")
Back to Journals » Substance Abuse and Rehabilitation » Volume 6
The pharmacology of neurokinin receptors in addiction: prospects for therapy
Authors Sandweiss A, Vanderah T
Received 25 June 2015
Accepted for publication 23 July 2015
Published 7 September 2015 Volume 2015:6 Pages 93—102
DOI https://doi.org/10.2147/SAR.S70350
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Li-Tzy Wu
Video abstract presented by Alexander J Sandweiss and Todd W Vanderah.
Views: 1620
Alexander J Sandweiss, Todd W Vanderah
Department of Pharmacology, College of Medicine, University of Arizona, Tucson, AZ, USA
Abstract: Addiction is a chronic disorder in which consumption of a substance or a habitual behavior becomes compulsive and often recurrent, despite adverse consequences. Substance p (SP) is an undecapeptide and was the first neuropeptide of the neurokinin family to be discovered. The subsequent decades of research after its discovery implicated SP and its neurokinin relatives as neurotransmitters involved in the modulation of the reward pathway. Here, we review the neurokinin literature, giving a brief historical perspective of neurokinin pharmacology, localization in various brain regions involved in addictive behaviors, and the functional aspects of neurokinin pharmacology in relation to reward in preclinical models of addiction that have shaped the rational drug design of neurokinin antagonists that could translate into human research. Finally, we will cover the clinical investigations using neurokinin antagonists and discuss their potential as a therapy for drug abuse.
Keywords: reward, substance p, alcohol, morphine, cocaine, dopamine
Introduction
Drugs of abuse such as opioids, cocaine, amphetamines, alcohol, and nicotine affect the reward pathway in unique ways, leading to the potential of addiction. In the United States, the cost of substance abuse to society is more than $700 billion per year, necessitating new strategies in the management of addiction.1 Particularly alarming is the rate of deaths due to heroin overdose, which has skyrocketed since 2010. The National Institute on Drug Abuse (NIDA) and Centers for Disease Control and Prevention attribute this increase in heroin usage and mortality to an inadvertent consequence of reducing the availability of prescription painkillers.2 While abstinence from drugs of abuse seems like the most logical strategy, this has proven to be only an illusory goal. Therefore, the FDA and NIDA have planned to change the requirements for new therapies designed as deterrents for drugs of abuse; a reduction in the use of drugs of abuse over the long term may be the more appropriate requirement for FDA approval. Neurokinins are a family of peptide transmitters involved in the reward pathway for each of the drugs of abuse, giving researchers a target to design new medications aimed at reducing the addictive profile of said drugs of abuse.
Substance p (SP) was the first member of the neurokinin family of peptides to be isolated, initially from equine intestine and brain in 1931, and shown to act as a vasodepressor.3 The subsequent decades of research implicated SP and its neurokinin cousins in numerous central nervous system (CNS) disorders including anxiety, depression, migraine, schizophrenia, and addiction. Here, we review the basic and clinical science of the last 80 years that have helped shape our current understanding of how neurokinins specifically alter the neuronal pathway involved in addiction. We will then introduce potential neurokinin-directed therapies that may have efficacy in clinical practice relating to addiction.
Historical overview of neurokinins
In 1931, Ulf Von Euler and John Gaddum were on an expedition of sorts, in search of the distribution of acetylcholine in various equine organs. They came across a previously unidentified substance that they were able to concentrate in a powdered form, thus naming it “substance p”.4 By the 1950s, SP was well accepted as a polypeptide located in the CNS, particularly concentrated in the thalamus, hypothalamus, basal ganglia, and tegmentum in addition to the dorsal root ganglia of the peripheral nervous system, mediating nociceptive transmission from the primary afferent.5,6 However, it was not until 1970 when Chang and Leeman were able to isolate, characterize, and sequence SP as an 11-amino-acid peptide that the neurokinin field really evolved.7 With the newly available antibodies to SP, immunohistochemical techniques allowed more precise characterization of SPergic neurons in the CNS. Even with the rather rudimentary techniques available in the 1970s (ie, no optogenetics), SPergic projections were specifically found traveling from habenula (Hb) to the interpeduncular nucleus (IPN) and ventral tegmental area (VTA) via the fasciculus retroflexus,8 the striatum to the substantia nigra via the striato-nigral pathway,9 and the nucleus accumbens (NAc) to the ventral pallidum (specifically the nucleus basalis magnocellularis),10 with further evidence supporting SP as a neurotransmitter with vesicular release (Figure 1).11 At roughly the same time, the importance of dopamine in the mesolimbic and mesocortical pathways on drug seeking behavior was coming to fruition.12,13 Most importantly, it seemed that dopaminergic projections from the VTA to the NAc and other regions facilitated reward.14 With the basic topography of SP signaling in place, the next step was determining its functionality in the mesolimbic system. Indeed, it was shown that SP could directly activate dopaminergic neurons in the VTA,15 but how was SP signaling to the postsynaptic neuron and were there other ligands of the same family in humans?
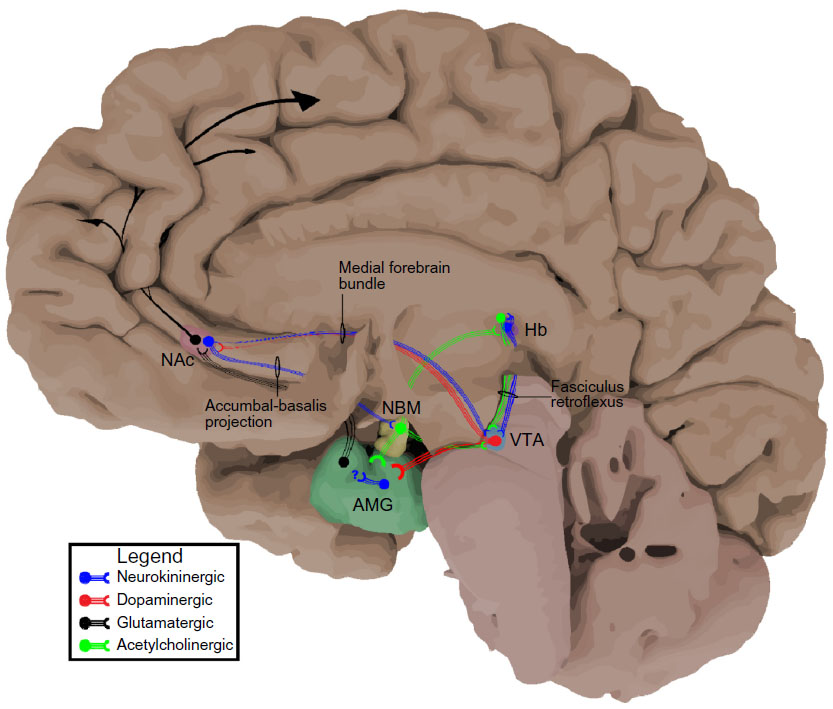 | Figure 1 Neurokininergic projections in the reward pathway. |
Basic neurokinin pharmacology
The ensuing era of neurokinin research revolved around the characterization of two more human neurokinins and their receptors. In 1983, neurokinin A and neurokinin B (NKA and NKB, respectively) were discovered and characterized, putting them in the same family as SP (the tachykinin family16) based on similar −CO2 terminal sequences.17 By 1984, all three neurokinin receptors had been proposed,18 followed by the permanent nomenclature: neurokinin-1 receptor (NK1R), neurokinin-2 receptor (NK2R), and neurokinin-3 receptor (NK3R) in 1986.19 Each ligand can bind and activate each receptor; however, they all have their preference owing to a graded affinity: SP preferentially activates NK1R, NKA preferentially activates NK2R, and NKB preferentially activates NK3R (Table 1).20 Cellular and molecular experiments linked neurokinin receptor activation to inositol phospholipid hydrolysis21 (later referred to as Gq coupling). Following receptor activation, the NK1R is rapidly internalized, leading to visual NK1R+ endosomal varicosities in the dendrites and somata of neurons, disappearing an hour later.22 This discovery made future cellular investigations into neurokinin pharmacology easier to trace.
