Back to Journals » Breast Cancer: Targets and Therapy » Volume 7
The network of pluripotency, epithelial–mesenchymal transition, and prognosis of breast cancer
Authors Voutsadakis IA ![]()
Received 10 June 2015
Accepted for publication 27 July 2015
Published 7 September 2015 Volume 2015:7 Pages 303—319
DOI https://doi.org/10.2147/BCTT.S71163
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Ioannis A Voutsadakis1,2
1Division of Medical Oncology, Department of Internal Medicine, Sault Area Hospital, Sault Ste Marie, ON, Canada; 2Division of Clinical Sciences, Northern Ontario School of Medicine, Sudbury, ON, Canada
Abstract: Breast cancer is the leading female cancer in terms of prevalence. Progress in molecular biology has brought forward a better understanding of its pathogenesis that has led to better prognostication and treatment. Subtypes of breast cancer have been identified at the genomic level and guide therapeutic decisions based on their biology and the expected benefit from various interventions. Despite this progress, a significant percentage of patients die from their disease and further improvements are needed. The cancer stem cell theory and the epithelial–mesenchymal transition are two comparatively novel concepts that have been introduced in the area of cancer research and are actively investigated. Both processes have their physiologic roots in normal development and common mediators have begun to surface. This review discusses the associations of these networks as a prognostic framework in breast cancer.
Keywords: stem cells, epithelial-to-mesenchymal transition, mesenchymal-to-epithelial transition
Introduction
Breast cancer is the most common malignancy of women and affects approximately one in eight women during their lifetime. Breast cancer is a heterogeneous disease, and prognosis may vary significantly in individual patients. Based on the expression of a few proteins by the cancer cells, estrogen receptor (ER), progesterone receptor (PR), Her2/Neu, and Ki67 antigen, breast cancers are divided into subtypes with clinical prognostic and therapeutic implications. These subtypes correlate well with genomic profiles that have been determined by whole-genome techniques.1 ER-positive breast cancers are the most common, representing approximately three-quarters of the total number of cases, and may also express PR to various degrees. They are further divided into two subtypes based on low or high expression of Ki67 antigen. These subtypes correspond to the genomically defined subtypes luminal A and luminal B, respectively. The remaining one-quarter of breast cancers is shared by two other subtypes, the Her2/Neu-overexpressing and the so-called triple-negative (not expressing ER, PR, or Her2/Neu). The clinical, prognostic, and therapeutic implications of breast cancer subtypes are well known. Hormone receptor-positive luminal cancers metastasize more often to bones, while Her2/Neu-overexpressing cancers tend to favor the liver, brain, and lungs, and triple-negative cancers metastasize more often to lungs and brain.2 Her2/Neu-overexpressing and triple-negative cancers tend to have a worse prognosis but respond better to chemotherapy than the luminal types.3 In contrast, luminal cancers are the only ones that respond to hormonal treatments, albeit to various degrees, with luminal B subtype being less hormone-sensitive, at least in the long run.4 Despite the ability to prognosticate along the above broad lines in clinical practice, a significant amount of uncertainty remains in determining prognosis of the individual cancer patient, a fact that impedes individualized therapeutic decisions. Thus, further efforts to improve prognosis determination in breast cancer would be helpful in the clinic. These would further refine the current prognostic classification and would be particularly useful for luminal and triple-negative cancers where it is evident that there exists significant intra-category variation. This paper will discuss two important cancer cell processes, pluripotency and epithelial–mesenchymal transition (EMT); their relationship; and the ability of proteins involved in them to predict prognosis in breast cancer patients.
Cancer stem cells
Normal adult tissues possess a small percentage of cells that retain the potential to self-renew and differentiate to cell types of their resident organ. These cells are called stem cells and serve in cell replacement and organ regeneration during the adult organism lifespan.5 Stem cells expressing pluripotency proteins have been identified in the normal human breast.6
According to the cancer stem cell hypothesis, cancer stem cells (or tumor-initiating cells), representing usually a small percentage of the total cancer cell population, are at the basis of tumor development and propagation. Cancer stem cells have properties of normal tissue stem cells and may derive from malignant transformation of these cells or from transformation of differentiated cells that acquire mutations allowing dedifferentiation and acquisition of tumor-initiating properties. Tumor-initiating properties are defined experimentally by the ability of the cells in question to be serially passaged in immunosuppressed mice and produce tumors, with heterogeneity of cells observed in the initial tumor. They are able to do so even when transplanted in mice at much lower numbers than the bulk of the tumor cells.7,8 The derivation of tumor stem cells from normal stem cells or differentiated cells is not mutually exclusive, and either may be true in different patients. In addition, it may explain the different subtypes, as neoplastic stem cells may retain epigenetic memory of their initial cell of provenance and differentiate back to it when producing the bulk cell population. For example, in breast cancer, such considerations would explain how transformation taking place in diverse normal cells would give rise to the different cancer phenotypes. Derivation from a normal stem cell may produce triple-negative cancers, while derivation from dedifferentiation of ductal cells may give rise to luminal subtypes. Stem cells have the phenotype CD44high/CD24low in human breast cancers.7 As few as 200 cells with this phenotype reconstitute tumors when implanted in mice, while more than 50×103 unsorted cells are needed.7 Experimental work has attempted to capture the provenance of different subtypes of breast cancer from different normal breast epithelial cells.9 Although more prevalent and easily identifiable by immunohistochemistry in triple-negative tumors, CD44+/CD24– cancer stem cells are common for all breast cancer subtypes.10
Transformed (cancer) stem cells contain mutations that lead to the acquisition of additional properties on top of the tumor-initiating cells’ properties described above which are essentially properties of normal tissue stem cells. These mutations endow them with the required abilities for cancer development and maintenance, including sustained proliferation and evasion from growth suppressors, resistance to apoptosis, induction of angiogenesis, invasion and metastasis potential, genome instability, and avoidance of immune recognition and destruction.11 Activating mutations in oncogenes or debilitating mutations in tumor suppressor genes are predicted to be stronger cancer promoters if they can establish the stem cell network and concomitantly favor the acquisition of cancer capabilities. Cancer capabilities overlap partially with stem cell properties but are no longer regulated by extrinsic signals. The presence of the survival-promoting pluripotency network in stem (or stem cell-like) cells would allow for a requirement of less additional genetic lesions in order for a cancer to be established.
Given that normal tissue and cancer stem cells have the potential to reconstitute the whole variety of the cells in their organ or of the bulk tumor cell population, they are characterized by plasticity reminiscent of pluripotent embryonal cells that can produce diverse tissues of the three embryonic layers (endoderm, mesoderm, and ectoderm) in development. Pluripotency is conferred by a network of pluripotency transcription factors and microRNAs (miRs), which have also been found to induce a state of pluripotency when exogenously expressed in differentiated cells (so-called induced pluripotent stem cells).12–14 Thus, pluripotency networks may be expected to be in place in cancer stem cells in order to endow them with the ability to self-renew indefinitely and to differentiate toward phenotypically distinct cells of the tumor tissue.
One of the programs present in stem cells of tissues and their embryonic and cancerous counterparts is EMT. EMT and the reverse process, mesenchymal–epithelial transition (MET), are developmentally derived programs that are reactivated in cancers and facilitate the acquisition of cancer properties such as invasiveness, motility, and metastasis.15
The embryonal pluripotency network and EMT are intertwined in normal development and in breast cancer cells.16,17 Given the importance of both processes in cancer and their relationship with each other and with stem cell properties, they deserve to be investigated as markers in breast cancer prognosis.
The network of pluripotency transcription factors and miRs and their expression in breast cancer
The pluripotency state, the ability of cells to remain undifferentiated but at the same time to retain the potential of differentiation to multiple tissues is obtained through the function of defined combinations of a few transcription factors and miRs that work together, establishing a network. These transcription factors include Oct4, Nanog, Sry containing box 2 (Sox2), Krüppel-like factor 4 (KLF4), LIN28, and c-myc. The miR-372/373 cluster of miRs also participates in pluripotency networks and has been reported to enable the induction of pluripotency without the need for additional transcription factors.18 Transcription factors of pluripotency and miRs are involved in establishing the pluripotency state during development in embryonic cells and during the backward reprogramming of adult differentiated cells, termed induced pluripotent stem cells, that have recently been obtained experimentally.12,13 Transcription factors of the network often work in concert on promoters of target genes to establish and maintain pluripotency, a characteristic of embryonic and immature cells that may differentiate toward any cell of the three embryonic layers. Besides inhibiting differentiation, the network of pluripotency promotes cell survival to ensure embryonic development.19 In addition, this program could be usurped by cancer stem cells which display a similarity to normal embryonal pluripotent cells and induced pluripotent stem cells in that they can reconstitute the tumor tissue. A discussion of transcription factors and miRs involved in pluripotency establishment and their expression in breast cancer follows in this section.
