Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
The origin, dynamics, and molecular evolution of transmissible cancers
Authors Jones E, Cheng Y, Belov K
Received 31 March 2015
Accepted for publication 4 June 2015
Published 21 September 2015 Volume 2015:5 Pages 317—326
DOI https://doi.org/10.2147/AGG.S61298
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr John Martignetti
Elizabeth A Jones, Yuanyuan Cheng, Katherine Belov
Faculty of Veterinary Science, University of Sydney, NSW, Australia
Abstract: Three transmissible cancers are known to have emerged naturally in the wild: canine transmissible venereal tumor (CTVT); Tasmanian devil facial tumor disease (DFTD); and a recently discovered leukemia-like cancer in soft-shell clams (Mya arenaria). These cancers have all acquired the ability to pass between individuals. DFTD emerged approximately 20 years ago and has decimated the Tasmanian devil population. CTVT arose over 10,000 years ago in an ancient breed of dog. The clam cancer is believed to have evolved at least 40 years ago. In this manuscript, we review CTVT and DFTD, the two transmissible mammalian cancers, and provide an overview of the leukemia-like cancer of clams. We showcase how genetics and genomics have enhanced our understanding of the unique biology, origins, and evolutionary histories of these rare cancers.
Keywords: transmissible cancer, devil facial tumor disease, DFTD, canine transmissible venereal tumor, origin, evolution
Introduction
Cancer is a group of diseases that arises due to the accumulation of mutations in oncogenes or tumor suppressor genes resulting in abnormal cell proliferation.1,2 In common cases, cancer is not contagious and only limited to the individual that it arises from. Yet, three cases of naturally occurring transmissible cancers have been found: canine transmissible venereal tumor (CTVT), a sexually transmitted cancer of dogs (Canis familiaris);3 Tasmanian devil facial tumor disease (DFTD) that is transmitted between Tasmanian devils (Sarcophilus harrisii) by biting;4,5 and a leukemia-like cancer in soft-shell clams (Mya arenaria).6 Here, we review CTVT and DFTD, the two cases of contagious cancers that have been extensively studied. We provide a brief summary of the clam cancer as, in April 2015, the first paper was published revealing its clonal origin. The genetic and genomic investigations into DFTD and CTVT will provide the framework for future research into clam cancer.
CTVT and DFTD tumor cells have acquired the ability to escape the confines of the host in which they originally arose and to spread between individuals by overcoming the host immune barriers that normally reject nonself cells.7 Both cancers have escaped the physical and evolutionary confines of a single host and embarked on their own unique evolutionary paths, making them excellent models to study tumor evolution. In the last decade, comparative immunology,8 cytogenetics,9,10 genetics, and genomics studies11–13 have allowed us to gain significant ground in characterizing these cancers, contributing to our understanding of their biology, origins, and evolutionary diversity. The sequencing of the dog genome, devil genome,11,12 CTVT,13 and DFTD12 genomes have particularly driven this surge in interest in these cancers. Here, we review the main findings of this research.
An overview of CTVT and DFTD
DFTD and CTVT have a number of similarities, but they also have distinctive histories, pathologies, and impacts on their host species. Here, we provide a short overview of these rare cancers (summary provided in Table 1).
 | Table 1 Comparison between CTVT and DFTD |
CTVT (also called Stickers sarcoma) was first reported in 19th century in London.14 It was the first tumor to be experimentally transmitted, and at the time was considered an exciting breakthrough in oncology.3 Since then, hundreds of CTVT cases have been reported and CTVT is currently distributed worldwide (reviewed in Das and Das3 and Strakova and Murchison14).
Sexual transmission during coitus is the main mode of CTVT transmission.3,15 Tumor cells can also be spread by licking and sniffing tumor-affected areas.3 CTVT affects sexually active dogs of all breeds and ages.3,16 It has been experimentally transferred to wolves, coyotes, red foxes, and jackals,15,17–20 but not to other common lab animals, demonstrating its specificity for Canidae. Despite CTVT’s potential to be transmitted to other canids experimentally, CTVT has never been reported to naturally occur in any wild dog population.14 This may be due to the absence of CTVT cases being reported in wild canids or, alternatively, that CTVT does not naturally cross the species barrier.
In both sexes, tumors often present as nodular masses found on the external genitalia.21,22 They may also be present in the absence of genital lesion, such as on the skin or oral mucosa.3,18,23,24 Tumors first appear as small (<5 mm diameter) hyperemic papules, which fuse to form large, hemorrhagic, friable, ulcerating, cauliflower-like lesions that can exceed 15 cm in diameter.3,21,22 CTVT transmission may be facilitated by the contact maintained between male and female genital mucosa, which is commonly injured during canid mating and postcoital tie.15
In vivo experiments showed that CTVT tumors undergo three distinct phases of growth: progressive, stable, and regressive.25–28 Healthy dogs that are experimentally inoculated with CTVT undergo high levels of spontaneous regression.15,25,26 Mortality and regression rates in stray dog and wild dog populations remain unknown. In cases where the tumors regress, dogs develop immunity for life, protecting them from consequent transplantation events.29–33 Transcriptome comparison between regressive phase and growth phase CTVT tumors showed a significant link between tumor regression and the upregulation of T-cell- and cytokine-mediated immune pathways.34 Immune involvement, including the cell-mediated and humoral immune response, is involved in tumor regression which, after a period of initial CTVT growth, tips the scale back in favor of the host (reviewed in Siddle and Kaufman8). CTVT metastasis is not common in healthy dogs, occurring in only 5%–17% of cases.35 However, in immunocompromised animals36 and puppies,37 a higher rate of metastasis is observed. CTVT treatment options include chemotherapy,22,38,39 radiotherapy,40 and surgical excision.41 Chemotherapy using vincristine is regarded to be the most successful and practical therapy.39,42
The Tasmanian devil is a marsupial carnivore that has been isolated to the Australian island state of Tasmania for over 12,000 years.43 DFTD was first observed at Mt William national park in the northeast region of Tasmania in 1996.4 DFTD is characterized by large ulcerating friable tumors found predominantly on the devil’s face, neck, and within the oral cavity.4,44 DFTD affects both sexes and is observed predominantly in sexually mature devils (>2 years old).44 DFTD is entirely species-specific and has not been found in any other marsupials.45
Biting is the main mechanism by which DFTD is transmitted,4,5 but cannibalism of DFTD-infected carrion or transmission via fomites may also result in the transmission of DFTD cells.5 Direct inoculation through biting is the more plausible transmission route due to the fact that DFTD cells have a short survival time outside the host.5 Biting commonly occurs when devils mate and during “jaw wrestling” when devils communally feed.44 Interestingly, a higher incidence of DFTD is seen in animals that “bite” rather than those that are bitten,5 suggesting that more aggressive devils may be more susceptible to contracting the disease.5 A higher incidence of primary tumor formation within the oral cavity, as opposed to external facial tumors, is observed and consistent with the aggressive biter “receiving” tumor cells orally from an infected submissive animal who has ulcerating facial tumors.5 Devils often present with oral cavity wounds and avulsions, which they commonly sustain through devil to devil interaction and their consumption of tough food items like bones. Such wounds may assist in DFTD cellular transmission by compromising the oral mucosal epithelial barrier.5
It has been hypothesized that if DFTD eliminates the more aggressive devils, a less aggressive devil phenotype may result.5 Alternatively, sexual selection may favor the more aggressive dominant devils who may achieve higher rates of paternity.5 To determine if DFTD-induced directional selection is occurring in devil populations, a longitudinal data collection study assessing aggressive and submissive behavioral traits and disease prevalence would be required.
