Back to Archived Journals » ChronoPhysiology and Therapy » Volume 4
The potential of light therapy in Parkinson's disease
Authors Johnstone D, Coleman K, Moro C, Torres N, Eells J, Baker GE, Ashkan K, Stone J, Benabid A, Mitrofanis J
Received 7 November 2013
Accepted for publication 10 December 2013
Published 18 February 2014 Volume 2014:4 Pages 1—14
DOI https://doi.org/10.2147/CPT.S57180
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Daniel M Johnstone,1 Kristina Coleman,2 Cécile Moro,3 Napoleon Torres,3 Janis T Eells,4 Gary E Baker,5,† Keyoumars Ashkan,6 Jonathan Stone,1 Alim-Louis Benabid,3 John Mitrofanis2
1Department of Physiology and Bosch Institute, 2Department of Anatomy and Histology, University of Sydney, Sydney, NSW, Australia; 3Clinatec LETI-DTBS, CEA Grenoble, France; 4Department of Biomedical Science, University of Wisconsin, Madison, WI, USA; 5Department of Optometry and Visual Science, City University, 6Department of Neurosurgery, King’s College Hospital, London, UK
†Gary E Baker passed away on 15 November 2011.
Abstract: Parkinson’s disease is a movement disorder with cardinal signs of resting tremor, akinesia, and rigidity. These manifest after a progressive death of many dopaminergic neurons of the midbrain. Unfortunately, the progression of this neuronal death has proved difficult to slow and impossible to reverse despite an intense search for the specific causes and for treatments that address the causes. There is a corresponding need to develop approaches that regulate the self-repair mechanisms of neurons, independent of the specific causes of the damage that leads to their death. Red to infrared light therapy (λ=600–1,070 nm) is emerging as an effective, repair-oriented therapy that is capable of stabilizing dying neurons. Initially a space-age anecdote, light therapy has become a treatment for tissue stressed by the known causes of age-related diseases: hypoxia, toxic environments, and mitochondrial dysfunction. Here we focus on several issues relating to the use of light therapy for Parkinson's disease: 1) What is the evidence that it is neuroprotective? We consider the basic science and clinical evidence; 2) What are the mechanisms of neuroprotection? We suggest a primary mechanism acting directly on the neuron’s mitochondria (direct effect) as well as a secondary, supportive mechanism acting indirectly through systemic systems (indirect effect); 3) Could this be effective in humans? We discuss the pros and cons of this treatment in humans, including the development of a new surgical method of delivery; and 4) What are the advantages of using light therapy? We explore the features that make this therapy a promising potential treatment. In summary, early evidence indicates that light regulates specific neuronal functions and is neuroprotective in animal models of Parkinson’s disease. The stage is set for detailed and rigorous explorations into its use on Parkinson’s disease patients, in particular, whether light slows the disease progression rather than simply mitigating signs.
Keywords: infrared, near infrared, neuroprotection, photobiomodulation, substantia nigra
Introduction
The current “gold standard” treatments for Parkinson’s disease are very effective at attenuating the motor signs, at least initially. However, they do not reliably slow the progression of the disease; neurons continue to die during the course of treatment. The discovery of new therapeutic approaches that offer neuroprotection against parkinsonian insult is therefore paramount. In this context, several recent studies in animal models of Parkinson’s disease, as well as other models of disease (eg, retinal degeneration, stroke, multiple sclerosis, Alzheimer’s disease), have reported that red to infrared light therapy (λ=600–1,070 nm; referred to forthwith as light therapy) can be neuroprotective. There is real potential for the development of light therapy as a treatment option for Parkinson’s disease patients – one that slows the ongoing neuronal death and progression of the disease.
Overview of Parkinson’s disease
Parkinson’s disease is a slow, progressive disorder that affects ~1% of people over the age of 55–60 years. It has distinct cardinal signs of resting (pill-rolling) tremor, lead-pipe rigidity (increased muscle tone), akinesia (difficulty in initiating and stopping movement), and/or bradykinesia (slowness of movement).1,2 These signs manifest as a consequence of degeneration of pigmented dopaminergic neurons in the substantia nigra pars compacta (SNc) of the basal ganglia; there is also some limited dopaminergic neuronal loss in other nuclei, including the ventral tegmental area and retrorubral field.1,3–5 Degeneration of the SNc dopaminergic neurons, thought to be in the range of 50%–85%,1,4 leads to a substantial reduction in the levels of dopamine in other regions of the basal ganglia, in particular the striatum. This, in turn, leads to the generation of abnormal neuronal activity in certain basal ganglia nuclei, for example, the subthalamic nucleus and the globus pallidus, which then manifests as the distinct signs of the disease.1,4,5
The disease has an insidious onset. There is evidence that it may be caused by exposure to a neurotoxin, for example, paraquat, rotenone, 6 hydroxydopamine, or methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).6–8 This exposure may have occurred well before the onset of clinical signs since compensatory mechanisms such as sprouting of new dopaminergic branches in striatum have been shown to delay clinical manifestation.9 Indeed, animal models that involve exposure to these and other toxins reproduce many of the parkinsonian signs and pathological features.6–8 There is also evidence, in a small number of cases (10%–15%), that defective genes (eg, parkin, PINK1, SNCA [that encodes the synaptic protein alpha-synuclein]) contribute to the development of Parkinson’s disease.10,11 Further, there are several transgenic animal models of the disease which, as with the toxin-induced models, display many Parkinson-like features.12,13
The mechanisms underpinning the degeneration of dopaminergic neurons, whether after toxic insult or genetic defect, have come under much scrutiny in recent years. It is clear that mitochondrial dysfunction plays a central role in the process.14 Mitochondria are the “engine rooms” of neurons – they produce energy in the form of adenosine triphosphate (ATP) that fuels neuronal function. Under certain conditions, for example in ageing or after toxic insult/genetic defect, there is a progressive accumulation of mutations in mitochondrial DNA that reduces mitochondrial efficiency and ATP yield. This process leads to an increase in toxic reactive oxygen species, generating oxidative stress and subsequent neuronal degeneration.14,15 A rather common feature of neurodegeneration is the development and accumulation of abnormal proteins, such as alpha-synuclein within the neurons. This protein is a major constituent of Lewy bodies, found in particular in Parkinson’s disease.16 Further exacerbating the degenerative process are glutamate excitotoxicity,17–20 inflammation,21 and glial cell activation.22 The glutamatergic inputs to the dopaminergic neurons, particularly those from the subthalamic nucleus, become overactive in Parkinson’s disease, and the excessive glutamate promotes mitochondrial defects within the dopaminergic neurons. Under normal conditions, glial cells, both microglia and astrocytes, support neuronal function, but in the adverse parkinsonian condition, they become reactive and toxic to neurons, generating a sustained local inflammation within the SNc.21,22
Current therapies for Parkinson’s disease
The current treatment option for most patients with Parkinson’s disease is dopamine replacement drug therapy, followed by surgery in selected patients. The basic strategy of each treatment is simple: dopamine drug therapy aims to replace the dopamine lost from the system, while surgery aims to correct the abnormal function of the basal ganglia circuitry caused by the loss of the dopamine. The surgical option is usually recommended to patients after the efficacy of drug treatment lessens (see Surgical treatment section below) or when the disease has progressed sufficiently. Both of these main treatments provide symptomatic relief (ie, treat the signs that characterize the disease).23 They will each be discussed briefly below.