Oct4 (alternatively named Oct3/4, OTF3, or POU5F1) is a homeodomain transcription factor of the POU family. It binds to DNA sequences through its POU domain.20 Its action in embryonic stem cells (ESCs) ensures their survival and maintenance of their pluripotentiality.21 These effects are accomplished in cooperation with other pluripotency transcription factors such as, notably, Sox2.22 Oct4 is downregulated by promoter methylation and its expression is suppressed in differentiated adult tissues.23 Various posttranslational modifications are important in controlling Oct4 activity. Phosphorylation of Oct4 by kinase Akt facilitates its interaction with Sox2 in promoters of target genes such as Oct4 itself and Nanog (Figure 1).24 In addition, this phosphorylation inhibits ubiquitination of Oct4 that leads to degradation by the proteasome and promotes dissociation of the unphosphorylated Oct4 from the Akt promoter where it acts as a transcription suppressor.24 Akt has a key position in transduction cascades downstream of several cell surface growth factors and, importantly for breast cancer, downstream of Her2/Neu. Regulation of Oct4 by Akt is only one example of how signaling cascades emanating from the cell surface affect pluripotency. Oct4 is expressed in less than 1% of cells in normal breast epithelium, but its expression is detected in 11.4% of cells in the lactating breast and further increases to more than 30% of cells in breast cancers diagnosed during lactation.25 These data support the role of Oct4 in breast physiology during a phase of generation of functional lactation units that is usurped in breast cancer.
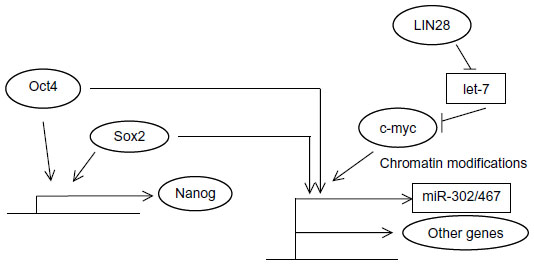 | Figure 1 Schematic representation of the cooperative action of pluripotency transcription factors on promoters of target genes. |
Sox2 is a transcription factor encoded by an intronless gene at human chromosome 3q26. It belongs to the Sox family, group SoxB1 (together with Sox1 and Sox3), and has a length of 317 amino acids.26,27 Sox factors display a high conservation along species.28 Sox2 binds to DNA through its HMG domain, and this binding leads to stereotactic repositioning of the minor groove of the DNA helix to allow accommodation of the transcription machinery.26 Besides being essential for epiblast development in early mammalian embryo, Sox2 plays a part in the maintenance of ESCs and progenitor cells in many tissues. Another common characteristic of Sox factors is that in general they do not elicit a transcription response alone, but depend on binding of partner transcription factors in adjacent DNA sites.29 Notable partners are Oct4 and Nanog, with several hundred promoters bound together by these factors in ESCs when studied by ChIP-on-chip or ChIP-seq.30,31 MCF-7 breast cancer cell line fraction with high Sox2 promoter activity (denoting high Sox2 expression) displays a high sphere-forming capacity and high expressions of CD44 and Nanog compared to the fraction with low Sox2 promoter activity.32 Sox2 is expressed in early breast cancer samples studied by immunohistochemistry.33 Its increased expression confers enhanced mammosphere formation in breast cancer cells in vitro, while knockdown of Sox2 prevented mammosphere formation and delayed xenograft tumor formation in mice.33 Another study confirmed Sox2 expression in a significant proportion of breast cancer pathology samples across tumor subtypes.34
Nanog is another core pluripotency network member. Similarly to Sox2, it is conserved across species.35 Nanog is a 305 amino acid transcription factor possessing a homeobox domain located centrally in the protein through which it binds DNA.36 It activates transcription through two transactivation domains located in the carboxy-terminus of the protein.36 Nanog has important effects in modulating the Oct4 transcriptome, and this translates to the transition phase of reprogramming leading from dedifferentiation to pluripotency.37 This is also due in part to its acting as a pioneer factor for Oct4.38 Pioneer factors are proteins bound to DNA that promote the interaction of partner transcription factors with subsets of their target promoters in a particular cell context where they are expressed. Nanog is also a target of the Oct4/Sox2 module of transcription and then participates with these two transcription factors in the activation of several target gene promoters (Figure 1).39 In breast cancer cells, although Nanog transfection by itself is not sufficient to transform cells, it can cooperate with other pathways to this effect.40 Forced expression of Nanog enhances proliferation of breast cancer cells while knockdown of Nanog inhibits them.41
All three transcription factors, Oct4, Sox2, and Nanog, are expressed in breast cancer cell lines, albeit to differing degrees.42 Nanog protein expression has been confirmed in the side population of the MCF-7 cell line,43 while another study showed that both Oct4 and Nanog were expressed in human breast carcinomas, in contrast to normal breast epithelial cells.44
c-myc is a basic helix-loop-helix (bHLH) transcription factor with a role in neoplastic transformation. The protein binds to DNA through its carboxyterminal DNA binding domain. The target DNA sequence is known as E-box and has the consensus CANNTG (where N is any nucleotide).45 c-myc was included in one of the gene factor “cocktails” that may reprogram adult cells into pluripotent stem cells.46 Mechanistically, c-myc contributes to pluripotency induction by promoting chromatin modifications that subsequently permit binding of additional factors (Figure 1). Moreover, its well-known effect in cell cycle promotion may favor the selection of rare cells in a population primed to become pluripotent.47 These mechanistic insights corroborate with the fact that there are actually no known genes that are regulated solely by c-myc and, on the other hand, approximately 15%–20% of vertebrate genes are regulated by c-myc.48 c-myc is involved in normal breast physiology, where it contributes to lobular proliferation and milk production during lactation.49 Its role in normal mammary progenitor cell maintenance is highlighted by the decreased ability of mutant glands (with a disabling mutation) to reconstitute mammary glands in experiments with cleared mammary fat pads in mice.49 The role of c-myc was extensively studied in breast cancer before its role in stem cell networks became known.50 c-myc is activated by ERα, Her2/Neu, and oncogene K-Ras downstream of it, as well as by the Wnt and Notch pathways. ERα and Her2/Neu are breast cancer-specific oncogenes activated in defined subsets, while the Wnt and Notch pathways are involved in development but also in carcinogenesis. In addition to being a target of β-catenin transcription in the Wnt pathway, c-myc provides a feedforward activation of the pathway by inhibiting the transcription of two inhibitors of Wnt signaling, DKK1 and SFRP1.51 C-myc protein is expressed in most clinical breast cancer specimens, as shown in a breast cancer patient series.52 Another evaluation using tumor microarrays from Her2/Neu-positive patients that had participated in a randomized trial showed high (more than 30% of cells) nuclear c-myc staining in 33% of patients and intermediate staining (10%–30% of cells) in 28% of patients, while 38% of patients had low (less than 10% of cells) nuclear c-myc staining.53 Nuclear staining correlated with cytoplasmic staining of c-myc.