Despite a competent immune system, devils do not illicit an immune response against the tumor.46–48 Devils are able to reject skin grafts from other devils, showing that they have the ability to mount a normal graft rejection response to transplanted devil tissues.49 DFTD transplantation experiments in mice have shown that DFTD cells are allogenic and stimulate an immune response in immunocompetent mice.50 This makes it unlikely that any other animal with a competent immune system would be susceptible to DFTD. The mechanisms by which DFTD cells circumnavigate the devils’ immune system are not completely understood.
DFTD is untreatable and it causes almost 100% mortality within 6–9 months from the first appearance of tumors.4,51 Devils often die due to starvation or metastasis, which occurs in 65% of cases.44 Devil populations have plummeted since DFTD emerged. Before DFTD was discovered, the devil population numbered approximately 150,000 devils.52 The overall population has now declined by 84%.53 In some areas, local population declines exceed 95%.54 DFTD has now spread to over 75% of the state, with the disease front expanding further every year.53
DFTD follows a frequency-dependent transmission pattern akin to a sexually transmitted disease. Even in low devil density populations, DFTD transmission occurs,54,55 making it unlikely that DFTD will die out. With no current vaccine or treatment options for DFDT, the survival of the devil is currently being facilitated by the reintroduction of animals that have been bred and raised in captive populations. The release and monitoring of the “insurance population” devils is an ongoing project that aims to prevent the extinction of devils in the wild.53 A major aim of the project is that captive devils are genetically representative of the species and that greater than 95% of the genetic diversity can be maintained over the course of the breeding program.53
Mode of tumor transmission – the allograft theory
CTVT and DFTD are contagious cancers and are passed between individuals as allografts. Several lines of evidence have been found to support this hypothesis.
The first suggestion that CTVT was a transmissible cancer came from early experimental work which demonstrated that tumor could be passed between dogs.3 Cytological banding analysis of the tumor karyotypes supported this,56–59 and cytogenetic evidence also suggested that like CTVT, DFTD was passed as an allograft.60 Compared to the normal somatic cells of their respective hosts, CTVT and DFTD tumors both have undergone significant chromosomal rearrangements,9,56–58 and the rearranged karyotypes are conserved between tumors collected from different hosts.56–58,60 The normal dog karyotype consists of 78 chromosomes, including 76 acrocentric and two metacentric sex chromosomes, X and Y, while CTVT cells sampled from across the world possessed consistent and unique cytogenetic profiles with 57–59 chromosomes, including 42 acrocentric and 15–17 metacentric chromosomes.56–59 The reduced chromosome number is attributed to fusion events of smaller chromosomes that form 16–18 bi-armed derivatives.58,61
The normal diploid chromosome number of the Tasmanian devil is 14, including six pairs of autosomes and the sex chromosomes XX or XY.60 DFTD tumors have 13 chromosomes and the addition of 4–5 small marker chromosomes. Upon first appearance, the tumor karyotype appears to contain full or partial loss of three autosomes and the sex chromosomes.60 However, the genetic material from the “missing” chromosomal regions is reshuffled in DFTD, and is predominantly located on the four marker chromosomes.9
Further proof that CTVT and DFTD were each derived from a single ancestral source was obtained by genetic analyses at specific loci between matched host and tumor samples, and among the tumors themselves. Genotyping at microsatellite, mitochondrial DNA, and canine major histocompatibility complex (MHC) loci revealed that CTVT tumors are consistently genetically distinct from their hosts with only a moderate amount of diversity between tumors.62,63 A long interspersed nuclear element (LINE) insert upstream of the v-myc myelocytomatosis viral oncogene (avian) (MYC) gene was identified in all examined CTVT tumors, but it is absent in the normal dog genome.64 Similarly, the clonal nature of DFTD has been demonstrated by multilocus genetic profiling,65 showing that the tumors share identical genotypes at all examined microsatellite loci43,65 and MHC genes,65 but are genetically distinct from their hosts.
The allograft theory for CTVT and DFTD is also supported by the results of more recent whole-genome sequencing11–13 and molecular cytogenetic9,10 analyses. These studies showed that the chromosome and genome profiles of both transmissible tumors are stable and highly conserved, akin to a monophyletic origin.
Immune evasion strategies of transmissible tumors
Although CTVT and DFTD tumors are transmitted as allografts between individuals, they do not immediately trigger normal graft rejection responses.7 In both cases, epigenetic mechanisms alter cell-surface MHC expression on the surface of tumors, allowing them to slip under the radar of the immune system.8,66–69 A key difference between the two cancers is that DFTD does not undergo spontaneous regression like CTVT does. The mechanisms that drive CTVT regression are still not completely understood. The expression of MHC genes is believed to be a critical driver of CTVT tumor cell clearance.
