")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
The Phenomenon of Gene Rearrangement is Frequently Associated with TP53 Mutations and Poor Disease-Free Survival in Hepatocellular Carcinoma
Authors He F, Song K , Guan G , Huo J, Xin Y, Li T, Liu C, Zhu Q, Fan N, Guo Y, Wu L
Received 1 April 2021
Accepted for publication 3 June 2021
Published 21 June 2021 Volume 2021:14 Pages 723—736
DOI https://doi.org/10.2147/PGPM.S313848
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Fu He,1,2 Kangjian Song,1,2 Ge Guan,3 Junyu Huo,1,2 Yang Xin,1 Tianxiang Li,3 Chao Liu,1 Qingwei Zhu,1,2 Ning Fan,1 Yuan Guo,1 Liqun Wu1
1Liver Disease Center, The Affiliated Hospital of Qingdao University, Qingdao, 266003, Shandong, People’s Republic of China; 2Department of Clinical Medicine, Qingdao University, Qingdao, 266071, Shandong, People’s Republic of China; 3Organ Transplant Center, The Affiliated Hospital of Qingdao University, Qingdao, 266003, Shandong, People’s Republic of China
Correspondence: Liqun Wu
Liver Disease Center, The Affiliated Hospital of Qingdao University, No. 59 Haier Road, Qingdao, 266003, Shandong Province, People’s Republic of China
Tel +86 18661809789
Email [email protected]
Purpose: Gene rearrangements (GRs) have been reported to be related to adverse prognosis in some tumours, but the relationship in hepatocellular carcinoma (HCC) remains less studied. The objective of our study was to explore the clinicopathological characteristics and prognosis of HCC patients (HCCs) with GRs (GR-HCCs).
Patients and Methods: This retrospective study included 297 HCCs who underwent hepatectomy and had their tumours sequenced by next-generation sequencing. Categorical variables between groups were compared by the chi-square test. The impact of variables on disease-free survival (DFS) and survival after relapse (SAR) was analysed by the Kaplan–Meier method and Cox regression.
Results: We observed four repetitive GR events in 297 HCCs: BRD9/TERT, ARID2/intergenic, CDKN2A/intergenic and OBSCN truncation. GR-HCCs frequently presented with low tumour differentiation, tumour necrosis, microvascular invasion, elevated AFP and gene mutations (TP53, NTRK3 and BRD9). The 1-, 2-, and 3-year cumulative DFS rates in GR-HCCs were 45.1%, 31.9%, 31.9%, respectively, which were significantly lower than those of GR-negative HCCs (NGR-HCCs) (72.5%, 57.9%, and 49.0%, respectively; P = 0.001). GR was identified as an independent risk factor for inferior DFS in HCCs (HR = 1.980, 95% CI = 1.246– 3.147; P = 0.004). However, there was no significant difference in SAR between GR-HCCs and NGR-HCCs receiving targeted therapy or immunotherapy.
Conclusion: GR is frequently associated with TP53 mutations and significantly affects DFS following radical resection for HCC. We recommend that GR-HCCs should be closely followed up as a high-risk group for postoperative recurrence.
Keywords: hepatocellular carcinoma, gene rearrangement, next-generation sequencing, prognosis, risk factors
Introduction
According to the latest global cancer statistics in 2020, primary liver cancer accounted for 4.7% of all new malignancies worldwide, and its mortality rate (8.3%) ranked third in tumour-specific mortality.1 Hepatocellular carcinoma (HCC) accounts for the majority of primary liver cancer cases, and liver transplantation (LT), liver resection (LR) and local ablation are currently effective treatments for early HCC.2 However, due to the complexity of the genetic background of HCC, the high recurrence rate after surgery is still a challenge for the prognosis of HCC patients (HCCs). Fortunately, with the large-scale adoption of next-generation sequencing (NGS), an increasing number of molecular alterations (TP53, TERT and CTNNB1 mutations, etc) that promote the development of HCC have been revealed.3 In addition, several clinical trials have shown improved overall survival in unresectable patients treated with molecular targeting drugs (sorafenib, lenvatinib, regorafenib, etc) and immune checkpoint inhibitors (nivolumab, pembrolizumab, etc).4 Regrettably, at present, there is still a lack of actionable and characteristic biomarkers for HCC to evaluate therapeutic efficacy and prognosis in clinical practice.
In recent years, however, gene rearrangements (GRs) or fusions have been confirmed as one of the driving factors of malignant tumours.5,6 GR occurs when some gene fragments change their original order of articulation through insertion, deletion, translocation, inversion, etc, and eventually rearrange themselves into a new transcription unit by a DNA repair mechanism.7 GRs can lead to a variety of consequences, including gene activation, silencing, or fusion. Some fusion proteins expressed by rearranged genes have become distinctive targets of treatment, which has led to a remarkable improvement in patient prognosis.8,9 Imatinib, dasatinib and nilotinib have been successively approved by the US Food and Drug Administration (FDA) for the treatment of leukaemia with BCR/ABL rearrangement.10 Crizotinib, ceritinib and alectinib have been approved one after another by the FDA for the treatment of non-small cell lung cancer (NSCLC) with ALK rearrangement.11 The value of GR is exemplified by the clinical benefits obtained by patients, and the approval of the aforementioned drugs has also greatly encouraged researchers to investigate the phenomenon of GR in other malignancies. Some GR events were described by The Cancer Genome Atlas in HCC;12,13 however, there are few studies on the clinical effects of GR on HCCs. Thus, we conducted a retrospective analysis based on data from 297 HCCs who underwent surgery in our hospital to explore the clinicopathological characteristics and prognosis of HCCs with GR (GR-HCCs) and then provide new ideas for the clinical management of HCC with different gene statuses.
Patients and Methods
Enrolled Cases
We collected the specimens and corresponding clinical materials of patients who underwent surgical resection for HCC in the Affiliated Hospital of Qingdao University from April 2016 to October 2020. A total of 297 patients were ultimately enrolled in our research study after excluding patients who had received preoperative treatment and patients with a history of primary malignancy in other tissues or organs. Inflammation grading and fibrosis staging of each specimen were determined by Scheuer’s method, and grades 3–4 or stages 3–4 were considered as advanced stages. Radical (R0) resection refers to the complete removal of the tumour and postoperative microscopic confirmation that the margins are free of tumour cells, no tumour lesions should be found by postoperative imaging examination, and the dynamic detection of tumour markers should indicate that the quantitative level gradually decreased to the normal range within two months after surgery.
NGS
Immunohistochemistry (IHC)
Programmed cell death 1 ligand 1 (PD-L1) IHC staining of all FFPE (4 μm thick) sections was completed using an automatic sample pretreatment instrument (PT link, Dako) and the EnVision™ FLEX Kit (Cat# K800221-2, Dako, DK) on the automated immunostainer Dako Autostainer Link 48 (Agilent Technologies Inc., CA, USA) according to the manufacturer’s instructions. In addition to the primary antibody (FLEX RTU, Dako), other important detection reagents were provided with the kits, including Peroxidase-Blocking Reagent (DM841), Monoclonal Mouse Anti-Human PD-L1 (clone 22C3, Cat# M3653), EnVision FLEX DAB + Chromogen (DM847), and EnVision FLEX Substrate Buffer (DM843). Placental tissue and phosphate buffered saline were used as positive and negative controls, respectively, in each staining run. All sections were interpreted by 2 trained diagnostic pathologists, and the expression of HCC PD-L1 protein was interpreted by the combined positive score (CPS), which is the number of PD-L1 stained cells (tumour cells, lymphocytes, and macrophages) divided by the total number of viable tumour cells and multiplied by 100.14 According to previous studies, the positive expression of PD-L1 was defined as CPS≥1, while CPS≥20 was defined as high expression.14,15
Systemic Treatment After Recurrence
Sorafenib (400 mg twice daily, Bayer HealthCare Pharmaceuticals Inc, Germany) was applied to patients with well-preserved liver function (Child-Pugh A) and with tumour staging (BCLC B-C), as well as to those who were unsuitable for local therapies. Patients who were intolerant to sorafenib and had positive expression of PD-L 1 protein or TMB >6 Muts/Mb (the median of our data) were treated with camrelizumab (an anti-PD-1 agent, Hengrui Pharmaceuticals Inc, China) at a dose of 200 mg every 3 weeks.