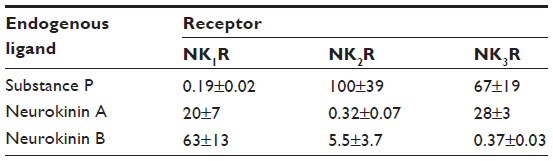 | Table 1 IC50 values of the three primary endogenous neurokinin ligands for the three primary neurokinin receptors |
Receptor localization is important in determining how the pharmacology affects a local neuronal circuit. Unfortunately for neuroscientists, the reward circuitry and accompanying pharmacology is quite complex. Further complicating the picture, G-protein-coupled receptors like the neurokinin family of receptors can act in both rapid (Ca2+ or Na+ induced cell activation) and delayed (transcriptional) ways. Validating this notion, the activation of the neurokinin family of receptors will ultimately lead to an increase in intracellular Ca2+, thus potentiating neuronal mechanisms of firing an action potential, in addition to activating the nuclear translocation of certain transcription factors including NF-κB.23 Additionally, recent evidence implicates swift activation of a Na+ leak channel, NALCN, as well as the closure of G-protein-linked inwardly rectifying K+ channels in the rapid SP-induced activation of neuronal action potentials.24,25
For years, neurokinin antagonist studies were made very difficult by the lack of penetration of the available ligands (ie, only peptidergic antagonists were available).26 The breakthrough came in 1991 when scientists at Pfizer discovered the first nonpeptide molecule with classical competitive antagonism at the NK1R.27 The identification of this compound led to the discovery of even more selective, structurally diverse, nonpeptidic NK1R antagonists at other pharmaceuticals, namely, Merck’s MK-869, which later became known in the clinic as the antiemetic aprepitant (EMEND®, Merck & Co, NJ, USA).28 While considerable time and money went into the possibility of aprepitant working as a standalone analgesic and/or antidepressant (without much success),29 the prospect of an NK1R antagonist for the treatment of addiction still remains viable. The rest of this review will cover the specifics of neurokinin pharmacology in addiction.
Neurokinins in the reward pathway
While SPergic cell bodies have been found in a number of CNS foci including the septal complex, nucleus tractus diagonalis (diagonal band of Broca), NAc, and Hb,30 with projections to various regions including the nucleus basalis magnocellularis, VTA, and IPN, each constituent seems to have a unique function in the limbic loop. A recent report links NK1R activation to μ-opioid receptor recycling, offering direct evidence for a neurokinin-mediated opioid resensitization.31 In fact, the neurokinin system is consistently found colocalized or in other ways affecting endogenous opioid, dopamine, and serotonergic signaling, thereby exerting its effect on affective and drug-seeking behavior.32,33
Ventral tegmental area
Important to the reward pathway and the study thereof, the VTA most notably sends dopaminergic projections to the NAc where dopamine release triggers euphoria or positive reinforcement.34 Critically, intra-VTA injections of both an NK1R agonist and NK3R agonist facilitate dopamine release in the NAc.35 Indeed, autoradiographic studies using the radiolabeled NK3R agonist [3H]senktide confirmed the presence of NK3Rs in the VTA in addition to the IPN and Hb.36 This complemented the previously studied NK1R localization in the VTA among other CNS locations.37 While in vitro electrophysiologic studies in the VTA demonstrated NK3Rs may mediate more of the excitatory effects of dopaminergic neurons while leaving a role for SP out,38 in vivo electrophysiologic recordings in the VTA confirmed that systemic administration of an NK1R antagonist was sufficient to block dopamine cell firing.39 In addition, studies demonstrated an increased firing rate of VTA dopaminergic neurons due to the application of SP.40 These apparent discrepancies in dopaminergic activity and neurokinin pharmacology may be in part due to the marked receptor heterogeneity of the VTA.41
The expression of various neurokinin receptor subtypes is rather diverse. For example, while there is somatodendritic expression of the NK1R in the cell membrane of dopaminergic and nondopaminergic neurons of the VTA,42 NK3Rs are often found in the cytoplasm of dopaminergic and nondopaminergic neurons of the VTA.43 Additionally, the NK3Rs found in the plasma membrane are frequently extrasynaptic. Furthermore, VTA glia exhibit substantially more NK3Rs than NK1Rs, suggesting the importance of the immune cells in the reward pathway (see “Involvement of the immune system in addiction” for more details). Overall, the expression of NK3Rs in the VTA is twice that of NK1Rs.44 Interestingly, NK3Rs, but not NK1R or NK2Rs, were found within the nuclear envelope of projection neurons of the VTA. This suggests the possibility of direct NK3R involvement in gene transcription. Indeed, ligand-dependent and -independent nuclear translocation of the NK3R in the VTA has been observed.45,46 The significance of these nuclear events in the reward pathway has not been fully elucidated.
The literature on the NK2R in the VTA is sparse; however, it has been shown that intravenous infusion of the selective NK2R antagonist SR-48968 did not alter basal dopaminergic firing rate in rats.47 Peculiarly, the acute administration of SR-48968 intraperitoneal (ip) increased the number of spontaneously active VTA DA neurons; however, this may be due to dosing differences or possible pharmacologically active metabolites. The intracellular interaction between neurokinin receptors has also been studied. When expressed by the same cell, NK1R activation sequesters β-arrestins in endosomes, impeding ligand-dependent NK3R endocytosis.48 The paucity of information regarding how heterologous interactions between neurokinin receptors affects the reward pathway indicates the necessity of future research in addiction.
