion, migration and invasion.9,10 However, thus far, the prognostic significance of CIRBP/INPP5K expression in gynecologic cancers has not been reported.
Alternative splicing (AS) and alternative polyadenylation (APA) are the important modes of post-transcriptional modification and regulation. AS occurs by rearranging the pattern of intron and exon elements that are joined by splicing to alter the mRNA coding sequence, and nearly 95% of the posttranscriptional translation events of human multiexon genes involve AS regulation.11 To date, the following terms have been coined for seven types of AS events: alternative 3ʹ acceptor site (AA), alternative 5ʹ donor site (AD), alternate promoter (AP), alternate terminator (AT), exon skipping (ES), mutually exclusive exon (ME), and retained intron (RI) events.12 During viral infections, the loss of m6A in CIRBP promotes alternative splicing; similarly, pathogenic INPP5K variants have been reported in families with congenital muscular dystrophy, intellectual disability, and cataract.13,14 Furthermore, abnormal AS is involved in tumor proliferation, metastasis, angiogenesis, anti-apoptosis, immune escape and cancer metabolism.
APA is a special form of alternative splicing. Differing from other AS events, the selection of different polyadenylation signal sites (PAS) in the 3ʹ-UTR during the maturation of premRNA can produce multiple mature mRNA subtypes from the same premRNA.15 Approximately 70% of human genes with multiple polyadenylation sites undergo APA. Therefore, APA can influence the occurrence and development of tumors by regulating the transcription and translation of tumor-associated genes.16 Nonetheless, the prognostic value of AS or APA events in CIRBP/INPP5K and their biological functions in gynecologic cancers remain largely unknown.
DNA methylation is an important epigenetic regulatory mechanism in eukaryotes responsible for modulating gene expression without DNA sequence alteration. The most well-studied epigenetic change occurs in CpG islands, and methylation changes occur in approximately 70% of mammalian promoters.17 Moreover, growing evidence suggests that alterations in DNA methylation are promising targets for the development of powerful prognostic and predictive biomarkers in cancer. Recent research shows that chronic hypoxia-induced CIRBP hypermethylation attenuates hypothermic cardioprotection via the downregulation of ubiquinone biosynthesis.18 Next, we evaluate the prognostic value of DNA methylation of CIRBP/INPP5K in gynecologic cancers.
We systematically analyzed the prognostic landscape of CIRBP and INPP5K from the perspectives of post-translational modifications and DNA methylation events. Our preliminary analysis found that the activation of T cells might be the main signaling pathway by which these genes modulate cancer progression.
Materials and Methods
Analysis of Prognostic Genes
Genes considered significant for an unfavorable or favorable prognosis in cancer were downloaded from the Human Pathology Atlas,19 which is an interactive open-access online database containing mRNA expression data based on TCGA database. The mRNA expression was considered significant when higher levels were associated with a worse outcome in gynecologic cancers based on a Kaplan-Meier survival plot with a p<0.001. For the OV cohorts, the prognostic value of CIRBP/INPP5K was analyzed using the Kaplan-Meier plotter (KM plotter).20 The KM plotter is another online database containing gene expression data and clinical outcomes derived from the Gene Expression Omnibus (GEO), TCGA, and Cancer Biomedical Informatics Grid, which contains more than 2200 OV patients.
Expression Analysis
The analysis of CIRBP/INPP5K expression was performed using the following two online software programs: Gene Expression Profiling Interactive Analysis (GEPIA) and ONCOMINE (www.oncomine.org).21 Gynecologic cancer matched TCGA and Genotype-Tissue Expression (GTEx) data were used, and profiling was performed according to the cancer type or pathological stage, and log2 (TPM+1) was applied for the log-scale GEPIA analysis. ONCOMINE includes 715 independent datasets and gene expression data and aims to facilitate discovery from genome-wide expression analyses. ONCOMINE was also employed to evaluate the CIRBP/INPP5K mRNA levels in gynecologic cancers.
Clinical Specimens
Patient studies were conducted in accordance with the Declaration of Helsinki. The study was approved by the Ethical Committee of the Peking university Third hospital. All individuals enrolled in this study provided informed consent in written form by signing with their names or fingerprints. Verbal informed consent was obtained from the patients who were illiterate, and their guardians were also asked to sign the informed consent form. All specimens were handled according to legal and ethical standards. For total RNA isolation, the freshly frozen cervical cancer tissues, epithelial ovary cancer tissues and endometrial cancer tissues were collected from the Peking university Third hospital. All tissue specimens were immediately frozen in liquid nitrogen after resection and stored at − 80 °C until later RNA extraction.
RNA Extraction and qRT-PCR
Total RNA was isolated from tissues or cell lines using TRIzol reagent (Takara, Japan). QRT-PCR was performed with the SYBR Premix Ex Taq (TAKARA, Dalian, China) using the 7500 fast Real-Time PCR system (Applied Biosystems, USA). The primer sequences of CIRBP are listed: Forward primer (5ʹ-3ʹ): AGGGCTGAGTTTTGACACCAA; Reverse primer (5ʹ-3ʹ): ACAAACCCAAATCCCCGAGAT.
Functional Enrichment Analysis
Gene Ontology (GO) terms in the biological process, cellular component, and molecular function categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways are commonly used to annotate genes, gene products and sequences to underlying biological phenomena. Metascape is an online program that precisely integrates several authoritative data resources, such as GO, KEGG, UniProt and Molecular Complex Detection (MCODE) algorithms, to perform pathway enrichment and biological process annotation and provide comprehensive and detailed information regarding each gene.22 Terms with a P-value<0.01, a minimum count of 3, and an enrichment factor>1.5 were collected and grouped into clusters based on their membership similarities. The protein-protein interaction enrichment analysis was performed using the BioGrid, InWeb_IM, and OmniPath databases, and densely connected network components were identified using an MCODE algorithm. The three best-scoring terms (by P-value) were retained as the functional description of the corresponding components.
Alternative Splicing Event Analysis
Profiles of AS events in gynecologic cancer patients were downloaded from the data portal TCGA SpliceSeq,23 which is an AS database constructed by applying SpliceSeq analysis methods to RNASeq samples from TCGA. The Percent-Spliced-In (PSI) value, which ranges from 0 to 100%, is a ratio of the normalized read counts indicating the inclusion of a transcript element over the total normalized reads per AS event. To generate the most reliable set of AS events possible, we applied a series of stringent filters to a percentage of samples with a PSI value ≥ 75 and an average PSI value ≥ 0.05.
Analysis of Gene Expression and Tumor-Infiltrating Immune Cells
To investigate the correlation between the expression of CIRBP/INPP5K and tumor infiltrating immune cells (B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells), we applied the TIMER web server (https://cistrome.shinyapps.io/timer/), which is a comprehensive resource for the systematical analysis of immune infiltrates across diverse cancer types available in TCGA database.