Follow-Up
All HCCs were followed up once a month in the first 3 months of the first year after surgery and then every 3 months until 2 years after surgery and every 6 months thereafter. All enrolled populations were followed up until 30 November 2020 or death. The follow-up items included laboratory tests (alpha-fetoprotein (AFP) and liver function tests) and imaging evaluations (upper abdominal ultrasonography or computed tomography (CT)). If HCC recurrence or metastasis was suspected, one or two of the following examination methods were selected for confirmation: enhanced CT, magnetic resonance imaging (MRI), Gd-EOB-DTPA-enhanced MRI, bone scan or positron emission tomography/CT (PET/CT). In addition, if the imaging manifestation of the lesion was atypical, ultrasound-guided biopsy might be required to further clarify the diagnosis.
The date of HCC recurrence or metastasis was the date of diagnosis by imaging. Disease-free survival (DFS) and survival after relapse (SAR) were calculated from the date of specimen acquisition and the date of recurrence or metastasis, respectively. The follow-up data of all patients were obtained through the HIS system of our hospital and telephone calls.
Statistical Analysis
TMB and mutation number were nonnormally distributed and are expressed as the median (interquartile range (IQR)); their differences between groups were compared by the Mann-Whitney U-test. Categorical variables between groups were compared by the Pearson’s chi-square test. DFS and SAR were analysed by the Kaplan–Meier method and compared by the Log rank test. Based on factors with a P value < 0.1 in univariate analysis, the independent risk factors for postoperative DFS in HCC were determined by a Cox proportional hazards regression model. Receiver operating characteristic (ROC) curves and the areas under the curve (AUCs) were calculated to evaluate the prognostic abilities of the risk factors. P values <0.05 (two-tailed) were considered to be statistically significant. The GR events observed in this study were demonstrated by a cluster heatmap (produced by R software 3.6.1). All data were statistically analysed by SPSS software (version 24.0, IBM Corp, NY, USA), Kaplan–Meier curves and ROC curves were drawn with GraphPad Prism software (version 8.3, San Diego, CA, USA).
Results
Baseline Characteristics
A total of 297 postoperative patients were included in this study, including 60 (20.2%) GR-HCCs and 237 (79.8%) GR-negative HCCs (NGR-HCCs). In this study, there were 253 males (85.2%) and 44 females (14.8%), with an average age of 56.3 ± 9.8 years. Compared with NGR-HCCs, GR-HCCs had higher levels of AFP (P = 0.041). Tumours in the GR-HCCs were less differentiated (P = 0.026) and more frequently accompanied by necrosis (P = 0.005) and microvascular invasion (MVI) (P = 0.023).
Overall, the PD-L1 positivity rate in this study was 47.5% (141/297). There were 156 patients (52.5%) with CPS<1, 117 patients (39.4%) with CPS 1–19, and 24 patients (8.1%) with CPS≥20. Nevertheless, the expression of PD-L1 protein showed no significant difference between GR-HCCs and NGR-HCCs. The detailed clinicopathological features of GR-HCCs and NGR-HCCs are shown in Table 1.
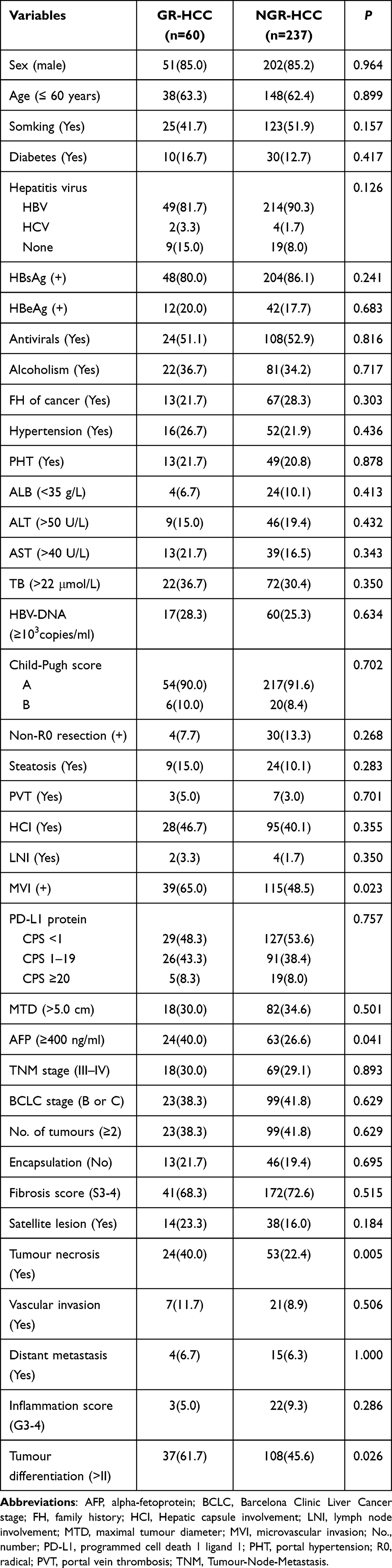 |
Table 1 Demographics and Clinical Characteristics of GR-HCCs and NGR-HCCs [n (%)] |
Profile of Gene Variations
A total of 81 GR events involving 115 genes were observed in 297 HCC samples. Repetitive GR events were defined as GR events detected in at least 2 samples where the breakpoints of genes from different samples were in the same region, such as promoter, exon, intron or intergenic regions, and we found 4 repetitive GR events in this study: BRD9/TERT (NC_000005.9:g.892748_1295893dup), CDKN2A/intergenic (NC_000001.10:g.90508893_90508894ins[NC_000009.11:g.21967750_21974544inv]), OBSCN truncation (NC_000001.10:g.228538022_232211839del), and ARID2/intergenic (NC_000012.11:g.46123275_46124923del). The differences in the above four repetitive events between GR-HCCs and NGR-HCCs were statistically significant (P < 0.05). In addition, genes that were present in at least 3 GR events were annotated as the most frequent genes involved in rearrangement events, including NTRK3 (n = 6), TERT (n = 4), CDKN2A (n = 4), CCND1 (n = 3), and BRD9 (n = 3). The detailed GR events observed in this study are presented in Figure 1.