MHC genes encode cell-surface molecules that bind and present foreign peptides to T-cells, and they therefore play a critical role in immunosurveillance, self-/nonself-recognition, and graft rejection.70,71 Complete MHC loss or downregulation of MHC expression is commonly observed in tumors72 as a mechanism to evade T-cell-mediated immune responses.73 Alteration of MHC expression profiles in tumors can be caused by genetic mutations or epigenetic changes in MHC genes or other genes associated with antigen processing and presentation.74
In CTVT, the progressive phase coincides with the downregulated expression of MHC molecules on tumor cells.68,69,75 Additionally, expression of the immunosuppressive cytokine transforming growth factor β (TGFβ), which suppresses T-cell and natural killer (NK) cell activities, also promotes tumor growth in this phase.76 During CTVT tumor regression, MHC expression increases significantly (from 5% to 30%–40% of cell expression).68,69,75,77 MHC restoration coincides with an influx of tumor-infiltrating leukocytes69,76,77 and high levels of the cytokines interferon gamma (IFNγ) and interleukin (IL)-6.75 These cytokines synergistically relieve the suppressive effect of TGFβ on NK cells and increase MHC expression.75,76
Immunohistochemistry analysis and gene expression profiling have revealed that the majority of DFTD cells do not express cell-surface MHC molecules, except in rare instances when DFTD cells are in close proximity to lymphocytes.67 The lack of MHC presentation appears to be the result of the epigenetic modification of genes involved in antigen processing beta-2 microglobulin (β2M), transporter 1 ATP binding cassette (TAP1), and transporter 1 ATP binding cassette (TAP2).67 In vitro experiments showed that the downregulation of these genes can be reversed, and that MHC expression restored after treatment with histone deacytlase inhibitor and cytokine IFNγ, which acts to upregulate transcription factors with deacytlase activity.67 This provides hope for the future development of a vaccine against the disease.
Origin and evolution of transmissible cancers
Despite the similar features stated earlier, CTVT and DFTD are highly distinct in their origin and evolutionary history.
DFTD
DFTD is a recently emerged disease,4,44 which was first seen in 1996. Transcriptome analysis suggests that it is derived from a Schwann cell or Schwann cell precursor.78 DFTD cells express the S-100 protein, vimentin (VIM), melan A (MLANA), neuron-specific enolase (NSE), chromogranin A (CHGA), and synaptophysin (SYP) genes, which are tissue-specific markers commonly expressed in neuroendocrine cells.44,79 DFTD cells express multiple genes associated with Schwann cell differentiation, including sex-determining region Y-box 10 (SOX10), sex-determining region Y-box 10 (SOX2), POU class 5 homebox 1 (POU5F1), and Jun proto-onco gene (JUN),78 as well as the myelin-specific genes, myelin protein zero (MPZ), periaxin (PRX), myelin basic protein (MBP), and peripheral myelin protein 22 (PMP22).78 Chromosome painting and gene copy number analysis revealed two copies of X chromosome marker genes in the DFTD genome9 and the absence of Y-borne genes such as sex-determining region Y (SRY),9,12 suggesting that the founder animal that DFTD arose in was a female. The DFTD genome also has a comparable number of X chromosome-derived single-base substitution variants to that of the female devil genome, and twice the number of variants of the male X chromosomes. This further supports that X-borne genes present in DFTD are derived from a female homologous pair, rather than being due to somatic X gene duplication events.12
Chromosome painting and gene mapping studies suggest that chromothripsis may have been the mutational event that led to the initial development of DFTD.9 Chromothripsis is the phenomenon by which vast chromosomal rearrangements within confined genomic regions, such as a single chromosome, occur in a single catastrophic breakage fusion event.80 Chromothripsis tends to occur early in cancer development, producing a large proportion of driver mutations that results in a rapid cellular transformation.81 In the case of DFTD, chromosome 1 and chromosome X have been extensively fragmented and rearranged.9
The rearranged DFTD karyotype is remarkably stable. Four major strains have been identified based on banding techniques.82 All strains retain this basal karyotype with minor cytogenetic differences, including a variable number of marker chromosomes (between 4 and 5) and the presence of Dmins (a mini chromosome presumed to contain oncogenes).82 So far, three major strains (strains 1, 2, and 3) have been characterized using molecular cytogenetics.9 All DFTD tumors retained large regions of homology between them, and few changes were seen across the majority of chromosomes.9 Strain 1 represents the first DFTD karyotype that was originally described by banding analysis.60 Strains 2 and 3 each contains two distinct substrains (strains 2b and 2c, and 3a and 3b). Chromosomes 4, 5, and X have been progressively rearranged during the evolution of DFTD strains.9 The general conservation of chromosomal rearrangement across DFTD strains suggests that mild chromosomal instability may be tolerated within some regions,9 whereas other regions may be under selective pressure to remain conserved.
As DFTD evolves, it is likely that some variations that are either beneficial or deleterious to tumor growth, and that transmission may accumulate. Indeed, different DFTD strains have different in vitro growth rates. The distinct biological characteristics of DFTD strains have important implications for strain persistence within the population, as well as for population impacts and long-term survival of the Tasmanian devil. Distinct geographic distribution patterns have been observed for different DFTD karyotypic strains.82 Mitochondrial and nuclear DFTD genotyping reveals that there are differences in the populations of DFTD in different regions of Tasmania.12 This has provided evidence for the evolutionary dynamics and clonal expansion of DFTD, as it has spread across the state.12
Across the Tasmanian mainland, several distinct DFTD tumor subclones exist, many of which have overlapping geographic ranges.12,82 DFTs have regionally distinct population structures, with certain clones displaying dominance in some regions. This dominance may be the result of neutral processes or due to selection that favors a particular DFTD type.12
In the Forestier Peninsula, an isolated Eastern coastal region of Tasmania, all DFTD tumors cluster in a single clonal lineage, making it likely that the DFTD tumor population in this area arose from a single DFTD subclone.12 The DFTD lineages found within this region remain isolated and have not spread any further across the mainland.12 One of the Forestier Peninsula DFTD lineages, and a rise in the tetraploid form of the strain 1 tumor, has temporally increased in prevalence in this region akin to a selective sweep.12,83 In a disease suppression trial that took place at this location,84 animals with tumors were culled, and it is believed that this activity could have led to the selection of tetraploid clones83 and subclone dominancy.
The long-term consequences of a rise in DFTD tetraploid strains are not yet known.85 Tetraploidy is known to provide cells with a slower growth rate.86,87 Indeed, tetraploid DFTD tumors display the slowest in vitro growth.82 This may extend the period of host survival and provide a longer window of opportunity for DFTD transmission and evolution.