KLF4 is a zinc finger transcription factor included in combinations of transcription factors able to induce pluripotency.46 The 470 amino acid KLF4 protein recognizes a GC-rich sequence that contains a CACCC element and an element called BTE (basic transcription element).54 In the fetus, KLF4 expression peaks in later parts of development, while in the adult organism it is expressed in skin, lung, and intestinal tissues. In breast cancer cells from tumors in transgenic mice and in human breast cancer cell lines, KLF4 is expressed in higher levels in the stem cell subset compared with the bulk cell population.55 Knockdown of KLF4 by shRNA decreased the cancer stem cell population percentage while, in contrast, forced expression of the protein increased the stem cell population.55 In addition, KLF4 promotes cytokine production in breast tumor cells, resulting in tumor progression through modifications of the tumor immune microenvironment.56
LIN28A and LIN28B are two paralogous RNA-binding proteins that are important regulators of development and pluripotency promotion,57 mainly by blocking the biogenesis of the differentiation-inducing let-7 family of miRs (Figure 1). let-7 family members target mRNAs such as LIN28 itself (establishing a double-negative feedback loop), c-myc, Sall4, interleukin 6, and K-Ras mRNAs.58 In breast cancer cells, LIN28 is upregulated by the Wnt/β-catenin/LEF1 pathway and suppresses let-7 to expand stem cells (Figure 1).59 The two LIN28 paralogs characterize different subsets of breast cancers, with LIN28A mostly expressed in Her2-positive cancers and LIN28B expressed predominantly in triple-negative cancers.60
In addition to the let-7 family, three other human miR families, the miR-372/373, the miR-302/467, and the miR-17-92 clusters, participate in the establishment of pluripotency. These clusters cooperate in very early embryonic development when pluripotency is established in the zygote after fertilization.61 The miR-302/467 cluster is a target of Oct4 and of c-myc transcription (Figure 1)62,63 and is able to reprogram human and mouse fibroblasts to pluripotency without transcription factors.18 Reprogramming using miR transfection arises as a more efficient method than using transfection with the Oct4/Sox2/KLF4/c-myc combination and does not require any additional factors, at least in human fibroblasts. Oct4 is upregulated by miR-302/467 cluster transfection, establishing a feedforward loop of pluripotency. In breast cancer, miR-302a downregulation has been associated with radio-resistance of unfractionated cells, and re-expression of the cluster was found to increase cell radio-sensitivity in vitro and in vivo in mice.64 miR-302a downregulation allowed higher expression of AKT1 and RAD52 proteins. Other members of the cluster were not changed in radiosensitive and resistant cells.64 This is in direct contrast to human head and neck cancer stem cells, where miR-302 induced by Oct4/Sox2/Nanog, following interaction with the ligated hyaluronan receptor variant CD44v3, induced increased clonogenicity and cisplatin resistance.65 Although these results are difficult to reconcile, it is interesting to note that the breast cancer cell experiments were carried out with phenotypically radio-resistant bulk cell lines without sorting. In contrast, the head and neck experiments, as mentioned, referred specifically to cells expressing the stem cell marker CD44v3.
The pluripotency-inducing miR-17-92 cluster is a target of induction by c-myc.66 Members of the cluster and of the paralogous cluster miR-106b-25 target PI3K pathway inhibitor PTEN, factors of the E2F transcription factor family, proteins of the TGFβ pathway, and the apoptotic bcl-2 family member BIM.67 These downregulations would lead to cell cycle and apoptosis inhibition, PI3K pathway activation, and inhibition of EMT. The effect on TGFβ pathway signaling and EMT is more complex, because the miR-106b-25 cluster has been reported to activate this signaling by targeting the inhibitor of the pathway Smad7.68 Members of the cluster have been reported to be upregulated in triple-negative breast cancers but also in ER-positive cancers where they target ERα mRNA.69,70
The above data paint a picture of the complexity of the network of pluripotency in general and in breast cancer in particular. Several transcription factors cooperate to establish this network, although, certainly, not all of them may be necessary at one time or in a particular cancer case. In addition, an optimal range in the level of expression of several factors is necessary for the operative establishment of the network and higher levels may paradoxically promote differentiation as is seen with Oct4 in ESCs.71
EMT factors and expression in breast cancer
EMT is a process during which an epithelial cell ceases to be part of an epithelial membrane, invades through the dissolving basement membrane, and moves on to the sub-epithelium of the tissue or further to distant organs. Several events take place in a cell undergoing EMT, including junction dissolution, loss of epithelial cell polarity, acquisition of a fibroblast-like shape, downregulation of epithelial markers, and upregulation of mesenchymal markers.72 EMT is a process that physiologically takes place during normal embryonic development and in adult tissue injury repair. In contrast to these two physiologic conditions where EMT serves normal functions, cancer is a pathologic condition where EMT takes place. Cancer cells undergoing EMT upregulate specific mesenchymal markers such as S100A4 (also called FSP1 [fibroblast-specific protein 1]), vimentin, type I collagen and its receptor kinase DDR2 (discoidin domain receptor tyrosine kinase 2), and cadherin N. β-catenin moves to the nucleus during EMT, and downregulation of epithelial adhesion proteins such as E-cadherin, zonula occludens 1 (ZO-1), cytokeratins, claudins, occludins, and basement membrane components collagen IV and laminin 1 ensues.73
EMT occurring in cancer may be incomplete both at the individual-cell and cell-population level, and only some of the EMT markers may be expressed in small subsets of cancer cells.74 Moreover, incomplete EMT, as, for example, seen during the process of collective migration, allows cells to detach from the epithelial site and acquire some mesenchymal features but still move as small groups of few cells without losing adhesions between the members of the group.15 Further evidence for the role of EMT as intrinsic to the malignant process is provided by the discovery that beyond specific EMT-inducing factors, such as the core EMT transcription factors Snail and Slug (also called Snail2), a multitude of general cancer regulating pathways are important EMT regulators. Examples specifically pertinent to breast cancer include ERα, BRCA1, and p53 proteins, which will be discussed in later sections. Furthermore, confirmation of EMT interconnection with pluripotency networks (which will also be further discussed in a later section) highlights the role of both as cancer-intrinsic processes. In parallel with the above discoveries and related to the importance of the incompleteness of the EMT process in cancer and the associated presence of pluripotency features, it has now become clear that the reverse process of MET is of equal importance for metastasis establishment in remote organs.75
Several pathways activated in cancer have the ability to activate a set of core EMT transcription regulators which eventually lead to E-cadherin downregulation and cell–cell adhesion dissolution. EMT core factors include Snail1 and Slug, ZEB1 (also known as TCF8 or δEF1) and ZEB2 (also known as SIP1 or Zfhx16), Twist, and TCF3 (also known as E47). TGFβ signaling is prominent in inducing EMT core transcription factors and EMT in breast cancer76 and, reciprocally, Snail and Slug induce components of the TGFβ pathway that contribute to the invasion component of EMT and establish a feedforward loop.77 TGFβ signaling activates an additional EMT promoter in breast cancer cells, Sox4 (member of the Sox C Sox factors group), which works through regulation of the Polycomb repressive complex 2 (PRC2) histone methyltransferase EZH2 (enhancer of zeste homologue 2).78 EZH2 executes the methylation of lysine 27 in histone 3, a marker of chromatin primed for transcription, involved in both EMT and pluripotency (Figure 2).
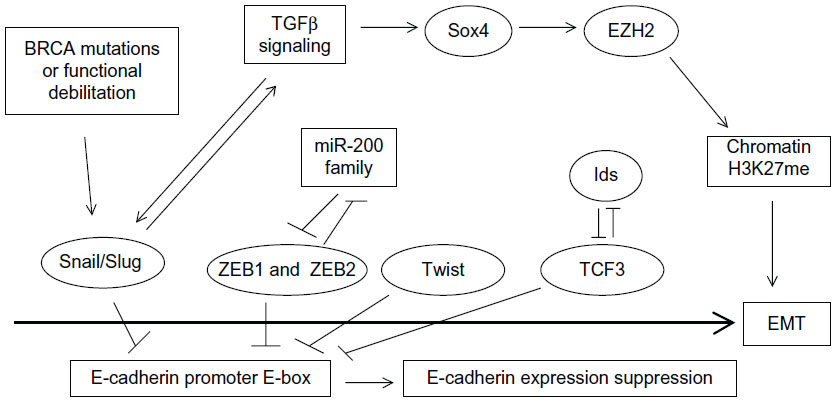 | Figure 2 The road to EMT. |
In breast cancer, the expression of EMT core factors supports various phases of EMT. For example, Snail1 is more important in the initiation phase, while Twist1 becomes essential later in the process.79 ZEB transcription factors are also later players required for EMT maintenance.80 EMT transcription factors have been most extensively studied in the basal-like subtype associated with BRCA1 mutations and, importantly, these mutations have been found to increase Slug stability (Figure 2).81 A brief discussion of EMT core transcription factors and their expression in breast cancer follows.