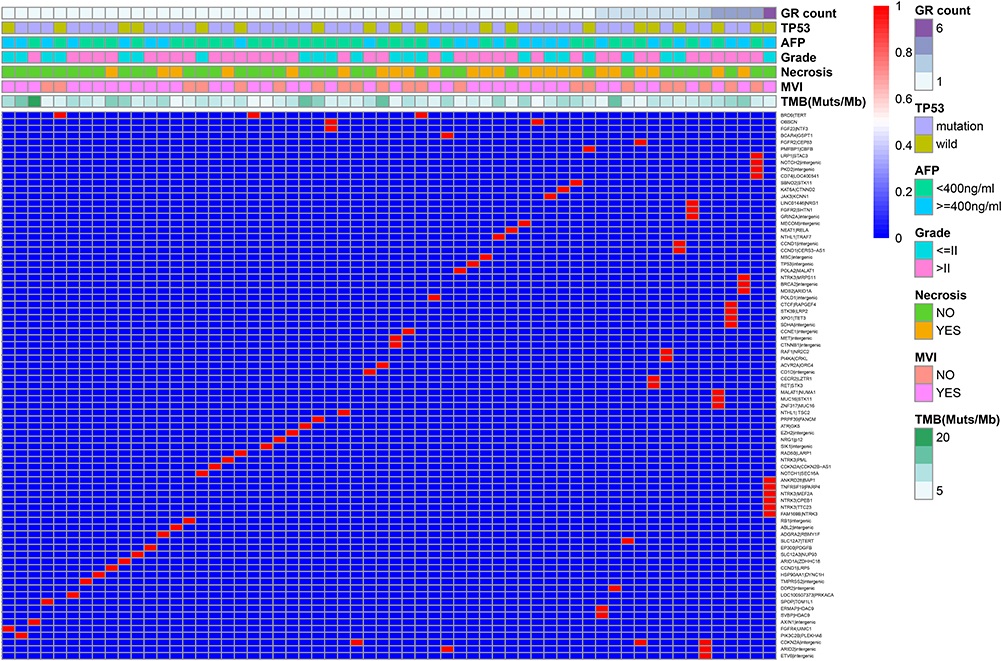 |
Figure 1 Detailed GR events observed in this research. The rows and columns represent gene rearrangement events and samples, respectively. The red mark in each column of the figure represents the rearrangement event of the sample. Clinicopathological information corresponding to each sample is displayed at the top of the figure, including the number of rearrangement events (GR count), TP53 mutation, AFP value, tumour differentiation (grade), tumour necrosis, microvascular invasion (MVI) and tumour mutation burden (TMB). The meaning of the various colours is shown on the far right side of the diagram. |
Furthermore, the top ten genes with the highest mutation incidences in this study were TP53 (163/297, 54.9%), TERT (127/297, 42.8%), CTNNB1 (65/297, 21.9%), AXIN1 (46/297, 15.5%), TSC2 (43/297, 14.5%), LRP1B (41/297, 13.8%), RB1 (38/297, 12.8%), SPTA1 (35/297, 11.8%), MUC16 (32/297, 10.8%), CCND1 (28/297, 9.4%), CDKN2A (28/297, 9.4%), and FAT3 (28/297, 9.4%). The mutation frequencies of TP53, NTRK3 and BRD9 in GR-HCCs were 66.7% (40/60), 8.3% (5/60), and 5.0% (3/60), respectively, which were significantly higher than those in NGR-HCCs (51.9%, 2.1%, and 0%, respectively) (P < 0.05). Details of TP53 mutations are summarized in Supplementary Table 1. In addition, we found that CCND1 and FGF19 often coamplified on chromosome 11q13.3 (χ2=228.6, P = 0.000). The relationships between GR events and the above high-frequency variant genes are shown in Table 2.
 |
Table 2 The Distribution of Genetic Variants in GR-HCCs and NGR-HCCs [n (%)] |
Prognosis of GR-HCCs and NGR-HCCs
The median follow-up time for this group of patients was 25.5 months (range, 6.1–55.8 months). Excluding patients with nonradical resection (34 cases) and patients with less than 6 months of follow-up (19 cases), a total of 244 patients (48 GR-HCCs and 196 NGR-HCCs) were finally included in the survival analysis.
The cumulative DFS rates of GR-HCCs at 1, 2, and 3 years were 45.1%, 31.9%, and 31.9%, respectively, which were significantly lower than those of NGR-HCCs (72.5%, 57.9%, and 49.0%, respectively) (P = 0.001). The probability of DFS in the NGR-HCC group dropped abruptly to zero at the end because one patient with the longest DFS (43.6 months) in the NGR-HCC group had pulmonary metastasis. Overall, the median DFS (mDFS) of GR-HCCs was significantly lower than that of NGR-HCCs (10.5 months vs 31.9 months, P = 0.001) (Figure 2).
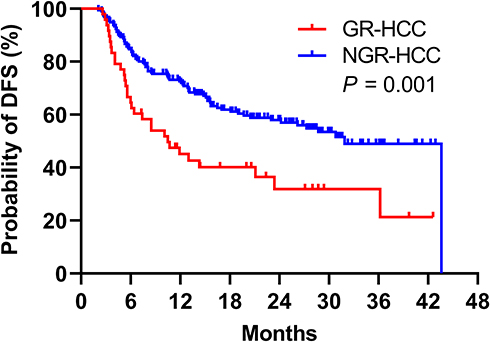 |
Figure 2 Kaplan–Meier plot for postoperative DFS of HCC. GR-HCCs showed significantly lower mDFS than NGR-HCCs (10.5 months vs 31.9 months, P = 0.001). Abbreviations: GR-HCCs, HCC patients with gene rearrangement; NGR-HCCs, HCC patients with gene rearrangement-negative; DFS, disease-free survival. |
Predictive Factors for DFS
Univariate analysis revealed that the following factors were associated with short DFS after R0 resection in HCCs: (a) clinical features, including Child-Pugh score and hypertension; (b) GR; and (c) tumour features, including number of lesions, maximum tumour diameter (MTD), necrosis, AFP, MVI, vascular invasion, hepatic capsule involvement (HCI), satellite lesions, Edmondson grade, Barcelona Clinic Liver Cancer (BCLC) stage, Tumour-Node-Metastasis (TNM) stage, and PD-L1 expression. Multivariate analysis indicated that AFP ≥ 400 ng/ml (P = 0.049) and GR (P = 0.004) were independent risk factors for adverse DFS after R0 resection for HCC (Table 3).
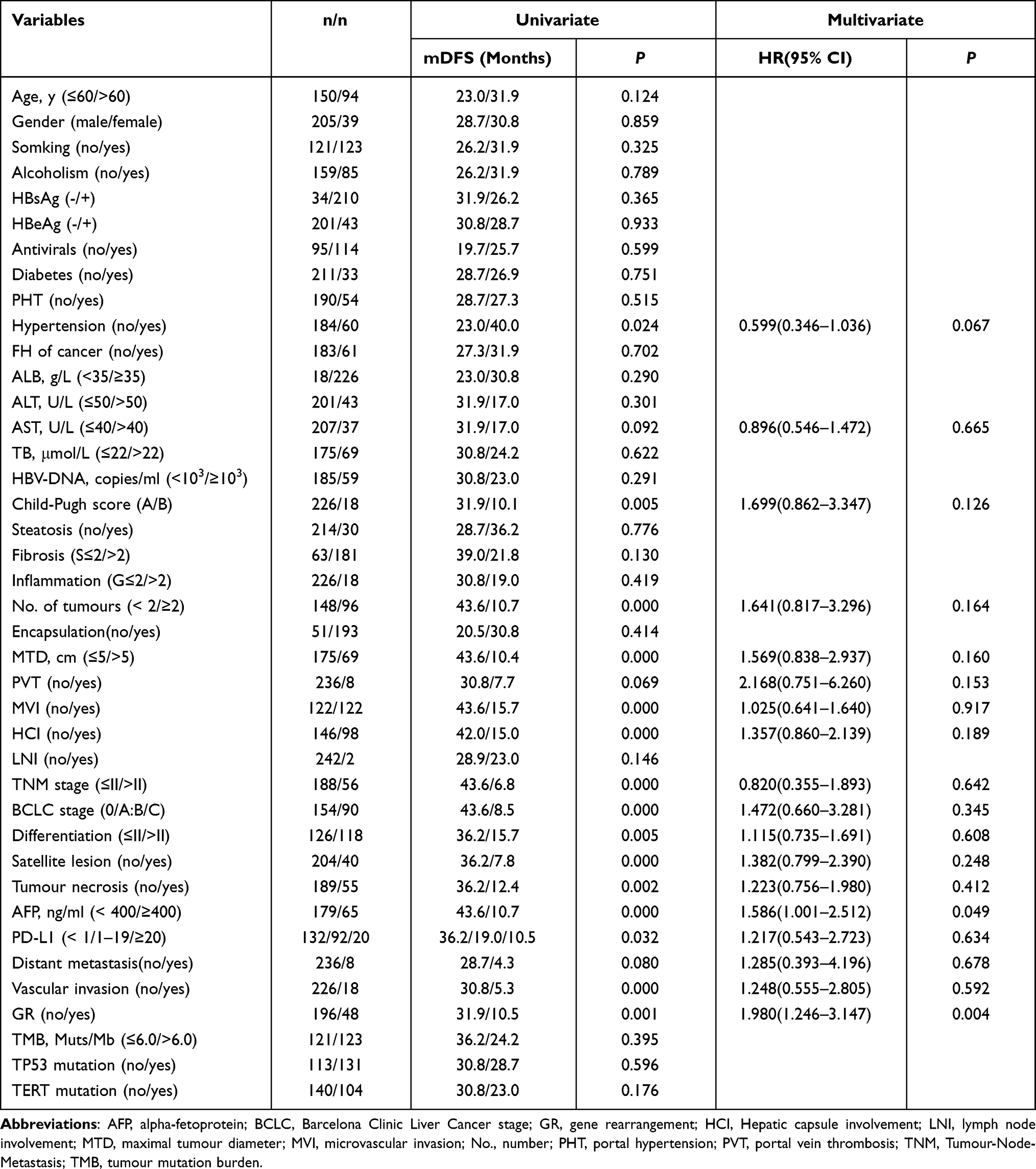 |
Table 3 Univariate and Multivariate Analysis of DFS Predictors in HCCs (n=244) |
For GR-HCCs, albumin (ALB) < 35 g/L, AFP ≥ 400 ng/ml, MTD > 5.0 cm, multiple tumours, tumour differentiation (III–IV), vascular invasion, MVI, portal vein thrombosis (PVT), satellite lesions, distant metastasis, BCLC stage (B/C), TNM stage (>II), PD-L1 expression, and CCND1/FGF19 coamplification were important factors leading to inferior DFS after R0 resection (P < 0.05); HCI (P=0.058) and MUC16 mutations (P=0.054) were also of marginal significance (as shown in Table 4). The survival curves for the abovementioned factors are displayed in Figure 3.