Dopamine drug therapy
Dopamine replacement drug therapy can work in one of three ways. First, it can increase the amount of dopamine available in the brain. Levodopa (L-Dopa) is a precursor to dopamine and is often used as first line treatment.24 While highly efficacious at reducing motor signs initially, its efficacy tapers with prolonged use. For example, over time (ie, 5–8 years), involuntary movements of the upper limbs (dyskinesias) develop when plasma levels of L-Dopa are high (“on” time) due to dysregulation of the striatal dopaminergic receptors. Second, some drugs – dopamine agonists such as bromocriptine and apomorphine – mimic the action of dopamine, activating dopamine receptors of neurons in the striatum directly. While generally not as effective as L-Dopa, dopamine agonists are associated with fewer motor complications (ie, dyskinesias) and may be the first-line treatment choice in younger patients.24,25 Finally, other drugs – monoamine oxidase type B inhibitors such as selegiline and rasagiline – help stop the breakdown of dopamine at the synapse, increasing its availability to the postsynaptic neurons.25
In general, these drug treatments have very good early symptomatic effects, but their longer-term neuroprotective or disease modifying effects are far from clear. For example, although drugs such as selegiline and rasagiline have been tested as putative neuroprotective agents in clinical trials, their ability to actually stop neuronal death and slow the pathology of the disease has yet to be demonstrated.26
Surgical treatment
Surgical intervention is reserved as a last-line treatment, after the efficacy of the drug therapy diminishes and side effects develop (eg, dyskinesias). The basic principle is to correct the abnormal activity of certain basal ganglia nuclei and motor thalamus, generated initially by the reduction of dopamine levels in the striatum. The nuclei targeted most commonly for intervention have been the motor nuclei of the thalamus, the globus pallidus, and in particular, the subthalamic nucleus, the efficacy of which extends to the “dopaminergic triad” of signs: tremor, akinesia, rigidity. Surgeons have used either destructive lesions or deep brain stimulation at high frequency (creating a “functional” lesion) to dampen the abnormal activity in these nuclei.27,28 These methods have been very effective in treating the motor signs of the disease in patients over long periods.27,28 Although there is some evidence in animal models that lesion or deep brain stimulation of the subthalamic nucleus is neuroprotective, by reducing glutamate excitotoxicity to the SNc, the evidence available in Parkinson’s disease patients is inconclusive.20,29 Due to its low morbidity, deep brain stimulation at high frequency is often proposed to patients earlier in their disease, when the medical treatment, although still efficient, does not provide them with satisfactory relief. The best time for surgery is before the patients have finished their professional activity or before their social and family life has been substantially altered.27,28
In summary, the primary limitation with the current treatments for Parkinson’s disease in humans is that there is little evidence for neuroprotection; the treatments seemingly have little effect on slowing the pathology and preventing the ongoing neuronal death. Thus, there is an aching gap in the current management of Parkinson’s disease.
Putative neuroprotective treatments
In various animal models of the disease and in vitro, there have been a large number of treatments – too many to mention in detail here – that have been shown to be neuroprotective or “disease modifying.” Unfortunately such treatments have, for the most part, not progressed well at the clinical level. They are either presently at a very early phase of clinical trial, with no clear outcome or they have yielded mixed results after trial.30 These treatments vary greatly in approach, for example, changing the phenotype of neurons (gene therapy, introduction of viral vectors),31 introducing trophic factors that increase neuronal survival (eg, glial cell line-derived neurotrophic factor),32 stimulating neurogenesis,33 increasing patient exercise programs,34 and finally, changing dietary intake, for instance, using antioxidants (eg, coenzyme Q10), fish oil, and vitamin E.35
Light therapy, specifically low-level laser therapy of red to infrared light, is an emerging, putative neuroprotective treatment that, as with the aforementioned treatments, is showing promise at the basic science level. It awaits rigorous exploration at the clinical level for Parkinson’s disease, but given its novel mode of application (see below; “What is light therapy and how does it offer neuroprotection”), we are very hopeful of positive outcomes for many patients. In the next section, we will explore in detail the basic science and clinical evidence for neuroprotection by light therapy in parkinsonian cases and consider its further use in Parkinson’s disease patients using a new method of application.
It should be noted that we use the term “light therapy” to refer specifically to red to infrared light (λ=600–1,070 nm). Light that includes wavelengths outside this range may actually be detrimental to cell survival. A recent study in rats has shown that continuous exposure to bright light, which includes a peak wavelength of ~440 nm, results in a reduction in the number of dopaminergic neurons in the SNc.36 Indeed, it has been suggested that excess bright light exposure (ie, “polluted” light) may be a cause of Parkinson’s disease.36
What is light therapy and how does it offer neuroprotection?
An intuitive reaction to the idea that light influences neuronal function and survival in the mammalian brain is that it is not possible, because the brain is encased in a cranium and meninges, covered with skin and muscle. Further, why would there be light-inducible protective mechanisms in tissue that is encased in such layers and hence presumably has little exposure to light? Perhaps the mechanisms evolved in epithelial tissues and therefore remain inducible in the neuroepithelium. In spite of these reservations, there is a growing body of evidence suggesting that neurons are in fact influenced by light and, in particular, that low-level laser therapy with red to infrared light induces neuroprotective responses. The neuroprotective efficacy of light has been demonstrated in animal models of retinal disease,37–41 traumatic brain injury,42–44 experimentally induced stroke,45–47 familial amyotrophic lateral sclerosis,48 multiple sclerosis,49 Parkinson’s disease,50–54 and Alzheimer’s disease.55–57 In humans, light therapy has been reported to improve cognition and emotional functions,58 together with performance in a range of clinical tests after ischemic stroke,59,60 brain trauma,61 depression,62 and in age-related macular degeneration.63 The fact that light therapy has been reported to be effective in so many different models of disease and in a range of neural systems suggests a common mechanism at play. Recent work indicates that light is effective in reducing neuronal death induced by apoptosis, but not necrosis.44 The pathway to apoptosis is likely to involve a critical decline in cellular energy production,64 which light may help to restore (see below, in this section). This mechanism may be common to all abovementioned conditions and is perhaps why light therapy has such broad potential applications.
The first applications of light therapy were with coherent-light sources (lasers), but effective therapy also has been reported recently with noncoherent light sources (light-emitting diodes [LED], eg, the WARP 10 LED; Quantum Devices, Barneveld, WI, USA).65–68 The optimal wavelengths required for therapeutic effects appear to lie within the range of 600–1,070 nm, with the longer wavelengths (eg, 810 nm, 830 nm, and 1,070 nm) having a greater depth of penetration through body tissues than the shorter ones within this range (eg, 670 nm),66,68 although the shorter ones have been used most commonly. Hence, unless stated otherwise, all the results below have been obtained using a 670 nm external LED device. The energy densities required for therapeutic effects vary greatly in the literature, ranging between ~1–60 joules/cm2 (and power intensities between ~1–750 mW/cm2), although the majority of studies have used densities <10 joules/cm2 and achieved positive outcomes.65–68 The reviews by Rojas and Gonzalez-Lima67 and Chung et al68 provide thorough outlines of light stimulation protocols used by previous studies on brain and peripheral tissues. There are also some guidelines set by the World Association of Laser Therapy in April 2010, but they apply to peripheral tissues, not brain. Some clear guidelines for use in brain are very much needed; as noted by Quirk et al,69 the sooner the “hodgepodge” of application ceases the better.