Snail1 and Slug are zinc finger-containing transcription factors of the C2H2 type (similarly to the previously discussed KLF4 pluripotency factor) that promote EMT by suppressing E-cadherin and other adhesion molecules. Snail1 is a more potent E-cadherin suppressor. Both proteins bind E-box sequences with the consensus 5′-CANNTG-3′ in the promoter of E-cadherin gene with their zinc fingers.82 Both Snail proteins as well as TCF3 are expressed in the branching sites during normal mammary morphogenesis and are necessary and sufficient for induction of the process.83 Snail1 and Slug protect cells in mammary tubules undergoing branching from apoptosis induced by p53 and BID.83 In addition, they promote estrogen production that supports ductal outgrowth by inducing expression of the aromatase enzyme.82 Snail1 also protects non-transformed human mammary epithelial cells from anoikis.84 Snail1 inhibition, on the other hand, correlates with downregulation of RhoA, a GTPase which promotes motility.85 Snail1 mRNA is expressed in a significant percentage (30%) of breast cancer specimens.86 Another study showed that both Snail1 and Slug proteins are expressed in higher levels in breast cancer tissues than in surrounding normal breast tissues.87 A report examining Snail expression across breast cancer subtypes found this transcription regulator to be overexpressed (defined as expression in more than 5% of cells) in 13% of ER-positive cancers and in 64% of ER-negative cancers.88
ZEB1 and ZEB2 are zinc finger transcription regulators that contain a homeodomain flanked by two zinc finger domains. Similarly to the two Snail proteins, they bind to DNA through E-boxes of target gene promoters such as E-cadherin, promoting EMT.89 Their function is inhibited by the miR-200 family of miRs, which bind multiple sites on the 3′-UTRs of their mRNAs and promote the inverse process, MET. There exists a double-negative feedback loop, as ZEB1 binds E-boxes in miR-200 promoters and suppresses their transcription (Figure 2). In addition, miR-200 family miRNAs are critical mediators of MET required during reprogramming to pluripotency and are targets of the Oct4/Sox2 couple of factors.90 In the breast cancer metastatic process, expression of ZEB transcription regulators has broader results than those conferred solely by suppression of E-cadherin, through suppression of the miR-200 family of miRs. When released from ZEB suppression, these miRs actively promote the reverse process, MET, in the metastatic site, at least partly, through suppression of transcription of secretome factor Sec23a.91 As a result of this suppression, secretion of proteins Igfbp4 and Tinagl1 is suppressed from tumor cells arriving in a metastatic site. These proteins contribute antimetastatic colonization signals to the stroma cells of the metastasis target organ and their suppression has the end result that the host organ of the metastasis may not mount a metastasis-inhibiting signal.91
Twist is a bHLH transcription factor with a role in mammary development.82 Twist molecules bind E-boxes in target gene promoters as dimmers. Twist1 was detected in a small subset (1%) of patients with breast cancer in circulating tumor cells.92 A study of the expression of Twist in breast cancer patients showed overexpression in approximately half of the patients.93 A similar percentage of the patients in this study showed high expressions of Snail and Slug.
TCF3, another bHLH transcription factor, is able to induce EMT in breast cancer cells by directly suppressing E-cadherin through binding to E-boxes of the promoter called E-pal and E3.94 The HLH factors of the Id family lacking a basic domain are inhibitors of TCF3 and modulate its effects on EMT creation (Figure 2). Despite this inhibition and the fact that TCF3 has been found to bind the E-cadherin promoter alone, Id proteins contribute to EMT maintenance in breast cancer and are expressed, together with TCF3, in human breast cancer samples, with a higher expression in the basal subtype compared to the luminal subtype.94 Similarly, in another study using a different antibody, Id expression was noted in several metaplastic breast carcinomas but not in carcinomas with the “usual” morphology.95 These data imply that an optimal level of TCF3 activity is required for maintenance of EMT, and Id proteins participate in the regulation of this activity.
The above data as a whole confirm that EMT core factors are expressed in subsets of breast carcinoma cells in human tumors and support the presence and importance of the EMT process in mammary carcinogenesis.
The pluripotency–EMT/MET connection in breast cancer
Stem cells are innately endowed with a transcriptional program that promotes their plasticity. As a result, they can undergo EMT and the reverse MET process. The two programs, pluripotency and EMT, are intertwined in normal mammary epithelium. Slug, for example, expressed in a subpopulation of basal cells of the duct, besides regulating EMT, is involved in enhanced mammosphere growth and suppression of apoptosis.96 After neoplastic transformation, EMT endows cancer stem cells with invasion and metastasis potential and MET endows them with the ability to establish metastases in remote organs.
A direct link between EMT and gain of stem cell markers and properties has been experimentally shown in breast cancer.16,17 When Snail or Twist were ectopically expressed in immortalized human mammary epithelial cells, these cells acquired a fibroblast-like, mesenchymal appearance; downregulated E-cadherin; and upregulated vimentin, N-cadherin, and fibronectin. In addition, they acquired a CD44high/CD24low phenotype consistent with the phenotype of normal and cancer mammary stem cells and displayed an increased ability to form mammospheres, a characteristic of stem cells.16 Reciprocally, endogenous cells with the stem cell phenotype CD44high/CD24low display a fibroblast-like morphology in culture and expression of mesenchymal markers. These results have been confirmed by using overexpression of Twist2 in non-transformed mammary cells and breast cancer cells.97 In addition, as few as 20 CD44high/CD24low breast cancer stem cells produced by overexpressing Twist are able to form tumors in immunocompromised mice.98 A mechanism of pluripotency promotion by Twist has been proposed in head and neck squamous cell carcinoma cells and involves upregulation of the RING finger protein BMI1 by Twist.99 Resistance to apoptosis in neoplastic cells undergoing EMT is accompanied by the acquisition of a stem cell phenotype also associated with drug resistance.16 Another group reported that transfection of the human mammary cell line MCF-10A with an oncogenic mutant K-Ras led to an increase of cells with both a stem cell phenotype and a mesenchymal morphology.17 Moreover, the addition of the EMT promoter TGFβ accelerated the effect of oncogenic K-Ras in inducing mesenchymal morphology and stem cell phenotype in primary mammary epithelial cells.
Additional evidence links pluripotency and EMT/MET in breast cancer by showing that member factors of the core program of either contribute to the other process or even that pathways outside the core programs promote both processes. For example, a study showed that when the core pluripotency transcription factors Oct4 and Nanog were overexpressed in stem cells sorted from the human breast cancer cell line BT-20, EMT transcription factors Snail and Slug and mesenchymal markers N-cadherin and vimentin were upregulated, while epithelial markers E-cadherin and cytokeratin 18 (CK-18) were downregulated.100 The opposite effects were observed when Oct4 and Nanog were knocked down by RNA interference. Oct4 has also been associated with MET through downregulation of TGFβ signaling components TGFβ3 and TGFβ RIII, leading to decreased Snail expression.101 In cooperation with Sox2, Oct4 activates the miR-200 cluster of miRs leading to ZEB mRNA repression (Figure 3).90 These data are consistent with a dual role of pluripotency factors promoting plasticity as opposed to a steady epithelial or mesenchymal state, depending on cooperations and cues from the cellular environment.