Some of the earliest investigations into neurokinin’s ability to functionally impel the reward pathway came in 1985 when Staubli and Huston49 showed that injection of SP into the medial forebrain bundle, the neuronal tract that connects the VTA to the NAc, resulted in positive conditioned place preference (CPP). With regard to specific drugs of abuse, microinjection of the SP analog DiMe-C7 induced reinstatement of cocaine-seeking behavior, which could be significantly reduced by the D1 receptor antagonist SCH23390.50
Nucleus accumbens
The NAc is often regarded as the limbic–motor interface receiving inputs from the VTA and amygdala, among other regions, and sending projections to the cortex, ventral pallidum, globus pallidus, and reciprocal projections to the VTA and amygdala.51 One study provided evidence that the NAc required input from both the VTA and the basolateral amygdala for excitation of NAc efferents.52
SP injected into the NAc by itself increases concentrations of extracellular DA but does not induce positive CPP.53 An SP antibody injected into the NAc prevents amphetamine-induced increase of extracellular DA in the NAc.54 Likewise, NAc administration of the NK1R antagonist L-733,060 significantly diminishes cocaine-induced DA release.55
Nucleus basalis magnocellularis-substantia innominata
Evidence for SPergic fibers projecting to the nucleus basalis magnocellularis (nucleus basalis of Meynert) comes from simple light microscopic images and immunohistochemical staining.56 Accordingly, the injection of SP or the C-terminal fragment of SP into the nucleus basalis magnocellularis resulted in a positive CPP, which the authors attributed to the positive reinforcing effects of the specific C-terminal sequence of SP.57 Of course, the C-terminal fragment is shared among all tachykinin ligands, so resolving which receptor subtype responsible was an obvious next step. With the use of selective agonists, they went on to show that this effect was mediated by both NK1R and NK3R activation.58 Furthermore, SP injection into the nucleus basalis magnocellularis increased extracellular dopamine content in the NAc,59 a barometer of positive reinforcement. The fact that the injection of the NK3R agonist amino-senktide into the nucleus basalis magnocellularis inhibits alcohol intake at first glance contradicts the aforementioned positive reinforcement.60 The authors speculated that alcohol may actually be mediating its effects on the reward pathway via NK3R, thereby rendering an NK3R agonist a substitute for the rewarding properties of alcohol.61 Whether or not alcohol engages the NK3R system in the nucleus basalis magnocellularis remains to be investigated.
NKB fibers have been traced from the dorsal AND ventral striatum to the substantia innominata.62 Pre-protachykinin-B, the mRNA for NKB, has also been found heavily concentrated in fibers from the lateral stripe of the striatum, a region just lateral to the shell of the NAc, to the nucleus basalis magnocellularis.63 The importance of these projections in reward is not well understood; however, it should be remembered that NKB is the most efficacious endogenous ligand for the NK3R in mammals.
Habenula
The Hb is an understudied brain region, let alone the role neurokinins play in its function. What is known is that the Hb white matter tracts tie it extensively to other regions of the limbic pathway, including the ventral pallidum and ventral midbrain. Moreover, SPergic and NKBergic cell bodies are indeed found in the medial Hb with axons projecting to the VTA and the adjacent IPN.64,65 The role of the VTA in reward is well described; the IPN also seems to contribute to positive reinforcement.66 The Hb has long been known to be involved in nicotine dependence and, more recently, in cocaine and morphine dependence as well.67 No studies to our knowledge have examined the direct role of habenular neurokinin antagonists on addictive behavior in animals; however, NK1R and NK3Rs were found to be involved in nicotine-induced excitation of habenular neurons.68
Amygdala
Neurokinin receptors have been found in relatively high concentrations in the amygdala of primates.69 Accordingly, SP microinjection into the central amygdala enhanced passive avoidance learning behaviors70 and generated CPP.71 While efferent projections from the amygdala are abundant and promiscuous, specific projections to regions of the limbic loop will be discussed here.
Regarding neurokinins in addiction, the smoking gun of sorts came in the seminal 2000 Nature paper by Murtra et al72 when they documented NK1R−/− exhibited a lack of morphine CPP. Knockouts additionally eliminated CPP to amphetamines but surprisingly retained a positive CPP to cocaine and food, indicating distinct mechanisms mediating reward for each of these natural and unnatural rewards.73 The role of NK1R in opioid reward was further corroborated using the self-administration paradigm in NK1R−/− mice.74 To better understand which neuroanatomical location may play the principle role in neurokinin-mediated opioid reward, SP-saporin (a ribosome inactivating toxin) was used to ablate NK1R expressing neurons in the amygdala. Indeed, mice with ablation of NK1R+ neurons in the amygdala demonstrated similar CPP scores for both morphine and saline.75 These observations support the role of the neurokinin system in facilitating opioid reward via amygdaloid processes, although it does not rule out the importance of other limbic regions. Importantly, the neurokinin knockout data are supported by the fact that intracerebral ventricular administration of an NK1R antagonist has no effect on cocaine self-administration,76 ruling out possible developmental confounders to the knockout mice. Although cocaine CPP was not altered in the NK1R−/− mice, reinstatement of cocaine is in fact supplemented by administration of the SP analog [Sar9Met(O2)11]-SP.77 This, at the very least, implicates the neurokinin system in cocaine reinstatement, albeit endogenous SP may not play a role as two separate NK1R antagonists were unable to inhibit cocaine reinstatement.
Alcohol reward may be mediated in the amygdala as well. SP levels were lower in the central amygdala of Indiana alcohol-preferring rats than nonpreferring rats as measured by SP mRNA.78 For this reason, the investigators microinfused SP into the central amygdala of alcohol-preferring rats, rendering the animals indifferent to alcohol consumption while sucrose seeking remained the same. The apparent paradox in neurokinin signaling and alcohol reward has been noted twice: SP injection in the nucleus basalis magnocellularis reduces alcohol consumption in Sardinian alcohol-preferring rats and SP injection in the central amygdala facilitates a similar effect in Indiana alcohol-preferring rats. The concern with these studies lies in the fact that the alcohol-preferring animals are selectively bred and do not represent the typical rodent or primate condition.79 These studies contradict the vast majority of studies in rodents and humans that indicate a neurokinin antagonist reduces alcohol preference (see “Neurokinin in the clinic” for more details).80
Clear evidence exists linking stress to alcoholic relapse.81 Mild and severe emotional stressors are sufficient to release SP in the medial amygdala.82 Naturally, linking stress-induced SP release to stress-induced alcohol reinstatement was studied. Expectedly, the NK1R antagonist, L822429, was adequate to prevent alcohol reinstatement in an alcohol self-administration paradigm in wild-type Wistar rats.83 A related study demonstrated efficacy of the NK1R antagonist in suppressing alcohol seeking in wild-type mice at baseline and in preventing escalation of voluntary alcohol intake.84 Corroborating this data, NK1R silencing with a microRNA directed at the receptors’ transcript reduced alcohol consumption in mice.85 The study noted attenuated NK1R expression in the hippocampus, the only subcortical area they examined for proof of action. To the contrary, ezlopitant, the NK1R antagonist developed for chemotherapy induced nausea and vomiting, exhibited little to no efficacy in reducing operant self-administration of alcohol in Long–Evans rats.86 The aforementioned inconsistency raises a valid point about nonconserved regions of the neurokinin receptors between humans and rodents, giving rise to potential obstacles in extrapolating preclinical models to the human condition.87
While stress has been shown to increase extracellular SP in the amygdala, it should be mentioned that SP and NKA have been found colocalized in neurons of the infundibulum of the CNS and myenteric plexus of enteric nervous system, a possibility that has not been specifically investigated in neurons of the amygdala.88,89 In fact, NK2Rs do appear in a significant concentration in the amygdala,90 and the NK2R antagonist SR48968 was sufficient to block stress-induced behaviors in mice and central neuronal markers of stress in rats.91
An analogous pathway observed in the alcohol reward system in relation to neurokinin pharmacology is that observed with corticotrophin-releasing factor (CRF).92 There is extensive research into the effects of CRF and other neuropeptides on addiction that are out of the scope of this review. In general, it is accepted in the addiction field that the stress response is mediated by several neurotransmitter systems including CRF and SP, thus precipitating undesirable outcomes such as relapse.