Alternatively, it is possible that tetraploid tumors, harboring higher rates of mutations, may allow the cells to better adapt and evolve.83 It could even make them more aggressive, metastatic, and drug resistant, as has been observed in some tetraploid human cancers.88,89
Future research will focus on whether DFTD is evolving in a particular trajectory. Perhaps increases in tetraploidy and a slowing down of growth rates may allow the tumor to become less aggressive. After all, if DFTD kills the entire species, it too will become extinct. The ideal situation would be for DFTD to co-evolve with the devil, and ultimately become a parasite, like CTVT.
CTVT
CTVT arose approximately 10,000–12,000 years ago, and it is the oldest clonal cell line in existence.13 A genome comparison with 86 dog breeds, wolves, and coyotes revealed that the primary tumor is likely to have arisen in an ancient dog breed that is most genetically similar to Alaskan malamutes or huskies, and is similar in size to a medium to large dog with an agouti or black coat.13 Based on a heterozygosity analysis of the tumor genome, the founder dog likely belonged to a relatively inbred population.13
It has been hypothesized that the inbred status of the dog from which CTVT arose may have been a factor that assisted in immune evasion of the original CTVT clone.13 Indeed, Tasmanian devils have low genetic diversity at microsatellite,43 nuclear,11 mitochondrial, and immune genes such as the MHC.90,91 The genetic diversity of the founder may therefore be a governing predisposing factor required for transmissible cancer to emerge and spread.
CTVT is believed to have evolved from a macrophage, as it expresses lysozyme (LYZ), alpha-1 antitripsin (SERPINA1),92 VIM,92 and NSE,93 which are all macrophage-related proteins. The histiocytic origin is also supported by the susceptibility of CTVT tumor cells to infection by Leishhmania infantum, a parasite that has a tropism for macrophage cells.23,94,95
Throughout its long evolutionary history, the CTVT genome has accumulated approximately 1.9 million somatic mutations and acquired thousands of copy number changes, retrotranposon insertions, and rearrangements.13 Evolution has resulted in two phylogenetically distinct subgroups of CTVT tumors, which have a considerable amount of lineage diversity between them.62,63 Genome sequencing and mutation analysis of two CTVT tumors showed that the two lineages have approximately 105,000 and 110,000 unique somatic mutation variants, respectively.13 Ancestral dating prediction suggested that the two CTVT variants likely last shared a common ancestor approximately 460 years ago, corresponding with the era of human exploration.13 Modern CTVT subgroups are believed to represent a recent global sweep of an ancient lineage that has successfully spread worldwide in recent times.13,63 This indicates that CTVT worldwide dissemination may have been assisted by human intervention.
CTVT may have once existed in a more virulent form and over time, CTVT may have evolved to becoming more benign. The natural regression of CTVT tumors may be an adaptive response to maintain host population viability.62 Indeed, in experimentally transferred CTVT, tumors follow a benign course with higher rates of regression and host recovery after subsequent generations.25 The ancient origin of CTVT and long propagation throughout successive hosts has provided ample opportunity for such adaption to occur. We do not know if DFTD will run a similar evolutionary trajectory to CTVT and in time adapt to coexist with devils. Considering the limited number of devils that are left in the wild, without devil reintroduction, the evolutionary potential of DFTD may be limited.
Like CTVT, where all extant CTVTs are found to be derived from two distinct lineages, over time, we may observe clonal expansion of specific DFTD strains and lineages across Tasmania.
Roles of oncogenes and tumor suppressor genes in transmissible cancers
In humans, over 500 genes96 have been associated with tumor progression and initiation across a range of different cancers. Genetic mutations, including copy number changes, truncations, gene fusions, and single-base substitutions, have been observed in a number of these genes in CTVT (Table 2) and DFTD (Table 3), and they may be of biological significance to their tumorigenic initiation and progression. A number of important tumor suppressor genes have been found mutated in the transmissible tumors (Tables 2 and 3). For instance, in CTVT, an amino acid substitution has occurred in the tumor protein 53 (TP53) gene,12,97 resulting in functional abnormality in the gene and its products.98 However, this mutation is found to be absent in some CTVT variants, and therefore not believed to be of germline origin.99 Another important mutation is the possible copy number loss of the neurofibromin 2 (NF2) gene in DFTD.9 Abnormality in this gene is commonly associated with brain tumors and schwannomas100,101 in humans. It remains to be confirmed if these genomic changes are associated with changes in gene expression and whether they have important functional roles in DFTD and CTVT.