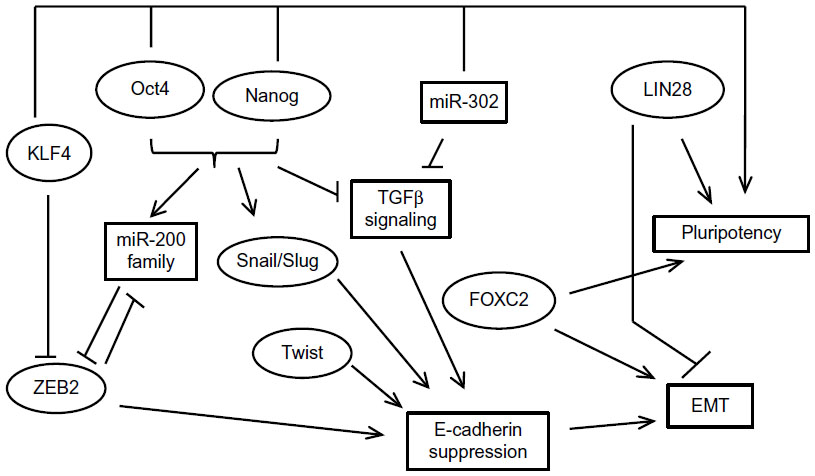 | Figure 3 An overview of the close relationship of the EMT and pluripotency networks. |
The reverse may be also true, given that EMT inducers may decrease the percentage of cells with a stem cell phenotype and suppress expression of markers associated with stemness, such as the ABC cassette transporter ABCG2 (also known as BCRP [breast cancer resistance protein]). This was shown in MCF-7 breast cancer cells which displayed decreased expression of this transporter and, as a result, a decreased percentage of side population after exposure to TGFβ and induction of EMT.102,103 Similarly, in another study, neutralization of TGFβ signaling by the introduction of a dominant negative version of TGFβ RII led to a decrease of the stem cell subset.104 Two additional interesting findings in the study were, firstly, that TGFβ1 mRNA production was higher in luminal-type human breast tumors compared with basal ones, a finding that agrees with the increased stem cell phenotype generally seen in basal breast cancers; and, secondly, that TGFβ downregulates Id1.104 This last finding agrees with the fact mentioned in the “EMT factors and expression in breast cancer” section that a higher expression of Id proteins has been observed in basal tumors,94 arguing for the need for an optimal balance of various agonists and antagonists for establishment of the required level of transcription in downstream genes that would lead to EMT or MET. In addition, these findings implicate a major EMT player, the TGFβ pathway, in the promotion of differentiation instead of stemness, at least in some settings.
Another pluripotency transcription factor that is involved in EMT is KLF4. In this case, data are controversial regarding the role of KLF4 as a breast tumor promoter or suppressor, but a clearer picture is beginning to arise. KLF4 antagonizes ZEB2 in the promoter of E-cadherin and thus upregulates this key epithelial junctional protein and promotes MET (Figure 3).105,106 Moreover, there are indirect regulations of EMT involving a complex influence of KLF4 on ER and p53 signaling, both of which are regulators of EMT, as will be discussed in later sections.107,108
In addition to enhancing mammosphere formation in MCF-7 breast cancer cells, forced expression of pluripotency-promoting RNA-binding protein LIN28 promoted MCF-7 migration in vitro as well as the expression of mesenchymal marker vimentin and downregulated E-cadherin.109 In contrast, knockdown of LIN28 in MDA-MB-231 breast cancer cells suppressed their migration. The pluripotency-associated miR-302 cluster is involved in MET by suppressing TGFβ RII mRNA translation, and this effect contributes to reprogramming of mouse and human fibroblasts to pluripotency.110,111
Besides the aforementioned studies of Snail and Twist, EMT-associated transcription factor FOXC2 is another example of a factor promoting both programs. FOXC2 mRNA is induced by both Snail and Twist and leads to expression of mesenchymal markers and a partial suppression of epithelial markers in breast cancer cells.112 FOXC2 was found to be expressed in specimens of human breast cancers, especially the basal phenotype, in contrast to normal mammary epithelium.112 In immortalized human mammary epithelial cells that have undergone EMT through forced expression of Snail, Twist, or TGFβ, FOXC2 was required for maintenance of the mesenchymal morphology.113 In addition, FOXC2 expression was higher in the stem cell phenotype CD44high/CD24low mammosphere-forming fraction of breast cancer cells compared to non-stem cells with the reverse phenotype. Suppression of FOXC2 with RNA interference reduced the fraction of cells with the stem cell phenotype in this model.113
Overall, both types of studies, those that associate EMT with pluripotency/stem cell phenotype and those that support the involvement of specific factors in both processes, argue for a closely intertwined network that regulates both processes. A so-called metastable state in between the epithelial and mesenchymal cell conditions during EMT or MET has been described and evokes the pluripotency state whence cells retain the ability to be easily tipped toward different fates, adjusting to their environment. Moreover, EMT factors, usually associated with stemness establishment, may in certain conditions promote differentiation toward the epithelial phenotype. This is particularly true for the TGFβ cascade and may explain the well-known dual role of this signaling in carcinogenesis.
ER and PR, cancer stem cells, and EMT in breast cancer
ER has a dual effect in breast cancer by promoting cancer initiation but being associated with less aggressive tumor biology in established cancers.114 Fundamentally, ER is associated with differentiation, as seen experimentally in human ESCs expressing ER and cultured with estrogens in vitro.115
Similarly to the hierarchy of breast epithelial cells in normal breast tissue, breast cancer stem cells do not express ER irrespective of cancer subtype (whether the bulk of the tumor cells express the receptor).116,117 This may be related to the fact that pluripotency factor Sox2 suppresses transcription of factor FOXA1, which is indispensable for both ER expression and function (Figure 4).118,119 In addition, Sox2 suppression of FOXA1 provides a connection of pluripotency with EMT, as FOXA1 is a transcription activator of E-cadherin expression.120 Another study suggested that ER signaling upregulates Oct4 through an estrogen response element in its promoter and promotes mammosphere formation in MCF-7 breast cancer cells.121 This would contribute to establishment of the pluripotency network which, in turn, would suppress ER expression through suppression of FOXA1 (Figure 4). When the network is established, Oct4 expression may become independent of ER. Reciprocally, in differentiated cells that stably express ER, additional regulators may be acting to neutralize the effect of ER on Oct4 promoter in order to avert its expression that would suppress ER. The overexpression of all three pluripotency factors, Oct4, Sox2, and Nanog, leads to reduction in the expression of ER, corroborating the observation that cancer stem cells from ER-positive cancers are ER-negative.122
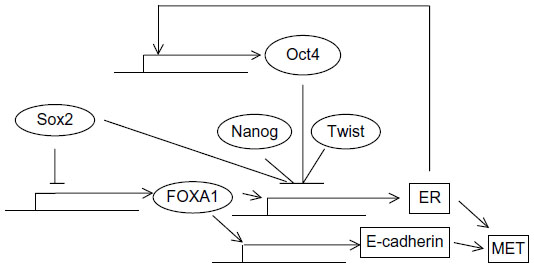 | Figure 4 Regulation of ER by pluripotency and EMT factors. |
Although the presence of ER-negative stem cells in ER-positive cancers does not establish the cell where transformation occurs, it does imply that effects of estrogen signaling on breast cancer stem cells take place through a paracrine mode or through alternative ERs expressed by stem cells. A paracrine role of ER in breast cancer stem cell expansion through induction of FGF signaling in neighboring cells has been reported.123 Breast cancer cells with acquired tamoxifen resistance, implying loss of ER expression or function or independence from this function, gain an EMT phenotype and display upregulated β-catenin transcription.124
Progesterone signaling is also involved in a paracrine stimulation of hormone receptor-negative cancer stem cells mediated by the RANKL–RANK ligand–receptor pair.125,126 Moreover, EMT has been observed following knockdown of ER in MCF-7 breast cancer cells by RNA interference.127 Upregulation of TGFβ and its receptors (TGFβ RI, II, and III), FGF1 and receptor FGFR1, EGFR, and PDGFA were observed during the transition (Figure 4). ER-negative cell lines express TGFβ RII and are able to bind the ligand in in vitro assays, in contrast to ER-positive breast cancer cell lines.128 In addition, primary breast cancer cells with the stem cell phenotype CD44+/CD24–/low were ER-negative and TGFβ RII-positive and had a more mesenchymal phenotype than CD44–/CD24+ cells that were ER-positive and did not express the TGFβ receptor.129 Of note, in the same study, these latter cells seemed to be derived by the former but presented additional genetic lesions and, although they displayed no stem cell markers, they had an abnormal localization of E-cadherin, suggesting that they were not normal epithelial cells. Reciprocally, the EMT-induced Twist was shown to bind E-boxes on the ER promoter and downregulate it through histone acetylation and DNA methylation.130 Cell lines with high Twist expression were noted to be negative for ER by Western blotting, while the reverse was true for cell lines with low Twist expression. Of note, promoter methylation is a factor contributing to ER silencing in breast cancer patient specimens.131
The intracellular environment, in the form of parallel pathways being present and functional, is clearly also of importance in the response to ER signaling. The hedgehog (HH) pathway, for example, may be activated by ER and contributes to both cancer stem cell development and EMT.132 The Notch signaling pathway is also activated by ER in MCF-7 breast cancer cells.133 Both Notch receptor and its ligand Jagged1 are upregulated after estradiol treatment of these cells. Given the well-known role of Notch signaling in pluripotency134 and EMT,73 its activation by ER would be expected to promote both, although this was not specifically investigated in the study.133 Polycomb complex member protein BMI1 is additionally a target of ER stimulation by estrogens and promotes stemness and EMT.135
Overall, ER signaling appears to have multiple effects in hormone receptor-positive breast cancer. In non-stem cells expressing the receptor, it acts inside the cell to suppress EMT and safeguard epithelial phenotype, but if parallel pathways such as HH and Notch are in place, it may promote stemness and EMT. In the small population of ER-negative stem cells, ER acts, through intermediate effectors, to promote their proliferation and expansion in a paracrine manner. As a result, inhibition of ER by anti-hormonal treatments may actually counterintuitively promote EMT in the bulk breast cancer cell population (except if parallel pathways are operational). These cells would be less able to metastasize because they lack the plasticity of the pluripotency network. In contrast, ER inhibition would tend to inhibit EMT in adjacent stem cells that have the pluripotency network in place and would be able to metastasize if EMT is induced in them. The implications of these considerations in ER-positive breast cancer therapeutics would lie in attempts to delay or reverse hormonal treatment resistance by inhibition of paracrine pathways, but also in attempts to dissect and prevent any EMT-promoting effect that hormone inhibition would have in the bulk tumor cells.