Frontal cortex
The literature on neurokinins in the cortex is more scant than other brain regions; nevertheless, cortical neurokinins seem to play an important role in the limbic system. As one of the terminal sites of mesencephalic dopaminergic projections, the frontal cortex has been shown to have increased dopamine metabolites (DOPAC) in response to stress (ie, footshock).93 This increase in cortical dopamine is correlated to periods of intoxication and craving, particularly with cocaine abuse.94 Pretreatment with the selective NK1R antagonist (S)-GR205171 ip was sufficient to prevent footshock-induced dopamine release in the cortex.95 In addition to stress, morphine injections ip increased SP levels in the cortex that subsequently significantly decreased due to the administration of the opioid antagonist, naloxone.96 The relative importance of the frontal cortex in neurokinin-mediated addiction is not well understood and warrants further exploration.
Involvement of the immune system in addiction
The role of the resident immune cells in the brain, the glia (astrocytes, microglia, and oligodendrocytes), has been emerging in the last 20 years as critical for normal neuronal signaling.97 Importantly, microglia and astrocytes have recently been implicated in addictive processes as activated microglia release “proinflammatory” cytokines that act at the neuronal synapse, strengthening the signal.98 Expanding on this notion, alcohol, cocaine, morphine, and amphetamines have all been indicted for their role in microglial activation with microglial activation proven to be critical to the maintenance of addictive behaviors.99 Critically, NK1Rs are located on microglia and are inducible by IL-1β in astrocytes.100,101 SP has been shown to activate NF-κB in microglia, which has a strong, yet neglected, role in the progression of addiction.102 In astrocytes, SP application induces a complex depolarization by modulating Cl− and K+ currents.103 Astrocytes are probably most notorious for their role in glutamate homeostasis, and so there is a high likelihood of the neurokinin system modulating extracellular glutamate in brain regions, including those of the limbic system. There is substantial information on both neurokinins and glia in the reward pathway, yet a dearth of information on the interaction between the two. It may end up representing one of the more promising avenues in addiction research.
Neurokinin in the clinic
We have outlined the neural and pharmacological basis for the use of neurokinin antagonists in addiction. To summarize, SP appears to be overexpressed after chronic administration of drugs of abuse and mediates some of the negative effects such as CPP and reinstatement. Here, the focus will be on the use of neurokinin antagonists specifically in humans and the potential success as a therapeutic. While SP has long been infamous as one of the primary pronociceptive neurotransmitters, an NK1R antagonist did not achieve appreciable analgesia as a standalone medication in patients suffering from pain.29 However, one of the first investigations into neurokinins in human disease with positive results demonstrated elevated cerebrospinal fluid levels of SP in psychiatric patients with depression or schizophrenia.104 With the development of radiolabeled substance p antagonists (SPAs), imaging of receptor localization and saturation in humans became possible with positron emission tomography.105 [18F]-SPA-RQ was taken up in the brain of healthy male volunteers in the regions already described that are involved in reward including the VTA, amygdala, Hb, and ventral striatum.106 The most notable differences from rats were the high density of NK1Rs in the cortex of humans and a greater NK1R/NK3R ratio in the VTA of humans.107,108
Functionally, the NK1R antagonist aprepitant has an effect on positive incentive in humans. In an experiment enlisting healthy volunteers of both sexes, monetary incentive delay was the paradigm used to determine if the NK1R antagonist could prevent NAc activation typical of incentive anticipation. Indeed, when subjects expected a monetary reward for completing a task in the study, aprepitant reduced NAc blood oxygenation-level-dependent (BOLD) contrast compared to control as seen on fMRI, indicating the attenuation of NAc activation.109
An association between various NK1R gene (TACR1) single nucleotide polymorphisms (SNPs) and alcoholism may exist. In a large sample of heavy drinkers (7 drinks per day on average), 5 SNPs of the TACR1 gene were predictive of BOLD activation as assessed by fMRI in response to alcohol cues.110 In a separate study, 1 SNP and 2 haplotypes (a specific combination of alleles on the same chromosome) of the TACR1 gene were associated with alcohol dependence.111 The significance of these studies is not well understood; however, they point to a link between a specific neurokinin genotype and alcohol-dependent phenotype that may have potential as a drug target. Of course, the NK1R is not the only SNP found to dysregulate the reward pathway as OPRM1 (μ-opioid receptor) has also been highlighted as a troublesome gene of interest.112 Much like carriers of certain OPRM1, SNPs are more sensitive to the effects of naltrexone on reducing alcohol cravings, so too should NK1R antagonists on specific TACR1 SNP-related addictions.113
Unexpectedly, in a clinical study examining the role of aprepitant on oxycodone abuse liability, the authors found the NK1R antagonist actually increased the abuse potential of oxycodone in patients who were already opioid drug abusers.114 Several explanations for the unanticipated outcomes were proposed, including the pharmacokinetic interaction between the two drugs. That is, aprepitant and oxycodone compete for metabolism by the enzyme CYP3A4, rendering higher concentrations of serum oxycodone than expected. The unfortunate pharmacokinetic profiles of many drugs have hindered their success in the past, despite promising pharmacodynamic actions on the biology of the system. Future studies on opioid dependence may require a novel neurokinin antagonist that is not involved in CYP3A4 metabolism, a requirement that will surely prove challenging though not impossible.
In alcohol-dependent humans who were recently detoxified, LY686017, a brain penetrant NK1R antagonist with high bioavailability was efficacious in suppressing spontaneous alcohol cravings as assessed by the Alcohol Urge Questionaire.115 When the alcohol-dependent subjects were then provoked with a combined stress test and alcohol-cue challenge, the treatment group still had reduced cravings for alcohol compared to controls. To the contrary, psychiatric patients with comorbid posttraumatic stress disorder (PTSD) and alcoholism experienced no reduction in symptoms of alcohol craving after administration of an NK1R antagonist.116 This may point to the fact that comorbidity with PTSD complicates the syndrome by adding another “stress”-related illness.
