Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
The noradrenergic paradox: implications in the management of depression and anxiety
Authors Montoya A, Bruins R, Katzman M, Blier P
Received 9 July 2015
Accepted for publication 10 December 2015
Published 1 March 2016 Volume 2016:12 Pages 541—557
DOI https://doi.org/10.2147/NDT.S91311
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Alonso Montoya,1 Robert Bruins,1 Martin A Katzman,2 Pierre Blier3
1Eli Lilly Canada Inc, 2START Clinic for the Mood and Anxiety Disorders, Toronto, 3Mood Disorders Research Unit, Institute of Mental Health Research, University of Ottawa, Ottawa, ON, Canada
Abstract: Both major depressive disorder and the anxiety disorders are major causes of disability and markedly contribute to a significant global burden of the disease worldwide. In part because of the significant socioeconomic burden associated with these disorders, theories have been developed to specifically build clinical treatment approaches. One such theory, the monoaminergic hypothesis, has led to the development of several generations of selective and nonselective inhibitors of transporters of serotonin and norepinephrine, with the goal of augmenting monoaminergic transmission. These efforts have led to considerable success in the development of antidepressant therapeutics. However, there is a strong correlation between enhanced noradrenergic activity and fear and anxiety. Consequently, some physicians have expressed concerns that the same enhanced noradrenergic activity that alleviates depression could also promote anxiety. The fact that the serotonergic and noradrenergic reuptake inhibitors are successfully used in the treatment of anxiety and panic disorders seems paradoxical. This review was undertaken to determine if any clinical evidence exists to show that serotonergic and noradrenergic reuptake inhibitors can cause anxiety. The PubMed, EMBASE, and Cochrane Library databases were searched, and the results limited to randomized, double-blind, placebo-controlled studies performed in nongeriatric adults and with clear outcome measures were reported. Based on these criteria, a total of 52 studies were examined. Patients in these studies suffered from depression or anxiety disorders (generalized and social anxiety disorders, panic disorder, and posttraumatic stress disorder). The large majority of these studies employed venlafaxine or duloxetine, and the remainder used tricyclic antidepressants, atomoxetine, or reboxetine. All the studies reported clinically significant alleviation of depressive and/or anxious symptoms by these therapeutics. In none of these studies was anxiety a treatment-emergent adverse effect. This review argues against the impression that enhanced generalized noradrenergic activity promotes the emergence of anxiety.
Keywords: anxiety, atomoxetine, desvenlafaxine, duloxetine, monoamine, norepinephrine reuptake inhibitor, norepinephrine transporter
Introduction
Major depressive disorder (MDD) continues to exert a tremendous socioeconomic cost worldwide. A 2013 analysis of data obtained from the Global Burden of Diseases, Injuries, and Risk Factors Study 2010 found that mental and substance abuse disorders accounted for 7.4% of the global burden of disease; MDD alone represented 40% of this burden.1 The anxiety disorders, which include generalized anxiety disorder (GAD), panic disorder, posttraumatic stress disorder (PTSD), social anxiety disorder, and simple phobias, follow MDD and represent 14.6% of the burden of disease attributed to mental health and substance abuse.1
The mid-1950s ushered in an era of intense interest in the treatment of mental disorders, thanks to the serendipitous discoveries of lithium’s ability to treat bipolar disorder and chlorpromazine’s ability to treat schizophrenia.2,3 Likewise, interest in the fundamental mechanisms underlying MDD and its management grew from two revolutionary observations that ultimately led to the formulation of a monoaminergic hypothesis of depressive disorders. The first of these findings took place with the development of iproniazid for the treatment of tuberculosis, in which depressed tuberculosis patients undergoing clinical trials with iproniazid were found to have an elevation in their mood. Subsequently, iproniazid became the first clinically useful antidepressant.4 Second, imipramine, a chemical congener of chlorpromazine, developed as an antipsychotic medication and later was revealed to have antidepressant properties during its clinical trials.4
Subsequent discoveries verified that iproniazid inhibited monoamine oxidase (MAO), while imipramine blocked the neuronal reuptake of serotonin (5-hydroxytryptamine [5-HT]) and norepinephrine (NE).4 Both of these mechanisms lead to increased concentrations of NE and 5-HT,4 with the MAO enzyme being important in the catabolism of NE and reuptake of 5-HT and NE acting to terminate the synaptic activity of these biogenic amines.5 Thus, the inhibition of the activity of the NE transporters (NETs) (Figures 1 and 2) and serotonin transporters (SERTs) or of MAO can prolong the duration during with which these neurotransmitters are available in the synaptic cleft.
 | Figure 1 Illustration of presynaptic and postsynaptic noradrenergic receptors. |
 | Figure 2 NETs and synaptic function in noradrenergic transmission. |
Contemporaneous studies in the mid-1950s with the antihypertensive agent reserpine suggested that it produced depression by the depletion of biogenic amines.4,6 Collectively, these observations led to the formation of the monoaminergic hypothesis of depression, which stated that depression was likely due to an absolute or relative deficiency of NE and 5-HT.7,8 Over the years, the monoaminergic hypothesis of depression has been revised to include changes in the sensitivity of noradrenergic and serotonergic receptors and to include a possible role for dopamine in depression.4,9,10
The monoaminergic hypothesis is the underlying basis for much of drug development aimed at the management of MDD. In addition to their antidepressant activities, the MAO inhibitors and tricyclic antidepressants have many adverse or undesired effects due to their interactions with several other neurotransmitter systems. Efforts to reduce these unwanted side effects led to the development of the “atypical” monoamine reuptake inhibitors and, more recently, the selective serotonin reuptake inhibitors ([SSRIs]; eg, fluoxetine, citalopram), selective NE reuptake inhibitors ([NERI]; eg, atomoxetine, reboxetine), and the mixed serotonergic and noradrenergic reuptake inhibitors (SNRIs; eg, venlafaxine, duloxetine, milnacipran, desvenlafaxine).4,9,11,12
The three monoamines – dopamine, 5-HT, and NE – have specific distributions in the central nervous system and consequently exert differential effects on several neurologic functions. The major sources of dopamine in the central nervous system are within neurons of the substantia nigra (A8 and A9) and the ventral tegmental area (VTA; A10).13 The projections from the substantia nigra to the striatum regulate motor control, and degradation of this dopaminergic function leads to Parkinsonism.13 The bulk of dopaminergic projections from the ventral tegmental area are to the nucleus accumbens and the prefrontal cortex (PFC) and constitute an important part of the reward and motivational circuitry.13 In addition, there are dopaminergic projections to the amygdala. However, the role of dopamine in depression is neither as well known nor as prominent as the contribution of 5-HT and NE. None of the currently used antidepressants were designed with the idea of blocking dopamine reuptake.