Tumor suppressor p53 as a major suppressor of pluripotency and EMT
p53, a well-known tumor suppressor and the most commonly mutated gene in cancers, is involved in inhibition of tumor development by promoting processes such as cell cycle arrest, apoptosis, and DNA damage repair. In addition, p53 regulates both EMT and pluripotency, being an inhibitor of EMT and a major block to pluripotency. p53 is a direct or indirect suppressor of the expression of pluripotency factors, an example of the former being direct suppression of Nanog and of the latter induction of miR-145 which suppresses Oct4 and Sox2.136 p53 is a transcriptional repressor of CD44 which is a transmembrane receptor protein and a hallmark of breast cancer stem cells.137 p53 is also a suppressor of EMT by inducing miRs of the miR-200 family that are suppressors of EMT transcription factors and also ligase HDM2 that ubiquitinates Slug leading to its degradation by the proteasome.138 In contrast, mutant p53 may promote EMT. In MCF-7 and MCF-10 cells, introduction of mutant p53 decreased the expression of E-cadherin and epithelial CK-18, promoted EMT, and altered cell polarity.139,140 In addition, mutant p53 leads to activation of transcription factor NF-κB, a prosurvival factor.141 A role of NF-κB activation is proposed in stem cells of ER-positive breast cancers. These stem cells, despite being hormone receptor-negative, may be stimulated by hormone receptor-positive cancer cells constituting the bulk of the tumor in a paracrine manner, as discussed also in the “ER and PR, cancer stem cells, and EMT in breast cancer” section.142 Hormone receptor-positive cells are stimulated by PR (an ER target gene) signaling to upregulate and secrete receptor activator of NF-κB ligand (RANKL), which then ligates its receptor RANK in adjacent hormone receptor-negative stem cells activating intracellular pathways of survival, among which the NF-κB pathway is a prominent one.143 Moreover, NF-κB promotes stem cell expansion, and its inhibition by I-κB inhibitor of NF-κB in mammary epithelial cells in transgenic mice delayed tumor development and suppressed tumor stem cell expansion.143 A role of NF-κB in EMT induction in mammary cancer cells is mediated through upregulation of transcription regulators Snail and Twist.144 Thus, mutant p53 may cooperate, through NF-κB activation, with hormone receptors in activation of survival pathways or substitute for the function of hormone receptors in hormone receptor-negative cancers.
In breast cancers, mutations of p53 are present with varying frequencies depending on the subtype. In luminal A carcinomas, an incidence of 17% was found, while in luminal B carcinomas, this incidence is 41% and, in ER-negative cancers, it is 50% in Her2-positive carcinomas and 88% in triple-negative.145 Paradoxically, p53 mutations have been associated with responses to doxorubicin/cyclophosphamide chemotherapy in contrast to patients with wild-type p53, in whom no complete responses were obtained with the same regimen. This may be explained by the program (cell cycle arrest versus apoptosis) that is executed following p53 activation in breast cancer cells. Cellular survival and senescence was observed in wild-type p53 breast cancers, while aberrant mitoses followed by apoptosis, possibly mediated by alternative non-p53-dependent pathways, was observed in p53-mutant breast cancers.145 The program activated by wild-type p53 is defined in part by various posttranslational modifications that modify its affinity for different promoters of target genes.146 An important modification favoring apoptosis over cell cycle arrest is phosphorylation at serine in position 46, which is mediated by kinases HIPK2 and DYRK2. Thus, availability and activity or lack thereof of these kinases may modulate the outcome of p53 activation in cancer cells. In addition to promoting p53-induced apoptotic programs, DYRK2 phosphorylates Snail in a serine residue at position 104 leading to a second phosphorylation by kinase GSK3β followed by ubiquitination by ligase βTrCP and degradation by the proteasome. As a result, downregulation of DYRK2 observed in breast cancers leads to Snail accumulation and EMT, promoting the invasive potential of these cancers.147 At the same time, it may contribute to treatment resistance by favoring the cell cycle arrest program of p53 instead of apoptosis.
Pluripotency and EMT factors and prognosis of breast cancer
Several studies have examined the prognostic value of the expression of pluripotency and EMT factors in human breast cancer.
In a series of 90 breast cancer patients, Oct4 expression positivity, defined as more than 10% of tumor cells expressing the protein in their nucleus, was associated with decreased disease-free (DFS) and overall (OS) survival in Cox univariate analysis, although it lost its statistical significance in multivariate analysis, possibly due to its significant association with established prognostic parameters such as lymph node positivity and grade.148 In another series, with 126 breast cancer patients across subtypes, using less stringent criteria of 1% of cells as the cutoff for positivity, both Oct4 and Nanog expression and their combination were associated with reduced OS.100
Copy number gain of chromosome 3q, where the Sox2 gene is located, is a predictor of recurrence in breast cancer patients, and elevated Sox2 expression is associated with tamoxifen resistance.149 Another study found Sox2 mRNA expression to be associated with reduced DFS but not OS in breast cancer patients.150 mRNA expression was correlated with protein expression by immunohistochemistry in all samples tested. In contrast to Sox2, levels of Nanog or KLF4 mRNA were not predictive of outcome in this study.150 An extensive study of over 500 breast cancers also confirmed the association of Sox2 expression by immunohistochemistry with poor DFS.151 In addition, Sox2 was negatively associated with ER and PR status and more commonly seen in metastatic lymph nodes than in the corresponding primary tumor site.