Neurokinin prospects for therapy
The neurokinin field indeed does seem poised to produce significant contributions to addiction research and therapy. We have outlined the role neurokinins play in the reward pathway, particularly via NK1Rs and NK3Rs. Accordingly, GlaxoSmithKline has a dual NK1R/NK3R antagonist, GSK1144814, in the pipeline for future clinical trials for psychiatric disorders.117 Vanda Pharmaceuticals acquired worldwide licensing for LY686017 (now called VLY-686) from Eli Lilly after the proof of concept studies in alcohol cravings mentioned earlier. Vanda is now attempting to commercialize and develop this compound “for all human conditions”, including an indication for substance abuse. Our pharmacology/chemistry group has created several opioid agonist/NK1R antagonist compounds that have efficacy in antinociception and do not produce CPP or increase extracellular dopamine content in the NAc.118–120
In addition to the new compounds in the pipeline, the original gold standard NK1R antagonist is still under investigation for its effects on substance abuse potential since it already has FDA approval for the clinic. A brief ClinicalTrials.gov search reveals that aprepitant is currently undergoing clinical trials for the evaluation of its effects on cannabis cravings in cannabis-dependent outpatients, comorbid alcoholic and cannabis-dependent patients, and in opioid-dependent patients. More compounds that selectively block the neurokinin system will undoubtedly materialize in the drug pipeline as preclinical and clinical studies further identify the role of the neurokinin system in drug addiction.
Disclosure
The authors report no conflicts of interest in this report.
References
Trends and Statistics. Available from http://www.drugabuse.gov/related-topics/trends-statistics#costs. Accessed July 28, 2015. | |
Volkow N. What is the Federal Government Doing to Combat the Opioid Abuse Epidemic? Bethesda, MD: NIDA: NIH; 2015. | |
V Euler US, Gaddum JH. An unidentified depressor substance in certain tissue extracts. J Physiol. 1931;72(1):74–87. | |
Gaddum JH, Schild H. Depressor substances in extracts of intestine. J Physiol. 1934;83(1):1–14. | |
Pernow B. Distribution of substance P in the central and peripheral nervous system. Nature. 1953;171(4356):746. | |
Yaksh TL, Jessell TM, Gamse R, Mudge AW, Leeman SE. Intrathecal morphine inhibits substance P release from mammalian spinal cord in vivo. Nature. 1980;286(5769):155–157. | |
Chang MM, Leeman SE. Isolation of a sialogogic peptide from bovine hypothalamic tissue and its characterization as substance P. J Biol Chem. 1970;245(18):4784–4790. | |
Mroz ED, Brownstein MJ, Leeman SE. Evidence for substance P in the habenulo-interpeduncular tract. Brain Res. 1976;113(3):597–599. | |
Kanazawa I, Emson PC, Cuello AC. Evidence for the existence of substance P-containing fibres in striato-nigral and pallido-nigral pathways in rat brain. Brain Res. 1977;119(2):447–453. | |
Napier TC, Mitrovic I, Churchill L, Klitenick MA, Lu XY, Kalivas PW. Substance P in the ventral pallidum: projection from the ventral striatum, and electrophysiological and behavioral consequences of pallidal substance P. Neuroscience. 1995;69(1):59–70. | |
Cuello AC, Jessell TM, Kanazawa I, Iversen LL. Substance P: localization in synaptic vesicles in rat central nervous system. J Neurochem. 1977;29(4):747–751. | |
Kelly PH, Iversen SD. Selective 6OHDA-induced destruction of mesolimbic dopamine neurons: abolition of psychostimulant-induced locomotor activity in rats. Eur J Pharmacol. 1976;40(1):45–56. | |
Spyraki C, Fibiger HC, Phillips AG. Attenuation of heroin reward in rats by disruption of the mesolimbic dopamine system. Psychophamacology. 1983;79(2–3):278–283. | |
Baik JH. Dopamine signaling in reward-related behaviors. Front Neural Circuits. 2013;7:152. | |
Stinus L, Kelley AE, Iversen SD. Increased spontaneous activity following substance P infusion into A10 dopaminergic area. Nature. 1978; 276(5688):616–618. | |
Erspamer V, Anastasi A. Polypeptides active on plain muscle in the amphibian skin. Paper presented at: Proceedings of the International Symposium: Hypotensive Peptides; October 25–29, 1965, 1966; Florence, Italy. | |
Kimura S, Okada M, Sugita Y, Kanazawa I, Munekata E. Novel neuropeptides, neurokinin-alpha and neurokinin-beta isolated from porcine spinal-cord. P Jpn Acad B Phys. 1983;59(4):101–104. | |
Buck SH, Burcher E, Shults CW, Lovenberg W, O’Donohue TL. Novel pharmacology of substance K-binding sites: a third type of tachykinin receptor. Science. 1984;226(4677):987–989. | |
Henry JL. Substance P and neurokinins. Paper presented at: Proceedings of “Substance P and Neurokinins-Montreal ’86.” A Satellite Symposium of the XXX International Congress of The International Union of Physiological Sciences; 1986; Montreal, Canada. | |
Regoli D, Drapeau G, Dion S, D’Orleans-Juste P. Pharmacological receptors for substance P and neurokinins. Life Sci. 1987;40(2):109–117. | |
Mantyh PW, Pinnock RD, Downes CP, Goedert M, Hunt SP. Correlation between inositol phospholipid hydrolysis and substance P receptors in rat CNS. Nature. 1984;309(5971):795–797. | |
Mantyh PW, Allen CJ, Ghilardi JR, et al. Rapid endocytosis of a G protein-coupled receptor: substance P evoked internalization of its receptor in the rat striatum in vivo. Proc Natl Acad Sci U S A. 1995; 92(7):2622–2626. | |
Steinhoff MS, von Mentzer B, Geppetti P, Pothoulakis C, Bunnett NW. Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol Rev. 2014;94(1):265–301. | |
Lu B, Su Y, Das S, et al. Peptide neurotransmitters activate a cation channel complex of NALCN and UNC-80. Nature. 2009;457(7230):741–744. | |
Koike-Tani M, Collins JM, Kawano T, et al. Signal transduction pathway for the substance P-induced inhibition of rat Kir3 (GIRK) channel. J Physiol. 2005;564(Pt 2):489–500. | |
Iversen L. Leslie Iversen. In: Squire LR, editor. The History of Neuroscience in Autobiography. Vol 6. Oxford: Oxford University Press; 2009:190–225. | |