Alternative Polyadenylation Analysis
To quantify the APA events based on a continuous RNA-seq signal, the Distal polyA site Usage Index (PDUI) percentage is measured by the percentage of transcripts with distal polyA sites among all transcripts. The greater the PDUI, the more distal the polyA site of a transcript, and vice versa. The PDUI values of the gynecologic cancer patients were obtained from the TC3A Portal, which provides visualization, analysis and downloading of large-scale cancer genomics APA data sets from TCGA.24
DNA Methylation
TCGA level 3 DNA methylation data were procured from TCGA using the Fire Browse platform (http://firebrowse.org/). Infinium HumanMethylation 450K BeadChip arrays (Illumina Inc, CA, USA) were used to examine genome-wide DNA methylation profiles in gynecologic cancers. The DNA methylation level at each CpG site is represented as a β-value, which is the ratio of the intensity of the methylated-probe signal to the total locus signal intensity ranging from 0 (no methylation) to 1 (100% methylation). Only data of patients with available clinical survival information were selected. UALCAN was utilized to estimate the effect of the DNA methylation level on the clinicopathologic features of the gynecologic cancers.25
Survival Analysis
The clinical information of gynecologic cancer patients was downloaded from the data portal TCGA via the Fire Browse platform. All statistical analyses were performed using the R statistical package (R version 4.0.0). Kaplan–Meier curves and a Log rank test were applied to compare the ability of the prognostic signatures to predict outcomes in patients with gynecologic cancers. Student’s t-test was used to compare the means of two groups.
Results
Functional Enrichment Analysis of Prognostic Genes
The functions of the prognostic genes were predicted by analyzing GO and KEGG in Metascape. Regarding CESC, high expression levels of 501 and 223 genes were associated with favorable and unfavorable clinical outcomes, respectively (Figure 1A). According to the KEGG analysis, these genes are mainly enriched in signaling pathways, such as the cell cycle, HIF-1 signaling, NF-kappa B signaling, primary immunodeficiency and amino acid metabolism (Figure S1A). The GO term enrichment analysis revealed the following: these genes are mainly involved in lymphocyte activation, apoptotic signaling, the generation of precursor metabolites and energy and cytokine-mediated signaling in the biological process category; oxidoreductase activity, protein N-terminus binding and carboxyl-O-methyltransferase activity in the molecular function category; and the alpha-beta T cell receptor complex, chromosomal region and nuclear lamina in the cellular component category (Figure S1B). Based on the protein–protein interaction (PPI) network and MCODE components of these prognostic genes, their biological functions are mainly related to ciliary basal body-plasma membrane docking, spliceosome and G1/S transition of the mitotic cell cycle (Figure S1C).
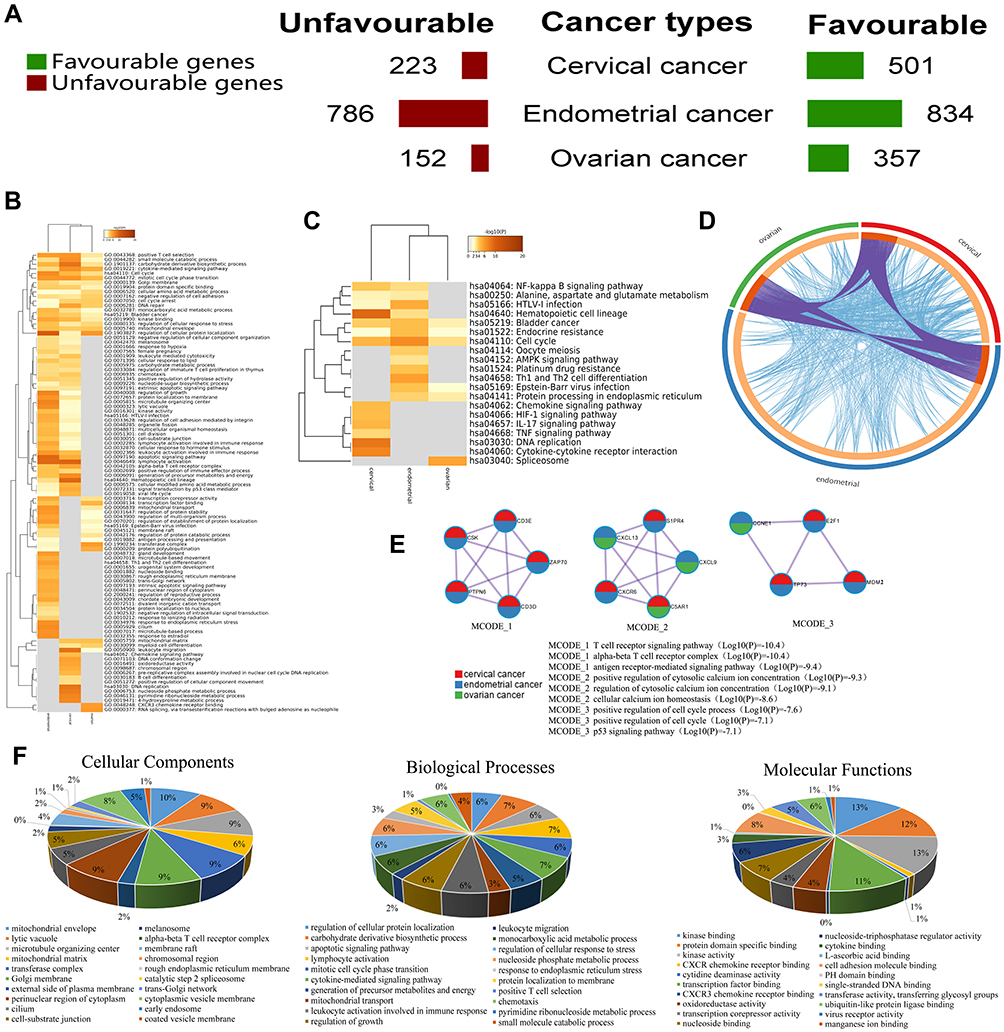 |
Figure 1 Meta-analysis of the functional enrichment of prognostic genes in gynecological cancers (Metascape). (A) The numbers of prognostic genes associated with the cancer type and favorable or unfavorable clinical outcomes are summarized. (B-C) Heatmap of enriched terms across the included prognostic genes colored by P-values. (D) The overlaps include the shared term level, and the blue curves link genes that belong to the same enriched ontology term. The inner circle represents the gene lists, and the hits are arranged along the arc. Genes that hit multiple lists are shown in dark orange, and genes unique to a list are shown in light orange. (E) Networks of enriched terms are presented as pie charts, and the pies are color-coded based on the identities of the gene lists. (F) GO terms identified in the GO analysis of the prognostic genes in the categories biological processes, cellular components, and molecular functions with 20 minimum P-values. |
Regarding UCEC, high expression levels of 834 and 786 genes were associated with favorable and unfavorable clinical outcomes, respectively (Figure 1A). Enrichment in the cell cycle, Th1 and Th2 cell differentiation, NF-kappa B signaling pathways, amino acid metabolism and fatty acid metabolism signaling pathways were mainly observed in the KEGG analysis (Figure S2A). The GO term enrichment analysis revealed the following: enrichment in mitotic cell cycle phase transition, regulation of the establishment of protein localization, T cell activation and positive regulation of programmed cell death in the biological process category; transcription factor binding, lipid binding and cyclin-dependent protein serine/threonine kinase inhibitor activity in the molecular function category; and lipid droplet, membrane raft and perinuclear region of the cytoplasm in the cellular component category (Figure S2B). The T cell receptor signaling pathway, mediator complex and protein targeting the ER were highlighted by the PPI network (Figure S2C) and MCODE analyses (Figure S2D).