The mechanisms that underpin the neuroprotective effects of light therapy are not entirely clear, but there appear to be two general modes of action (Figure 1). By far the most commonly studied mode of action is light acting directly on the neurons themselves, triggering intrinsic trophic factors. The light “boosts” mitochondrial function by increasing electron transfer in the respiratory chain and activating photoacceptors, such as cytochrome oxidase, resulting in increased ATP production65–68 and a reduction in apoptosis (see above, in this section). A second, emerging hypothesis relating to mode of action is that light may also trigger a more indirect, systemic response. Several studies have reported remote, often bilateral, effects on body tissues after local light application to, for example, skin wounds.70 Intriguingly, neuroprotection of the mouse brain has been demonstrated after application of light to the dorsum of the animal, with no direct irradiation of the head.71,72 While the mechanism remains unknown, it could involve the stimulation of one or more circulating molecules or cell types. One possibility is the stimulation of immune cells, for example mast cells and macrophages, could help neuroprotect cells in the brain.49,68,73 There may also be effects on inflammatory mediators, as light therapy is associated with downregulation of proinflammatory cytokines (γ-interferon, α-tumor necrosis factor) and upregulation of anti-inflammatory cytokines (interleukin-4, interleukin-10).49 In addition, bone marrow-derived stem cells may also be involved; a series of studies has demonstrated that light exposure increases proliferation of c-kit-positive cells in the bone marrow and that, following myocardial infarction in rats, these cells are mobilized and recruited specifically to the site of damage where they contribute to a reduction in myocardial infarct size and ventricular dilatation.74,75 These cells, together with immune cells, may release trophic factors such as nerve growth factor and vascular endothelial growth factor that improve the function of dying (apoptotic) cells and aid in their survival.74,76 Such an indirect mechanism is similar to the so-called “abscopal” effect (Figure 1), where localized treatment of a tumor leads to not only a shrinking of the local tumor but also to shrinking of tumors far from the treated area.77 Finally, several groups have suggested that light promotes functional recovery by stimulating ventricular stem cells, promoting neurogenesis and neuronal migration.47,60,68
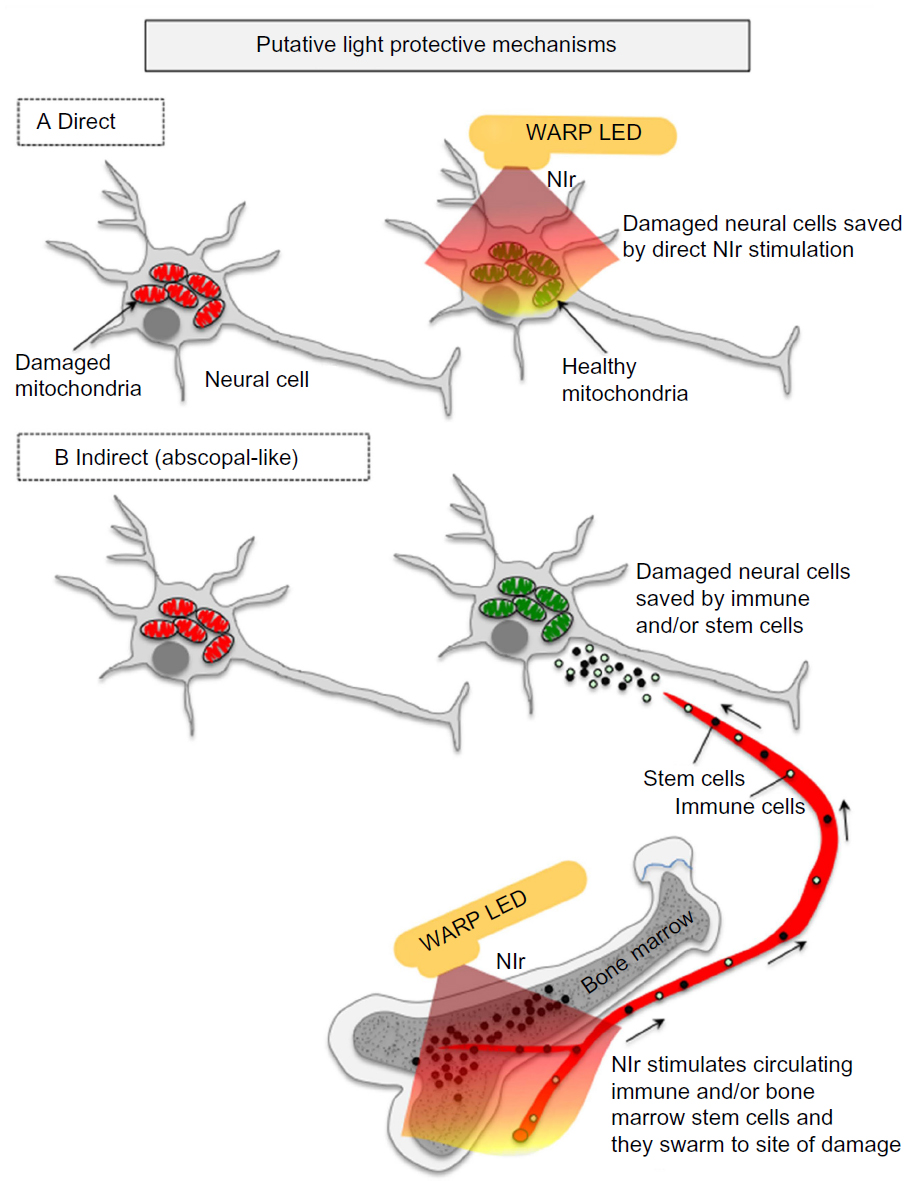 | Figure 1 The putative light protective mechanisms in the brain. |
It remains to be determined which of these mechanisms, direct or indirect, offers the neuroprotection, but we note that they are not mutually exclusive and that both may contribute to the process.71,72 ,78 As a working hypothesis, direct stimulation of the mitochondria, which is supported by the most compelling evidence (as described above), is likely to form the primary mechanism of protection by light therapy, while the indirect stimulation of immune and/or stem cells may form a secondary and supportive mechanism. Some early results in an animal model of Parkinson’s disease indicate that, although there is some neuroprotection of the brain after remote application of light therapy (eg, dorsum of body), the neuroprotection is not as great as when light therapy is applied directly to the head.71,72,78 In other words, neuroprotection is achieved with both direct and indirect irradiation, but the direct irradiation is the more effective.
Basic science and clinical evidence for neuroprotection by light therapy in Parkinson’s disease
The view from the dish: the in vitro evidence
The first studies to report the neuroprotective effects of light treatment were in vitro. Light therapy was shown to reduce neuronal death, increase ATP content, and decrease levels of oxidative stress among rat striatal and cortical neurons after exposure to the parkinsonian toxins rotenone and 1-methyl-4-phenylpyridium (MPP+).45,51 In cultures of human neuroblastoma cells engineered to overexpress alpha-synuclein, light therapy has been reported to increase mitochondrial function and reduce oxidative stress after MPP+ exposure.79,80 Further, mitochondrial movement along axons in hybrid cells bearing mitochondrial DNA from Parkinson’s disease patients improves substantially after light therapy (810 nm, laser), with movement restored to near control levels.81
The view from the animal model: the in vivo evidence
Cell survival
The pioneering in vitro findings led inevitably to in vivo explorations. To date, the impact of light therapy on dopaminergic neuronal survival has been examined in the well-known MPTP mouse model as well as in a transgenic mouse model (K369I) of Parkinson’s disease. In both acute53,62,71,78,79 and chronic54 MPTP models, light therapy saves many dopaminergic neurons from death; in many cases, the numbers of neurons in the light-treated animals are near control levels. Further, results are similar whether the therapy is applied at the same time or well after the insult, indicating that light therapy both protects healthy neurons against an insult and also rescues damaged neurons (self-repair) after the insult.54 The rescue of neurons is particularly relevant to the clinical reality of the parkinsonian condition, where individuals first suffer insult (and dopaminergic neuronal death) and then receive therapeutic intervention. In terms of mechanism, the protection and rescue of neurons by light is presumably due to an increase in ATP production. The increase in ATP before insult may help protect neurons from damage, while reducing the decline in ATP after the insult may help to rescue the dying neurons.65–68 In the K369I transgenic mouse model of frontotemporal dementia, which also shows a chronic and progressive degeneration of dopaminergic neurons in the SNc and parkinsonian signs,13 light therapy decreases oxidative stress and hyperphosphorylated tau and increases dopaminergic neuronal survival in the SNc.80
Degree of neuroprotection
A common feature evident in most neuroprotection studies is that beneficial outcomes are far greater when the therapy is commenced earlier in the disease process.20,82 In other words, there is greater capacity to save neurons when there is less prior neuronal degeneration. This is certainly the case with light therapy. In the acute MPTP model, there is a greater proportion of neurons saved by light therapy following a milder MPTP regime compared to a stronger one.53,72 There may be a stage in the disease process, presumably at very late stages (or after a very strong toxin dose) and extensive neuronal death (eg, 70%–80%), when the neuroprotective effect of light therapy, as well as other treatments, becomes limited. Hence, the earlier the diagnosis and initiation of treatment, the better the neuronal survival and likely clinical outcome.