Serotonergic neurons arise from the raphe nuclei, principally from the nucleus raphe dorsalis and the nucleus raphe magnus. There are extensive serotonergic projections to the amygdala, nucleus accumbens, medial forebrain bundle, PFC, thalamus, and hypothalamus.14,15 Accordingly, 5-HT is intimately involved in the regulation of limbic function and is found in many regions that are associated with motivation as well as emotional and stress responses. The SSRI antidepressants (eg, fluoxetine, fluvoxamine, paroxetine, sertraline, citalopram, and escitalopram), which target the reuptake of 5-HT and enhance serotonergic transmission, achieved considerable success against MDD.16 However, they are not fully effective in many patients. Moreover, although the SSRIs can alleviate MDD, they also can still leave troublesome residual symptoms, such as difficulty sleeping, anxiety, impaired cognition, and pain.11,17 Interestingly, these residual symptoms have been reported to be relieved in some cases by the use of an SNRI.11,12 Moreover, the use of an SNRI alone has been shown, in some cases, to improve treatment outcomes in patients with depression compared to SSRIs. For example, switching patients with MDD who were unresponsive to fluoxetine to duloxetine produced improvements in depression.18,19 These observations suggest that alterations in 5-HT transmission alone may be insufficient in some people to fully treat MDD and point to the likelihood of complex interactions among the monoaminergic transmission systems in modulating mood and a sense of well-being.11,12 In support of this possibility, it is important to emphasize that SNRIs should be used at regimens that reliably engage NET to obtain optimal benefits. In the case of venlafaxine, it is 225 mg/day, and for duloxetine, it is 120 mg/day.20–22
Most noradrenergic projections in the brain arise from the locus coeruleus (LC; ie, A6) and the remainder from the pontine (ie, A4, A5, and A7 or Kölliker-Füse nuclei) and the A1 and A2 nuclei of the lateral tegmental area.5 The noradrenergic innervations of the hippocampus, neocortex, and cerebellum originate almost exclusively from the LC. Other regions, such as the hypothalamus and the amygdala, receive overlapping innervations from the LC via the dorsal noradrenergic bundle and the A5 and A7 through the ventral noradrenergic bundle.5 These nuclei also send projections to medullary and midbrain regions that regulate autonomic functions, such as the hypothalamus, as well as to the limbic structures, such as the amygdala.5,23 Moreover, neurons of the LC are hyperactive during conditions of anxiety and opiate withdrawal. Excitation of the LC is implicated in the enhanced activity of the amygdala and manifestations of stress, fear, and anxiety responses.5,23
Since enhanced central noradrenergic activity is associated with the activation of fear and anxiety circuitries, one may surmise that manipulations that enhance noradrenergic transmission would promote these behaviors. This mechanism has led to the perception among some clinical practitioners that SNRIs and NERIs might be anxiogenic, at least during the initial phase of therapy.11,24,25 However, the SNRIs are employed with considerable success in the treatment of MDD and anxiety disorders, and there is thus far no evidence to support the notion that these therapeutic agents promote anxiety if gradually titrated. This apparent disconnect between enhanced noradrenergic transmission without enhancing anxiety is sometimes referred to as the “noradrenergic paradox.”11 We undertook the present review of the literature to determine if there is indeed a risk of precipitating anxiety in patients with mental illness, especially MDD, and anxiety disorders, and being treated with SNRIs or NERIs. The relative preference for NET and SERT of some clinically employed SNRIs and NERIs is summarized in Table 1.
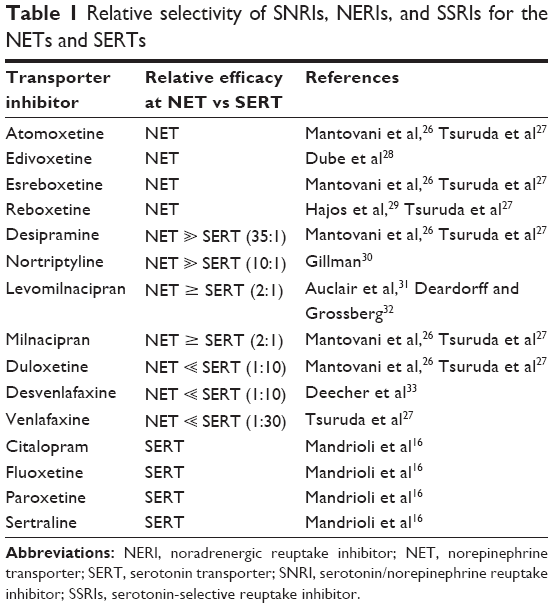 | Table 1 Relative selectivity of SNRIs, NERIs, and SSRIs for the NETs and SERTs |
Methods
Search of the literature
Literature searches were performed with PubMed, EMBASE, and the Cochrane Library on February 27, 2014. The search was updated on August 13, 2015. This review follows guidelines proposed by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses Group.34,35 Original articles, including research abstracts, were included. In those instances in which an aggregate study and the original studies in the aggregate were found, the combined study was excluded and the original studies were included to avoid duplication and to preserve the integrity of the original findings.
Search strategy
The search strategy employed is summarized in Figure 3. The search was performed with the generic and the preregistration designation when available. When several spellings were possible, all permutations of the drug names were used. The search was performed specifically for SNRI and NERI drugs, including atomoxetine, edivoxetine, esreboxetine, reboxetine, milnacipran, levomilnacipran, desipramine, nortriptyline, desvenlafaxine, duloxetine, and venlafaxine. Alternate spellings and company drug codes were also used. The searches by drug name were combined with searches for depressive disorder and for anxiety and anxiety disorder. Wildcard characters and the “exp” shortcut, to “explode” the search subject headings, were used to maximize search results. The search results were then filtered for randomized trials and for placebo.
 | Figure 3 Search strategy. |
Inclusion/exclusion criteria
The abstracts obtained from the searches were screened to confirm that they described placebo-controlled, randomized clinical trials and that the trials were related to the treatment of anxiety or mentioned their effect on anxiety when used in treatment for MDD or anxiety disorders. Searches covered the period from 1950 (PubMed), 1974 (EMBASE), or 1948 (Cochrane Library) to the date the search was performed. Studies with participants younger than 18 years or older than 65 years (when available) or in which “elderly” participants were indicated were excluded. Studies in which no definitive treatment outcome measures were given were also excluded, as these studies would not clearly establish whether anxiety was diminished, increased, or occurred de novo during the course of the trial. Consequently, only studies that included an applicable rating scale were included. The inclusion/exclusion criteria are summarized in Table 2. As this review was not a meta-analysis of the data, no assessment of risk of bias in individual studies or across studies was undertaken. Moreover, the statistical methods used to declare statistical significance were not relevant to this review.
 | Table 2 Literature search |
Outcome measures
Accepted, clinically valid outcome measures were considered in this review. These outcome measures address the anxiolytic/anxiogenic responses to therapy. Most are considered to be valid and reliable for assessing the improvement in symptoms and severity of anxiety. The incidence of anxiety as a treatment-emergent adverse event was also noted. The outcome measures considered for the purposes of the review are the following:
- Hamilton Anxiety Scale (HAMA) total score
- HAMA psychic anxiety factor score
- HAMA somatic factor score
- Hospital Anxiety and Depression Scale (HADS)
- Liebowitz Social Anxiety Scale (LSAS)
- Panic Disorder Severity Scale (PDSS)
- Covi Anxiety Scale
- Panic and Anticipatory Anxiety Scale
- Phobia Scale
- Hopkins Symptom Checklist-90
- Sheehan Panic Attack And Anxiety Scale
- 17-item Clinician-Administered PTSD Scale
- State–Trait Anxiety Score
- State–Trait Anxiety Inventory
Heterogeneity between the trials
The strength of this review was the inclusion of evidence from all published clinical trials that fell within the search. Consequently, the included trials assessed different interventions and drug dosages. The treatment success was assessed at different time points, and short- and long-term trials were included. Only the endpoints are reported here. The primary and secondary endpoints were considered to be of equal importance. Medication dosages could be fixed or flexible.
Results
The combined searches of the PubMed, EMBASE, and Cochrane databases yielded a total of 263 articles related to transporter inhibitors and depressive or anxiety disorders. A systematic examination of the search results resulted in the culling of 57 duplicate articles and the removal of several because they failed to meet inclusion criteria, as summarized in Table 2. Three articles were removed because they addressed a combination of clinical trials that were also published separately. The original, independent studies were retained for the review. Ultimately, 52 reports were used in the present review.