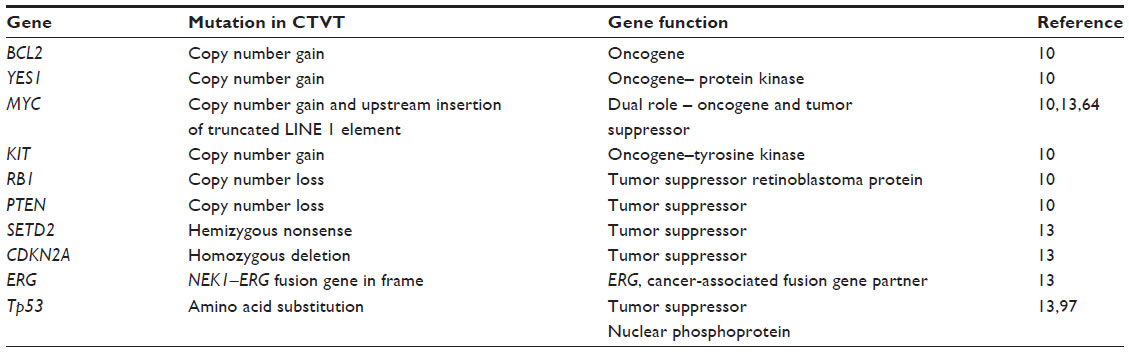 | Table 2 Putative driver gene mutations in CTVT |
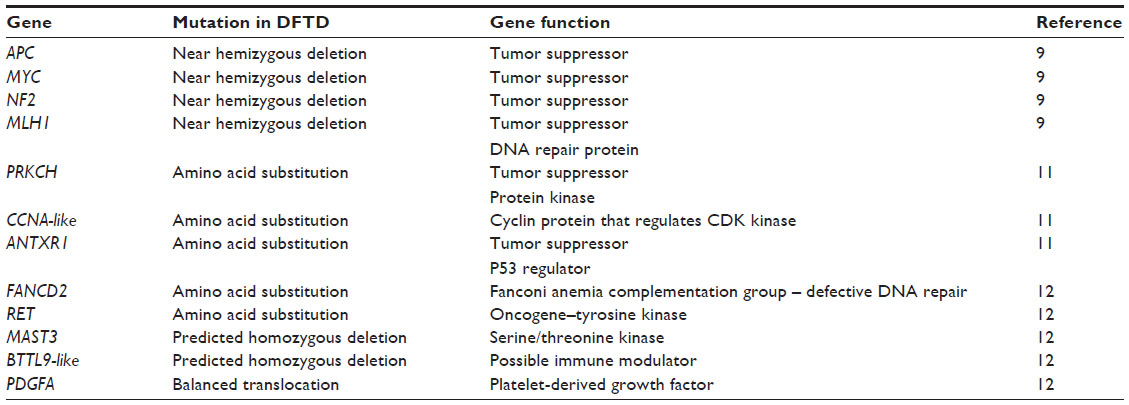 | Table 3 Putative driver gene mutations in DFTD |
Transmissible cancers in other species
In humans, the transmission of cancer between unrelated individuals is rare due to histocompatibility barriers. Examples of human transmissible cancers include mother to fetus transmission via placenta102 and organ donor to transplant recipient via artificial transmission.103 The rare cases of cancer transmission and progression are often associated with genetic similarity, lack of immune surveillance,104 or host immunosuppression.103
The only other transmissible cancer reported in mammals is the Syrian hamster sarcoma.105 This sarcoma is limited to an inbred lab population of hamsters that were derived from a single family line. The tumor is transmitted by feeding tumor tissue, cannibalism, and mosquitoes.105–108
It was recently shown that a leukemia-like cancer in soft-shell clams (M. arenaria)6 that has been devastating clam populations since the 1970s109,110 is actually transmissible. In the clam cancer, like CTVT and DFTD, the tumor genotypes are identical and distinct from those of their hosts.6 The long terminal repeat (LTR) retrotransposon element Steamer shows a correlation to disease expression111 and has been implicated as a potential tumorigenic driver of this cancer.6 Healthy clams have less than ten genome copies of Steamer compared to leukemic clams that have between 150 and 300 copies throughout their genome.6 The sessile nature of clams renders the transfer of tumor cells by physical contact not possible, which is in stark contrast to the primary transmission route of CTVT and DFTD. As filter feeders, the ingestion of leukemic tumor cells that have been shown to readily survive in the water is proposed to be the way in which tumor cells are transferred through the water. The clams affected by this leukemia are located along the North Atlantic coast and separated by hundreds of miles. Clam populations in the United States and Canada have distinct cancer subpopulations with significant divergence, indicating that the clam cancer cell lineages are continuing to evolve.6 It remains unknown whether this cancer has the potential to spread to other bivalves.6 Other clams, mussels, oysters, and cockles are known to have transmissible neoplasias. The possibility that many of these may represent independent contagious cancer lineages is now being questioned. Unlike the well-defined MHC graft rejection response in mammals, a self-/nonself-immune rejection in molluscs has never been defined.6 This may make molluscs vulnerable to allograft transfer,6 and raises significant conservation concerns for species where direct contact to spread of such neoplasia is facilitated by an aquatic environment.
Conclusion
CTVT, DFTD, and the leukemia-like clam cancer are currently the only known contagious cancers to emerge in wild populations. CTVT and DFTD have remained remarkably stable as they passed through successive generations of their respective hosts. Genetics and genomics research has aided in our understanding of these cancers by revealing how these cancers emerged and have successfully evaded immune responses to pass from animal to animal. A number of genetic alterations in genes that may have tumorigenic potential have been identified, and future research will focus on developing a deeper understanding of the function of these genes within each cancer. The genomic sequences of dogs, devils, and their tumors have provided outstanding resources for the development of genomics techniques to study in detail the evolutionary trajectories of these cancers. With time, we will learn whether contagious cancers will ultimately become more benign and be able to be rejected by the host, as in CTVT, or whether they are capable of driving a species to extinction, as was initially predicted in DFTD, but which has not yet eventuated. The recent discovery of the transmissible clam cancer has revoked CTVT and DFTD’s status as the only transmissible cancers to have emerged naturally in the wild. Indeed, it is likely that in the future, we may see a rise in the occurrence of contagious cancer, which makes it vital for us to strive to understand their evolutionary and biological dynamics. Genomics technologies will allow us to do that.
Acknowledgments
KB is funded by an ARC Future Fellowship. Our research is funded by the Australian Research Council. We thank the Save the Tasmanian Devil Program, the Zoo and Aquarium Association, and our collaborators for collaborating with us on this exciting and important conservation project.
Disclosure
The authors report no conflicts of interests in this work.