Regarding OV, high expression levels of 357 genes were associated with favorable clinical outcomes, and 152 genes were associated with unfavorable clinical outcomes (Figure 1A). The KEGG signaling pathways of these prognostic genes were mainly enriched in the spliceosome, pathways in cancer, primary immunodeficiency, p53 signaling pathway and butanoate metabolism (Figure S3A). In the GO term enrichment analysis, these genes were mainly enriched in the regulation of cell adhesion, cell cycle arrest, positive regulation of T cell tolerance induction and cellular amino acid metabolic process in the biological process category. The molecular function categories of these genes were mainly transcription coregulator activity, kinase binding and NF-kappa B signaling pathways, and the cellular component categories were the mitochondrial envelope, Golgi membrane and MHC class I peptide loading complex (Figure S3B). The PPI network and MCODE component analyses showed that the biological functions of these genes are mainly related to CXCR3 chemokine receptor binding, proteasome-mediated ubiquitin-dependent protein catabolic process and RNA splicing via transesterification reactions with bulged adenosine as nucleophile (Figure S3C).
Metascape provides an automated meta-analysis tool to assist biologists in dissecting multiple gene lists. Thus, we also examined the functional enrichment of the prognostic genes in the above three gynecologic cancers. A heatmap of the enriched terms across the three prognostic gene lists is depicted in Figure 1B and C. The integrated signaling pathways include the cell cycle, positive T cell selection, cytokine-mediated signaling pathway, cellular amino acid metabolic process and negative regulation of cell adhesion. In addition, the overlaps among these three gene lists are illustrated in a Circos plot (Figure 1D). According to the integrated PPI network and MCODE component analyses, the biological functions are mainly related to the T cell receptor signaling pathway, positive regulation of cytosolic calcium ion concentration and positive regulation of the cell cycle process (Figure 1E). In the integrated GO term enrichment analysis, these three groups of genes were mainly enriched as follows: the cytokine-mediated signaling pathway, lymphocyte activation, small molecule catabolic process, regulation of growth and monocarboxylic acid metabolic process in biological processes; transferase activity, L-ascorbic acid binding and cytokine binding in molecular functions; and membrane raft, alpha-beta T cell receptor complex and perinuclear region of the cytoplasm in cellular components (Figure 1F).
Based on the above analysis, in total, 70 genes were simultaneously identified as prognostic genes in gynecologic cancers (Figure S4A). Interestingly, these genes show enrichment in T cell activation, p53 signaling, DNA repair, regulation of cell cycle G2/M phase transition and ammonium ion metabolic process (Figure S4B-D); thus, the elucidation of these signaling pathways could help contribute to improving patient prognosis.
Expression Levels and Prognostic Values of CIRBP/INPP5K
We compared the transcriptional expression of 70 prognostic genes between gynecologic cancer and normal tissues using the GEPIA dataset. Only the mRNA expression levels of CIRBP and INPP5K were simultaneously significantly lower in the gynecologic cancer tissues than the corresponding normal tissues (Figure 2A-B). Furthermore, we analyzed the relationship between the transcriptional levels of CIRBP/INPP5K and the tumor stage in patients with gynecologic cancer using the GEPIA dataset. The mRNA expression of CIRBP was significantly and negatively associated with the tumor stage of UCEC, whereas the level of INPP5K transcription was not significantly associated with the tumor stage of gynecologic cancers.
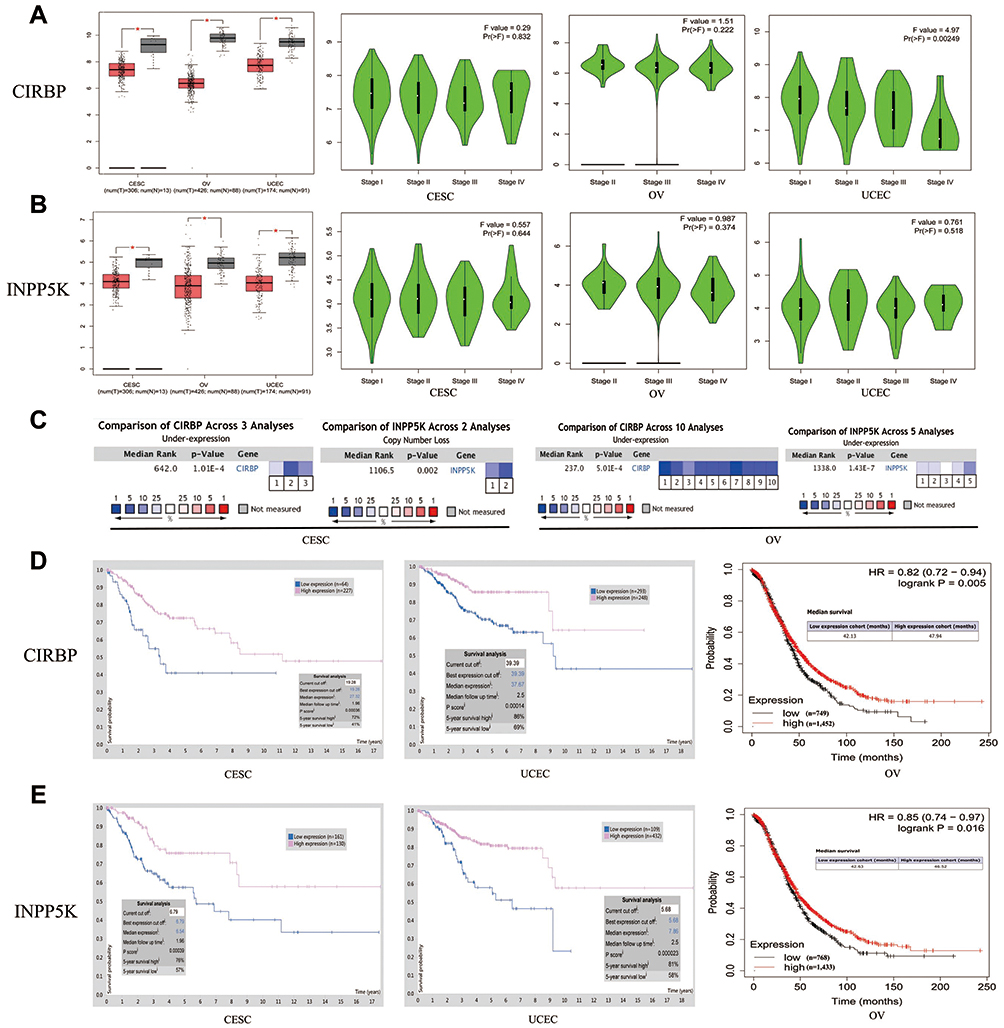 |
Figure 2 Expression |
We further confirmed the differential expression levels of CIRBP and INPP5K between the cancer and normal tissue samples by using the ONCOMINE database (Figure 2C and Table S1), which revealed that the mRNA expression of CIRBP was downregulated in the patients with CESC (meta-analysis of 3 datasets) or OV (meta-analysis of 10 datasets). Additionally, the transcriptional levels of INPP5K were significantly lower in the patients with OV in the five datasets, and the DNA copy number of INPP5K was significantly lower in the patients with CESC in TCGA datasets. In Scotto’s dataset,26 INPP5K was downregulated in cervical squamous cell carcinoma with a fold change of 2.880 and a P–value of 0.003. Unfortunately, data related to CIRBP/INPP5K expression in EC are not available in the ONCOMINE database.