Neuroprotection among other dopaminergic neurons
Other groups of dopaminergic neurons, for example those in the retina, periaqueductal grey matter, and zona incerta-hypothalamus, are not as likely to be saved by light therapy after MPTP insult.53,54,83 The mechanisms that save dopaminergic neurons in the SNc from MPTP insult (see above; “What is light therapy and how does it offer neuroprotection”) are thus less effective in the other dopaminergic neurons. Clinically, light therapy may be more effective in the treatment of the central motor signs of the disease caused by SNc neuronal loss (eg, akinesia) than the nonmotor symptoms caused by a loss of other dopaminergic neurons (eg, sleep-wakefulness and vision).83,84
Functional restoration
Not only does light therapy protect against the degeneration of dopaminergic neurons, but it also appears to restore functional activity to those neurons that are saved. For example, light therapy has been shown to correct abnormal neuronal activity generated by the parkinsonian condition.85 Using Fos immunohistochemistry (a well-established measure of neuronal activity), the overactivity of neuronal firing in the subthalamic nucleus and zona incerta (two key basal ganglia nuclei) characteristic of parkinsonian cases has been reported to be reduced substantially after light therapy. This reduction does not quite reach control levels, indicating that the restoration is partial, and has been attributed to the surviving SNc dopaminergic neurons being functionally active, continuing to produce and release dopamine at their terminals in the striatum.85 These early functional results could be built upon by further electrophysiological and pharmacological explorations.
Behavioral improvements
The restoration of functional activity in the basal ganglia by the surviving dopaminergic neurons manifests in improved motor behavior. Previous studies have reported that light therapy improves various parameters of locomotive behavior, for example mobility and velocity, after MPTP treatment.52,86,79 It also delays disease progression and reduces the severity of the disease phenotype in transgenic mice expressing the A53T human alpha-synuclein mutation.87 In the MPTP-treated cases, the beneficial effects of light therapy are not immediate, being seen only after several doses of light over a period of 2–3 days.52,79 The mitochondria of the dopaminergic cells, after several treatments, may have been stimulated further to increase their activity and ATP production,65–68 thereby being better prepared to protect against the MPTP toxicity.79
Does pigmentation limit benefits of light therapy?
Moro et al79 have shown that the beneficial effects of light therapy after MPTP insult were, somewhat surprisingly, more evident in an albino mouse strain (Balb/c) than they were in a pigmented mouse strain (C57BL/6).79 When compared to albino mice, pigmented mice had a smaller increase in dopaminergic neuronal number and no improvement in locomotor activity in the light-treated compared to the untreated MPTP cases. They found that there was less penetration of light through fur overlying the cranium and brain in the pigmented mice compared to the albinos. Hence, they assumed that the melanin in the fur absorbed most of the light before it reached the brain.79 However, unlike the Moro et al study, DeSmet et al,86 and Whelan et al,52 reported improvements in mobility and velocity of movement in MPTP-treated pigmented mice (C57BL/6) after light treatment. One possible explanation for the differences in these studies is the use of a greater total light energy in the DeSmet et al and Whelan et al studies (8 and 30 joules/cm2, respectively) relative to the Moro study (2 joules/cm2). The greater energy presumably allowed for greater penetration of light to the brain and hence neuroprotection. In summary, pigmentation may limit penetration of light into the brain, but this can be overcome by using high energy doses of light or shaving the pigmented hair/fur before application.
Dose of light: how much does a neuron need for self-repair or protection?
This issue has proved surprisingly difficult to define. At first thought, one would give as much light as possible to the neurons, so they can better resist the toxic insult or disease process by increasing ATP production (see above; “What is light therapy and how does it offer neuroprotection”). However, empirical evidence suggests that this may not be the optimal strategy.67 Using a new optical fiber device that is implanted inside the brain to deliver light intracranially (see below), neuronal survival against MPTP insult was just as effective, if not marginally better, when light was applied in short pulses (eg, 360 seconds) compared to continuously (eg, 6 days). This phenomenon has been noted in other systems; light applied in short bursts (810 nm, laser) is more effective in treating traumatic brain injury and stroke than if applied continuously.41,43,60 Conversely, there are examples of single doses of light not being sufficient to elicit a neuroprotective effect, and that several doses over a period of 2–3 days are needed.52,68,79 It appears that light therapy has a threshold; the neurons need a certain level of exposure to reap the beneficial effects, but after that level is reached these benefits taper off.67,88 Its mechanism of action on mitochondrial activity, for example, has been likened to a “switch”, but the mechanism of this switch is not known (Glen Jeffery, University College London, personal communication May 2013). There is clearly much left to discover regarding the mechanism of action of light therapy.
The view from the human: the clinical evidence
Given these promising experimental results in animal models of the disease, there have been surprisingly few reports on the impact of light therapy in Parkinson’s disease patients. In fact, there have been no major clinical trials published to date, although two appear to be in progress through the GAAD Medical Research Institute and Quietmind Foundation. The Quietmind Foundation trial89 provides a linked to a YouTube video showing a Parkinson’s disease patient displaying improved movement and reduced tremor after transcranially directed application of light therapy (1,072 nm, laser), but few details are provided. There is a recent noncontrolled and nonrandomized clinical report indicating improvements in speech, cognition, freezing episodes, and gait after transcranial light therapy in parkinsonian patients.90 There are also some clinical reports suggesting improvements in parkinsonian “symptoms” in the majority of patients after light application utilizing an intranasal device.91–93 Finally, there is a rather serendipitous finding in one Parkinson’s disease patient who was treated with light (660 nm, laser) for a dental problem; this patient was reported to display a reduction in his parkinsonian signs following the light treatment.94 Hence, although these anecdotal and casual clinical observations are encouraging, they lack detail and rigor. Further, it remains far from clear whether light therapy is neuroprotective (stopping neuronal death) and/or symptomatic (treating the motor signs) in humans. If neuroprotective, the saving of neurons would ultimately manifest in symptomatic improvements, yet it may turn out that light therapy is symptomatic only, with no neuroprotective effects. (This issue is explored further in the next section; “Could light therapy work in Parkinson’s disease patients”). There is a need for a major, systematic, long-term clinical trial on the neuroprotective and symptomatic effects of light therapy on a large number of patients. Such a trial is certainly warranted given the scientific findings gleaned from experimental animals, together with the fact that light therapy is safe and simple to use (see below; “What would be the advantages of using light therapy”).
In summary, the evidence from basic science, gathered from a number of different laboratories, indicates that light therapy is to a certain extent neuroprotective, restores functional activity, and improves movement in various animal models of Parkinson’s disease. However, the clinical evidence is far sparser, prompting the need for a systematic, large-scale clinical trial of light therapy in patients.
Could light therapy work in Parkinson’s disease patients?