References
Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13(11):795–806. | |
Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–334. | |
Das U, Das AK. Review of canine transmissible venereal sarcoma. Vet Res Commun. 2000;24(8):545–556. | |
Hawkins CE, Baars C, Hesterman H, et al. Emerging disease and population decline of an island endemic, the Tasmanian devil Sarcophilus harrisii. Biol Conserv. 2006;131(2):307–324. | |
Hamede RK, McCallum H, Jones M. Biting injuries and transmission of Tasmanian devil facial tumour disease. J Anim Ecol. 2013; 82(1):182–190. | |
Metzger MJ, Reinisch C, Sherry J, Goff SP. Horizontal transmission of clonal cancer cells causes leukemia in soft-shell clams. Cell. 2015; 161(2):255–263. | |
Siddle HV, Kaufman J. A tale of two tumours: comparison of the immune escape strategies of contagious cancers. Mol Immunol. 2013; 55(2):190–193. | |
Siddle HV, Kaufman J. Immunology of naturally transmissible tumours. Immunology. 2015;144(1):11–20. | |
Deakin JE, Bender HS, Pearse AM, et al. Genomic restructuring in the Tasmanian devil facial tumour: chromosome painting and gene mapping provide clues to evolution of a transmissible tumour. PLoS Genet. 2012;8(2):e1002483. | |
Thomas R, Rebbeck C, Leroi AM, Burt A, Breen M. Extensive conservation of genomic imbalances in canine transmissible venereal tumors (CTVT) detected by microarray-based CGH analysis. Chromosome Res. 2009;17(7):927–934. | |
Miller W, Hayes VM, Ratan A, et al. Genetic diversity and population structure of the endangered marsupial Sarcophilus harrisii (Tasmanian devil). Proc Natl Acad Sci U S A. 2011;108(30):12348–12353. | |
Murchison EP, Schulz-Trieglaff OB, Ning Z, et al. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell. 2012;148(4):780–791. | |
Murchison EP, Wedge DC, Alexandrov LB, et al. Transmissible dog cancer genome reveals the origin and history of an ancient cell lineage. Science. 2014;343(6169):437–440. | |
Strakova A, Murchison EP. The changing global distribution and prevalence of canine transmissible venereal tumour. BMC Vet Res. 2014;10:168. | |
Cohen, D. The canine transmissible venereal tumor: a unique result of tumor progression. Adv Cancer Res. 1985;43:75–112. | |
Batamuzi EK, Kassuku AA, Agger JF. Risk factors associated with canine transmissible venereal tumour in Tanzania. Prev Vet Med. 1992; 13(1):13–17. | |
Cockrill JM, Beasley JN. Transmission of transmissible venereal tumor of the dog to the coyote. Am J Vet Res. 1979;40(3):409–410. | |
Higgins DA. Observations on the canine transmissible venereal tumour as seen in the Bahamas. Vet Rec. 1966;79(3):67–71. | |
Sticker A. Transplantables rundzellensarkom des hundes. Z Krebsforsch. 1906;4:227–314. German. | |
Wade MD. An experimental investigation of infective sarcoma of the dog, with a consideration of its relationship to cancer. J Pathol. 1908; 12(2):384–425. | |
Mello Martins MI, Ferreira de Souza F, Gobello C. The canine transmissible venereal tumor: etiology, pathology, diagnosis and treatment. In: Concannon PW, England G, Verstegen J III, Linde-Forsberg C, editors. Recent Advances in Small Animal Reproduction. Ithica, NY: International Veterinary Information Service; 2005. | |
Brown NO, Calvert C, MacEwen EG. Chemotherapeutic management of transmissible venereal tumors in 30 dogs. J Am Vet Med Assoc. 1980;176(10 Pt 1):983–986. | |
Albanese F, Poli A, Millanta F, Abramo F. Primary cutaneous extragenital canine transmissible venereal tumour with Leishmania-laden neoplastic cells: a further suggestion of histiocytic origin? Vet Dermatol. 2002;13(5):243–246. | |
Ndiritu CG, Mbogwa SW, Sayer PD. Extragenitally located transmissible venereal tumor in dogs. Mod Vet Pract. 1977;58(11):940–946. | |
Karlson AG, Mann FC. The transmissible venereal tumor of dogs: observations on forty generations of experimental transfers. Ann N Y Acad Sci. 1952;54(6):1197–1213. | |
Epstein RB, Bennett BT. Histocompatibility typing and course of canine venereal tumors transplanted into unmodified random dogs. Cancer Res. 1974;34(4):788–793. | |
Chu RM, Sun TJ, Yang HY, et al. Heat shock proteins in canine transmissible venereal tumor. Vet Immunol Immunopathol. 2001;82(1–2):9–21. | |
Hill DL, Yang TJ, Wachtel A. Canine transmissible venereal sarcoma: tumor cell and infiltrating leukocyte ultrastructure at different growth stages. Vet Pathol. 1984;21(1):39–45. | |
Cohen D. The transmissible venereal tumor of the dog – a naturally occurring allograft? A review. Isr J Med Sci. 1978;14(1):14–19. | |
Gonzalez CM, Griffey SM, Naydan DK, et al. Canine transmissible venereal tumour: a morphological and immunohistochemical study of 11 tumours in growth phase and during regression after chemotherapy. J Comp Pathol. 2000;122(4):241–248. | |
Mizuno S, Fujinaga T, Tajima M, Otomo K, Koike T. Role of lymphocytes in dogs experimentally re-challenged with canine transmissible sarcoma. Nihon Juigaku Zasshi. 1989;51(1):86–95. | |
Bennett BT III. Transplantation Immunogenetics of the Canine Transmissible Venereal Tumor [dissertation]. Chicago, IL: University of Illinois at Chicago, Health Sciences Center; 1974. | |
Cohen D. Detection of humoral antibody to the transmissible venereal tumour of the dog. Int J Cancer. 1972;10(1):207–212. | |
Chiang HC, Liao AT, Jan TR, et al. Gene-expression profiling to identify genes related to spontaneous tumor regression in a canine cancer model. Vet Immunol Immunopathol. 2013;151(3–4):207–216. | |
Rogers KS. Transmissible venereal tumor. Compend Contin Educ Vet. 1997;19:1036–1045. | |
Cohen D. The biological behaviour of the transmissible venereal tumour in immunosuppressed dogs. Eur J Cancer. 1973;9(4):253–258. | |
Yang TJ, Jones JB. Canine transmissible venereal sarcoma: transplantation studies in neonatal and adult dogs. J Natl Cancer Inst. 1973;51(6):1915–1918. | |
Javanbakht J, Pedram B, Taheriyan MR, et al. Canine transmissible venereal tumor and seminoma: a cytohistopathology and chemotherapy study of tumors in the growth phase and during regression after chemotherapy. Tumour Biol. 2014;35(6):5493–5500. | |