A correlation with poor prognosis was suggested for Nanog in a microarray study, while no prognostic significance was attributed to Oct4, Sox2, or c-myc in this study.152 This study examined protein expression, a fact that may explain the contradictory results with the previously mentioned study that examined mRNA.150
KLF4 expression was also examined in the above study and was determined to be, in contrast to Nanog expression, a marker of good prognosis.152 In contrast, another study maintained that, in early breast cancers (stage I and IIA), increased nuclear accumulation of KLF4, as determined by immunohistochemistry, was associated with a statistically significant worse prognosis, an effect that was not observed in more advanced stages.153
c-myc expression was examined by immunohistochemistry in a breast cancer series of 206 patients and was shown to have little independent value as a prognostic marker compared with established markers.154 Nevertheless, cytoplasmic expression of c-myc in central areas of tumors was associated with cases being already metastatic at diagnosis, implying aggressiveness. Interestingly, in this study, nuclear expression of c-myc was inversely correlated with ER expression.154 Another analysis of Her2-positive patients confirmed that higher (>30%) nuclear staining for c-myc had no prognostic significance and these patients had similar outcomes compared to Her2-positive patients with lower levels of c-myc nuclear staining and benefited equally by the addition of trastuzumab to their treatment.155 In contrast to these data, in patients with sporadic breast cancers and BRCA1 gene functional insufficiency due to promoter methylation or downregulation at the protein level, c-myc overexpression was associated with adverse clinical outcomes.156 Two other studies examined c-myc gene amplification and its prognostic significance in breast cancer. An investigation in a series of 181 node-negative, mostly (two-thirds) small (T1), ER-positive, postmenopausal breast cancers showed, using differential polymerase chain reaction, that c-myc was amplified in approximately 20% of patients and the amplified cases had a worse DFS but no difference in OS compared with non-amplified counterparts.157 The other study, using CISH, found a lower percentage of c-myc amplification of 5%–10% across breast cancer subtypes, and this amplification predicted worse OS.158
LIN28 overexpression in breast cancer cells leads to Her2 upregulation and has been correlated with poor prognosis in breast cancer patients.159 In addition, LIN28B was associated with aggressive clinicopathologic characteristics, such as lymph node positivity and expression of the proliferation marker Ki67, in a series of 190 breast cancer patients.160
An association of higher expression of EMT factors with a shorter relapse-free period was shown specifically in a study of ER-positive patients.93 High coexpression of Snail and Twist could identify patients with worse prognosis in this tissue microarray study. In a more recent extensive tissue microarray study of 1,043 breast cancer patients, nuclear Snail expression correlated with known clinicopathologic prognostic factors such as stage, grade, lymph node status, and ER expression.88 High Snail expression was an adverse prognostic marker for OS in luminal B, Her2-positive, and basal-like cancers, but not in the luminal A subtype.88 Another study that used previously published microarray data confirmed that higher Snail mRNA expression was associated with decreased 5-year relapse-free survival in breast cancer patients.161
Slug in cooperation with Sox9 (a major factor mediating sexual development) has been found to convert mammary epithelial cells to stem cells with increased ability of mammosphere formation and tumor development in mice.162 The two factors, Slug and Sox9, when coexpressed in breast cancer patients, conferred decreased survival compared with patients expressing only one or none of the two factors.162
High transcription factor Twist1 mRNA expression was associated with decreased DFS and OS in patients with ER-positive lymph node-negative breast cancer.163 Another report showed that both high Twist and high Slug mRNA expression were directly associated with poor outcomes.87 In contrast to previously discussed data, higher Snail expression was associated with better outcomes in this study.
Discordant data should be viewed with the perspective of tumor heterogeneity in mind. As the cancer stem cell theory states, not all cancer cells in the bulk or different areas of a given tumor have the same gene expression and tumor propagation potential at a given time. To ascertain this, a study comparing Snail, Slug, and Twist expression from primary breast cancers and lymph node metastases showed that expression of these genes correlated with OS only when studied in the lymph nodes but not in the primaries.164 In addition, patients that displayed a conversion of Snail from negative in the primary tumors to positive in their lymph node metastases had a significantly worse DFS and OS compared to patients that did not convert.164
Integration of pluripotency and EMT programs in breast cancer prognosis and therapy
Although, as discussed above, the close relationship of EMT and pluripotency programs has been revealed in cancer in general and in breast cancer in particular,16,17,165 the molecular connections of pluripotency/plasticity and EMT/MET transitions at the level of their respective core transcription factors and epigenetic modifications have only started to be specifically addressed. The pluripotency network establishes bispecific chromatin methylations on important developmental genes that keep them suppressed but retain them in a primed state ready for future expression or for more permanent suppression upon receiving additional input. The EMT core network may act on this environment to skew the balance toward EMT, and later on in the metastatic site back toward MET, taking advantage of the plasticity conferred by the established pluripotency network on cancer stem cells.166 This model implies that the EMT network will work together with the pluripotency network to endow cancer stem cells with metastatic potential (a hallmark of cancer), while in cells with no pluripotency network in place, the effect would not be the establishment of metastatic disease because these cells would lack the required plasticity and would be unable to undergo a MET (Figure 5). This requirement also predicts that the strongest oncogenic events would promote both pathways in parallel. An additional observation supporting the close interrelationship of the two networks comes from examination of promoter sequences upstream of transcription start sites of genes encoding for the human transcription factors of the pluripotency and EMT networks. These preliminary results show that all promoter sequences possess multiple putative binding sites for other transcription factors of the two networks (Voutsadakis, unpublished data, 2015). The importance of both programs for metastatic dissemination has recently been investigated in circulating tumor cells which have been shown to coexpress markers of both processes.167 The fact that such cells are circulating in the blood of cancer patients is an initial proof of the pathophysiologic importance of the two programs acting in concert and provides opportunities for both prognostication and treatment of breast cancer and other cancers.
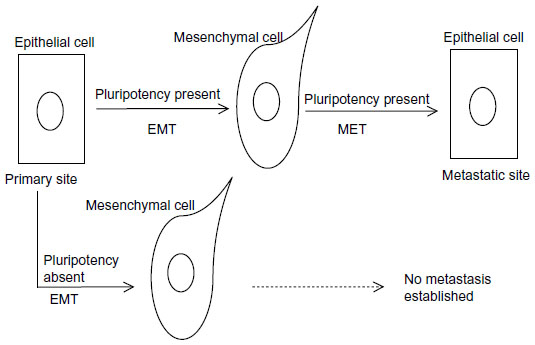 | Figure 5 A proposed model of the cooperation of pluripotency and EMT/MET processes in metastasis establishment. |
Given the multiple factors that are needed to act in a coordinated manner in order to establish the pluripotency and EMT networks, it may be necessary to examine several of them in a multiplex platform similar to the ones currently in clinical use to derive the full prognostic value of the presence of the two networks in breast cancer cells. In this type of prognostic panel building, already published genomic sets with clinical annotation could be of significant help. For example, a search of such a set of breast cancer patients using the online tool survExpress (http://bioinformatica.mty.itesm.mx:8080/Biomatec/SurvivaX.jsp)168 shows that a panel of transcription factors from the pluripotency and EMT programs performs quite similarly in prediction of OS to the panel of genes included in the Oncotype Dx test when tested in a set of breast cancers published by The Cancer Genome Atlas (Voutsadakis, unpublished data, 2015). Further refinements of such panels may guide future developments of even better prognostic tools for validation and use in the clinic.
Disclosure
The author reports no conflicts of interest in this work.