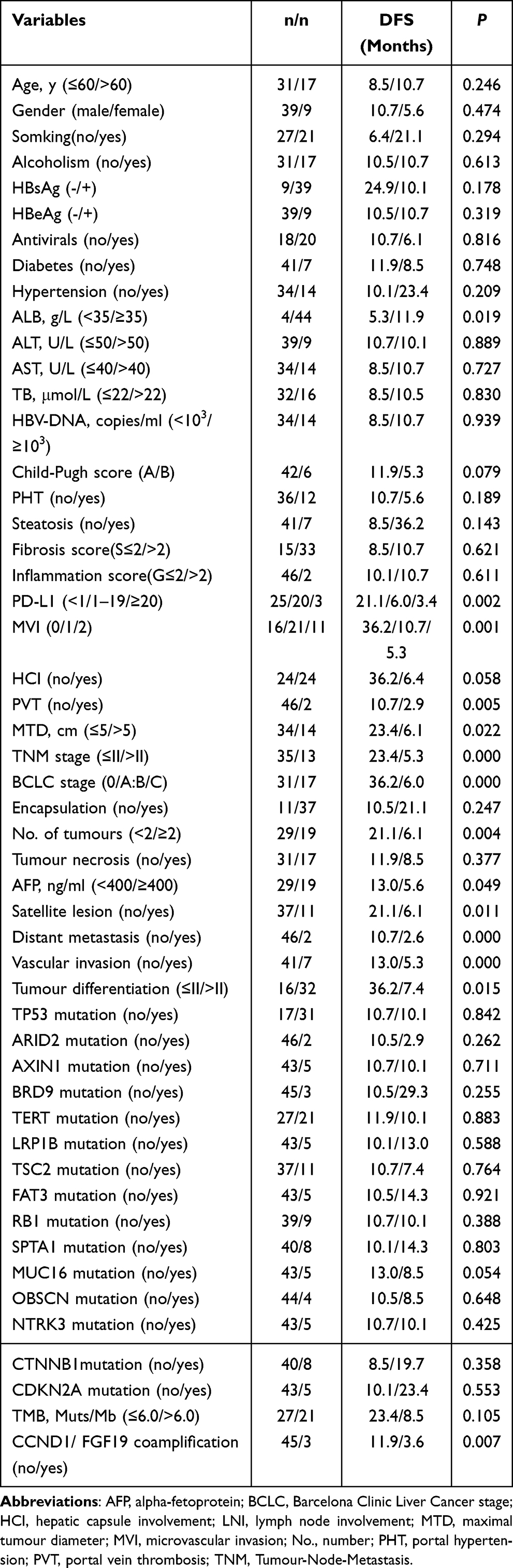 |
Table 4 Factors Affecting DFS After Radical Treatment in GR-HCCs |
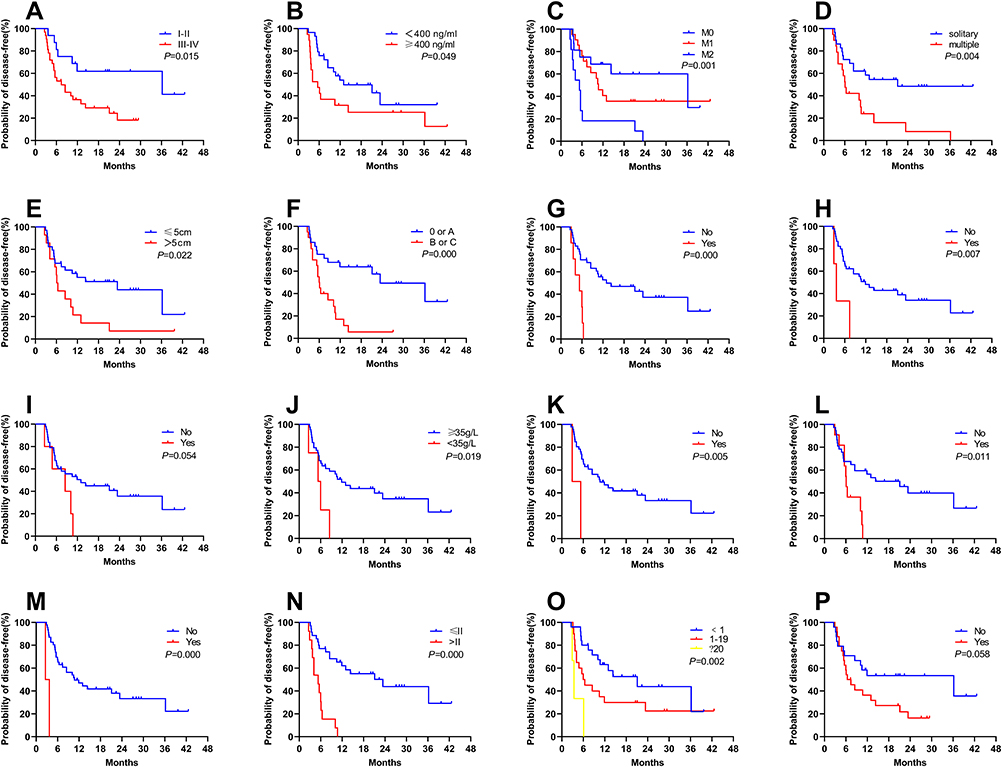 |
Figure 3 Survival curves of factors influencing DFS in GR-HCCs. (A) Tumour differentiation (grade III–IV), (B) serum AFP values ≥ 400ng/ml, (C) microvascular invasion, (D) multiple tumours, (E) maximal tumour diameter > 5.0cm, (F) BCLC stage (B or C), (G) Vascular invasion, (H) CCND1/FGF19 coamplification, (I) MUC16 mutation, (J) ALB < 35g/L, (K) portal vein thrombosis, (L) satellite lesion, (M) distant metastasis, (N) TNM stage (>II), (O) PD-L1 expression, and (P) hepatic capsule involvement were important factors leading to inferior DFS in GR-HCCs. |
The Prognostic Performance of the Predictive Factors for DFS
The predictive power of the above factors for predicting recurrence was compared by ROC curve analysis. The AUCs of GR and AFP for predicting postoperative recurrence of HCC were 0.572 (95% CI: 0.500–0.645) and 0.598 (95% CI: 0.526–0.670), respectively (Figure 4A). Nevertheless, the AUCs of MTD, multiple tumours, BCLC stage (B/C), and TNM stage (>II) for predicting postoperative recurrence of GR-HCC were 0.680 (95% CI: 0.530–0.831), 0.715 (95% CI: 0.568–0.863), 0.729 (95% CI: 0.586–0.871), and 0.710 (95% CI: 0.567–0.852), respectively (P < 0.05) (Figure 4B). The AUCs of all predictors are listed in Table 5.