Said R, Silva L, Albuquerque A, Sousa-Neta E, Lavinsky M. Efficacy and side effects of vincristine sulphate treatment on canine transmissible venereal tumour. Paper presented at: Proceedings of the 34th World Small Animal Veterinary Congress; 2009; Sao Paulo, Brazil; WSAVA2009. | |
Thrall DE. Orthovoltage radiotherapy of canine transmissible venereal tumors. Veterinary Radiology. 1982;23(5):217–219. | |
Idowu AL. A retrospective evaluation of four surgical methods of treating canine transmissible venereal tumour. Journal of Small Animal Practice. 1984;25(4):193–198. | |
Nak D, Nak Y, Canqul IT, Tuna B. A clinico-pathological study on the effect of vincristine on transmissible venereal tumour in dogs. J Vet Med A Physiol Pathol Clin Med. 2005;25(7):366–370. | |
Jones ME, Paetkau D, Geffen E, Moritz C. Genetic diversity and population structure of Tasmanian devils, the largest marsupial carnivore. Mol Ecol. 2004;13(8):2197–2209. | |
Loh R, Bergfeld J, Hayes D, et al. The pathology of devil facial tumor disease (DFTD) in Tasmanian Devils (Sarcophilus harrisii). Vet Pathol. 2006;43(6):890–895. | |
McCallum H. Tasmanian devil facial tumour disease: lessons for conservation biology. Trends Ecol Evol. 2008;23(11):631–637. | |
Woods GM, Kreiss A, Belov K, Siddle HV, Obendorf DL, Muller HK. The immune response of the Tasmanian devil (Sarcophilus harrisii) and devil facial tumour disease. EcoHealth. 2007;4(3):338–345. | |
Kreiss A, Fox N, Bergfeld J, Quinn SJ, Pyecroft S, Woods GM. Assessment of cellular immune responses of healthy and diseased Tasmanian devils (Sarcophilus harrisii). Dev Comp Immunol. 2008;32(5):544–553. | |
Kreiss A, Wells B, Woods GM. The humoral immune response of the Tasmanian devil (Sarcophilus harrisii) against horse red blood cells. Vet Immunol Immunopathol. 2009;130(1–2):135–137. | |
Kreiss A, Cheng Y, Kimble F, et al. Allorecognition in the Tasmanian devil (Sarcophilus harrisii), an endangered marsupial species with limited genetic diversity. PLoS One. 2011;6(7):e22402. | |
Pinfold TL, Brown GK, Bettiol SS, Woods GM. Mouse model of devil facial tumour disease establishes that an effective immune response can be generated against the cancer cells. Front Immunol. 2014;5:251. | |
Lachish S, Jones M, McCallum H. The impact of disease on the survival and population growth rate of the Tasmanian devil. J Anim Ecol. 2007;76(5):926–936. | |
Threatened Species Scientific Committee [webpage on the Internet]. Commonwealth listing advice on Sarcophilus harrisii (Tasmanian devil). Parkes ACT, Australia: Australian Government, Department of the Environment; 2006. Available from: http://www.environment.gov.au/biodiversity/threatened/species/sarcophilus-harrisii.html. Accessed February 15, 2015. | |
Department of Primary Industries, Parks, Water and Environment. DRAFT Recovery Plan for the Tasmanian Devil (Sarcophilus harrisii). Hobart, Australia: Department of Primary Industries, Parks, Water and Environment; 2010. | |
McCallum H, Tompkins DM, Jones M, et al. Distribution and impacts of Tasmanian devil facial tumor disease. EcoHealth. 2007;4(3):318–325. | |
McCallum H, Jones M, Hawkins C, et al. Transmission dynamics of Tasmanian devil facial tumor disease may lead to disease-induced extinction. Ecology. 2009;90(12):3379–3392. | |
Barski G, Cornefert-Jensen F. Cytogenetic study of Sticker venereal sarcoma in European dogs. J Natl Cancer Inst. 1966;37(6):787–797. | |
Oshimura M, Sasaki M, Makino S. Chromosomal banding patterns in primary and transplanted venereal tumors of the dog. J Natl Cancer Inst. 1973;51(4):1197–1203. | |
Fujinaga T, Yamashita M, Yoshida MC, et al. Chromosome analysis of canine transmissible sarcoma cells. Zentralbl Veterinarmed A. 1989; 36(7):481–489. | |
Mukaratirwa S, Gruys E. Canine transmissible venereal tumour: cytogenetic origin, immunophenotype, and immunobiology. A review. Vet Q. 2003;25(3):101–111. | |
Pearse AM, Swift K. Allograft theory: transmission of devil facial-tumour disease. Nature. 2006;439(7076):549. | |
Makino S. Some epidemiologic aspects of venereal tumors of dogs as revealed by chromosome and DNA studies. Ann N Y Acad Sci. 1963;108:1106–1122. | |
Murgia C, Pritchard JK, Kim SY, Fassati A, Weiss RA. Clonal origin and evolution of a transmissible cancer. Cell. 2006;126(3):477–487. | |
Rebbeck CA, Thomas R, Breen M, Leroi AM, Burt A. Origins and evolution of a transmissible cancer. Evolution. 2009;63(9):2340–2349. | |
Katzir N, Rechavi G, Cohen JB, et al. “Retroposon” insertion into the cellular oncogene c-myc in canine transmissible venereal tumor. Proc Natl Acad Sci U S A. 1985;82(4):1054–1058. | |
Siddle HV, Kreiss A, Eldridge MD, et al. Transmission of a fatal clonal tumor by biting occurs due to depleted MHC diversity in a threatened carnivorous marsupial. Proc Natl Acad Sci U S A. 2007; 104(41):16221–16226. | |
Cohen D, Shalev A, Krup M. Lack of beta 2-microglobulin on the surface of canine transmissible venereal tumor cells. J Natl Cancer Inst. 1984;72(2):395–401. | |
Siddle HV, Kreiss A, Tovar C, et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc Natl Acad Sci U S A. 2013;110(13):5103–5108. | |
Yang TJ, Chandler JP, Dunne-Anway S. Growth stage dependent expression of MHC antigens on the canine transmissible venereal sarcoma. Br J Cancer. 1987;55(2):131–134. | |
Hsiao YW, Liao KW, Hung SW, Chu RM. Effect of tumor infiltrating lymphocytes on the expression of MHC molecules in canine transmissible venereal tumor cells. Vet Immunol Immunopathol. 2002; 87(1–2):19–27. | |
Trowsdale J. The MHC disease and selection. Immunol Lett. 2011; 137(1–2):1–8. | |
Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001. The major histocompatibility complex and its functions. | |
Garcia-Lora A, Algarra I, Gaforio JJ, Ruiz-Cabello F, Garrido F. Immunoselection by T lymphocytes generates repeated MHC class I-deficient metastatic tumor variants. Int J Cancer. 2001;91(1):109–119. | |
Maudsley DJ, Pound JD. Modulation of MHC antigen expression by viruses and oncogenes. Immunol Today. 1991;12(12):429–431. | |