Furthermore, we assessed the correlation between CIRBP and INPP5K expression and different clinical characteristics of patients with gynecologic cancers. The TP53 tumor suppressor gene is mutated in approximately half of all human cancers, allowing evasion of tumor cell death and rapid tumor progression.27 The results obtained based on the cohort from TCGA showed that TP53 mutations (P<0.01) were significantly associated with the expression of CIRBP and INPP5K in UCEC (Figure S5A). In UCEC, CIRBP expression was correlated with the histological subtype and patient weight, and INPP5K expression was correlated with the histological subtype and patient menopause status (Figure S5B-D). This information provides a new direction for further research investigating the mechanism of CIRBP/INPP5K in the development of UCEC.
The patients were classified into two groups based on the expression level of CIRBP/INPP5K using the best expression cut-off, yielding the maximal difference in survival between the two groups at the lowest log-rank P-value. The Kaplan–Meier survival curves are presented in Figure 2D–E. Decreased mRNA levels of CIRBP and INPP5K were strongly associated with a poor overall survival in the gynecologic cancer patients (P<0.05).
Association of CIRBP/INPP5K Expression with Tumor Purity and Immune Infiltration
The tumor microenvironment consists of tumor cells, stromal cells, and infiltrating immune cells. We utilized TIMER to explore the potential associations between the expression of CIRBP/INPP5K and both the tumor purity and infiltration of immune cells. Tumor purity is an important factor affecting immune infiltration in clinical tumor samples.28 Interestingly, CIRBP and INPP5K were negatively associated with the tumor purity, and positive associations were observed between these two genes and the infiltration of B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells (Figure 3A–B). The expression of CIRBP/INPP5K, CD4+ T cells and CD8+ T cells are the major risk factors of survival in UCEC patients according to the cox regression analysis (Figure 3C). These findings strongly suggest that CIRBP/INPP5K plays a specific role in immune infiltration in gynecologic cancers, especially infiltration of T cells in UCEC.
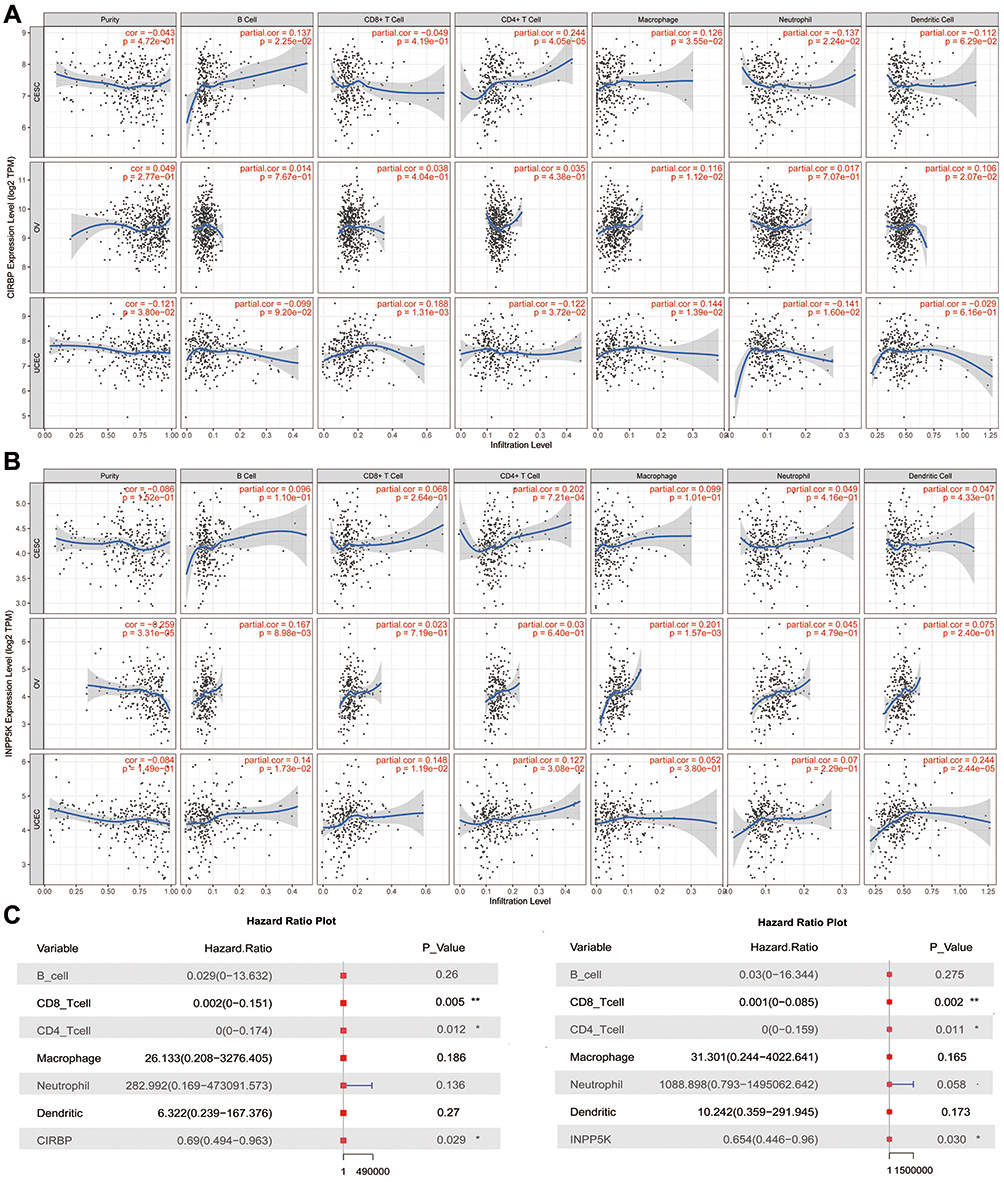 |
Figure 3 Association between CIRBP/INPP5K expression and immune infiltration in gynecologic cancers. (A) CIRBP. (B) INPP5K. (C) The results of the multivariate Cox regression analysis are presented with a Forest plot. P<0.05 denotes significance. Each dot represents a sample in TCGA dataset. *, P < 0.05; **, P < 0.01. |
Given the association between CIRBP/INPP5K expression and T cells’ prognostic value in UCEC, we subsequently investigated the correlations between CIRBP/INPP5K and immune marker sets of different functional T cells, include Th1 cells, Th2 cells, Tfh cells, Th17 cells, Tregs, and exhausted T cells, in UCEC in the TIMER databases (Table 1). After the correlation adjustment by purity, the results revealed that the INPP5K expression level was significantly correlated with most immune marker sets of different T cells in UCEC. However, the CIRBP expression level was significantly correlated with only 5 gene markers in UCEC (Table 1).