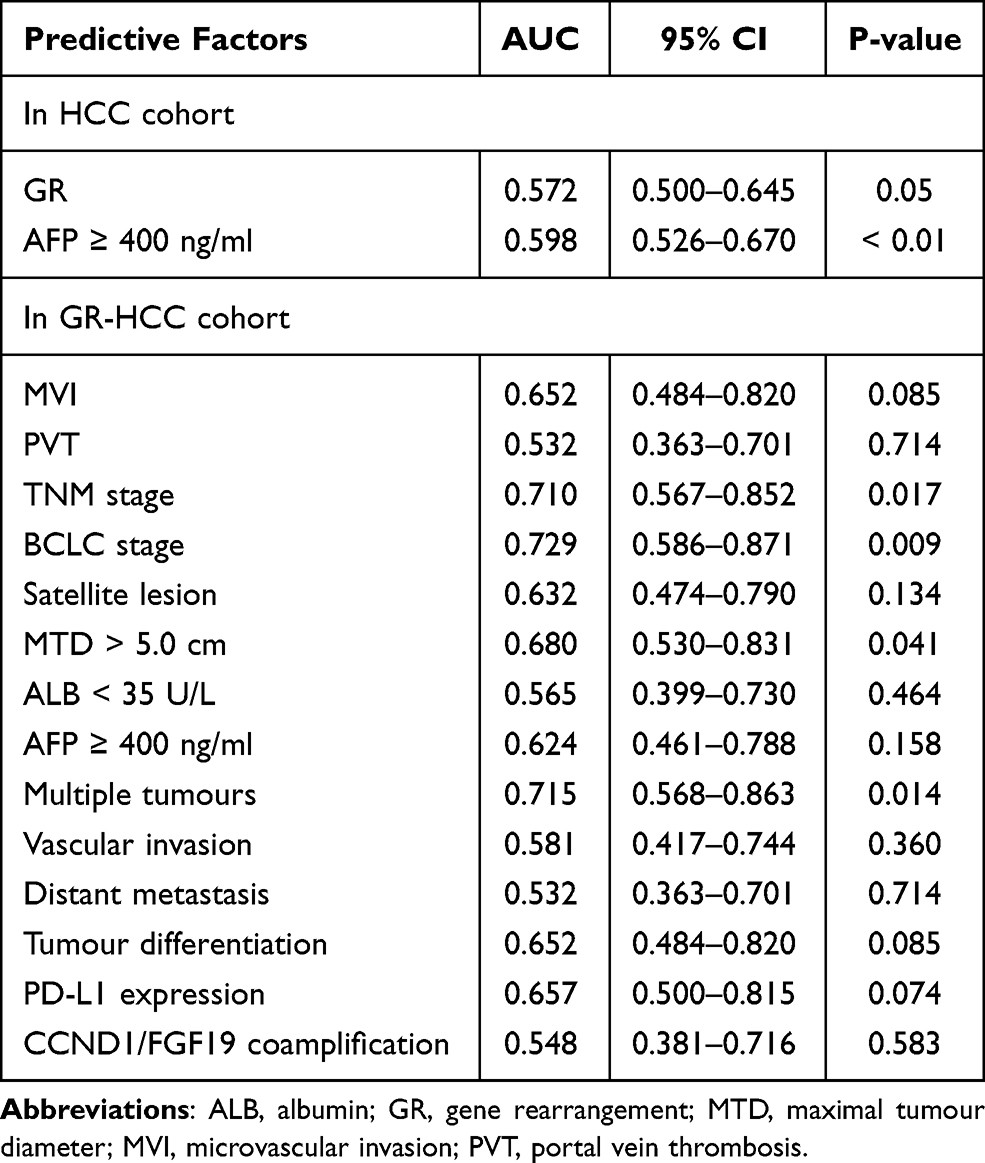 |
Table 5 Prognostic Performance of Predictors of DFS |
 |
Figure 4 Receiver operating characteristic curves of factors predicting DFS for HCC (A) and GR-HCC (B). |
Therapies and Survival After Recurrence
A total of 113 patients (46.3%, 113/244) presented with recurrence during our follow-up period, including 31 GR-HCCs and 82 NGR-HCCs. The median SAR (mSAR) of 113 patients with recurrent HCC (r-HCCs) was 32.4 months (95% CI: 18.42–46.4 months). There was no statistically significant difference in the mSAR between GR-HCCs (not reached) and NGR-HCCs (32.4 months, 95% CI: 21.48–43.32 months) (χ2 = 0.58, P = 0.4).
Among the 113 r-HCCs, 38 patients (7 GR-HCCs and 31 NGR-HCCs) received targeted therapy, and the cumulative SAR rates appeared to be higher in r-GR-HCCs at 1 and 2 years (75% and 75%, respectively) than in r-NGR-HCCs (69% and 56%, respectively), but the difference was not statistically significant (P = 0.284) (Figure 5A). In addition, 17 r-HCC patients (4 GR-HCCs and 13 NGR-HCCs) in this study underwent immunotherapy, and r-GR-HCCs showed slightly higher cumulative SAR rates at 1 and 2 years (100% and 100%, respectively) than r-NGR-HCCs (92% and 78%, respectively), but the difference was not statistically significant (P = 0.464) (Figure 5B).