The big question that still remains is whether light therapy can be effective (neuroprotective and/or symptomatic) in humans. The answer is perhaps not simple. If the primary mechanism of action of light is direct mitochondrial stimulation of the damaged or diseased neurons (see above; “What is light therapy and how does it offer neuroprotection”), then there may be an issue with its therapeutic potential for Parkinson’s disease patients. While light penetration and dissipation are not a major consideration when there are few or no tissue barriers, as in the culture dish,37,45,95,96 the retina,38–41 or even in the mouse SNc (4–5 mm inside the skull),53,54 such issues may become prohibitive when light is required to penetrate a thick bony cranium, meninges, skin, and a large mass of intervening brain parenchyma (80–100 mm inside the skull) in order to reach the human SNc (Figure 2). Indeed, almost all the current clinical studies reporting the beneficial effects of light therapy in humans have been in cases where the target region is in the cortex, lying only 8–10 mm below the cranium,60 whether in patients suffering trauma (633 nm and 870 nm, LED),61 stroke (810 nm, laser),59,60 or depression (810 nm, LED).62 Structures lying much deeper in the human brain, such as the SNc, may be beyond the range of the externally applied light therapy.
 | Figure 2 Penetration of light through brain and surrounding tissues. |
This issue has been examined by several previous studies. It has been estimated that light reaches a depth of 20–30 mm from the cortical surface in rabbits (810 nm, laser),45 monkeys (670 nm, laser; personal observations), and humans (810 nm, laser),97 while a similar distance of ~30 mm has been reported along the spinal cord in rats (810 nm, laser).73 However, there would be considerable attenuation and loss of power intensity from the bony surface and through the neural parenchyma. It has been reported that there is ~90% attenuation of light signal inside the cranium of mice.53 Similar sets of values have been reported recently for human cadaver specimens.98 Further, De Taboada et al46 estimated that there would be a power density drop of >99% from the cortex to a depth of 18 mm in rats. In a recent report by Abdo et al99 in rats, the authors noted significant attenuation of light signal, with a power reduction of ~90% at ~1 mm depth from the cortex. Moro et al78 have noted that, although a light signal is detectable from across 10 mm of brain tissue, its intensity at this point is <1% of that emitted from the source. They estimated a 65% reduction of signal across each millimeter of brain.78 Hence, over a distance of 80–100 mm – the predicted distance between any externally applied light and the SNc in humans – the light signal would be extremely weak, perhaps undetectable.65,78,97
Taken together, these data indicate that transcranial light, even at the longer wavelengths and higher energy doses, may not reach deeper brain regions in effective doses, particularly regions deeper than ~30 mm from cranial surface. This may present a limitation in the use of light as a long-term and reliable neuroprotective treatment in humans, particularly those suffering from Parkinson’s disease.60,78 While there may be some neuroprotection by light therapy via the indirect systemic effect, we suggest that it may not solely be enough to have a substantial impact in the SNc. In order for the light to have maximum neuroprotective effect, it may be necessary to place the light source within ~30 mm of the diseased SNc neurons, within the brain itself, to stimulate the mitochondria directly. This could potentially also stimulate an indirect systemic effect; for example, by activating circulating immune cells in nearby blood vessels, providing the secondary, supportive neuroprotective mechanism of action.
Hence, there appears to be a need to develop effective methods of delivering strong light signal to deeper brainstem structures in humans. To this end, we are in the process of developing a novel intracranial light-optical fiber device to deliver light in regions near the SNc in order for diseased neurons to receive sufficient signal and subsequent neuroprotection (Figure 3). Our initial results in mice have shown that the light does not cause any toxic damage around the implant sites in the brain, and that the intracranially applied light is indeed neuroprotective to dopaminergic SNc neurons against MPTP insult.78 We are currently using monkeys to undertake our first implants within the third ventricle, a site that lies close to the SNc (5–10 mm) and one that has been used for deep brain stimulation.100 In addition, this site is ideal because Parkinson’s disease is often a bilateral disease,1,2,4,5 and the placement of the optical fiber within the midline third ventricle ensures that the SNc of both sides will be irradiated. We are hopeful that our results in rodents and monkeys will provide the template for future clinical trials using this device in Parkinson’s disease patients.
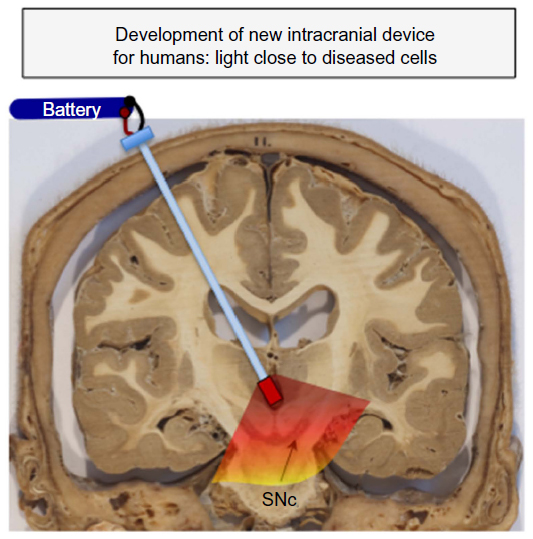 | Figure 3 Development of new intracranial device for humans. |
Although light applied from an external source may not reach the SNc in humans and hence, we argue, have a limited neuroprotective effect, previous studies have reported that light still reaches the cortex and has at least symptomatic effects (Figure 2; see above in same section). If Parkinson’s disease patients were to receive transcranial light therapy from an external source, the abnormal activity of motor cortex101–103 may be restored to normal (eg, by decreasing overactivity and increasing underactivity in the different areas), thereby leading to improvements in movement. Further, if motor cortical activity is returned to normal, and because motor cortex projects heavily to the SNc, the activity of SNc might also return to normal, leading ultimately to a relief of motor signs. In this scenario, light therapy would be symptomatic, but not necessarily neuroprotective; its impact would be on part of the neural circuitry affected by the loss of dopaminergic neurons, rather than on the diseased dopaminergic neurons themselves. Hence, future clinical trials using an external source must carefully consider, perhaps by following patients over a number of years and using functional imaging methods, whether any improvements they observe are purely symptomatic or indeed due to neuroprotection of dopaminergic neurons.
In summary, although there may be symptomatic treatment, it appears unlikely that light therapy, when applied extracranially, can be neuroprotective to the human SNc. If, however, applied intracranially, in regions very close to the SNc, the diseased neurons may receive strong light signal and hence be neuroprotected. These issues can be addressed through carefully designed clinical trials.
What would be the advantages of using light therapy?
There would be several key advantages for the use of light therapy over current treatments for Parkinson’s disease. First and foremost, light therapy has the potential to be neuroprotective. A growing body of basic science evidence indicates that light therapy slows or stops the pathology of the disease (see above; “What is light therapy and how does it offer neuroprotection”), a feat not achieved by dopamine drug therapy, the current mainstay of treatment. There are some substances (eg, coenzyme Q10 and melatonin)104,105 or methods (eg, deep brain stimulation at high frequency)20 that have been shown to be neuroprotective in experimental animals, and light therapy certainly fits into this category. These are promising indications for a neuroprotective function in humans.
Second, light therapy is safe, and there are no reported side effects. Previous studies using external (eg, WARP-LED) or internal (eg, optical fiber device) methods to deliver light therapy at power intensities ranging from ~1–700 mW/cm2 have reported no adverse effect on brain tissue structure and function (810 nm, laser and 670 nm, LED).61,68,69,78,106–108 While there is one report of some neuronal damage and negative behavioral outcomes in mice subjected to exceptionally high power intensity of 750 mW/cm2,106 approximately one hundred times higher than the dose required to elicit a therapeutic response (eg, <10 mW/cm2), in general the impact of light on all body tissues examined thus far has been overwhelmingly positive.61,65–69,78,96,106–108 Hence, these data indicate that when light is applied at therapeutic doses (and even well above these doses) it has little or no adverse effect on body tissues; there appears to be a large safety margin for this treatment. Unlike many of the current therapies used for Parkinson’s disease, particularly after prolonged use, light therapy has been reported to have no major side effects, even at doses well above the therapeutic window.61,68,97
Third, treatment with light therapy would be simple. For effective neuroprotection of the SNc, the patient would require a minimally invasive surgical stereotactic procedure for the insertion of a light-optical device within the brain. This device would be linked to a battery source and pacemaker device (as with patients receiving deep brain stimulation),28 applying the light to the SNc when required. The procedural risks would be comparable to those of single electrode deep brain stimulation.