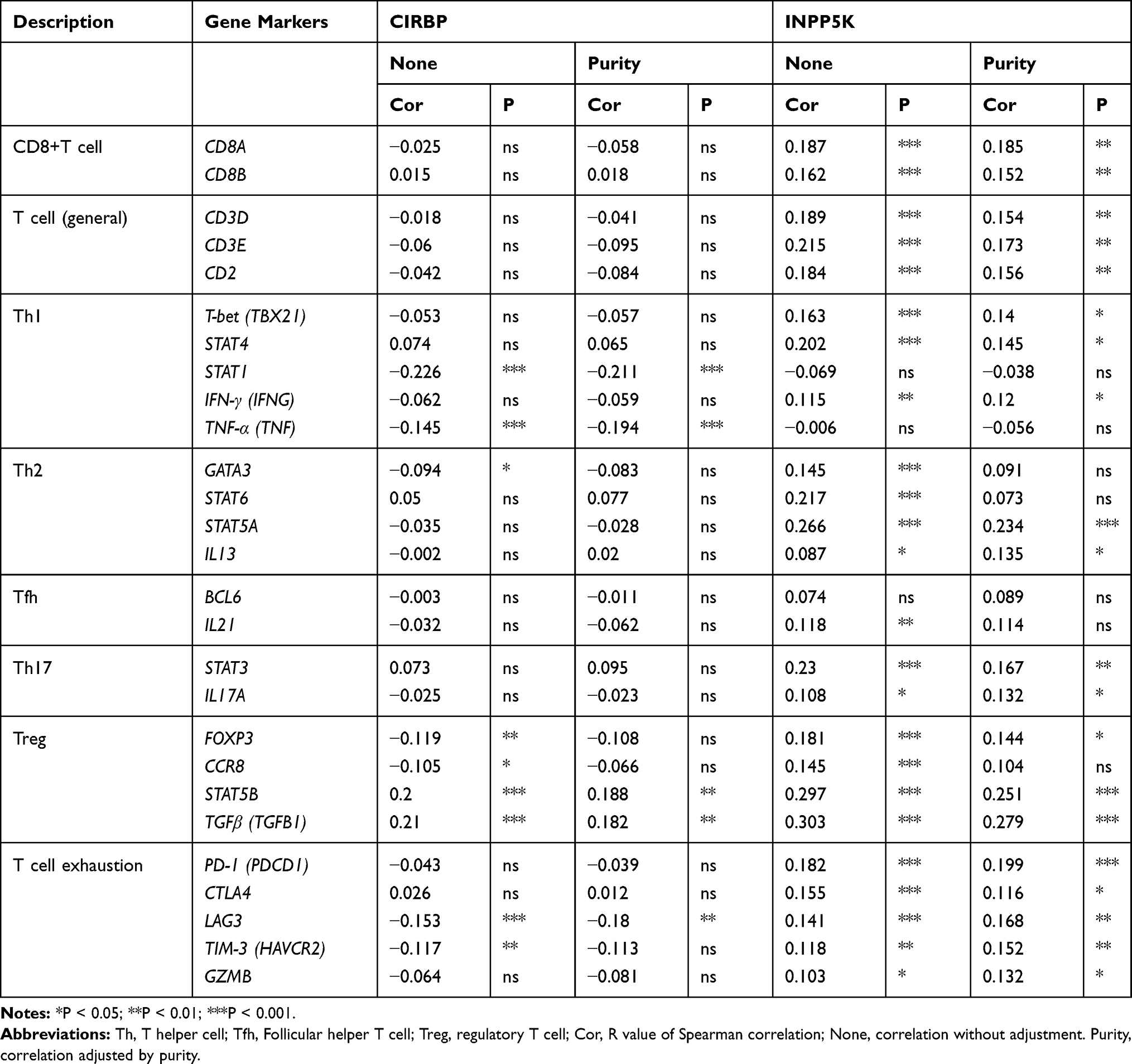 |
Table 1 Correlation Analysis Between CIRBP/INPP5K and Relate Genes and Markers of T Cells in UCEC |
Specifically, we identified significant correlations between INPP5K and marker genes of CD8+ T cells and T cell exhaustion, including CD8A, CD8B, PD-1, CTLA4, LAG3, TIM-3 and GZMB (Table 1). PD-1 is an immune checkpoint receptor that serves as a potent mechanism by which tumors escape from host immune responses.29 A clinical trial has confirmed that the PD-1–blocking antibody pembrolizumab possesses durable antitumor activity in patients with PD-L1–positive advanced endometrial cancer.30 Similarly, the checkpoint molecule TIM-3 is a target for emerging immunotherapies and has been shown to be commonly expressed in endometrial carcinomas.31 Interestingly, PD-1 and TIM-3 are both strongly positively correlated with INPP5K expression, suggesting that high INPP5K expression plays an important role in T cell exhaustion. Therefore, the above results further confirm the findings that INPP5K is specifically correlated with immune infiltrating T cells in UCEC, suggesting that INPP5K plays a vital role in immune escape in the gynecologic cancer microenvironment.
CIRBP Expression is Downregulated in Cancers Tissues
Given the above analysis, CIRBP with the lower P-value in prognostic prediction, had been verified to involve into the inflammation and cancers. Therefore, CIRBP was selected to perform further study. To validate the expression of CIRBP, we measured the CIRBP expression levels in gynecologic cancers tissues and corresponding normal tissues, respectively. As shown in Figure 4A, the expression of CIRBP in gynecologic cancers tissues was markedly decreased compared with normal tissues. Furthermore, we found that Low CIRBP expression was detected in a variety of reproductive tumor cells, including cervical cancer cells (SiHa and HeLa), ovarian cancer cells (EFO-21) and endometrial cancer cells (AN3-CA) using Cell Atlas website (Figure S6A). Immunofluorescence assay was used to demonstrate that CIRBP was localized to the nucleoplasm (Figure S6B). This may provide evidence for CIRBP acting as a translational activator in gynecologic cancers progression. These data indicated that CIRBP may play a protective role in gynecologic cancers progression. Furthermore, CIRBP expression was detected in many kinds of immune cells, including CD8+ T cell (Figure S6C).
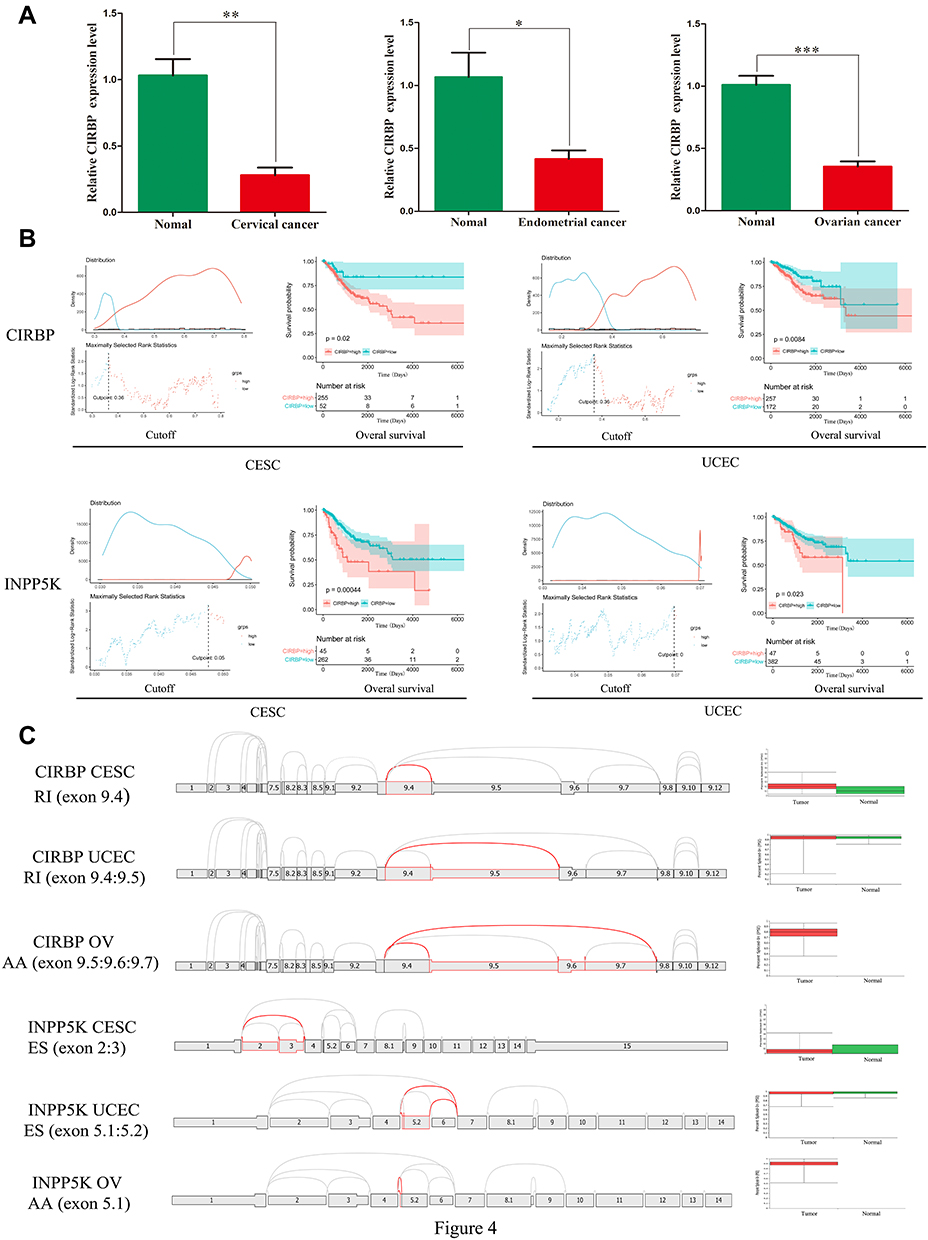 |
Figure 4 Prognostic value of the DNA methylation levels and splice graph of the prognostic AS events in CIRBP and INPP5K in CESC and UCEC patients. (A) The expression of CIRBP in gynecological cancer tissues and normal tissues (n=5/5). (B) Patients were divided into high (red) and low (blue) β-value groups based on the best cut-off values using the R package. (C) Splice graph of the prognostic AS events (left) and the difference in PSI values between gynecologic cancer and normal tissues (right). *, P < 0.05; **, P < 0.01; ***, P < 0.001. |
Prognostic Value of the DNA Methylation of CIRBP/INPP5K
We also analyzed the association between the DNA methylation of CIRBP and INPP5K and the clinical features of gynecologic cancers using the UALCAN web tool. CIRBP DNA methylation was correlated with the histological subtype, tumor grade, TP53 mutation and patient age in UCEC (Figure S7A-D). Interestingly, CIRBP DNA methylation was also significantly correlated with the patients’ weight (body mass index, Figure S7E), indicating that the methylation of this gene may be involved in metabolic processes in UCEC as mentioned in the functional analysis section. In CESC, CIRBP DNA methylation was also correlated with the histological subtype (Figure S7F). Similarly, INPP5K DNA methylation was correlated with the histological subtype, patients’ weight and nodal metastasis in CESC (Figure S8A-C) and histological subtype, tumor grade and TP53 mutation in UCEC (Figure S8D-F). These results indicate that the DNA methylation of INPP5K may participate in cancer progression.