The distribution of retrieved publications sorted by disorder and by treatment intervention is summarized in Table 3. The included trials contained nine trials relating to depressive disorders, particularly MDD, and the remainder of the studies addressed the anxiety disorders, including GAD, as well as generalized social anxiety disorder, panic disorders, and PTSD and multisomatoform disorder. These conditions, along with treatments and outcome measures, are summarized in Table 4.
 | Table 3 Search results sorted by disorder and intervention |
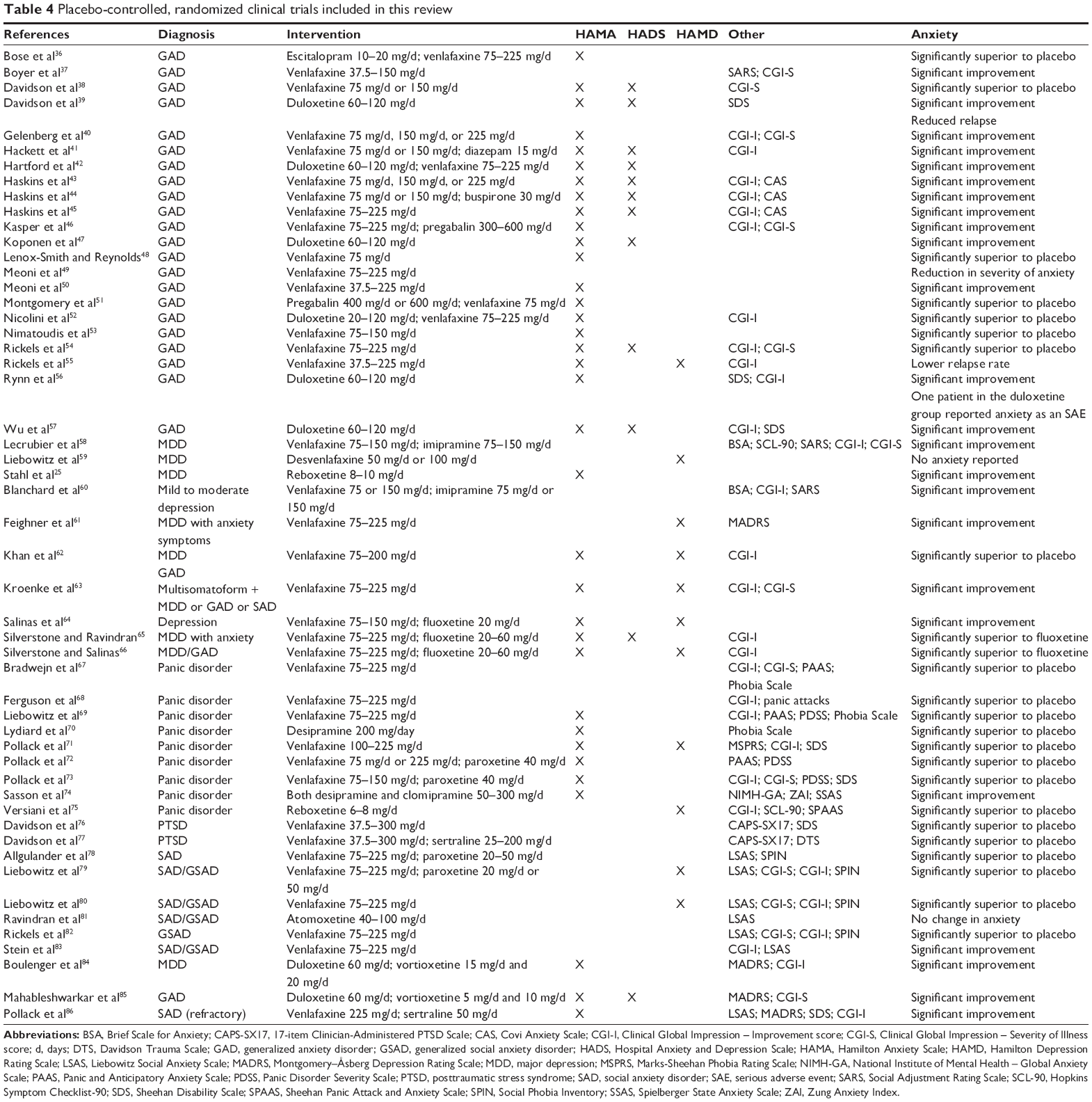 | Table 4 Placebo-controlled, randomized clinical trials included in this review |
Duloxetine
Several clinical trials have shown that duloxetine improves anxiety scores, as determined by HAMA or HADS, in patients with GAD. Flexible-dose56,57 and fixed-dose47 short-term (9–15 weeks) trials showed that duloxetine produced significantly (P≤0.05) greater improvements in the HAMA total score, HAMA psychic anxiety factor score, and individual HAMA items for anxious mood or tension, as well as the HADS total score when compared to the placebo-treated cohorts. Moreover, duloxetine was well tolerated and significantly (P≤0.05) improved remission and sustained improvement rates compared with placebo.47,56,57 Likewise, long-term (26 weeks) duloxetine treatment showed efficacy in preventing or delaying the relapse of GAD in patients showing an initial response.39 Patients treated with placebo significantly (P≤0.001) worsened on the HAMA total score, HAMA psychic anxiety and somatic factors, and HADS anxiety subscale when compared to the duloxetine-treated group.39 The patients receiving duloxetine showed a significantly greater remission rate (68% vs 39%; P≤0.001) than placebo recipients.39 Notably in one study,56 one patient with GAD and receiving duloxetine reported anxiety as a serious adverse event.
In a pair of 10-week clinical trials, the effect of duloxetine (60–120 mg/day) against GAD was compared to placebo and to an active comparator venlafaxine (75–225 mg/day).42,52 Treatment with either duloxetine or venlafaxine improved core psychic and somatic anxiety symptoms in adults with GAD.42,52 In both studies, duloxetine or venlafaxine produced significant improvements on the HAMA total score, the HAMA psychic anxiety factor score, and the HAMA somatic factor score compared with placebo (P<0.01).42,52 In one trial, both duloxetine and venlafaxine also produced significant improvements in the HADS total score relative to placebo (P≤0.01).42 In that study, remission criteria were met by the venlafaxine-treated patients (30%; P<0.05) but not duloxetine-treated patients (23%) at a rate significantly greater than placebo-treated patients (19%). However, the sustained improvement rates were significantly (P<0.01) greater in both active treatment groups (55% for duloxetine and 54% for venlafaxine) compared with the placebo group (39%).42 These trials demonstrated that both duloxetine and venlafaxine are effective antianxiety agents as determined by standard clinical measures of anxiety in patients with GAD.87 Duloxetine (60 mg/day) was used as the comparator in two studies of vortioxetine, a novel antidepressant with multiple mechanisms of action in studies on MDD and GAD.84,85 Duloxetine produced significant reductions (ie, improvement) in the HAMA anxiety score in depressed patients with anxiety.84 In the study of patients with GAD,85 duloxetine, unlike vortioxetine, was significantly superior to placebo in the primary efficacy endpoint, which was a reduction in HAMA scores, as well as the secondary endpoint of reduced HADS scores. There were no reports of increased anxiety in patients in either of these two studies. However, during the second week of discontinuation of duloxetine, at least 10% of patients reported worsening of preexisting symptoms, including nervousness or anxiety, agitation, and insomnia.84
Venlafaxine
Like duloxetine, venlafaxine (37.5–225 mg/day) consistently demonstrated superior efficacy compared to placebo in improving anxiety symptoms as determined by HAMA total scores and other measures of anxiety.37,40,43,45,48,53,55 Short-term and long-term studies with venlafaxine showed improved HAMA total scores and anxious mood individual item scores, which were significantly different with the higher (225 mg) dose.43,45 Moreover, 150 mg and 225 mg of venlafaxine significantly improved the HAMA psychic anxiety factor and tension individual item scores.43,45 Significant improvements were also noted on HADS and the Covi Anxiety Scale scores.43,45 Relapse, defined by a HAMA score of ≥16, occurred more frequently with placebo compared to venlafaxine.55 The relapse rates were 9.8% for venlafaxine and 53.7% for placebo (P<0.001).55 Similar significant improvements on the HAMA psychic anxiety factor scores were seen with an 8-week study with venlafaxine.53
In a long-term study (28 weeks), response rates were significantly increased (69%; P<0.001) for patients receiving venlafaxine (75 mg/day, 150 mg/day, or 225 mg/day) compared to those receiving placebo (42%–46%).40 In that study, venlafaxine was significantly superior to placebo, according to the HAMA total and the HAMA psychic anxiety factor scores (P<0.001).40 Likewise, short-term (8 weeks) and long-term evaluations performed in patients with different levels of social impairment due to anxiety showed significant (P<0.05) improvements with venlafaxine.37
Several trials included active comparators in the study of antianxiety effects of venlafaxine. In one short-term (8 weeks) study, venlafaxine (75–225 mg/day) or escitalopram (10–20 mg/day) both reportedly improved HAMA total scores, although only venlafaxine was significantly (P<0.05) different from placebo.36 A different study comparing venlafaxine to diazepam found that diazepam (15 mg/day), but not venlafaxine (75 mg/day or 150 mg/day), was different from placebo on the HAMA total score.41 In another study, venlafaxine was found to be significantly (P<0.05) superior to buspirone on the HADS anxiety subscale.38 A second study also demonstrated the superiority of venlafaxine over buspirone when measured on the HAMA psychic anxiety factor score.44