It should be noted that a potential disadvantage of light therapy is that it may not be effective in treating the nonmotor symptoms of the disease. As discussed, results in animal models indicate that dopaminergic neurons in regions outside of the SNc are less likely to be protected by light treatment after parkinsonian insult. However, these symptoms are minor compared to the striking motor signs of the disease.
In summary, light therapy compares favorably with, and has notable advantages over, the current treatments of Parkinson’s disease. It is fast developing into a treatment option that is not only safe and simple but moreover helps arrest the disease neuropathology.
Conclusions and implications of future therapy
Although very much in its infancy, with the bulk of results still at the basic science “proof of concept” stage, red to infrared light therapy has the potential to develop into a viable treatment option for patients with Parkinson’s disease (and other neurodegenerative diseases). Light therapy would offer patients the advantage of neuroprotection, something that dopamine replacement drug therapy does not do. If light therapy was applied at early stages, for example at first diagnosis, it could potentially slow the progression of the disease by rescuing the critical neurons from damage and death. Consequently, over time, the greater survival of neurons would lessen the clinical signs of tremor, akinesia, and/or rigidity. Light therapy may not only be effective in slowing the progression of the disease, but also in treating the signs. Further, light therapy, because of its lack of side effects, is amenable to use in conjunction with other treatments. For example, patients may have light therapy with a reduced dose of dopamine drug therapy as a first line treatment. This would then prolong the efficacy of the drug therapy and ultimately delay the use of surgery and deep brain stimulation. There is so much to do in the further development of this treatment, but the therapeutic (neuroprotective and symptomatic) possibilities are many and the potential outcomes very exciting.
Acknowledgments
We are forever grateful to Tenix corp, Salteri family, Sir Zelman Cowen Universities Fund, Foundation Philanthropique Edmond J Safra, France Parkinson, Michael J Fox Foundation, Fight for Sight, International Retinal Research Foundation, and the French National Research Agency (ANR Carnot Institute) for funding our work. We thank Sharon Spana, Rat Venceslas, Vincente Di Calogero, Christophe Gaude, Caroline Meunier, and the Leti-DTBS staff for excellent technical assistance for many of the experiments. Many thanks also to Glen Jeffery (University College London) for his critical analysis of our manuscript. We dedicate this work to our dear friend and esteemed colleague, Gary Baker, who passed away during the early stages of the preparation of this review.
Disclosure
The authors report no conflict of interest concerning the use of light therapy. KC, CM, NT, DMJ, JTE, KA, JS, ALB, and JM are members of staff at their respective institutions. DMJ is supported by a National Health and Medical Research Council of Australia (NHMRC) Early Career Fellowship. JS is a director of Clear Sight Clear Mind (CSCM) Pty Ltd. All authors contributed to the writing of the manuscript. The authors report no other conflicts of interest in this work.
References
Bergman H, Deuschl G. Pathophysiology of Parkinson’s disease: from clinical neurology to basic neuroscience and back. Mov Disord. 2002;17(Suppl 3):S28–S40. | |
Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–376. | |
Rinne JO. Nigral degeneration in Parkinson’s disease. Mov Disord. 1993;(8 Suppl 1):S31–S35. | |
Parent A. Carpenter’s Human Neuroanatomy. 9th ed. Baltimore, MD: Williams and Wilkins; 1996. | |
Blandini F, Nappi G, Tassorelli C, Martignoni E. Functional changes of the basal ganglia circuitry in Parkinson’s disease. Prog Neurobiol. 2000;62(1):63–88. | |
Schober A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 2004;318(1):215–224. | |
Blesa J, Phani S, Jackson-Lewis V, Przedborski S. Classic and new animal models of Parkinson’s disease. J Biomed Biotechnol. 2012;2012:845618. | |
Bové J, Perier C. Neurotoxin-based models of Parkinson’s disease. Neuroscience. 2012;211:51–76. | |
Bezard E, Dovero S, Imbert C, Boraud T, Gross CE. Spontaneous long-term compensatory dopaminergic sprouting in MPTP-treated mice. Synapse. 2000;38(3):363–368. | |
Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7(4):306–318. | |
Corti O, Brice A. Mitochondrial quality control turns out to be the principal suspect in parkin and PINK1-related autosomal recessive Parkinson’s disease. Curr Opin Neurobiol. 2013;23(1):100–108. | |
Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34(4):521–533. | |
Ittner LM, Fath T, Ke YD, et al. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc Natl Acad Sci U S A. 2008;105(41):15997–16002. | |
Muqit MM, Abou-Sleiman PM, Saurin AT, et al. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem. 2006;98(1):156–169. | |
Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31(14):3038–3062. | |
Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9(1):13–24. | |
Albin RL, Greenamyre JT. Alternative excitotoxic hypotheses. Neurology. 1992;42(4):733–738. | |
Beal MF, Brouillet E, Jenkins BG, et al. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13(10):4181–4192. | |
Piallat B, Benazzouz A, Benabid AL. Subthalamic nucleus lesion in rats prevents dopaminergic nigral neuron degeneration after striatal 6-OHDA injection: behavioural and immunohistochemical studies. Eur J Neurosci. 1996;8(7):1408–1414. | |
Wallace BA, Ashkan K, Heise CE, et al. Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP-treated monkeys. Brain. 2007;130(Pt 8):2129–2145. | |
Whitton PS. Neuroinflammation and the prospects for anti-inflammatory treatment of Parkinson’s disease. Curr Opin Investig Drugs. 2010;11(7):788–794. | |
McGeer PL, McGeer EG. Glial reactions in Parkinson’s disease. Mov Disord. 2008;23(4):474–483. | |
Jankovic P, Poewe W. Therapies in Parkinson’s disease. Curr Opin Neurol. 2012;25(4):433–447. | |
Schapira AH. Present and future drug treatment for Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2005;76(11):1472–1478. | |
Worth PF. How to treat Parkinson’s disease in 2013. Clin Med. 2013; 13(1):93–96. | |
Hart RG, Pearce LA, Ravina BM, Yaltho TC, Marler JR. Neuroprotection trials in Parkinson’s disease: systematic review. Mov Disord. 2009; 24(5):647–654. | |
Ashkan K, Wallace B, Bell BA, Benabid AL. Deep brain stimulation of the subthalamic nucleus in Parkinson’s disease 1993–2003: where are we 10 years on? Br J Neurosurg. 2004;18(1):19–34. | |
Benabid AL, Chabardes S, Mitrofanis J, Pollak P. Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson’s disease. Lancet Neurol. 2009;8(1):67–81. | |
Charles PD, Gill CE, Davis TL, Konrad PE, Benabid AL. Is deep brain stimulation neuroprotective if applied early in the course of PD? Nat Clin Pract Neurol. 2008;4(8):424–426. | |
Stocchi F, Olanow CW. Obstacles to the development of a neuroprotective therapy for Parkinson’s disease. Mov Disord. 2013;28(1):3–7. | |
Kordower JH, Bjorklund A. Trophic factor gene therapy for Parkinson’s disease. Mov Disord. 2013;28(1):96–109. | |