Ting JP, Baldwin AS. Regulation of MHC gene expression. Curr Opin Immunol. 1993;5(1):8–16. | |
Hsiao YW, Liao KW, Chung TF, Liu CH, Hsu CD, Chu RM. Interactions of host IL-6 and IFN-gamma and cancer-derived TGF-beta1 on MHC molecule expression during tumor spontaneous regression. Cancer Immunol Immunother. 2008;57(7):1091–1104. | |
Hsiao YW, Liao KW, Hung SW, Chu RM. Tumor-infiltrating lymphocyte secretion of IL-6 antagonizes tumor-derived TGF-beta 1 and restores the lymphokine-activated killing activity. J Immunol. 2004; 172(3):1508–1514. | |
Pérez J, Day MJ, Mozos E. Immunohistochemical study of the local inflammatory infiltrate in spontaneous canine transmissible venereal tumour at different stages of growth. Vet Immunol Immunopathol. 1998; 64(2):133–147. | |
Murchison EP, Tovar C, Hsu A, et al. The Tasmanian devil transcriptome reveals Schwann cell origins of a clonally transmissible cancer. Science. 2010;327(5961):84–87. | |
Loh R, Hayes D, Mahjoor A, O’Hara A, Pyecroft S, Raidal S. The immunohistochemical characterization of devil facial tumor disease (DFTD) in the Tasmanian Devil (Sarcophilus harrisii). Vet Pathol. 2006;43(6):896–903. | |
Jones MJ, Jallepalli PV. Chromothripsis: chromosomes in crisis. Dev Cell. 2012;23(5):908–917. | |
Forment JV, Kaidi A, Jackson SP. Chromothripsis and cancer: causes and consequences of chromosome shattering. Nat Rev Cancer. 2012; 12(10):663–670. | |
Pearse AM, Swift K, Hodson P, et al. Evolution in a transmissible cancer: a study of the chromosomal changes in devil facial tumor (DFT) as it spreads through the wild Tasmanian devil population. Cancer Genet. 2012;205(3):101–112. | |
Ujvari B, Pearse AM, Swift K, et al. Anthropogenic selection enhances cancer evolution in Tasmanian devil tumours. Evol Appl. 2014; 7(2):260–265. | |
Lachish S, McCallum H, Mann D, Pukk CE, Jones ME. Evaluation of selective culling of infected individuals to control Tasmanian devil facial tumor disease. Conserv Biol. 2010;24(3):841–851. | |
Ujvari B, Pearse AM, Peck S, et al. Evolution of a contagious cancer: epigenetic variation in Devil Facial Tumour Disease. Proc Biol Sci. 2013;280(1750):20121720. | |
Otto SP. The evolutionary consequences of polyploidy. Cell. 2007;131(3):452–462. | |
Cavalier-Smith T. Nuclear volume control by nucleoskeletal DNA, selection for cell volume and cell growth rate, and the solution of the DNA C-value paradox. J Cell Sci. 1978;34:247–278. | |
Castedo M, Coquelle A, Vitale I, et al. Selective resistance of tetraploid cancer cells against DNA damage-induced apoptosis. Ann N Y Acad Sci. 2006;1090:35–49. | |
Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011;27:585–610. | |
Siddle HV, Marzec J, Cheng Y, Jones M, Belov K. MHC gene copy number variation in Tasmanian devils: implications for the spread of a contagious cancer. Proc Biol Sci. 2010;277(1690):2001–2006. | |
Cheng Y, Sanderson C, Jones M, Belov K. Low MHC class II diversity in the Tasmanian devil (Sarcophilus harrisii). Immunogenetics. 2012; 64(7):525–533. | |
Mozos E, Méndez A, Gómez-Villamandos JC, Martín De Las Mulas J, Pérez J. Immunohistochemical characterization of canine transmissible venereal tumor. Vet Pathol. 1996;33(3):257–263. | |
Marchal T, Chabanne L, Kaplanski C, Rigal D, Magnol JP. Immunophenotype of the canine transmissible venereal tumour. Vet Immunol Immunopathol. 1997;57(1–2):1–11. | |
Carreira VS, Ferrari HF, Langohr IM, et al. Leishmania sp. amastigotes identification in canine transmissible venereal tumor. Case Reports in Veterinary Medicine. 2014;(2014):1–4. | |
Catone G, Marino G, Poglayen G, Gramiccia M, Ludovisi A, Zanghi A. Canine transmissible venereal tumour parasitized by Leishmania infantum. Vet Res Commun. 2003;27(7):549–553. | |
Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. | |
Choi YK, Kim CJ. Sequence analysis of canine LINE-1 elements and p53 gene in canine transmissible venereal tumor. J Vet Sci. 2002; 3(4):285–292. | |
Moro JV, Tinucci-Costa M, Silveira ACT, Gerardi DG, Alessi AC. Reactivity of p53 protein in canine transmissible venereal tumor. Arquivo Brasileiro de Medicina Veterinária e Zootecnia. 2010;62(2):318–323. | |
Vázquez-Mota N, Simón-Martínez J, Córdova-Alarcon E, Lagunes L, Fajardo R. The T963C mutation of TP53 gene does not participate in the clonal origin of canine TVT. Vet Res Commun. 2008;32(2):187–191. | |
Hilton DA, Hanemann CO. Schwannomas and their pathogenesis. Brain Pathol. 2014;24(3):205–220. | |
Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet. 2009;373(9679):1974–1986. | |
Myron Kauffman H, McBride MA, Cherikh WS, Spain PC, Marks WH, Roza AM. Transplant tumor registry: donor related malignancies. Transplantation. 2002;74(3):358–362. | |
Welsh JS. Contagious cancer. Oncologist. 2011;16(1):1–4. | |
Isoda T, Ford AM, Tomizawa D, et al. Immunologically silent cancer clone transmission from mother to offspring. Proc Natl Acad Sci U S A. 2009;106(42):17882–17885. | |
Brindley DC, Banfield WG. A contagious tumor of the hamster. J Natl Cancer Inst. 1961;26:949–957. | |
Banfield WG, Woke PA, Mackay CM, Cooper HL. Mosquito transmission of a reticulum cell sarcoma of hamsters. Science. 1965; 148(3674):1239–1240. | |
Copper HL, Mackay CM, Banfield WG. Chromosome studies of a contagious reticulum cell sarcoma of the Syrian hamster. J Natl Cancer Inst. 1964;33:691–706. | |
Ashbel R. Spontaneous transmissible tumors in the Syrian hamster. Nature. 1945;155:607. | |
Brown RS, Wolke RE, Saila SB, Brown CW. Prevalence of neoplasia in 10 New England populations of the soft-shell clam (Mya arenaria). Ann N Y Acad Sci. 1978;298:522–534. | |
Yevich PP, Barszcz CA. Neoplasia in soft-shell clams (My a arenaria) collected from oil-impacted sites. Ann N Y Acad Sci. 1978;298:409–426. | |
Arriagada G, Metzger MJ, Muttray AF, et al. Activation of transcription and retrotransposition of a novel retroelement, Steamer, in neoplastic hemocytes of the mollusk Mya arenaria. Proc Natl Acad Sci U S A. 2014;111(39):14175–14180. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.