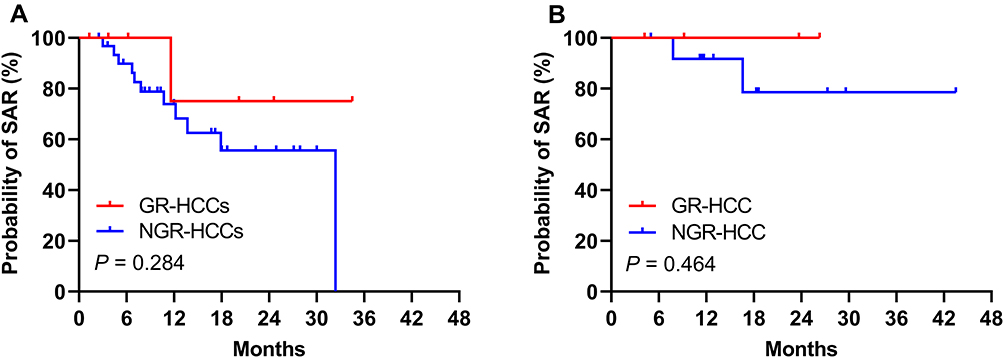 |
Figure 5 Kaplan–Meier plot for SAR of recurrent HCC patients according to different treatments. (A) Targeted therapy, (B) immunotherapy. |
Discussion
Initially, the phenomenon of GR was mainly observed in haematologic disorders, such as leukaemia16 and lymphoma.17 With the continuous maturity of sequencing technology, many studies found that GRs were also present in a variety of solid tumours, such as the RET/CCDC6 rearrangement in thyroid cancer,8 the TMPRSS2/ERG rearrangement in prostate cancer,9 the BCOR/CCNB3 rearrangement in osteosarcoma,18 ALK rearrangements in renal cell carcinoma,19 and the EML4/ALK rearrangement in NSCLC.20 Most of these rearranged genes played a carcinogenic role. GRs also exist in malignant liver tumours. FGFR2 fusion or rearrangement was found in approximately 10% to 16% of intrahepatic cholangiocarcinoma patients, and the FGFR kinase inhibitor pemigatinib was approved by the FDA for the clinical treatment of the aforementioned patients in April 2020.21 The DNAJB1-PRKCA rearrangement has also been frequently reported in fibroblastic HCC;22 however, the specificity of GR in ordinary HCC remains unclear.12,23
Based on whole-genome sequencing, Fernandez-Banet et al observed the first recurrent rearrangement event (ABCB11/LRP2) in 88 HCC cases and found that similar events frequently induced the aberrant overexpression of genes on the 3ʹ side of the 3ʹ breakpoint in DNA.12 However, Zhu et al identified 43 recurrent rearrangement events in HCC from their own database and four public RNA sequencing (RNA-seq) databases via STAR software, three (HLA-DPB2-HLA-DRB1, CDH23-HLA-DPB1, and C15orf57-CBX3) of which were interpreted as disease-related.13 Due to the limited sample size and the use of different analysis strategies, the above events were not identified in our study. However, some of the rearrangement events identified in our study may have an important role in HCC. We identified 4 repetitive GR events (BRD9/TERT, ARID2/intergenic, CDKN2A/intergenic and OBSCN truncation) in 60 GR-HCCs. TERT activation is necessary to maintain the unlimited proliferative capacity of tumour cells. Bromodomain-containing protein 9 (BRD9) has been shown to promote the growth and metastasis of HCC cells by activating the TUFT1/AKT pathway.24 We found that the breakpoint of BRD 9 was located in the 3ʹ UTR, the breakpoint of TERT was located in its upstream promoter sequence, and this variant retained the complete coding region of TERT. It has been reported in the literature that the expression level of TERT mRNA in liposarcoma samples with other TERT rearrangements (such as TRIO-TERT) is more than 100 times higher than that in samples without TERT rearrangements,25 and the above study indicated that the BRD9/TERT rearrangement may lead to sustained high expression of TERT mediated by the BRD9 promoter. Cyclin-dependent kinase inhibitor 2A (CDKN2A) encodes two cell cycle inhibitory proteins, p16INK4a and p14ARF. CDKN2A deficiency leads to the inactivation of Rb and p53 function, which in turn leads to uncontrolled cell proliferation.26 We found that the breakpoint in the first intron of CDKN2A resulted in abnormal mRNA splicing, abnormalities or functional deletion of the p16INK4a protein. AT-rich interaction domain 2 (ARID2) encodes a protein that is a member of the DNA-binding protein family, a subunit of the PBAF (SWI/SNF-B) chromosome remodelling complex, which maintains the stability of PBAF and is involved in transcriptional regulation and chromatin structural modifications.27 We observed that the ARID2/intergenic rearrangement resulted in the deletion of the promoter region, transcription start site and exons 1–2 of ARID2, which might lead to abnormal ARID2 protein function. Moreno et al indicated through functional experiments that ARID2 deficiency affected DNA repair and enhanced the sensitivity of cells to DNA-damaging agents.28 The truncation of OBSCN resulted in the deletion of amino acid residues (6106–7968) from obscurin, and the deletion region contained part of the Ig-like structural domain, protein kinase structural domain and fibronectin type III structural domain. Obscurin is a protein (720 kDa) encoded by OBSN, which belongs to the family of giant sarcomeric signalling proteins. Knockdown of OBSCN significantly affects the growth and biological properties of epithelial cells, such as cell adhesion and intercellular communication, leading to increased cellular carcinogenesis.29 Thus, BRD9/TERT, ARID2/intergenic, CDKN2A/intergenic and OBSCN truncation may be involved in tumourigenesis, and further validation is needed in HCC.
Although the genetic characteristics of GRs in HCC have been described in previous studies, reports on the clinical impact of GRs on HCC are still scarce. Therefore, we performed this research and found that GR-HCCs had lower tumour differentiation (P = 0.026), higher serum AFP levels (P = 0.041), and higher incidences of tumour necrosis (P = 0.005) and MVI (P = 0.023) than NGR-HCCs. Previous studies have found that GRs can lead to gene fusions and the aberrant expression of some oncogenes or tumour suppressor genes7,13 and then induce the oncogenic activation of the WNT,23 RAS/MAPK and PI3K/AKT signalling pathways,30 resulting in the low differentiation and rapid proliferation of tumours. The rapid proliferation of tumours often causes relative hypoxia, microangiogenesis and vascular invasion around the tumour. Once the tumour proliferates faster than microangiogenesis, tumour necrosis will occur due to relative ischaemia.
PD-L1, a transmembrane protein expressed on the membranes of immune cells and tumour cells, can inhibit immune cells and promote the development of tumours by binding to the PD-1 receptor on the surface of immune cells. The combination of atezolizumab (an immunosuppressive agent targeting PD-L1) and bevacizumab was approved by the FDA for first-line treatment in patients with unresectable HCC in May 2020. Therefore, confirmation of PD-L1 expression is potentially valuable for HCC. To date, the FDA has approved four PD-L1 assays (22C3, 28–8, SP142 and SP263) as companion or complementary diagnostics of cancer immunotherapy. Shi et al found that three assays (22C3, 28–8 and SP263) were highly consistent in terms of PD-L1 scoring in HCC.15 This study used the 22C3 assay to assess PD-L1 expression, and the positive rate of PD-L1 expression was 47.5%, which was higher than the results of Huang et al (19%, 78/411).31 A meta-analysis revealed that the expression of PD-L1 in HCC was significantly correlated with low histological grade and poor survival.32 Although this study found that GR-HCCs often presented with poorly differentiated tumours, there was no significant difference in the expression of PD-L1 between GR-HCCs and NGR-HCCs. These differences may be mainly attributed to different threshold settings and whether the positive immune cells expressing PD-L1 in tumour areas are calculated. Notably, it has been reported that pathologists are highly inconsistent when evaluating the expression of PD-L1 at a lower cut-off.15 Therefore, it is suggested that trained pathologists are used and that the samples are deglycosylated before staining to estimate the expression of PD-L1 protein more accurately.
In addition, we found that GR-HCCs were frequently accompanied by TP53 mutations. The predominant type of TP53 mutation in this study was SNV, with C:G>T:A and G:C>T:A transitions being the most common forms. 52.8% of TP53 mutations showed loss of function (LOF), 22.6% of TP53 mutations showed gain of function (GOF), 22.1% of TP53 mutations were of unknown significance, and 2.5% of TP53 mutations were nonsense (see Supplementary Table 1). The specific mechanism of GRs combined with TP53 mutation in HCC is still unclear, but this phenomenon also exists in other tumours, such as leukaemia,33 NSCLC,34 and paediatric medulloblastoma.35 We assume that this may be due to GRs causing mutations in the tumour suppressor gene TP53 or TP53 mutations leading to increased chromosomal instability, which in turn creates GRs.35 Although TP53 mutations have been reported to be associated with poor prognosis in HCC in previous studies, our study failed to confirm this finding. This may be related to the fact that we identified TP53 mutations only at the DNA level rather than based on the expression or function of the p53 protein. Because some genetic mutations (eg, MDM2, MDM4, PPM1D, or CDKN2A) other than TP53 mutations can also inactivate the p53 protein in tumours, resulting in wild-type TP53 tumours exhibiting the same malignancy as TP53-mutated tumours.36 This is one of the important reasons for the inconsistency between TP53 mutation status and clinical outcome in many studies. Moreover, several studies have reported that concurrent TP53 mutations in ALK-rearranged NSCLC predict little benefit from systemic therapy.37 Unfortunately, we were unable to validate this phenomenon in GR-HCC due to the small sample size. We also found that GR-HCCs were frequently accompanied by NTRK3 and BRD9 mutations, however, only a small number of cases with NTRK3 (n=7) and BRD9 (n=3) mutations were observed, and this result remains to be further validated.