In patients with MDD and comorbid anxiety, venlafaxine was significantly superior to the SSRI fluoxetine against anxiety, as assessed by HAMA.65 Venlafaxine was shown, in several placebo-controlled trials, to improve mood in depressed patients, as assessed by Hamilton Depression Rating Scale (HAMD), and to improve anxiety scores, assessed by HAMA, in depressed patients with comorbid anxiety or in primary care patients with multisomatoform disorder with anxiety.61,63,88 In a study of patients with MDD, venlafaxine was superior to the comparator imipramine in the Social Adjustment Rating Scale total score and Social Adjustment Rating Scale items “social/leisure” and “extended family.”58 Both venlafaxine and imipramine improved mood, as reflected in Montgomery–Åsberg Depression Rating Scale and Clinical Global Impression (CGI) scores.58 Moreover, venlafaxine improved LSAS, CGI – Improvement, CGI – Severity, and Social Phobia Inventory scores in patients with social anxiety disorder (SAD)79,80 and improved outcomes in the social and work life of these patients.82 Studies of patients with PTSD also showed that venlafaxine, as well as the active comparators sertraline and paroxetine, improved hyperarousal, reexperiencing/intrusion, and avoidance/numbing.76,77 Venlafaxine was found to be effective against panic disorder in two recent large-scale studies.72,73 In one study, 653 patients with panic disorder with or without agoraphobia were randomized to receive 75 mg/day (163 patients) or 225 mg/day (167 patients) of extended-release venlafaxine and 40 mg/day of paroxetine (161 patients) or placebo (162 patients) for 12 weeks.72 In the second study, a total of 664 patients with panic disorder with or without agoraphobia received 75 mg/day (166 patients) or 150 mg/day (168 patients) of extended-release venlafaxine and 40 mg/day of paroxetine (166 patients) or placebo (163 patients) for 12 weeks.73 In both studies, patients with a primary diagnosis of depression or GAD were excluded.72,73 The primary outcome measure in both studies was the percentage of patients free from full-symptom panic attacks at the end of the treatment period, and secondary outcome measures were changes in baseline in the PDSS, frequency of panic attacks, anticipatory anxiety (as assessed by Panic and Anticipatory Anxiety Scale), and HAMA. Venlafaxine was significantly superior to placebo at all doses on the primary outcome measure and on most of the secondary outcome measures, as was paroxetine.72,73 Moreover, 225 mg/day of venlafaxine significantly increased the percentage of patients free from panic attacks and significantly improved PDSS scores when compared to paroxetine.72 In a recent randomized study, patients with SAD that was refractory to treatment with sertraline were switched to venlafaxine (225 mg/day), sertraline with clonazepam, or sertraline with placebo.86 Venlafaxine and sertraline plus placebo both produced similar (ie, 46%) levels of response, defined as an LSAS score of ≤50%.86 There were no reports of increased anxiety scores in LSAS or HAMA with these patients.
Atomoxetine
In a small study of 27 patients (14 with atomoxetine and 13 with placebo) with SAD, atomoxetine showed a significant (P=0.007) time effect against anxiety as assessed by LSAS, but the time-by-treatment (P=0.91) interaction was not significant, suggesting atomoxetine may not be effective against SAD alone.81
Reboxetine
Like atomoxetine, reboxetine is a selective inhibitor of NET.29 In a placebo-controlled, parallel-group, double-blind 8-week clinical trial of 82 patients (40 placebo and 42 reboxetine) with panic disorder, reboxetine treatment resulted in significantly fewer panic attacks and phobic symptoms than did placebo.75 Likewise, treatment with desipramine or clomipramine also significantly (P<0.001) reduced the frequencies of panic attacks and improved behavioral ratings on several anxiety scales (National Institute of Mental Health – Global Anxiety Scale, Zung Anxiety Scale [Raw and Index], and the Spielberger Anxiety Scale).74 In an earlier 12-week, placebo-controlled study of patients with panic disorder with and without agoraphobia, desipramine treatment significantly reduced the number of panic attacks and improved anxiety ratings on the HAMA and global phobia ratings.70
The randomized, placebo-controlled clinical trials that met the selection criteria for inclusion in the present review describe studies aimed at several psychiatric conditions and employ several reuptake inhibitors with varying levels of selectivity for NET and SERT. However, all these studies were designed to assess the levels of anxiety using commonly employed, standardized, and validated clinical instruments such as HAMA and HAMD. It is important to emphasize that in these studies, none of the active drugs were found to elicit any anxiogenic effect or did they increase anxiety scores. Rather, the anxiety scores in these studies were reduced or left unchanged after treatment with SNRIs or NERIs.
Discussion
The concern that SNRIs may be anxiogenic arises from some observations suggesting that enhanced noradrenergic transmission could be associated with anxiety and agitation.11,24,25 Several animal studies showed that stress and anxiety are associated with increases in released NE, especially in the hypothalamus, amygdala, and LC.89 Furthermore, noradrenergic stimulation of the paraventricular nucleus of the hypothalamus has activated the hypothalamic–pituitary–adrenal axis, thereby eliciting stress responses and anxiety.23 Moreover, enhanced hypothalamic activity can further stimulate noradrenergic regions (eg, LC).23,90 Stress and anxiety may be mediated in part by noradrenergic inputs to the paraventricular nucleus from the LC and the medullary A1 and A2 nuclei.23,91,92 Additionally, the enhanced activity of the LC and noradrenergic-mediated excitation of the amygdala are also associated with behavioral signs of anxiety in animal models.23
Studies performed in rodents and monkeys suggest that cognitive function follows an inverted U-shaped function with regard to noradrenergic activity in the PFC.93,94 Optimal functioning of the PFC occurs when moderate levels of NE are present activating the α2-adrenergic receptors and strengthening the preferred network inputs.93,94 With drowsiness or fatigue, there is reduced noradrenergic release into the PFC, reducing the activity of the preferred networks and resulting in impaired cognition and executive function. In contrast, during the periods of stress, there is an excessive release of NE into the PFC, activating the α1- and β-adrenergic receptors, leading to a collapse of network functioning and impairment of working memory.93,94 Stress and PTSD are associated with highly elevated levels of NE in the PFC and impaired cognitive function. Furthermore, antagonists of the α1- and β-adrenergic receptors are useful in treating PTSD and stress-induced impairment of cognitive flexibility.93,94 Collectively, these observations present a strong theoretical rationale for the suggestion that enhancing noradrenergic transmission with NET-selective inhibitors could enhance manifestations of anxiety. Yet, reviews of numerous clinical trials,25 including the present review, indicate that these antidepressants do not promote anxiety, a situation that has been called the “noradrenergic paradox.”11
However, the interactions among the noradrenergic nuclei and with the serotonergic and dopaminergic regions with respect to hypothalamic and cortical functioning are exceedingly complex.5,11,12,23,90,94 Release of NE from noradrenergic terminals may be regulated by dopaminergic and serotonergic inputs, as well as presynaptic α2-adrenergic autoreceptors (Figure 4). Moreover, the different dopaminergic and serotonergic receptor subtypes are excitatory or inhibitory.94–97 Consequently, alterations in expression and/or availability of one of these transmitters may be offset by compensatory changes in the functioning of the remaining transmitters.