The association between the level of CIRBP/INPP5K methylation and survival was investigated using the survival package in R. Regarding CIRBP, the hypomethylated samples showed a better prognosis than the hypermethylated samples in CESC and UCEC (P<0.05, Figure 4B). Similarly, the overall survival of the CESC and UCEC patients with high INPP5K DNA methylation was significantly shorter than that of the patients with low INPP5K DNA methylation (P<0.05, Figure 4B). These results indicate that CIRBP/INPP5K DNA methylation is related to poor prognoses in CESC and UCEC. Unfortunately, data relate to CIRBP/INPP5K DNA methylation in OV were not available.
Prognostic Value of Alternative Splicing Events in CIRBP/INPP5K
Integrated mRNA splice variant profiles were curated with detailed AS event data and the clinical outcomes of gynecologic cancer patients. For each type of AS event, the gynecologic cancer patients were dichotomized into low- and high-PSI subgroups based on the best cut-off PSI value using the R package. The top significant survival-associated CIRBP/INPP5K AS events calculated by the log-rank P-value were identified as candidate prognosis-related AS events. As presented in Figure 4C (left), a prognostic AS event graph was used to describe the splicing patterns of CIRBP and INPP5K in gynecologic cancers. Although no statistically significant difference was observed in the prognostic AS events of CIRBP/INPP5K between the gynecologic cancer and normal tissues, we observed a trend toward differential splicing patterns (Figure 4C right).
In the survival analysis, all prognostic models built with different types of AS events demonstrated significant power to predict the outcome of gynecologic cancer patients. Regarding CIRBP, an RI event (exon 9.4; P=0.024) with a high PSI value in CESC, an RI event (exon 9.4:9.5; P=1e-04) with a high PSI value in UCEC and an AA event (exon 9.5:9.6:9.7; P=0.0016) with a low PSI value in OV showed better performance in predicting an adverse outcome (Figure 5A). Regarding INPP5K, an ES event (exon 2:3; P=0.014) in CESC, an ES event (exon 5.1:5.2; P=0.00069) in UCEC and an AA event (exon 5.1; P=0.0042) in OV with a high PSI value were observed to predict a better outcome (Figure 5B).
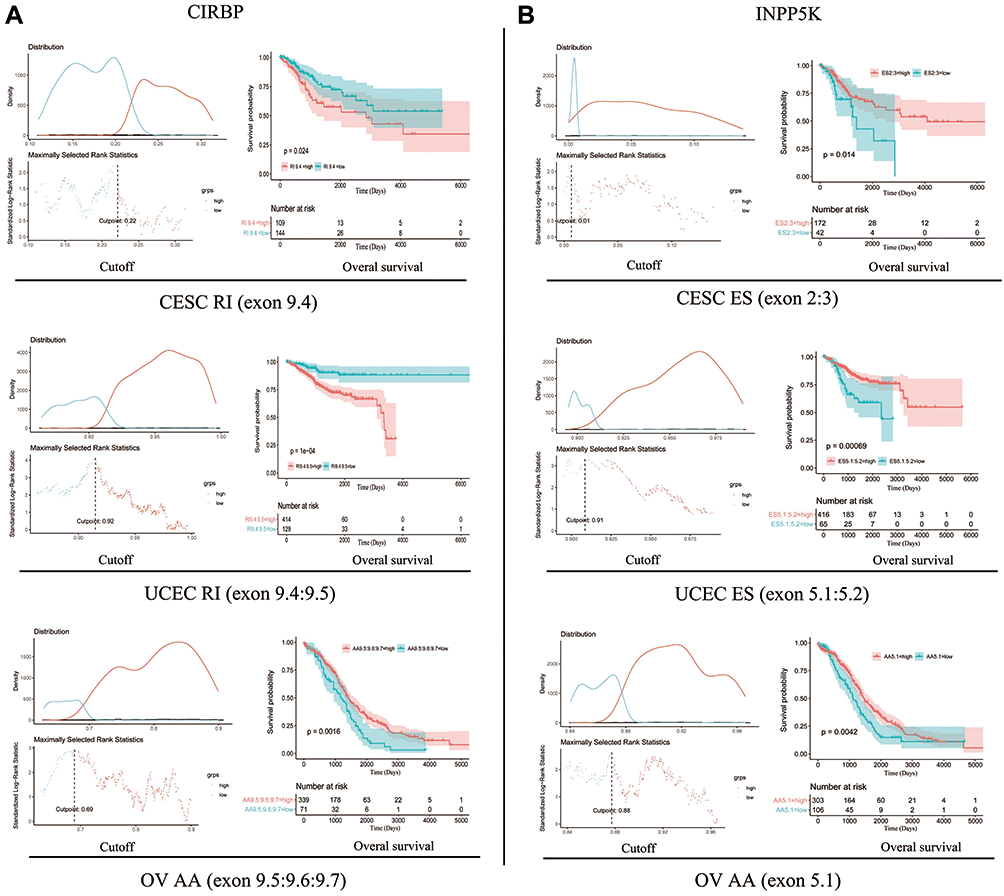 |
Figure 5 Prognostic value of AS events of CIRBP and INPP5K in gynecological cancer patients (Kaplan–Meier plotter). (A) Prognostic value of AS events of CIRBP. (B) Prognostic value of AS events of INPP5K. Patients were divided into high (red) and low (blue) PSI value groups based on the best cut-off values using the R package. |
Prognostic Value of Alternative Polyadenylation Events in CIRBP/INPP5K
The advantage of APA for tumor progression is the induction of oncogenes through 3ʹ-UTR shortening. To further explore the clinical impact of APA events involving CIRBP and INPP5K, we performed a Kaplan-Meier survival analysis of RNA sequencing data of gynecologic cancer patients in TCGA (Figure S9). The results showed that a CIRBP APA event with a low PDUI value in CESC (P=0.0011) and OV (P<0.05) was associated with a poor prognosis. The same result was found following an INPP5K APA event in OV (P<0.05). Nonetheless, INPP5K APA in CESC and UCEC, and CIRBP APA in UCEC were not significantly associated with the patients’ prognosis.