As previously mentioned, HCC patients have high recurrence rates after R0 treatment. Although a number of previous studies revealed some risk factors associated with postoperative recurrence,38–40 we found that GR-HCCs showed significantly lower DFS than NGR-HCCs (10.5 months vs 31.9 months, P = 0.001) and identified that GR (P = 0.004) and AFP (P = 0.049) were independent risk factors for poor DFS after R0 resection for HCC. We further revealed that MTD, multiple tumours, vascular invasion, satellite lesions, distant metastasis, PVT, ALB, AFP, BCLC stage (B/C), TNM stage (>II), tumour differentiation (grade III/IV), MVI, PD-L1 protein expression, and FGF19/CCND1 coamplification were important risk factors for adverse DFS after R0 resection in GR-HCCs (P < 0.05). GRs reflect the high instability of tumour genomes,34,41 which could give rise to tumourigenesis by generating gene fusions and causing abnormal gene expression. CCND1 amplification is a positive regulatory element in the G1 to S phase of the cell cycle and usually acts synergistically with other oncogenes or suppressor genes to induce tumour development.42 Our data revealed a significant correlation between CCND1 amplification and FGF19 amplification (P=0.000). Fibroblast growth factor 19 (FGF19) is an oncogene that can play a driving role in carcinogenesis through its receptor (FGFR4), and its amplification has been reported to be independently associated with poor survival and tumour recurrence in HCCs.43 Sawey et al confirmed that co-overexpression of CCND1 and FGF19 was more tumourigenic than single gene overexpression.44 Therefore, multiple mutations with synergistic effects rather than any single dominant mutation are more likely to be a potential risk for postoperative recurrence in GR-HCCs. Meanwhile, we recommend that patients with gene variations, especially those with GRs, should be closely followed up as a high-risk group for postoperative recurrence.
Although GR-HCCs exhibited poorer DFS than NGR-HCCs, no significant difference in SAR was observed between GR-HCCs and NGR-HCCs. We considered that this might be related to the regular postoperative follow-up of patients, early detection of recurrence, and timely access to individualized treatment. Similar to the findings of Tan et al,45 we did not observe a significant improvement in SAR of GR-HCCs in r-HCCs treated with immunotherapy. This may be related to the LOF in TP53, which promotes the recruitment of suppressor regulatory T (Treg) cells in tumours and attenuates CD4+ T helper 1 (Th1) and CD8+ T cell responses in vivo.46 In addition, it has been reported that some epithelial malignancies with abnormal activation of tyrosine kinase caused by GR respond well to targeted drugs, such as neurotrophic receptor tyrosine kinase (NTRK) rearrangement.20,30,47 However, our study found that there was no significant difference between GR-HCCs and NGR-HCCs in response to targeted therapy. We speculated that this might be related to the type of tumour, specific mutation targets, the small size of our sample and the duration of follow-up.
The limitations of this study lie in its single-centre design and retrospective nature. Our study only included patients from Asian, and detailed genetic comparisons were not performed between populations with different aetiologies and ethnicities, so there might be potential bias in our study with respect to patient populations. Additional experiments are needed to reveal the functional significance of the GR events identified from our research. Large-scale clinical trials are needed in GR-HCCs to screen populations that would benefit from targeted therapies.
Conclusions
Our work identified four repetitive GR events (BRD9/TERT, ARID2/intergenic, CDKN2A/intergenic and OBSCN truncation) in HCC. We found that GR-HCCs were frequently accompanied by TP53 mutations, NTRK3 mutations, and BRD9 mutations and had lower tumour differentiation, higher serum AFP levels and higher incidences of tumour necrosis and MVI than NGR-HCCs. In addition, GR was identified as an independent risk factor for DFS after R0 resection for HCC. Therefore, we recommend that GR-HCCs should be closely followed up as a high-risk group for postoperative recurrence. In addition, our study revealed that there was no significant difference between GR-HCCs and NGR-HCCs in the response to targeted therapy and immunotherapy. Our research provides new information on the phenomenon of GR in HCC. These findings may expand our understanding of rearrangement events in HCC and contribute to the clinical management of HCC.
Abbreviations
AFP, alpha-fetoprotein; BCLC, Barcelona Clinic Liver Cancer stage; CAP, the College of American Pathologists; CLIA, the Clinical Laboratory Improvement Amendments; DFS, disease-free survival; FDA, the US Food and Drug Administration; FH, family history; GRs, gene rearrangements; GR-HCCs, HCC patients with gene rearrangement; HBsAg, hepatitis B virus surface antigen; HCC, hepatocellular carcinoma; HCCs, hepatocellular carcinoma patients; r-HCCs, recurrent hepatocellular carcinoma patients; HCI, Hepatic capsule involvement; IQR, inter-quartile range; LNI, lymph node involvement; MDT, multidisciplinary team; MTD, maximal tumour diameter; MVI, microvascular invasion; NGR-HCCs, HCC patients with gene rearrangement-negative; NGS, next-generation sequencing; No., number; NSCLC, non-small cell lung cancer; PD-L1, programmed cell death 1 ligand 1; PHT, portal hypertension; PVT, portal vein thrombosis; SAR, survival after relapse; SLT, salvage liver transplantation; TMB, tumour mutation burden; TNM, Tumour-Node-Metastasis.
Ethics Approval and Informed Consent
This study was conducted in accordance with the criteria of the Declaration of Helsinki, and was sanctioned by the Research Ethics Committee of the Affiliated Hospital of Qingdao University (approval number: QDFYKYLLL-20,161,212). The informed consent was obtained from all patients.
Acknowledgments
Thanks to OrigiMed for providing guidance on NGS technology in this study.
Funding
There is no funding to report.