Snider RM, Constantine JW, Lowe JA III, et al. A potent nonpeptide antagonist of the substance P (NK1) receptor. Science. 1991;251(4992):435–437. | |
Kramer MS, Cutler N, Feighner J, et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science. 1998;281(5383):1640–1645. | |
Hill R. NK1 (substance P) receptor antagonists – why are they not analgesic in humans? Trends Pharmacol Sci. 2000;21(7):244–246. | |
Ljungdahl A, Hokfelt T, Nilsson G. Distribution of substance P-like immunoreactivity in the central nervous system of the rat – I. Cell bodies and nerve terminals. Neuroscience. 1978;3(10):861–943. | |
Bowman SL, Soohoo AL, Shiwarski DJ, Schulz S, Pradhan AA, Puthenveedu MA. Cell-Autonomous Regulation of Mu-Opioid Receptor Recycling by Substance P. Cell Rep. 2015;10(11):1925–1936. | |
Santarelli L, Gobbi G, Blier P, Hen R. Behavioral and physiologic effects of genetic or pharmacologic inactivation of the substance P receptor (NK1). J Clin Psychiatry. 2002;63(Suppl 11):11–17. | |
Mantyh PW. Neurobiology of substance P and the NK1 receptor. J Clin Psychiatry. 2002;63(Suppl 11):6–10. | |
Wise RA, Koob GF. The development and maintenance of drug addiction. Neuropsychopharmacology. 2014;39(2):254–262. | |
Elliott PJ, Mason GS, Stephens-Smith M, Hagan RM. Behavioural and biochemical responses following activation of midbrain dopamine pathways by receptor selective neurokinin agonists. Neuropeptides. 1991;19(2):119–126. | |
Dam TV, Escher E, Quirion R. Visualization of neurokinin-3 receptor sites in rat brain using the highly selective ligand [3H]senktide. Brain Res. 1990;506(1):175–179. | |
Mantyh PW, Gates T, Mantyh CR, Maggio JE. Autoradiographic localization and characterization of tachykinin receptor binding sites in the rat brain and peripheral tissues. J Neurosci. 1989;9(1):258–279. | |
Seabrook GR, Bowery BJ, Hill RG. Pharmacology of tachykinin receptors on neurones in the ventral tegmental area of rat brain slices. Eur J Pharmacol. 1995;273(1–2):113–119. | |
Minabe Y, Emori K, Toor A, Stutzmann GE, Ashby CR Jr. The effect of the acute and chronic administration of CP 96,345, a selective neurokinin1 receptor antagonist, on midbrain dopamine neurons in the rat: a single unit, extracellular recording study. Synapse. 1996;22(1):35–45. | |
Korotkova TM, Brown RE, Sergeeva OA, Ponomarenko AA, Haas HL. Effects of arousal- and feeding-related neuropeptides on dopaminergic and GABAergic neurons in the ventral tegmental area of the rat. Eur J Neurosci. 2006;23(10):2677–2685. | |
Sanchez-Catalan MJ, Kaufling J, Georges F, Veinante P, Barrot M. The antero-posterior heterogeneity of the ventral tegmental area. Neuroscience. 2014;282C:198–216. | |
Lessard A, Pickel VM. Subcellular distribution and plasticity of neurokinin-1 receptors in the rat substantia nigra and ventral tegmental area. Neuroscience. 2005;135(4):1309–1323. | |
Lessard A, Grady EF, Bunnett NW, Pickel VM. Predominant surface distribution of neurokinin-3 receptors in non-dopaminergic dendrites in the rat substantia nigra and ventral tegmental area. Neuroscience. 2007;144(4):1393–1408. | |
Lessard A, Savard M, Gobeil F Jr, Pierce JP, Pickel VM. The neurokinin-3 (NK3) and the neurokinin-1 (NK1) receptors are differentially targeted to mesocortical and mesolimbic projection neurons and to neuronal nuclei in the rat ventral tegmental area. Synapse. 2009;63(6):484–501. | |
Hether S, Misono K, Lessard A. The neurokinin-3 receptor (NK3R) antagonist SB222200 prevents the apomorphine-evoked surface but not nuclear NK3R redistribution in dopaminergic neurons of the rat ventral tegmental area. Neuroscience. 2013;247:12–24. | |
Sladek CD, Stevens W, Levinson SR, Song Z, Jensen DD, Flynn FW. Characterization of nuclear neurokinin 3 receptor expression in rat brain. Neuroscience. 2011;196:35–48. | |
Minabe Y, Ashby CR Jr. Effect of the acute and chronic administration of the selective neurokinin2 receptor antagonist SR 48968 on midbrain dopamine neurons in the rat: an in vivo extracellular single cell study. Synapse. 1997;25(2):196–204. | |
Schmidlin F, Dery O, Bunnett NW, Grady EF. Heterologous regulation of trafficking and signaling of G protein-coupled receptors: beta-arrestin-dependent interactions between neurokinin receptors. Proc Natl Acad Sci U S A. 2002;99(5):3324–3329. | |
Staubli U, Huston JP. Central action of substance P: possible role in reward. Behav Neural Biol. 1985;43(1):100–108. | |
Placenza FM, Fletcher PJ, Rotzinger S, Vaccarino FJ. Infusion of the substance P analogue, DiMe-C7, into the ventral tegmental area induces reinstatement of cocaine-seeking behaviour in rats. Psychopharmacology. 2004;177(1–2):111–120. | |
Mogenson GJ, Wu M, Jones DL. Locomotor activity elicited by injections of picrotoxin into the ventral tegmental area is attenuated by injections of GABA into the globus pallidus. Brain Res. 1980;191(2):569–571. | |
Ambroggi F, Ishikawa A, Fields HL, Nicola SM. Basolateral amygdala neurons facilitate reward-seeking behavior by exciting nucleus accumbens neurons. Neuron. 2008;59(4):648–661. | |
Schildein S, Agmo A, Huston JP, Schwarting RK. Intraaccumbens injections of substance P, morphine and amphetamine: effects on conditioned place preference and behavioral activity. Brain Res. 1998; 790(1–2):185–194. | |
Elliott PJ, Nemeroff CB, Kilts CD. Evidence for a tonic facilitatory influence of substance P on dopamine release in the nucleus accumbens. Brain Res. 1986;385(2):379–382. | |
Loonam TM, Noailles PA, Yu J, Zhu JP, Angulo JA. Substance P and cholecystokinin regulate neurochemical responses to cocaine and methamphetamine in the striatum. Life Sci. 2003;73(6):727–739. | |
Beach TG, Tago H, McGeer EG. Light microscopic evidence for a substance P-containing innervation of the human nucleus basalis of Meynert. Brain Res. 1987;408(1–2):251–257. | |
Hasenohrl RU, Gerhardt P, Huston JP. Positively reinforcing effects of the neurokinin substance P in the basal forebrain: mediation by its C-terminal sequence. Exp Neurol. 1992;115(2):282–291. | |