 | Figure 4 Monoaminergic systems and potential interactions. |
The role of adrenergic receptors stimulated by released NE is also critical (Figure 1). The α2-adrenergic receptor, with subtypes α2A, α2B, and α2C, is an inhibitory Gi-coupled receptor, whereas the α1-adrenergic receptor is generally an excitatory Gq-coupled receptor, although it can also mediate inhibitory responses.98 The β1- and β3-adrenergic receptors are excitatory Gs-coupled receptors, whereas the β2-adrenergic receptor may couple to either Gs or Gi.98 Electrical stimulation of the LC resulted in an initial suppression in the firing rate of dorsal hippocampus pyramidal neurons, which was blocked by the α1-adrenoceptor antagonist prazosin.99 The α2 receptor can act as a presynaptic autoreceptor regulating further release of NE or as a postsynaptic inhibitory receptor. Since α2-adrenergic receptors have greater affinity for NE relative to the α1- and β-adrenergic receptors, the low-level release of NE inhibits neuronal activity through the α2-adrenergic receptors.5,93,94 As NE levels are increased, the activation of stimulatory α1- and of β-adrenergic receptors mediates enhanced neuronal activity, which may mediate anxiety, stress, and PTSD.5,93,94 Conversely, α2-adrenergic receptor activation is anxiolytic, and clonidine has shown efficacy in panic disorder and PTSD.100–102
Importantly, although patients with PTSD or panic disorders show heightened responsiveness to NE, their baseline noradrenergic function is normal.100 This observation is consistent with the phasic release of NE in response to a stimulus, as perhaps a perceived threat, that mediates vigilance, and anxiety or stress in an extreme condition, and baseline, tonic NE release that underlies normal functioning (Figure 5).11 Individuals with depression, accompanied by fatigue, drowsiness, and impaired cognitive function, likely have low baseline levels of tonic NE, which is associated with reduced α2-adrenergic receptor-mediated inhibitory tone.11,12,103 Consequently, the appearance of a threatening stimulus or an acute stressor would result in a marked increase in neuronal firing in the absence of the dampening mechanism, thus manifesting as heightened anxiety.11,12,103 NET inhibition could elevate basal NE to normal levels, increasing the basal inhibitory tone and dampening stress responses.
 | Figure 5 Tonic and phasic noradrenergic activity. |
Finally, one must consider that the clinical benefits of antidepressants in general are not immediate in onset, but occur after a considerable latency of 2–3 weeks.104 This latency to effect indicates that the antidepressant clinical benefit is likely due to neuroadaptive changes. For example, prolonged NET inhibition results in desensitization of the presynaptic α2-adrenergic autoreceptors, thus enhancing NE release.11,12,103,105 In contrast, the somatodendritic α2-adrenergic receptors regulating postsynaptic neurons are not desensitized by prolonged NET inhibition.11,12,105
The appearance of “jitteriness syndrome,” indicated by jitteriness, shakiness, increased anxiety, and insomnia, reported after the use of tricyclic antidepressants in the treatment of anxiety or panic disorders is consistent with a noradrenergic hypothesis of anxiety.106 However, the SSRIs have also caused jitteriness syndrome or an initial worsening of anxiety, suggesting a mechanism independent of, or only partly dependent on, noradrenergic transmission, and thus do not support a noradrenergic hypothesis for the anxiogenic potential.107
Although the underlying mechanisms of depressive disorders are still not well understood, recent advances in therapeutics continue to support the monoaminergic hypothesis of depression. However, some concern has been raised that NET inhibition, by virtue of elevated NE levels, may be anxiogenic. Numerous careful, randomized, placebo-controlled clinical trials targeting the anxiety disorders have not borne out this concern. There are several mechanisms involving noradrenergic regulation and receptor sensitization that help explain why the noradrenergic paradox is not so paradoxical after all. Importantly, the studies reviewed strongly suggest that SNRI/NERI drugs are likely to provide relief to patients with MDD associated with anxiety disorders or anxious distress.
Although the studies considered in the present review provide considerable evidence that the use of SNRIs or NERIs is unlikely to evoke anxiogenic effects, there are some limitations that must be considered. Because the present review is not a meta-analysis, it does not include the attendant rigorous statistical analyses, and thus can make no definitive statements regarding statistical significance of the observations, other than those reported within the original studies themselves. An additional limitation of the study is that the inclusion criteria were fairly rigorous, limiting the inclusion of reports to those that included drugs approved for use against depression and/or anxiety. The use of antidepressants in children is still controversial and not widely accepted, and consequently, reports including individuals who are younger than 18 years were not included. It is also acknowledged that several studies exist showing a potential benefit of SNRIs and/or NERIs in a number of disorders, but these studies were not included since this class of drugs is not generally approved for these conditions. Moreover, pain syndromes, such as diabetic neuropathy and fibromyalgia, are successfully managed with these classes of drugs; however, the interrelationships among chronic pain states, depression, and anxiety are complex and interdependent. Consequently, including pain studies to the present review risks obscuring, rather than clarifying, the potential contribution of the reuptake monoaminergic inhibitors to the alleviation of conditions of anxiety. Also notable is that a large part of the reported studies in this review address venlafaxine, which might raise concerns that the interpretations presented here may not have a broad basis. However, the included studies do include a number of other SNRIs/NERIs, as do the additional references that were used in support of the argument, and therefore support a broad interpretation of the evidence that noradrenergic reuptake inhibitors are not likely to provoke anxiety.
Conclusion
Some practitioners of psychiatry, and even some anxiety specialists, believe that SNRIs may be anxiogenic because of enhancement of noradrenergic activity.24 However, no existing systematic analyses thus far support this notion. In the present review, we examined the anxiogenic or anxiolytic effects of SNRIs or NERIs given to patients with psychiatric conditions and in which anxiety was measured with commonly used standard clinical instruments. Crucially, none of the 52 randomized, controlled clinical trials that were included in this review indicated any evidence of an anxiogenic effect resulting from treatment with an SNRI or NERI. To the contrary, there was clear evidence of anxiolytic effects with the NET-selective inhibitors atomoxetine and reboxetine.70,74,75,81,108
This conclusion is well illustrated by the observation that reboxetine, which is a highly selective NET inhibitor, was significantly better than placebo in reducing the incidence of panic attacks in patients with panic disorder as well as improving phobic symptoms, anticipatory anxiety, and social disability.75 In a pooled analysis of nine studies of patients with MDD expressing the symptoms of anxiety, agitation, and insomnia, reboxetine was significantly better than placebo in relieving these specific symptoms.25 It was no different than placebo in the incidence of treatment of emergent anxiety.25 Similarly, the mixed SNRI venlafaxine was superior to placebo in numerous studies cited here in improving anxiety scores in patients with anxiety disorders as well as in patients with anxiety coincident with MDD. Other studies had shown that milnacipran reduced anxiety in patients with schizophrenic and anxiodepressive disorders.11 Collectively, these studies along with the controlled trials presented in this review clearly demonstrate that NET inhibition is not a risk factor in eliciting anxiogenic responses.
Acknowledgments
Angela Lorio, Teri Tucker, and Michael Ossipov of inVentiv Health Clinical, LLC provided editorial and writing assistance. Eli Lilly and Company contracted inVentiv Health Clinical, LLC, for writing and editorial services.