Discussion
The present study revealed for the first time the prognostic significance of CIRBP and INPP5K in the evaluation of gynecologic cancers from the perspectives of DNA methylation, APA and AS and its correlation with patient clinicopathological data using a public database. We found that CIRBP/INPP5K expression was significantly downregulated in gynecologic cancers, and we assessed the major biological functions and molecular mechanisms through which CIRBP/INPP5K might be involved. Moreover, we investigated whether DNA methylation and APA function as prognostic biomarkers in gynecologic cancers and identified prognostic AS events in CIRBP/INPP5K.
Based on a comprehensive Metascape bioinformatics analysis, T cell activation was the major and most significant functional enrichment event in all three gynecological cancers. T cells maintain the capacity to proliferate, persist, and mediate the complete destruction of tumor deposits. However, tumors can evade immune surveillance by creating hostile microenvironments that perturb T cell metabolism and effector function. In general, tumor-infiltrating T cells, particularly CD8+ T cells, are positive prognostic markers of multiple solid tumors.32 The dysfunction of T cells in cancers may be due to the mechanisms by which the upregulation of PD-1 on T cells or its ligand PD-L1 occur. A recent Phase II clinical trial was performed to test the safety and efficacy of nivolumab, which is an anti–PD-1 antibody that blocks PD-1 signaling, against platinum-resistant OV and showed that the patients were able to tolerate the subsequent four courses of nivolumab with a disease control rate of 45% in all 20 patients.33 Surprisingly, CIRBP activates T cells through a TLR4-dependent mechanism and leads to the upregulation of proinflammatory gene expression in both CD4+ and CD8+ T cells during sepsis.34 The above results further confirm the value of our study and reveal a novel mechanism by which CIRBP participates in gynecological tumor progression through the regulation of T cells.
AS participates in the control of the major hallmarks of cancer toward more aggressive invasive cancer phenotypes,35 and AS can serve as a biomarker of overall survival in cancers. For example, a nine-AS event signature was identified as a predictor of survival in CESC through a Cox proportional hazards regression model including ES events.36 Compared with the prognostic model constructed by a single AS event, the risk score (All) including ES events better predicted UCEC prognosis.37 Among the OV patients, most survival-associated AS events, including AA events, were found to be favorable prognostic factors (HR<1), and all top 20 AD and RI events indicated a good prognosis.38 Our findings are consistent with the above studies reporting that AA, RI and ES events in CIRBP/INPP5K may be novel prognostic indicators. In summary, we built powerful prognostic predictors for gynecologic cancer patients and uncovered interesting splicing networks that might reveal the underlying mechanisms.
APA phenomena are rather common as more than half of the genes in the human genome have APA sites.39 APA, which induces the shortening of the 3ʹ-UTR, has emerged as a prevalent feature associated with cancer progression. In recent years, a growing body of research has shown that APA can act as a powerful molecular biomarker with prognostic potential in cancers. High APA scores correlate with an advanced tumor stage and a poor prognosis (P<0.001) in lung cancer.40 In our study, CIRBP with low APA scores predicted a poor prognosis in CESC (P=0.0011). Overall, the details of APA events in gynecologic cancers require further in-depth investigation.
Regarding the clinical value of DNA methylation in cancer, approximately 14 DNA methylation-based biomarker assays have been translated into commercially available assays.41 Studies have investigated whether DNA methylation affects epithelial ovarian cancer risk by regulating the expression of a neighboring gene based on genetic data of nearly 63,000 women of European descent.42 Several studies have also verified that the hypermethylation of CIRBP is induced by chronic hypoxia and, thus, might modulate HIF-1alpha expression by binding the 3ʹ-UTR of its mRNA to increase its stability.18,43 In our study, the functional enrichment analysis of the prognostic genes identified the involvement of CIRBP in cell metabolism and DNA methylation as prognostic molecular biomarkers in gynecology cancer. This finding provides a theoretical basis for our further research investigating the mechanism by which CIRBP DNA methylation modulates gynecological tumor progression.
Conclusion
To the best of our knowledge, the current study is the first to perform a systematic identification and analysis of survival-associated genes from the perspectives of AS, APA and DNA methylation in gynecology cancers. Our results indicate that the mRNA expression levels of CIRBP and INPP5K are significantly downregulated and associated with the clinical outcomes of patients with CESC, UCEC and OV. Further enrichment analysis confirmed that these prognostic genes possibly modulate cancer progression by inducing T cell activation. CIRBP/INPP5K expression is positively associated with immune infiltration, and CIRBP and INPP5K are major risk factors of survival in UCEC patients. More importantly, survival-associated AS events, APA events and DNA methylation may serve as potential prognostic biomarkers and targets for gynecological cancers and provide valuable clues for further validation.
Abbreviations
AS, alternative splicing; APA, aolyadenylation; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; UCEC, uterine corpus endometrial carcinoma; OV, ovarian serous cystadenocarcinoma; PD1, programmed cell death-1; TCGA, The Cancer Genome Atlas; CIRBP, cold-inducible RNA-binding protein; INPP5K, inositol polyphosphate-5-phosphatase K; AA, alternative 3ʹ acceptor site; AD, alternative 5ʹ donor site; AP, alternate promoter; AT, alternate terminator; ES, exon skipping; ME, mutually exclusive exon; RI, retained intron; GEPIA, Gene Expression Profiling Interactive Analysis; MCODE, molecular complex detection; PSI, the percent-spliced-in value; PDUI, the distal polyA site usage index percentage.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81902648) and the China Postdoctoral Science Foundation (2018 M641115). We thank TCGA database for providing the platforms and contributing the valuable datasets.