Nikolaus S, Huston JP, Hasenohrl RU. Reinforcing effects of neurokinin substance P in the ventral pallidum: mediation by the tachykinin NK1 receptor. Eur J Pharmacol. 1999;370(2):93–99. | |
Boix F, Sandor P, Nogueira PJ, Huston JP, Schwarting RK. Relationship between dopamine release in nucleus accumbens and place preference induced by substance P injected into the nucleus basalis magnocellularis region. Neuroscience. 1995;64(4):1045–1055. | |
Ciccocioppo R, Panocka I, Polidori C, De Caro G, Regoli D, Massi M. Stimulation of tachykinin NK-3 receptors in the nucleus basalis magnocellularis reduces alcohol intake in rats. Peptides. 1997;18(9):1349–1355. | |
Ciccocioppo R, Panocka I, Polidori C, Froldi R, Angeletti S, Massi M. Mechanism of action for reduction of ethanol intake in rats by the tachykinin NK-3 receptor agonist aminosenktide. Pharmacol Biochem Behav. 1998;61(4):459–464. | |
Furuta T, Mori T, Lee T, Kaneko T. Third group of neostriatofugal neurons: neurokinin B-producing neurons that send axons predominantly to the substantia innominata. J Comp Neurol. 2000;426(2):279–296. | |
Zhou L, Furuta T, Kaneko T. Neurokinin B-producing projection neurons in the lateral stripe of the striatum and cell clusters of the accumbens nucleus in the rat. J Comp Neurol. 2004;480(2):143–161. | |
Velasquez KM, Molfese DL, Salas R. The role of the habenula in drug addiction. Front Hum Neurosci. 2014;8:174. | |
Burgunder JM, Young WS III. Neurokinin B and substance P genes are co-expressed in a subset of neurons in the rat habenula. Neuropeptides. 1989;13(3):165–169. | |
Antolin-Fontes B, Ables JL, Gorlich A, Ibanez-Tallon I. The habenulo-interpeduncular pathway in nicotine aversion and withdrawal. Neuropharmacology. 2015;96(Pt B):213–222. | |
Glick SD, Ramirez RL, Livi JM, Maisonneuve IM. 18-Methoxycoonaridine acts in the medial habenula and/or interpeduncular nucleus to decrease morphine self-administration in rats. Eur J Pharmacol. 2006;537(1–3):94–98. | |
Dao DQ, Perez EE, Teng Y, Dani JA, De Biasi M. Nicotine enhances excitability of medial habenular neurons via facilitation of neurokinin signaling. J Neurosci. 2014;34(12):4273–4284. | |
Nagano M, Saitow F, Haneda E, Konishi S, Hayashi M, Suzuki H. Distribution and pharmacological characterization of primate NK-1 and NK-3 tachykinin receptors in the central nervous system of the rhesus monkey. Br J Pharmacol. 2006;147(3):316–323. | |
Kertes E, Laszlo K, Berta B, Lenard L. Effects of substance P microinjections into the globus pallidus and central nucleus of amygdala on passive avoidance learning in rats. Behav Brain Res. 2009;198(2):397–403. | |
Kertes E, Laszlo K, Berta B, Lenard L. Positive reinforcing effects of substance P in the rat central nucleus of amygdala. Behav Brain Res. 2009;205(1):307–310. | |
Murtra P, Sheasby AM, Hunt SP, De Felipe C. Rewarding effects of opiates are absent in mice lacking the receptor for substance P. Nature. 2000;405(6783):180–183. | |
Murtra P, Sheasby A, Hunt SP, De Felipe C. Loss of Rewarding Effects of Morphine and Amphetamine, But Not Cocaine, in Mice with Disruption of the Substance P Receptor (NK1) Gene. New Orleans, LA: Society for Neuroscience; 2000. | |
Ripley T. Lack of self-administration and behavioural sensitisation to morphine, but not cocaine, in mice lacking NK1 receptors. Neuropharmacology. 2002;43(8):1258–1268. | |
Gadd CA, Murtra P, De Felipe C, Hunt SP. Neurokinin-1 receptor-expressing neurons in the amygdala modulate morphine reward and anxiety behaviors in the mouse. J Neurosc. 2003;23(23):8271–8280. | |
Placenza FM, Fletcher PJ, Vaccarino FJ, Erb S. Effects of central neurokinin-1 receptor antagonism on cocaine- and opiate-induced locomotor activity and self-administration behaviour in rats. Pharmacol Biochem Behav. 2006;84(1):94–101. | |
Placenza FM, Vaccarino FJ, Fletcher PJ, Erb S. Activation of central neurokinin-1 receptors induces reinstatement of cocaine-seeking behavior. Neurosci Lett. 2005;390(1):42–47. | |
Yang AR, Yi HS, Mamczarz J, June HL Jr, Hwang BH, June HL Sr. Deficits in substance P mRNA levels in the CeA are inversely associated with alcohol-motivated responding. Synapse. 2009;63(11):972–981. | |
Lumeng L, Hawkins TD, Li TK. New strains of rats with alcohol preference and nonpreference. In: Ronald G. Thurman JRW, Henry Drott, Britton Chance, editors. Alcohol and Aldehyde Metabolizing Systems: Intermediary Metabolism and Neurochemistry. Vol 3. New York: Academic Press, Inc.; 1977:537–544. | |
Higley AE, Koob GF, Mason BJ. Treatment of alcohol dependence with drug antagonists of the stress response. Alcohol Res. 2012;34(4):516–521. | |
Sinha R. How does stress lead to risk of alcohol relapse? Alcohol Res. 2012;34(4):432–440. | |
Ebner K, Rupniak NM, Saria A, Singewald N. Substance P in the medial amygdala: emotional stress-sensitive release and modulation of anxiety-related behavior in rats. Proc Natl Acad Sci U S A. 2004;101(12):4280–4285. | |
Schank JR, Pickens CL, Rowe KE, et al. Stress-induced reinstatement of alcohol-seeking in rats is selectively suppressed by the neurokinin 1 (NK1) antagonist L822429. Psychopharmacology. 2011;218(1):111–119. | |
Thorsell A, Schank JR, Singley E, Hunt SP, Heilig M. Neurokinin-1 receptors (NK1R:s), alcohol consumption, and alcohol reward in mice. Psychopharmacology. 2010;209(1):103–111. | |
Baek MN, Jung KH, Halder D, et al. Artificial microRNA-based neurokinin-1 receptor gene silencing reduces alcohol consumption in mice. Neurosci Lett. 2010;475(3):124–128. | |
Steensland P, Simms JA, Nielsen CK, Holgate J, Bito-Onon JJ, Bartlett SE. The neurokinin 1 receptor antagonist, ezlopitant, reduces appetitive responding for sucrose and ethanol. PLoS One. 2010;5(9):e12527. | |
Jensen CJ, Gerard NP, Schwartz TW, Gether U. The species selectivity of chemically distinct tachykinin nonpeptide antagonists is dependent on common divergent residues of the rat and human neurokinin-1 receptors. Mol Pharmacol. 1994;45(2):294–299. | |
Hrabovszky E, Borsay BA, Racz K, et al. Substance P immunoreactivity exhibits frequent colocalization with kisspeptin and neurokinin B in the human infundibular region. PloS One. 2013;8(8):e72369. | |