Disclosure
This work was sponsored and funded by Eli Lilly and Company, Indianapolis, IN, USA and/or any of its subsidiaries. Alonso Montoya is employed by Eli Lilly and Company and/or any of its subsidiaries and Robert Bruins is employed by Eli Lilly Canada, Inc. Pierre Blier received grant support, participated in advisory boards, and/or received honorarium for giving lectures from the following: AstraZeneca, Bristol-Myers Squibb, Canadian Institutes of Health Research, Eli Lilly, Forest, Janssen, Lundbeck, Merck, Ontario Brain Institute, Otsuka, Pierre Fabre Médicaments, Servier, Shire, and Sunovion. Martin A Katzman has consulted for Actavis, AstraZeneca, Bedrocan Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Forrest Genuine Health, GlaxoSmithKline, Janssen-Ortho, Lundbeck, Organon, Pfizer, Purdue, Shire, Solvay, and Wyeth; has received research funding from Canadian Institutes of Health Research, Canadian Foundation for Innovation, Canadian Psychiatric Research Foundation, Centre for Addiction and Mental Health Foundation, SickKids Foundation; and has received honoraria from Actavis, AstraZeneca, Bedrocan Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Forrest, The Hecht Foundation, Genuine Health, GlaxoSmithKline, Janssen-Ortho, Lundbeck, Organon, Pfizer, Purdue, Shire, Solvay, and Wyeth. The authors report no other conflicts of interest in this work.
References
Whiteford HA, Degenhardt L, Rehm J, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382:1575–1586. | ||
Ban TA. The role of serendipity in drug discovery. Dialogues Clin Neurosci. 2006;8:335–344. | ||
Blier P, de Montigny C. Serotonin and drug-induced therapeutic responses in major depression, obsessive-compulsive and panic disorders. Neuropsychopharmacology. 1999;21(2 suppl):91S–98S. | ||
Lopez-Munoz F, Alamo C. Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr Pharm Des. 2009;15:1563–1586. | ||
Szabadi E. Functional neuroanatomy of the central noradrenergic system. J Psychopharmacol. 2013;27:659–693. | ||
Lemieux G, Davignon A, Genest J. Depressive states during Rauwolfia therapy for arterial hypertension; a report of 30 cases. Can Med Assoc J. 1956;74:522–526. | ||
Bunney WE Jr, Davis JM. Norepinephrine in depressive reactions. A review. Arch Gen Psychiatry. 1965;13:483–494. | ||
Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122:509–522. | ||
Slattery DA, Hudson AL, Nutt DJ. Invited review: the evolution of antidepressant mechanisms. Fundam Clin Pharmacol. 2004;18:1–21. | ||
Heninger GR, Delgado PL, Charney DS. The revised monoamine theory of depression: a modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry. 1996;29:2–11. | ||
Blier P, Briley M. The noradrenergic symptom cluster: clinical expression and neuropharmacology. Neuropsychiatr Dis Treat. 2011;7(suppl 1):15–20. | ||
Blier P. Neurotransmitter targeting in the treatment of depression. J Clin Psychiatry. 2013;74(suppl 2):19–24. | ||
Bjorklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202. | ||
Waselus M, Valentino RJ, Van Bockstaele EJ. Collateralized dorsal raphe nucleus projections: a mechanism for the integration of diverse functions during stress. J Chem Neuroanat. 2011;41:266–280. | ||
Hornung JP. The human raphe nuclei and the serotonergic system. J Chem Neuroanat. 2003;26:331–343. | ||
Mandrioli R, Mercolini L, Saracino MA, Raggi MA. Selective serotonin reuptake inhibitors (SSRIs): therapeutic drug monitoring and pharmacological interactions. Curr Med Chem. 2012;19:1846–1863. | ||
Conradi HJ, Ormel J, de Jonge P. Presence of individual (residual) symptoms during depressive episodes and periods of remission: a 3-year prospective study. Psychol Med. 2011;41:1165–1174. | ||
Perahia DG, Quail D, Desaiah D, Corruble E, Fava M. Switching to duloxetine from selective serotonin reuptake inhibitor antidepressants: a multicenter trial comparing 2 switching techniques. J Clin Psychiatry. 2008;69:95–105. | ||
Perahia DG, Quail D, Desaiah D, Montejo AL, Schatzberg AF. Switching to duloxetine in selective serotonin reuptake inhibitor non- and partial-responders: effects on painful physical symptoms of depression. J Psychiatr Res. 2009;43:512–518. | ||
Sagman D, McIntosh D, Lee MS, et al. Attributes of response in depressed patients switched to treatment with duloxetine. Int J Clin Pract. 2011;65:73–81. | ||
Debonnel G, Saint-Andre E, Hebert C, de MC, Lavoie N, Blier P. Differential physiological effects of a low dose and high doses of venlafaxine in major depression. Int J Neuropsychopharmacol. 2007;10:51–61. | ||
Vincent S, Bieck PR, Garland EM, et al. Clinical assessment of norepinephrine transporter blockade through biochemical and pharmacological profiles. Circulation. 2004;109:3202–3207. | ||
Itoi K, Sugimoto N. The brainstem noradrenergic systems in stress, anxiety and depression. J Neuroendocrinol. 2010;22:355–361. | ||
Roy-Byrne P. Treatment in nonresponsive patients with social anxiety: back to the future with benzodiazepines. Am J Psychiatry. 2014;171:1–4. | ||
Stahl SM, Mendels J, Schwartz GE. Effects of reboxetine on anxiety, agitation, and insomnia: results of a pooled evaluation of randomized clinical trials. J Clin Psychopharmacol. 2002;22:388–392. | ||
Mantovani M, Dooley DJ, Weyerbrock A, Jackisch R, Feuerstein TJ. Differential inhibitory effects of drugs acting at the noradrenaline and 5-hydroxytryptamine transporters in rat and human neocortical synaptosomes. Br J Pharmacol. 2009;158:1848–1856. | ||
Tsuruda PR, Yung J, Martin WJ, Chang R, Mai N, Smith JA. Influence of ligand binding kinetics on functional inhibition of human recombinant serotonin and norepinephrine transporters. J Pharmacol Toxicol Methods. 2010;61:192–204. | ||
Dube S, Dellva MA, Jones M, et al. A study of the effects of LY2216684, a selective norepinephrine reuptake inhibitor, in the treatment of major depression. J Psychiatr Res. 2010;44:356–363. | ||
Hajos M, Fleishaker JC, Filipiak-Reisner JK, Brown MT, Wong EH. The selective norepinephrine reuptake inhibitor antidepressant reboxetine: pharmacological and clinical profile. CNS Drug Rev. 2004;10:23–44. | ||
Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol. 2007;151:737–748. | ||
Auclair AL, Martel JC, Assie MB, et al. Levomilnacipran (F2695), a norepinephrine-preferring SNRI: profile in vitro and in models of depression and anxiety. Neuropharmacology. 2013;70:338–347. | ||
Deardorff WJ, Grossberg GT. A review of the clinical efficacy, safety and tolerability of the antidepressants vilazodone, levomilnacipran and vortioxetine. Expert Opin Pharmacother. 2014;15:2525–2542. | ||
Deecher DC, Beyer CE, Johnston G, et al. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther. 2006;318:657–665. | ||
Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009;6:e1000100. | ||
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:e1000097. | ||
Bose A, Korotzer A, Gommoll C, Li D. Randomized placebo-controlled trial of escitalopram and venlafaxine XR in the treatment of generalized anxiety disorder. Depress Anxiety. 2008;25:854–861. | ||
Boyer P, Mahe V, Hackett D. Social adjustment in generalised anxiety disorder: a long-term placebo-controlled study of venlafaxine extended release. Eur Psychiatry. 2004;19:272–279. | ||