Disclosure
The authors have declared that they have no conflicts of interest for this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Zeng H, Zheng R, Guo Y, et al. Cancer survival in China, 2003-2005: a population-based study. Int J Cancer. 2015;136(8):1921–1930. doi:10.1002/ijc.29227
3. Wang M, Fan W, Ye M, et al. Molecular profiles and tumor mutational burden analysis in Chinese patients with gynecologic cancers. Sci Rep. 2018;8(1):8990. doi:10.1038/s41598-018-25583-6
4. Murali R, Grisham RN, Soslow RA. The roles of pathology in targeted therapy of women with gynecologic cancers. Gynecol Oncol. 2018;148(1):213–221. doi:10.1016/j.ygyno.2017.11.020
5. Nishiyama H, Itoh K, Kaneko Y, Kishishita M, Yoshida O, Fujita J. A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J Cell Biol. 1997;137(4):899–908. doi:10.1083/jcb.137.4.899
6. Wellmann S, Buhrer C, Moderegger E, et al. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J Cell Sci. 2004;117(Pt 9):1785–1794. doi:10.1242/jcs.01026
7. Yang R, Weber DJ, Carrier F. Post-transcriptional regulation of thioredoxin by the stress inducible heterogenous ribonucleoprotein A18. Nucleic Acids Res. 2006;34(4):1224–1236. doi:10.1093/nar/gkj519
8. Dong R, Zhu T, Benedetti L, et al. The inositol 5-phosphatase INPP5K participates in the fine control of ER organization. J Cell Biol. 2018;217(10):3577–3592. doi:10.1083/jcb.201802125
9. Wang R, Jiao Y, Li Y, et al. The prediction and prognostic significance of INPP5K expression in patients with liver cancer. Biomed Res Int. 2020;2020:9519235.
10. Ooms LM, Horan KA, Rahman P, et al. The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem J. 2009;419(1):29–49.
11. Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10(11):741–754. doi:10.1038/nrm2777
12. Sammeth M, Foissac S, Guigo R. A general definition and nomenclature for alternative splicing events. PLoS Comput Biol. 2008;4(8):e1000147. doi:10.1371/journal.pcbi.1000147
13. Gokhale NS, McIntyre A, Mattocks MD, et al. Altered m(6)A modification of specific cellular transcripts affects flaviviridae infection. Mol Cell. 2020;77(3):542–555. doi:10.1016/j.molcel.2019.11.007
14. Yousaf S, Sheikh SA, Riazuddin S, Waryah AM, Ahmed ZM. INPP5K variant causes autosomal recessive congenital cataract in a Pakistani family. Clin Genet. 2018;93(3):682–686. doi:10.1111/cge.13143
15. Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18(1):18–30. doi:10.1038/nrm.2016.116
16. Van Etten JL, Nyquist M, Li Y, et al. Targeting a single alternative polyadenylation site coordinately blocks expression of androgen receptor mRNA splice variants in prostate cancer. Cancer Res. 2017;77(19):5228–5235. doi:10.1158/0008-5472.CAN-17-0320
17. Liu S, Hu C, Luo Y, Yao K. Genome-wide DNA methylation profiles may reveal new possible epigenetic pathogenesis of sporadic congenital cataract. EPIGENOMICS. 2020;12(9):771–788. doi:10.2217/epi-2019-0254
18. Liu Y, Xing J, Li Y, et al. Chronic hypoxia-induced Cirbp hypermethylation attenuates hypothermic cardioprotection via down-regulation of ubiquinone biosynthesis. Sci Transl Med. 2019;11(489):eaat8406. doi:10.1126/scitranslmed.aat8406
19. Uhlen M, Zhang C, Lee S, et al. A pathology atlas of the human cancer transcriptome. SCIENCE. 2017;357(6352):eaan2507. doi:10.1126/science.aan2507
20. Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer. 2012;19(2):197–208. doi:10.1530/ERC-11-0329
21. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. doi:10.1093/nar/gkx247
22. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi:10.1038/s41467-019-09234-6
23. Ryan M, Wong WC, Brown R, et al. TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res. 2016;44(D1):D1018–D1022. doi:10.1093/nar/gkv1288
24. Feng X, Li L, Wagner EJ, Li W. TC3A: the cancer 3ʹ UTR atlas. Nucleic Acids Res. 2018;46(D1):D1027–D1030. doi:10.1093/nar/gkx892
25. Chandrashekar DS, Bashel B, Balasubramanya S, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. NEOPLASIA. 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
26. Scotto L, Narayan G, Nandula SV, et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer. 2008;47(9):755–765. doi:10.1002/gcc.20577
27. Bykov V, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89–102. doi:10.1038/nrc.2017.109
28. Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4(1):2612. doi:10.1038/ncomms3612
29. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci. 2002;99(19):12293–12297. doi:10.1073/pnas.192461099
30. Ott PA, Bang YJ, Berton-Rigaud D, et al. Safety and antitumor activity of pembrolizumab in advanced programmed death ligand 1-positive endometrial cancer: results from the kEYNOTE-028 study. Journal of Clinical Oncology. 2017;35(22):2535–2541. doi:10.1200/JCO.2017.72.5952
31. Moore M, Ring KL, Mills AM. TIM-3 in endometrial carcinomas: an immunotherapeutic target expressed by mismatch repair-deficient and intact cancers. Modern Pathol. 2019;32(8):1168–1179. doi:10.1038/s41379-019-0251-7
32. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi:10.1038/nrc3245
33. Hamanishi J, Mandai M, Ikeda T, et al. Safety and antitumor activity of Anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2015;33(34):4015–4022. doi:10.1200/JCO.2015.62.3397
34. Bolognese AC, Sharma A, Yang W, Nicastro J, Coppa GF, Wang P. Cold-inducible RNA-binding protein activates splenic T cells during sepsis in a TLR4-dependent manner. Cell Mol Immunol. 2018;15(1):38–47. doi:10.1038/cmi.2016.43
35. Venables JP, Klinck R, Koh C, et al. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol. 2009;16(6):670–676. doi:10.1038/nsmb.1608
36. Ouyang D, Yang P, Cai J, Sun S, Wang Z. Comprehensive analysis of prognostic alternative splicing signature in cervical cancer. Cancer Cell Int. 2020;20:221. doi:10.1186/s12935-020-01299-4
37. Wang C, Zheng M, Wang S, et al. Whole genome analysis and prognostic model construction based on alternative splicing events in endometrial cancer. Biomed Res Int. 2019;2019:2686875.
38. Zhu J, Chen Z, Yong L. Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol Oncol. 2018;148(2):368–374. doi:10.1016/j.ygyno.2017.11.028
39. Tian B, Hu J, Zhang H, Lutz CS. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33(1):201–212. doi:10.1093/nar/gki158
40. Ichinose J, Watanabe K, Sano A, et al. Alternative polyadenylation is associated with lower expression of PABPN1 and poor prognosis in non-small cell lung cancer. Cancer Sci. 2014;105(9):1135–1141. doi:10.1111/cas.12472
41. Koch A, Joosten SC, Feng Z, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. 2018;15(7):459–466. doi:10.1038/s41571-018-0004-4
42. Yang Y, Wu L, Shu X, et al. Genetic data from nearly 63,000 women of european descent predicts DNA methylation biomarkers and epithelial ovarian cancer risk. Cancer research. 2019;79(3):505–517. doi:10.1158/0008-5472.CAN-18-2726
43. Lu M, Ge Q, Wang G, et al. CIRBP is a novel oncogene in human bladder cancer inducing expression of HIF-1alpha. Cell Death Dis. 2018;9(10):1046. doi:10.1038/s41419-018-1109-5
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.