Deacon CF, Agoston DV, Nau R, Conlon JM. Conversion of neuropeptide K to neurokinin A and vesicular colocalization of neurokinin A and substance P in neurons of the guinea pig small intestine. J Neurochem. 1987;48(1):141–146. | |
Nagano M, Oishi T, Suzuki H. Distribution and pharmacological characterization of primate NK-2 tachykinin receptor in the central nervous system of the rhesus monkey. Neurosci Lett. 2011;503(1):23–26. | |
Steinberg R, Alonso R, Griebel G, et al. Selective blockade of neurokinin-2 receptors produces antidepressant-like effects associated with reduced corticotropin-releasing factor function. J Pharmacology Exp Ther. 2001;299(2):449–458. | |
Leggio L, Cardone S, Ferrulli A, et al. Turning the clock ahead: potential preclinical and clinical neuropharmacological targets for alcohol dependence. Curr Pharm Des. 2010;16(19):2159–2118. | |
Herman JP, Guillonneau D, Dantzer R, Scatton B, Semerdjian-Rouquier L, Le Moal M. Differential effects of inescapable footshocks and of stimuli previously paired with inescapable footshocks on dopamine turnover in cortical and limbic areas of the rat. Life Sci. 1982;30(25):2207–2214. | |
Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159(10):1642–1652. | |
Hutson PH, Patel S, Jay MT, Barton CL. Stress-induced increase of cortical dopamine metabolism: attenuation by a tachykinin NK1 receptor antagonist. Eur J Pharmacol. 2004;484(1):57–64. | |
Morley JE, Yamada T, Walsh JH, et al. Morphine addiction and withdrawal alters brain peptide concentrations. Life Sci. 1980;26(26):2239–2244. | |
Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277(5332):1684–1687. | |
Kovacs KJ. Microglia and drug-induced plasticity in reward-related neuronal circuits. Front Mol Neurosci. 2012;5:74. | |
Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun. 2011;25(Suppl 1):S4–S12. | |
Guo CJ, Douglas SD, Gao Z, et al. Interleukin-1beta upregulates functional expression of neurokinin-1 receptor (NK-1R) via NF-kappaB in astrocytes. Glia. 2004;48(3):259–266. | |
Rasley A, Bost KL, Olson JK, Miller SD, Marriott I. Expression of functional NK-1 receptors in murine microglia. Glia. 2002;37(3):258–267. | |
Yang J, Yu J, Jia X, et al. Inhibition of nuclear factor-kappaB impairs reconsolidation of morphine reward memory in rats. Behav Brain Res. 2011;216(2):592–596. | |
Backus KH, Berger T, Kettenmann H. Activation of neurokinin receptors modulates K+ and Cl- channel activity in cultured astrocytes from rat cortex. Brain Res. 1991;541(1):103–109. | |
Rimon R, Le Greves P, Nyberg F, Heikkila L, Salmela L, Terenius L. Elevation of substance P-like peptides in the CSF of psychiatric patients. Biol Psychiatry. 1984;19(4):509–516. | |
Hargreaves R. Imaging substance P receptors (NK1) in the living human brain using positron emission tomography. J Clin Psychiatry. 2002;63:18–24. | |
Hietala J, Nyman MJ, Eskola O, et al. Visualization and quantification of neurokinin-1 (NK1) receptors in the human brain. Mol Imaging Biol. 2005;7(4):262–272. | |
Whitty CJ, Paul MA, Bannon MJ. Neurokinin receptor mRNA localization in human midbrain dopamine neurons. J Comp Neurol. 1997;382(3):394–400. | |
Okumura M, Arakawa R, Ito H, et al. Quantitative analysis of NK1 receptor in the human brain using PET with 18F-FE-SPA-RQ. J Nucl Med. 2008;49(11):1749–1755. | |
Saji K, Ikeda Y, Kim W, et al. Acute NK(1) receptor antagonist administration affects reward incentive anticipation processing in healthy volunteers. Int J Neuropsychopharmacol. 2013;16(7):1461–1471. | |
Blaine S, Claus E, Harlaar N, Hutchison K. TACR1 genotypes predict fMRI response to alcohol cues and level of alcohol dependence. Alcohol Clin Exp Res. 2013;37(Suppl 1):E125–E130. | |
Seneviratne C, Ait-Daoud N, Ma JZ, Chen G, Johnson BA, Li MD. Susceptibility locus in neurokinin-1 receptor gene associated with alcohol dependence. Neuropsychopharmacology. 2009;34(11):2442–2449. | |
Heilig M, Goldman D, Berrettini W, O’Brien CP. Pharmacogenetic approaches to the treatment of alcohol addiction. Nat Rev Neurosci. 2011;12(11):670–684. | |
Anton RF, Oroszi G, O’Malley S, et al. An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the Combined Pharmacotherapies and Behavioral Interventions for Alcohol Dependence (COMBINE) study. Arch Gen Psychiatry. 2008;65(2):135–144. | |
Walsh SL, Heilig M, Nuzzo PA, Henderson P, Lofwall MR. Effects of the NK1 antagonist, aprepitant, on response to oral and intranasal oxycodone in prescription opioid abusers. Addict Biol. 2013;18(2):332–343. | |
George DT, Gilman J, Hersh J, et al. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science. 2008;319(5869):1536–1539. | |
Kwako LE, George DT, Schwandt ML, et al. The neurokinin-1 receptor antagonist aprepitant in co-morbid alcohol dependence and posttraumatic stress disorder: a human experimental study. Psychopharmacology. 2015;232(1):295–304. | |
Ridler K, Gunn RN, Searle GE, et al. Characterising the plasma-target occupancy relationship of the neurokinin antagonist GSK1144814 with PET. J Psychopharmacol. 2014;28(3):244–253. | |
Largent-Milnes TM, Yamamoto T, Nair P, et al. Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. Br J Pharmacol. 2010;161(5):986–1001. | |
Yamamoto T, Nair P, Jacobsen NE, et al. The importance of micelle-bound states for the bioactivities of bifunctional peptide derivatives for delta/mu opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2008;51(20):6334–6347. | |
Sandweiss A, Stark J, Largent-Milnes, TM, et al. An opioid agonist/NK1 antagonist inhibits nociception in an animal model of neuropathic pain while lacking accumbens dopamine release. International Narcotics Research Conference; 2015; Phoenix, AZ. | |
Ingi T, Kitajima Y, Minamitake Y, Nakanishi S. Characterization of ligand-binding properties and selectivities of three rat tachykinin receptors by transfection and functional expression of their cloned cDNAs in mammalian cells. J Pharmacol Exp Ther. 1991;259(3):968–975. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.