Maruyama W, Naoi M. “70th Birthday Professor Riederer’’ induction of glial cell line-derived and brain-derived neurotrophic factors by rasagiline and (-)deprenyl: a way to a disease-modifying therapy? J Neural Transm. 2013;120(1):83–89. | |
Arias-Carrión O, Yamada E, Freundlieb N, et al. Neurogenesis in substantia nigra of parkinsonian brains? J Neural Transm Suppl. 2009;(73):279–285. | |
Grazina R, Massano J. Physical exercise and Parkinson’s disease: influence on symptoms, disease course and prevention. Rev Neurosci. 2013;24(2):139–152. | |
Kones R. Parkinson’s disease: mitochondrial molecular pathology, inflammation, statins, and therapeutic neuroprotective nutrition. Nutr Clin Pract. 2010;25(4):371–389. | |
Romeo S, Viaggi C, Di Camillo D, et al. Bright light exposure reduces TH-positive dopamine neurons: implications of light pollution in Parkinson’s disease epidemiology. Sci Rep. 2013;3:1395. | |
Eells JT, Henry MM, Summerfelt P, et al. Therapeutic photobiomodulation for methanol-induced retinal toxicity. Proc Natl Acad Sci U S A. 2003;100(6):3439–3444. | |
Natoli R, Zhu Y, Valter K, Bisti S, Eells J, Stone J. Gene and noncoding RNA regulation underlying photoreceptor protection: microarray study of dietary antioxidant saffron and photobiomodulation in rat retina. Mol Vis. 2010;16:1801–1822. | |
Natoli R, Valter K, Barbosa M, et al. 670 nm photobiomodulation as a novel protection against retinopathy of prematurity: evidence from oxygen induced retinopathy models. PLoS One. 2013;8(8):e72135. | |
Albarracin R, Valter K. 670 nm red light preconditioning supports Müller cell function: evidence from the white light-induced damage model in the rat retina. Photochem Photobiol. 2012;88(6):1418–1427. | |
Begum R, Powner MB, Hudson N, Hogg C, Jeffery G. Treatment with 670 nm light up regulates cytochrome C oxidase expression and reduces inflammation in an age-related macular degeneration model. PloS One. 2013;8(2):e57828. | |
Ando T, Xuan W, Xu T, et al. Comparison of therapeutic effects between pulsed and continuous wave 810-nm wavelength laser irradiation for traumatic brain injury in mice. PLoS One. 2011;6(10):e26212. | |
Oron A, Oron U, Streeter J, et al. Near infrared transcranial laser therapy applied at various modes to mice following traumatic brain injury significantly reduces long-term neurological deficits. J Neurotrauma. 2012;29(2):401–407. | |
Quirk BJ, Torbey M, Buchmann E, Verma S, Whelan HT. Near-infrared photobiomodulation in an animal model of traumatic brain injury: improvements at the behavioral and biochemical levels. Photomed Laser Surg. 2012;30(9):523–529. | |
Lapchak PA, Wei J, Zivin JA. Transcranial infrared laser therapy improves clinical rating scores after embolic strokes in rabbits. Stroke. 2004;35(8):1985–1988. | |
De Taboada L, Ilic S, Leichliter-Martha S, Oron U, Oron A, Streeter J. Transcranial application of low-energy laser irradiation improves neurological deficits in rats following acute stroke. Lasers Surg Med. 2006;38(1):70–73. | |
Oron A, Oron U, Chen J, et al. Low-level laser therapy applied transcranially to rats after induction of stroke significantly reduces long-term neurological deficits. Stroke. 2006;37(10):2620–2624. | |
Moges H, Vasconcelos OM, Campbell WW, et al. Light therapy and supplementary Riboflavin in the SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis (FALS). Lasers Surg Med. 2009; 41(1):52–59. | |
Muili KA, Gopalakrishnan S, Meyer SL, Eells JT, Lyons JA. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by photobiomodulation induced by 670 nm light. PLoS One. 2012;7(1):e30655. | |
Liang HL, Whelan HT, Eells JT, Wong-Riley MT. Near-infrared light via light-emitting diode treatment is therapeutic against rotenone- and 1-methyl-4-phenylpyridinium ion-induced neurotoxicity. Neuroscience. 2008;153(4):963–974. | |
Ying R, Liang HL, Whelan HT, Eells JT, Wong-Riley MT. Pretreatment with near-infrared light via light-emitting diode provides added benefit against rotenone- and MPP+-induced neurotoxicity. Brain Res. 2008;1243:167–173. | |
Whelan H, Desmet K, Buchmann E, et al. Harnessing the cell’s own ability to repair and prevent neurodegenerative disease. SPIE Newsroom. 2008;2008:1–3. | |
Shaw VE, Spana S, Ashkan K, et al. Neuroprotection of midbrain dopaminergic cells in MPTP-treated mice after near-infrared light treatment. J Comp Neurol. 2010;518(1):25–40. | |
Peoples CL, Spana S, Ashkan K, et al. Photobiomodulation enhances nigral dopaminergic cell survival in a chronic MPTP mouse model of Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(5):469–476. | |
Michalikova S, Ennaceur A, van Rensburg R, Chazot PL. Emotional responses and memory performance of middle-aged CD1 mice in a 3D maze: effects of low infrared light. Neurobiol Learn Mem. 2008;89(4):480–488. | |
De Taboada L, Yu J, El-Amouri S, et al. Transcranial laser therapy attenuates amyloid-β peptide neuropathology in amyloid-β protein precursor transgenic mice. J Alzheimers Dis. 2011;23(3):521–535. | |
Grillo SL, Duggett NA, Ennaceur A, Chazot PL. Non-invasive infra-red therapy (1072 nm) reduces β-amyloid protein levels in the brain of an Alzheimer’s disease mouse model, TASTPM. J Photochem Photobiol B. 2013;123:13–22. | |
Barrett DW, Gonzalez-Lima F. Transcranial infrared laser stimulation produces beneficial cognitive and emotional effects in humans. Neuroscience. 2013;230:13–23. | |
Lampl Y, Zivin JA, Fisher M, et al. Infrared laser therapy for ischemic stroke: a new treatment strategy: results of the NeuroThera Effectiveness and Safety Trial-1 (NEST-1). Stroke. 2007;38(6):1843–1849. | |
Lapchak PA. Taking a light approach to treating acute ischemic stroke patients: transcranial near-infrared laser therapy translational science. Ann Med. 2010;42(8):576–586. | |
Naeser MA, Saltmarche A, Krengel MH, Hamblin MR, Knight JA. Improved cognitive function after transcranial, light-emitting diode treatments in chronic, traumatic brain injury: two case reports. Photomed Laser Surg. 2011;29(5):351–358. | |
Schiffer F, Johnston AL, Ravichandran C, et al. Psychological benefits 2 and 4 weeks after a single treatment with near infrared light to the forehead: a pilot study of 10 patients with major depression and anxiety. Behav Brain Funct. 2009;5:46. | |
Merry G, Devenyi R, Dotson R, Markowitz S, Reyes S. Treatment of dry age-related macular degeneration with photobiomodulation. Presented in: Proceedings of the 9th World Association of Laser Therapy Congress; September 28–30, 2012; Gold Coast (Australia). Paper P928C0072. | |
Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. Mitochondrial control of cellular life, stress, and death. Circ Res. 2012;111(9):1198–1207. | |
Desmet KD, Paz DA, Corry JJ, et al. Clinical and experimental applications of NIR-LED photobiomodulation. Photomed Laser Surg. 2006;24(2):121–128. | |
Hamblin MR, Demidova TN. Mechanisms of low level light therapy. Proc SPIE. 2006;6140:614001. | |
Rojas JC, Gonzalez-Lima F. Low-level light therapy of the eye and brain. Eye and Brain. 2011;3:49–67. | |
Chung H, Dai T, Sharma SK, Huang YY, Carroll JD, Hamblin MR. The nuts and bolts of low-level laser (light) therapy. Ann Biomed Eng. 2012;40(2):516–533. | |
Quirk BJ, Whelan HT. Near-infrared irradiation photobiomodulation: the need for basic science. Photomed Laser Surg. 2011;29(3):143–144. | |
Braverman B, McCarthy RJ, Ivankovich AD, Forde DE, Overfield M, Bapna MS. Effect of helium-neon and infrared laser irradiation on wound healing in rabbits. Lasers Surg Med. 1989;9(1):50–58. | |