Davidson JR, DuPont RL, Hedges D, Haskins JT. Efficacy, safety, and tolerability of venlafaxine extended release and buspirone in outpatients with generalized anxiety disorder. J Clin Psychiatry. 1999;60: 528–535. | ||
Davidson JR, Wittchen HU, Llorca PM, et al. Duloxetine treatment for relapse prevention in adults with generalized anxiety disorder: a double-blind placebo-controlled trial. Eur Neuropsychopharmacol. 2008;18:673–681. | ||
Gelenberg AJ, Lydiard RB, Rudolph RL, Aguiar L, Haskins JT, Salinas E. Efficacy of venlafaxine extended-release capsules in nondepressed outpatients with generalized anxiety disorder: a 6-month randomized controlled trial. JAMA. 2000;283:3082–3088. | ||
Hackett D, Haudiquet V, Salinas E. A method for controlling for a high placebo response rate in a comparison of venlafaxine XR and diazepam in the short-term treatment of patients with generalised anxiety disorder. Eur Psychiatry. 2003;18:182–187. | ||
Hartford J, Kornstein S, Liebowitz M, et al. Duloxetine as an SNRI treatment for generalized anxiety disorder: results from a placebo and active-controlled trial. Int Clin Psychopharmacol. 2007;22:167–174. | ||
Haskins JT, Aguiar L, Pallay A, Rudolph RL. Effects of extended release (ER) venlafaxine on anxiety in patients with major depression. Presentation at 11th European College of Neuropsychopharmacology Congress. Paris: 1998. | ||
Haskins JT, Rudolph RL, Aguiar L, Entsuah R. Double-blind, placebo-/comparator-controlled study of once daily venlafaxine XR (V-XR) and buspirone (Bsp) in outpatients with generalized anxiety disorder (GAD). Presentation at 11th European College of Neuropsychopharmacology Congress. Paris: 1998. | ||
Haskins JT, Rudolph RL, Aguiar L, Entsuah R, Salinas A. Venlafaxine XR (V-XR) is an efficacious short- and long-term treatment for generalized anxiety disorder (GAD). Presentation at 11th European College of Neuropsychopharmacology Congress. Paris: 1998. | ||
Kasper S, Herman B, Nivoli G, et al. Efficacy of pregabalin and venlafaxine-XR in generalized anxiety disorder: results of a double-blind, placebo-controlled 8-week trial. Int Clin Psychopharmacol. 2009;24:87–96. | ||
Koponen H, Allgulander C, Erickson J, et al. Efficacy of duloxetine for the treatment of generalized anxiety disorder: implications for primary care physicians. Prim Care Companion J Clin Psychiatry. 2007;9:100–107. | ||
Lenox-Smith AJ, Reynolds A. A double-blind, randomised, placebo controlled study of venlafaxine XL in patients with generalised anxiety disorder in primary care. Br J Gen Pract. 2003;53:772–777. | ||
Meoni P, Salinas E, Brault Y, Hackett D. Pattern of symptom improvement following treatment with venlafaxine XR in patients with generalized anxiety disorder. J Clin Psychiatry. 2001;62:888–893. | ||
Meoni P, Hackett D, Lader M. Pooled analysis of venlafaxine XR efficacy on somatic and psychic symptoms of anxiety in patients with generalized anxiety disorder. Depress Anxiety. 2004;19:127–132. | ||
Montgomery SA, Tobias K, Zornberg GL, Kasper S, Pande AC. Efficacy and safety of pregabalin in the treatment of generalized anxiety disorder: a 6-week, multicenter, randomized, double-blind, placebo-controlled comparison of pregabalin and venlafaxine. J Clin Psychiatry. 2006;67:771–782. | ||
Nicolini H, Bakish D, Duenas H, et al. Improvement of psychic and somatic symptoms in adult patients with generalized anxiety disorder: examination from a duloxetine, venlafaxine extended-release and placebo-controlled trial. Psychol Med. 2009;39:267–276. | ||
Nimatoudis I, Zissis NP, Kogeorgos J, Theodoropoulou S, Vidalis A, Kaprinis G. Remission rates with venlafaxine extended release in Greek outpatients with generalized anxiety disorder. A double-blind, randomized, placebo controlled study. Int Clin Psychopharmacol. 2004;19:331–336. | ||
Rickels K, Pollack MH, Sheehan DV, Haskins JT. Efficacy of extended-release venlafaxine in nondepressed outpatients with generalized anxiety disorder. Am J Psychiatry. 2000;157:968–974. | ||
Rickels K, Etemad B, Khalid-Khan S, Lohoff FW, Rynn MA, Gallop RJ. Time to relapse after 6 and 12 months’ treatment of generalized anxiety disorder with venlafaxine extended release. Arch Gen Psychiatry. 2010;67:1274–1281. | ||
Rynn M, Russell J, Erickson J, et al. Efficacy and safety of duloxetine in the treatment of generalized anxiety disorder: a flexible-dose, progressive-titration, placebo-controlled trial. Depress Anxiety. 2008;25:182–189. | ||
Wu WY, Wang G, Ball SG, Desaiah D, Ang QQ. Duloxetine versus placebo in the treatment of patients with generalized anxiety disorder in China. Chin Med J (Engl). 2011;124:3260–3268. | ||
Lecrubier Y, Bourin M, Moon CA, et al. Efficacy of venlafaxine in depressive illness in general practice. Acta Psychiatr Scand. 1997;95:485–493. | ||
Liebowitz MR, Manley AL, Padmanabhan SK, Ganguly R, Tummala R, Tourian KA. Efficacy, safety, and tolerability of desvenlafaxine 50 mg/day and 100 mg/day in outpatients with major depressive disorder. Curr Med Res Opin. 2008;24:1877–1890. | ||
Blanchard E, Hackett D, Danjou P, Nicholas T. A randomized double-blind comparison of venlafaxine, imipramine and placebo in general practice patients with mild moderate depression. Presentation at 8th European College of Neuropsychopharmacology Congress. Venice: 1995. | ||
Feighner JP, Entsuah AR, McPherson MK. Efficacy of once-daily venlafaxine extended release (XR) for symptoms of anxiety in depressed outpatients. J Affect Disord. 1998;47:55–62. | ||
Khan A, Upton GV, Rudolph RL, Entsuah R, Leventer SM. The use of venlafaxine in the treatment of major depression and major depression associated with anxiety: a dose-response study. Venlafaxine Investigator Study Group. J Clin Psychopharmacol. 1998;18:19–25. | ||
Kroenke K, Messina N, Benattia I, Graepel J, Musgnung J. Venlafaxine extended release in the short-term treatment of depressed and anxious primary care patients with multisomatoform disorder. J Clin Psychiatry. 2006;67:72–80. | ||
Salinas E, Nicholas T, Hackett DA. A randomised, double-blind, comparison of the efficacy and safety of venlafaxine (75 to 150 mg/day) versus fluoxetine (20 mg/day) in depressed outpatients. Presentation at 8th European College of Neuropsychopharmacology Congress. Venice: 1995. | ||
Silverstone PH, Ravindran A. Once-daily venlafaxine extended release (XR) compared with fluoxetine in outpatients with depression and anxiety. Venlafaxine XR 360 Study Group. J Clin Psychiatry. 1999;60:22–28. | ||
Silverstone PH, Salinas E. Efficacy of venlafaxine extended release in patients with major depressive disorder and comorbid generalized anxiety disorder. J Clin Psychiatry. 2001;62:523–529. | ||
Bradwejn J, Ahokas A, Stein DJ, Salinas E, Emilien G, Whitaker T. Venlafaxine extended-release capsules in panic disorder: flexible-dose, double-blind, placebo-controlled study. Br J Psychiatry. 2005;187:352–359. | ||
Ferguson JM, Khan A, Mangano R, Entsuah R, Tzanis E. Relapse prevention of panic disorder in adult outpatient responders to treatment with venlafaxine extended release. J Clin Psychiatry. 2007;68:58–68. | ||
Liebowitz MR, Asnis G, Mangano R, Tzanis E. A double-blind, placebo-controlled, parallel-group, flexible-dose study of venlafaxine extended release capsules in adult outpatients with panic disorder. J Clin Psychiatry. 2009;70:550–561. | ||