Disclosure
The authors have no conflict of interests related to this publication.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Forner A, Reig M, Bruix JJL. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. doi:10.1016/S0140-6736(18)30010-2
3. European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
4. De Mattia E, Cecchin E, Guardascione M, et al. Pharmacogenetics of the systemic treatment in advanced hepatocellular carcinoma. World J Gastroenterol. 2019;25(29):3870–3896. doi:10.3748/wjg.v25.i29.3870
5. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7(4):233–245. doi:10.1038/nrc2091
6. Dai X, Theobard R, Cheng H, Xing M, Zhang J. Fusion genes: a promising tool combating against cancer. Biochim Biophys Acta Rev Cancer. 2018;1869(2):149–160. doi:10.1016/j.bbcan.2017.12.003
7. Mani RS, Chinnaiyan AM. Triggers for genomic rearrangements: insights into genomic, cellular and environmental influences. Nat Rev Genet. 2010;11(12):819–829. doi:10.1038/nrg2883
8. Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15(6):371–381. doi:10.1038/nrc3947
9. Pederzoli F, Bandini M, Marandino L, et al. Targetable gene fusions and aberrations in genitourinary oncology. Nat Rev Urol. 2020;17(11):613–625. doi:10.1038/s41585-020-00379-4
10. Wei G, Rafiyath S, Liu D. First-line treatment for chronic myeloid leukemia: dasatinib, nilotinib, or imatinib. J Hematol Oncol. 2010;3(1):47. doi:10.1186/1756-8722-3-47
11. Kong X, Pan P, Sun H, et al. Drug discovery targeting Anaplastic Lymphoma Kinase (ALK). J Med Chem. 2019;62(24):10927–10954. doi:10.1021/acs.jmedchem.9b00446
12. Fernandez-Banet J, Lee NP, Chan KT, et al. Decoding complex patterns of genomic rearrangement in hepatocellular carcinoma. Genomics. 2014;103(2–3):189–203. doi:10.1016/j.ygeno.2014.01.003
13. Zhu C, Wu L, Lv Y, et al. The fusion landscape of hepatocellular carcinoma. Mol Oncol. 2019;13(5):1214–1225. doi:10.1002/1878-0261.12479
14. Kulangara K, Zhang N, Corigliano E, et al. Clinical utility of the combined positive score for programmed death ligand-1 expression and the approval of pembrolizumab for treatment of gastric cancer. Arch Pathol Lab Med. 2019;143(3):330–337. doi:10.5858/arpa.2018-0043-OA
15. Shi L, Zhang SJ, Chen J, et al. A comparability study of immunohistochemical assays for PD-L1 expression in hepatocellular carcinoma. Mod Pathol. 2019;32(11):1646–1656. doi:10.1038/s41379-019-0307-8
16. Rudkin CT, Hungerford DA, Nowell PC. DNA contents of chromosome Ph1 and chromosome 21 in human chronic granulocytic leukemia. Science. 1964;144(3623):1229–1231. doi:10.1126/science.144.3623.1229
17. Molyneux EM, Rochford R, Griffin B, et al. Burkitt’s lymphoma. Lancet. 2012;379(9822):1234–1244. doi:10.1016/S0140-6736(11)61177-X
18. Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44(4):461–466. doi:10.1038/ng.1107
19. Pal SK, Bergerot P, Dizman N, et al. Responses to alectinib in ALK-rearranged papillary renal cell carcinoma. Eur Urol. 2018;74(1):124–128. doi:10.1016/j.eururo.2018.03.032
20. Rosell R, Karachaliou N, Wolf J, Ou SH. ALK and ROS1 non-small-cell lung cancer: two molecular subgroups sensitive to targeted therapy. Lancet Respir Med. 2014;2(12):966–968. doi:10.1016/S2213-2600(14)70259-0
21. Hoy SM. Pemigatinib: first approval. Drugs. 2020;80(9):923–929. doi:10.1007/s40265-020-01330-y
22. Xu L, Hazard FK, Zmoos AF, et al. Genomic analysis of fibrolamellar hepatocellular carcinoma. Hum Mol Genet. 2015;24(1):50–63. doi:10.1093/hmg/ddu418
23. Longerich T, Endris V, Neumann O, et al. RSPO2 gene rearrangement: a powerful driver of β-catenin activation in liver tumours. Gut. 2019;68(7):1287–1296. doi:10.1136/gutjnl-2018-317632
24. Dou C, Sun L, Wang L, et al. Bromodomain-containing protein 9 promotes the growth and metastasis of human hepatocellular carcinoma by activating the TUFT1/AKT pathway. Cell Death Dis. 2020;11(9):730. doi:10.1038/s41419-020-02943-7
25. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5(1):4846. doi:10.1038/ncomms5846
26. Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378(2):F115–177. doi:10.1016/s0304-419x(98)00017-1
27. Oba A, Shimada S, Akiyama Y, et al. ARID2 modulates DNA damage response in human hepatocellular carcinoma cells. J Hepatol. 2017;66(5):942–951. doi:10.1016/j.jhep.2016.12.026
28. Moreno T, Monterde B, Gonzalez-Silva L, et al. ARID2 deficiency promotes tumor progression and is associated with higher sensitivity to chemotherapy in lung cancer. Oncogene. 2021;40(16):2923–2935. doi:10.1038/s41388-021-01748-y
29. Shriver M, Stroka KM, Vitolo MI, et al. Loss of giant obscurins from breast epithelium promotes epithelial-to-mesenchymal transition, tumorigenicity and metastasis. Oncogene. 2015;34(32):4248–4259. doi:10.1038/onc.2014.358
30. Passiglia F, Caparica R, Giovannetti E, et al. The potential of neurotrophic tyrosine kinase (NTRK) inhibitors for treating lung cancer. Expert Opin Investig Drugs. 2016;25(4):385–392. doi:10.1517/13543784.2016.1152261
31. Huang CY, Wang Y, Luo GY, et al. Relationship between PD-L1 expression and CD8+ T-cell immune responses in hepatocellular carcinoma. J Immunother. 2017;40(9):323–333. doi:10.1097/CJI.0000000000000187
32. Li XS, Li JW, Li H, Jiang T. Prognostic value of programmed cell death ligand 1 (PD-L1) for hepatocellular carcinoma: a meta-analysis. Biosci Rep. 2020;40:4. doi:10.1042/BSR20200459
33. Lanza C, Gaidano G, Cimino G, et al. Distribution of TP53 mutations among acute leukemias with MLL rearrangements. Genes Chromosomes Cancer. 1996;15(1):48–53. doi:10.1002/(SICI)1098-2264(199601)15:1<48::AID-GCC7>3.0.CO;2-4
34. Alidousty C, Baar T, Martelotto LG, et al. Genetic instability and recurrent MYC amplification in ALK-translocated NSCLC: a central role of TP53 mutations. J Pathol. 2018;246(1):67–76. doi:10.1002/path.5110
35. Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148(1–2):59–71. doi:10.1016/j.cell.2011.12.013
36. Donehower LA, Soussi T, Korkut A, et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 2019;28(5):1370–1384 e1375. doi:10.1016/j.celrep.2019.07.001
37. Kron A, Alidousty C, Scheffler M, et al. Impact of TP53 mutation status on systemic treatment outcome in ALK-rearranged non-small-cell lung cancer. Ann Oncol. 2018;29(10):2068–2075. doi:10.1093/annonc/mdy333
38. Peng SY, Chen WJ, Lai PL, Jeng YM, Sheu JC, Hsu HC. High alpha-fetoprotein level correlates with high stage, early recurrence and poor prognosis of hepatocellular carcinoma: significance of hepatitis virus infection, age, p53 and beta-catenin mutations. Int J Cancer. 2004;112(1):44–50. doi:10.1002/ijc.20279
39. Han JH, Kim DG, Na GH, et al. Evaluation of prognostic factors on recurrence after curative resections for hepatocellular carcinoma. World J Gastroenterol. 2014;20(45):17132–17140. doi:10.3748/wjg.v20.i45.17132
40. Marasco G, Colecchia A, Colli A, et al. Role of liver and spleen stiffness in predicting the recurrence of hepatocellular carcinoma after resection. J Hepatol. 2019;70(3):440–448. doi:10.1016/j.jhep.2018.10.022
41. Yoshihara K, Wang Q, Torres-Garcia W, et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2015;34(37):4845–4854. doi:10.1038/onc.2014.406
42. Sato Y, Itoh F, Hareyama M, et al. Association of cyclin D1 expression with factors correlated with tumor progression in human hepatocellular carcinoma. J Gastroenterol. 1999;34(4):486–493. doi:10.1007/s005350050301
43. Kang HJ, Haq F, Sung CO, et al. Characterization of hepatocellular carcinoma patients with FGF19 amplification assessed by fluorescence in situ hybridization: a Large Cohort Study. Liver Cancer. 2019;8(1):12–23. doi:10.1159/000488541
44. Sawey ET, Chanrion M, Cai C, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell. 2011;19(3):347–358. doi:10.1016/j.ccr.2011.01.040
45. Tan AC, Seet AOL, Lai GGY, et al. Molecular characterization and clinical outcomes in RET-rearranged NSCLC. J Thorac Oncol. 2020;15(12):1928–1934. doi:10.1016/j.jtho.2020.08.011
46. Blagih J, Zani F, Chakravarty P, et al. Cancer-specific loss of p53 leads to a modulation of myeloid and T cell responses. Cell Rep. 2020;30(2):481–496 e486. doi:10.1016/j.celrep.2019.12.028
47. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer. 2018;17(1):58. doi:10.1186/s12943-018-0782-4
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.