Stone J, Johnstone DM, Mitrofanis J. The helmet experiment in Parkinson’s disease: an observation of the mechanism of neuroprotection by near infra-red light. Presented in: Proceedings of the 9th World Association of Laser Therapy Congress; September 28–30, 2012; Gold Coast (Australia). Paper P928C0072. | |
Johnstone DM, Moro C, el Massri N, Torres N, Jaeger XD, Reinhart F, Purushothuman S, Benabid AL, Stone J, Mitrofanis J. Indirect application of near infrared light induces neuroprotection in a mouse model of Parkinson’s disease – an abscopal neuroprotective effect. In preparation. 2014. In press. | |
Byrnes KR, Waynant RW, Ilev IK, et al. Light promotes regeneration and functional recovery and alters the immune response after spinal cord injury. Lasers Surg Med. 2005;36(3):171–185. | |
Tuby H, Maltz L, Oron U. Modulations of VEGF and iNOS in the rat heart by low level laser therapy are associated with cardioprotection and enhanced angiogenesis. Lasers Surg Med. 2006;38(7):682–688. | |
Tuby H, Maltz L, Oron U. Induction of autologous mesenchymal stem cells in the bone marrow by low-level laser therapy has profound beneficial effects on the infarcted rat heart. Lasers Surg Med. 2011;43(5):401–409. | |
Hou ST, Jiang SX, Smith RA. Permissive and repulsive cues and signalling pathways of axonal outgrowth and regeneration. Int Rev Cell Mol Biol. 2008;267:125–181. | |
Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925–931. | |
Moro C, el Massri N, Torres N, et al. Photobiomodulation inside the brain: a novel method of applying near-infrared light intracranially and its impact on dopaminergic cell survival in MPTP-treated mice. J Neurosurgery. Epub October 25, 2014. | |
Moro C, Torres N, el Massri N, et al. Photobiomodulation preserves behaviour and midbrain dopaminergic cells from MPTP toxicity: evidence from two mouse strains. BMC Neurosci. 2013;14:40. | |
Purushothuman S, Nandasena C, Johnstone DM, Stone J, Mitrofanis J. The impact of near-infrared light on dopaminergic cell survival in a transgenic mouse model of parkinsonism. Brain Res. 2013;1535:61–70. | |
Trimmer PA, Schwartz KM, Borland MK, De Taboada L, Streeter J, Oron U. Reduced axonal transport in Parkinson’s disease cybrid neurites is restored by light therapy. Mol Neurodegener. 2009;4:26. | |
Ashkan K, Wallace BA, Mitrofanis J, et al. SPECT imaging, immunohistochemical and behavioural correlations in the primate models of Parkinson’s disease. Parkinsonism Relat Disord. 2007;13(5):266–275. | |
Peoples C, Shaw VE, Stone J, Jeffery G, Baker GE, Mitrofanis J. Survival of dopaminergic amacrine cells after near-infrared light treatment in MPTP-treated mice. ISRN Neurol. 2012;2012:850150. | |
Lu J, Jhou TC, Saper CB. Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci. 2006;26(1):193–202. | |
Shaw VE, Peoples C, Spana S, et al. Patterns of cell activity in the subthalamic region associated with the neuroprotective action of near-infrared light treatment in MPTP-treated mice. Parkinson’s Dis. 2012;2012:296875. | |
DeSmet K, Buchmann E, Henry M, et al. Near-infrared light as a possible treatment option for Parkinson’s disease and laser eye injury. Proc SPIE. 2009;7165:7 16503. | |
Quirk BJ, Desmet KD, Henry M, et al. Therapeutic effect of near infrared (NIR) light on Parkinson’s disease models. Front Biosci (Elite Ed). 2012;4:818–823. | |
Lanzafame RJ, Stadler I, Kurtz AF, et al. Reciprocity of exposure time and irradiance on energy density during photoradiation on wound healing in a murine pressure ulcer model. Lasers Surg Med. 2007;39(6):534–542. | |
Quietmind Foundation. Quietmind Foundation clinical Trial. Available from: http://www.quietmindfdn.org/. Marvin Berman. 2013. http://www.youtube.com/watch?v=9X-hjgay7pg. Accessed January16, 2012. | |
Maloney R, Shanks S, Maloney J. The application of low-level laser therapy for the symptomatic care of late stage Parkinson’s disease: a non-controlled, non-randomized study. April 14–15, 2010. Am Soc Laser Med Surg Abs. 2010;185. | |
Li Q, Song L, Guo K, Yu Y, Ma S, Shen L. [The effect of endonasal low energy He-Ne laser treatment of Parkinson’s disease on CCK-8 content in blood]. Chin J Neurol. 1999;32:364. Chinese. | |
Xu C, Lu C, Wang L, Li Q. [The effects of endonasal low energy He-Ne laser therapy on antioxydation of Parkinson’s disease]. Prac J Med Pharm. 2003;11:816–817. Chinese. | |
Zhao G, Guo K, Dan J. [36 case analysis of Parkinson’s disease treated by endonasal low energy He-Ne laser]. Acta Academiae medicinae Qingdao Universitatis. 2003;39:398. Chinese. | |
Burchman MA. Using Photobiomodulation on a severe Parkinson’s patient to enable extractions, root canal treatment and partial denture fabrication. J Laser Dent. 2011;19:297–300. | |
Wong-Riley MT, Liang HL, Eells JT, et al. Photobiomodulation directly benefits primary neurons functionally inactivated by toxins: role of cytochrome c oxidase. J Biol Chem. 2005;280(6):4761–4771. | |
Eells JT, DeSmet K, Kirk DK, et al. Photobiomodulation for the treatment of retinal injury and retinal degenerative diseases. Proceed Light-Activated Tissue Regeneration Therapy Conference. 2008;12:39–51. | |
Zivin JA, Albers GW, Bornstein N, et al; NeuroThera Effectiveness and Safety Trial-2 Investigators. Effectiveness and safety of transcranial laser therapy for acute ischemic stroke. Stroke. 2009;40(4):1359–1364. | |
Jagdeo JR, Adams LE, Brody NI, Siegel DM. Transcranial red and near infrared light transmission in a cadaveric model. PLoS One. 2012;7(10):e47460. | |
Abdo A, Sahin M. NIR Light Penetration Depth in the Rat Peripheral Nerve and Brain Cortex. In: Conference Proceedings of the IEEE. Eng Med Biol Soc. 2007:1723–1725. doi:10.1109/IEMBS.2007. 4352642. | |
Torres N, Chabardes S, Piallat B, Devergnas A, Benabid AL. Body fat and body weight reduction following hypothalamic deep brain stimulation in monkeys: an intraventricular approach. Int J Obes (Lond). 2012;36(12):1537–1544. | |
Samuel M, Ceballos-Baumann AO, Blin J, et al. Evidence for lateral premotor and parietal overactivity in Parkinson’s disease during sequential and bimanual movements. A PET study. Brain. 1997; 120(Pt 6):963–976. | |
Sabatini U, Boulanouar K, Fabre N, et al. Cortical motor reorganization in akinetic patients with Parkinson’s disease: a functional MRI study. Brain. 2000;123(Pt 2):394–403. | |
Haslinger B, Erhard P, Kämpfe N, et al. Event-related functional magnetic resonance imaging in Parkinson’s disease before and after levodopa. Brain. 2001;124(Pt 3):558–570. | |
LeWitt PA. Neuroprotection for Parkinson’s disease. J Neural Transm Suppl. 2006;71:113–122. | |
Ma J, Shaw VE, Mitrofanis J. Does melatonin help save dopaminergic cells in MPTP-treated mice? Parkinsonism Relat Disord. 2009;15(4):307–314. | |
Ilic S, Leichliter S, Streeter J, Oron A, DeTaboada L, Oron U. Effects of power densities, continuous and pulse frequencies, and number of sessions of low-level laser therapy on intact rat brain. Photomed Laser Surg. 2006;24(4):458–466. | |
McCarthy TJ, De Taboada L, Hildebrandt PK, Ziemer EL, Richieri SP, Streeter J. Long-term safety of single and multiple infrared transcranial laser treatments in Sprague-Dawley rats. Photomed Laser Surg. 2010;28(5):663–667. | |
Tata DB, Waynant RW. Laser therapy: a review of its mechanism of action and potential medical applications. Laser Photonics Rev. 2011;5(1):1–12. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.