Lydiard RB, Morton WA, Emmanuel NP, et al. Preliminary report: placebo-controlled, double-blind study of the clinical and metabolic effects of desipramine in panic disorder. Psychopharmacol Bull. 1993;29:183–188. | ||
Pollack MH, Worthington JJ, Otto MW, et al. Venlafaxine for panic disorder: results from a double-blind, placebo-controlled study. Psychopharmacol Bull. 1996;32:667–670. | ||
Pollack MH, Mangano R, Entsuah R, Tzanis E, Simon NM, Zhang Y. A randomized controlled trial of venlafaxine ER and paroxetine in the treatment of outpatients with panic disorder. Psychopharmacology (Berl). 2007;194:233–242. | ||
Pollack MH, Lepola U, Koponen H, et al. A double-blind study of the efficacy of venlafaxine extended-release, paroxetine, and placebo in the treatment of panic disorder. Depress Anxiety. 2007;24:1–14. | ||
Sasson Y, Iancu I, Fux M, Taub M, Dannon PN, Zohar J. A double-blind crossover comparison of clomipramine and desipramine in the treatment of panic disorder. Eur Neuropsychopharmacol. 1999;9:191–196. | ||
Versiani M, Cassano G, Perugi G, et al. Reboxetine, a selective norepinephrine reuptake inhibitor, is an effective and well-tolerated treatment for panic disorder. J Clin Psychiatry. 2002;63:31–37. | ||
Davidson J, Baldwin D, Stein DJ, et al. Treatment of posttraumatic stress disorder with venlafaxine extended release: a 6-month randomized controlled trial. Arch Gen Psychiatry. 2006;63:1158–1165. | ||
Davidson J, Rothbaum BO, Tucker P, Asnis G, Benattia I, Musgnung JJ. Venlafaxine extended release in posttraumatic stress disorder: a sertraline- and placebo-controlled study. J Clin Psychopharmacol. 2006;26:259–267. | ||
Allgulander C, Mangano R, Zhang J, et al. Efficacy of venlafaxine ER in patients with social anxiety disorder: a double-blind, placebo-controlled, parallel-group comparison with paroxetine. Hum Psychopharmacol. 2004;19:387–396. | ||
Liebowitz MR, Gelenberg AJ, Munjack D. Venlafaxine extended release vs placebo and paroxetine in social anxiety disorder. Arch Gen Psychiatry. 2005;62:190–198. | ||
Liebowitz MR, Mangano RM, Bradwejn J, Asnis G; SAD Study Group. A randomized controlled trial of venlafaxine extended release in generalized social anxiety disorder. J Clin Psychiatry. 2005;66:238–247. | ||
Ravindran LN, Kim DS, Letamendi AM, Stein MB. A randomized controlled trial of atomoxetine in generalized social anxiety disorder. J Clin Psychopharmacol. 2009;29:561–564. | ||
Rickels K, Mangano R, Khan A. A double-blind, placebo-controlled study of a flexible dose of venlafaxine ER in adult outpatients with generalized social anxiety disorder. J Clin Psychopharmacol. 2004;24:488–496. | ||
Stein MB, Pollack MH, Bystritsky A, Kelsey JE, Mangano RM. Efficacy of low and higher dose extended-release venlafaxine in generalized social anxiety disorder: a 6-month randomized controlled trial. Psychopharmacology (Berl). 2005;177:280–288. | ||
Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29:138–149. | ||
Mahableshwarkar AR, Jacobsen PL, Chen Y, Simon JS. A randomised, double-blind, placebo-controlled, duloxetine-referenced study of the efficacy and tolerability of vortioxetine in the acute treatment of adults with generalised anxiety disorder. Int J Clin Pract. 2014;68:49–59. | ||
Pollack MH, Van Ameringen M, Simon NM, et al. A double-blind randomized controlled trial of augmentation and switch strategies for refractory social anxiety disorder. Am J Psychiatry. 2014;171:44–53. | ||
Allgulander C, Nutt D, Detke M, et al. A non-inferiority comparison of duloxetine and venlafaxine in the treatment of adult patients with generalized anxiety disorder. J Psychopharmacol. 2008;22:417–425. | ||
Feighner JP, Entsuah R, McPherson MK. Effects of extended release (ER) venlafaxine on anxiety in patients with major depression. Presentation at 8th European College of Neuropsychopharmacology Congress. Venice: 1995. | ||
Tanaka M, Yoshida M, Emoto H, Ishii H. Noradrenaline systems in the hypothalamus, amygdala and locus coeruleus are involved in the provocation of anxiety: basic studies. Eur J Pharmacol. 2000;405:397–406. | ||
McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. | ||
Itoi K. Ablation of the central noradrenergic neurons for unraveling their roles in stress and anxiety. Ann N Y Acad Sci. 2008;1129:47–54. | ||
Itoi K, Helmreich DL, Lopez-Figueroa MO, Watson SJ. Differential regulation of corticotropin-releasing hormone and vasopressin gene transcription in the hypothalamus by norepinephrine. J Neurosci. 1999;19:5464–5472. | ||
Hains AB, Arnsten AF. Molecular mechanisms of stress-induced prefrontal cortical impairment: implications for mental illness. Learn Mem. 2008;15:551–564. | ||
Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–422. | ||
Guiard BP, El Mansari M, Blier P. Prospect of a dopamine contribution in the next generation of antidepressant drugs: the triple reuptake inhibitors. Curr Drug Targets. 2009;10:1069–1084. | ||
Guiard BP, El Mansari M, Blier P. Cross-talk between dopaminergic and noradrenergic systems in the rat ventral tegmental area, locus ceruleus, and dorsal hippocampus. Mol Pharmacol. 2008;74:1463–1475. | ||
Guiard BP, El Mansari M, Merali Z, Blier P. Functional interactions between dopamine, serotonin and norepinephrine neurons: an in-vivo electrophysiological study in rats with monoaminergic lesions. Int J Neuropsychopharmacol. 2008;11:625–639. | ||
Kobilka BK. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci. 2011;32:213–218. | ||
Curet O, de Montigny C. Electrophysiological characterization of adrenoceptors in the rat dorsal hippocampus. II receptors mediating the effect of synaptically released norepinephrine. Brain Res. 1988;475:47–57. | ||
Bremner JD, Krystal JH, Southwick SM, Charney DS. Noradrenergic mechanisms in stress and anxiety: II. Clinical studies. Synapse. 1996;23:39–51. | ||
Charney DS, Woods SW, Krystal JH, Nagy LM, Heninger GR. Noradrenergic neuronal dysregulation in panic disorder: the effects of intravenous yohimbine and clonidine in panic disorder patients. Acta Psychiatr Scand. 1992;86:273–282. | ||
Southwick SM, Bremner JD, Rasmusson A, Morgan CA 3rd, Arnsten A, Charney DS. Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol Psychiatry. 1999;46:1192–1204. | ||
Szabo ST, Blier P. Response of the norepinephrine system to antidepressant drugs. CNS Spectr. 2001;6:679–684. | ||
Millan MJ. Multi-target strategies for the improved treatment of depressive states: conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther. 2006;110:135–370. | ||
Crews FT, Smith CB. Presynaptic alpha-receptor subsensitivity after long-term antidepressant treatment. Science. 1978;202:322–324. | ||
Pohl R, Yeragani VK, Balon R, Lycaki H. The jitteriness syndrome in panic disorder patients treated with antidepressants. J Clin Psychiatry. 1988;49:100–104. | ||
Gollan JK, Fava M, Kurian B, et al. What are the clinical implications of new onset or worsening anxiety during the first two weeks of SSRI treatment for depression? Depress Anxiety. 2012;29:94–101. | ||
Adler LA, Liebowitz M, Kronenberger W, et al. Atomoxetine treatment in adults with attention-deficit/hyperactivity disorder and comorbid social anxiety disorder. Depress Anxiety. 2009;26:212–221. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.