References
Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. | |
Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28(20):3271–3277. | |
Carey LA, Dees EC, Sawyer L, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13(8):2329–2334. | |
Prat A, Parker JS, Fan C, et al. Concordance among gene expression-based predictors for ER-positive breast cancer treated with adjuvant tamoxifen. Ann Oncol. 2012;23(11):2866–2873. | |
Simons BD, Clevers H. Strategies for homeostatic stem cell self-renewal in adult tissues. Cell. 2011;145(6):851–862. | |
Villadsen R, Fridriksdottir AJ, Rønnov-Jessen L, et al. Evidence for a stem cell hierarchy in the adult human breast. J Cell Biol. 2007;177(1):87–101. | |
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | |
Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;15(1):555–567. | |
Ince TA, Richardson AL, Bell GW, et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell. 2007;12(2):160–170. | |
Honeth G, Bendahl PO, Ringnér M, et al. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10:R53. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. | |
Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007; 318(5858):1917–1920. | |
Marson A, Levine SS, Cole MF, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134(3):521–533. | |
Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15(2):117–134. | |
Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. | |
Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3(8):e2888. | |
Anokye-Danso F, Trivedi CM, Juhr D, et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8(4):376–388. | |
Nichols J, Zevnik B, Anastassiadis K, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95(3):379–391. | |
Stefanovic S, Pucéat M. [The dual role of OCT4]. Med Sci (Paris). 2010;26(4):411–416. French. | |
Kehler J, Tolkunova E, Koschorz B, et al. Oct4 is required for primordial germ cell survival. EMBO Rep. 2004;5(11):1078–1083. | |
Stefanovic S, Abboud N, Désilets S, Nury D, Cowan C, Pucéat M. Interplay of Oct4 with Sox2 and Sox17: a molecular switch from stem cell pluripotency to specifying a cardiac fate. J Cell Biol. 2009; 186(5):665–673. | |
Deb-Rinker P, Ly D, Jezierski A, Sikorska M, Walker PR. Sequential DNA methylation of the Nanog and Oct-4 upstream regions in human NT2 cells during neuronal differentiation. J Biol Chem. 2005;280(8):6257–6260. | |
Lin Y, Yang Y, Li W, et al. Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol Cell. 2012;48(4):627–640. | |
Hassiotou F, Hepworth AR, Beltran AS, et al. Expression of the pluripotency transcription factor OCT4 in the normal and aberrant mammary gland. Front Oncol. 2013;3:79. | |
Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12(1):15–30. | |
Liu K, Lin B, Zhao M, et al. The multiple roles for Sox2 in stem cell maintenance and tumorigenesis. Cell Signal. 2013;25(5):1264–1271. | |
Weina K, Utikal J. SOX2 and cancer: current research and its implications in the clinic. Clin Transl Med. 2014;3:19. | |
Rizzino A. Concise review: the Sox2-Oct4 connection: critical players in a much larger interdependent network integrated at multiple levels. Stem Cells. 2013;31(6):1033–1039. | |
Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122(6):947–956. | |
Chen X, Xu H, Yuan P, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133(6):1106–1117. | |
Liang S, Furuhashi M, Nakane R, et al. Isolation and characterization of human breast cancer cells with SOX2 promoter activity. Biochem Biophys Res Commun. 2013;437(2):205–211. | |
Leis O, Eguiara A, Lopez-Arribillaga E, et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012; 31(11):1354–1365. | |
Lengerke C, Fehm T, Kurth R, et al. Expression of the embryonic stem cell marker SOX2 in early-stage breast carcinoma. BMC Cancer. 2011;11:42. | |
Theunissen TW, Silva JC. Switching on pluripotency: a perspective on the biological requirement of Nanog. Philos Trans R Soc Lond B Biol Sci. 2011;366(1575):2222–2229. | |
Wang ML, Chiou SH, Wu CW. Targeting cancer stem cells: emerging role of Nanog transcription factor. Onco Targets Ther. 2013;6:1207–1220. | |
Silva J, Nichols J, Theunissen TW, et al. Nanog is the gateway to the pluripotent ground state. Cell. 2009;138(4):722–737. | |
Navarro P, Avner P. When X-inactivation meets pluripotency: an intimate rendezvous. FEBS Lett. 2009;583(11):1721–1727. | |
Shi G, Jin Y. Role of Oct4 in maintaining and regaining stem cell pluripotency. Stem Cell Res Ther. 2010;1(5):39. | |
Lu X, Mazur SJ, Lin T, Appella E, Xu Y. The pluripotency factor nanog promotes breast cancer tumorigenesis and metastasis. Oncogene. 2014;33(20):2655–2664. | |
Liao WY, Liaw CC, Huang YC, et al. Cyclohexylmethyl flavonoids suppress propagation of breast cancer stem cells via downregulation of NANOG. Evid Based Complement Alternat Med. 2013;2013:170261. | |
Ling GQ, Chen DB, Wang BQ, Zhang LS. Expression of the pluripotency markers Oct3/4, Nanog and Sox2 in human breast cancer cell lines. Oncol Lett. 2012;4(6):1264–1268. | |
Jeter CR, Badeaux M, Choy G, et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009;27(5):993–1005. | |
Ezeh UI, Turek PJ, Reijo RA, Clark AT. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer. 2005;104(10):2255–2265. | |
Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. | |
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131(5):861–872. | |
Knoepfler PS. Why myc? An unexpected ingredient in the stem cell cocktail. Cell Stem Cell. 2008;2(1):18–21. | |
Sodir NM, Evan GI. Nursing some sense out of Myc. J Biol. 2009;8:77. | |
Stoelzle T, Schwarb P, Trumpp A, Hynes NE. c-Myc affects mRNA translation, cell proliferation and progenitor cell function in the mammary gland. BMC Biol. 2009;7:63. | |
Xu J, Chen Y, Olopade OI. MYC and breast cancer. Genes Cancer. 2010;1(6):629–640. | |
Cowling VH, D’Cruz CM, Chodosh LA, Cole MD. c-Myc transforms human mammary epithelial cells through repression of the Wnt inhibitors DKK1 and SFRP1. Mol Cell Biol. 2007;27(14):5135–5146. | |
Elkak AE, Meligonis G, Salhab M, et al. hTERT protein expression is independent of clinicopathological parameters and c-Myc protein expression in human breast cancer. J Carcinog. 2005;4:17. | |
Dueck AC, Reinholz MM, Geiger XJ, et al. Impact of c-MYC protein expression on outcome of patients with early-stage HER2+ breast cancer treated with adjuvant trastuzumab NCCTG (alliance) N9831. Clin Cancer Res. 2013;19(20):5798–5807. | |
Ghaleb AM, Yang VW. The pathobiology of Krüppel-like factors in colorectal cancer. Curr Colorectal Cancer Rep. 2008;4(2):59–64. | |
Yu F, Li J, Chen H, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30(18):2161–2172. | |
Yu F, Shi Y, Wang J, Li J, Fan D, Ai W. Deficiency of Kruppel-like factor KLF4 in mammary tumor cells inhibits tumor growth and pulmonary metastasis and is accompanied by compromised recruitment of myeloid-derived suppressor cells. Int J Cancer. 2013;133(12):2872–2883. | |
Thornton JE, Gregory RI. How does Lin28 let-7 control development and disease? Trends Cell Biol. 2012;22(9):474–482. | |
Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635–647. | |
Cai WY, Wei TZ, Luo QC, et al. The Wnt-β-catenin pathway represses let-7 microRNA expression through transactivation of Lin28 to augment breast cancer stem cell expansion. J Cell Sci. 2013;126(Pt 13):2877–2889. | |
Piskounova E, Polytarchou C, Thornton JE, et al. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell. 2011; 147(5):1066–1079. | |
Svoboda P, Flemr M. The role of miRNAs and endogenous siRNAs in maternal-to-zygotic reprogramming and the establishment of pluripotency. EMBO Rep. 2010;11(8):590–597. | |
Liu H, Deng S, Zhao Z, et al. Oct4 regulates the miR-302 cluster in P19 mouse embryonic carcinoma cells. Mol Biol Rep. 2011;38(3):2155–2160. | |
Judson RL, Babiarz J, Venere M, Blelloch R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat Biotechnol. 2009;27(5):459–461. | |
Liang Z, Ahn J, Guo D, Votaw JR, Shim H. MicroRNA-302 replacement therapy sensitizes breast cancer cells to ionizing radiation. Pharm Res. 2013;30(4):1008–1016. | |
Bourguignon LY, Wong G, Earle C, Chen L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J Biol Chem. 2012;287(39):32800–32824. | |
O’Donnell KA, Wentzel FA, Zeller KI, Dang CV, Mendell JT. C-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005; 435(7043):839–843. | |
Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013;20(12):1603–1614. | |
Smith AL, Iwanaga R, Drasin DJ, et al. The miR-106b-25 cluster targets Smad7, activates TGF-β signaling, and induces EMT and tumor initiating cell characteristics downstream of Six1 in human breast cancer. Oncogene. 2012;31(50):5162–5171. | |
Farazi TA, Horlings HM, Ten Hoeve JJ, et al. MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res. 2011;71(13):4443–4453. | |
Leivonen SK, Mäkelä R, Ostling P, et al. Protein lysate microarray analysis to identify microRNAs regulating estrogen receptor signaling in breast cancer cell lines. Oncogene. 2009;28(44):3926–3936. | |
Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24(4):372–376. | |
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. | |
Voutsadakis IA. The ubiquitin-proteasome system and signal transduction pathways regulating epithelial mesenchymal transition of cancer. J Biomed Sci. 2012;19:67. | |
Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119(6):1429–1437. | |
Guo F, Parker Kerrigan BC, Yang D, et al. Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J Hematol Oncol. 2014;7:19. | |
Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15(2):169–190. | |
Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS One. 2011;6(10):e26514. | |
Tiwari N, Tiwari VK, Waldmeier L, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. 2013;23(6):768–783. | |
Tran DD, Corsa CA, Biswas H, Aft RL, Longmore GD. Temporal and spatial cooperation of Snail1 and Twist1 during epithelial-mesenchymal transition predicts for human breast cancer recurrence. Mol Cancer Res. 2011;9(12):1644–1657. | |
de Herreros AG, Peiró S, Nassour M, Savagner P. Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J Mammary Gland Biol Neoplasia. 2010;15(2):135–147. | |
Proia TA, Keller PJ, Gupta PB, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011;8(2):149–163. | |
Foubert E, De Craene B, Berx G. Key signalling nodes in mammary gland development and cancer. The Snail1-Twist1 conspiracy in malignant breast cancer progression. Breast Cancer Res. 2010;12:206. | |
Lee KA, Gjorevski N, Boghaert E, Radisky DC, Nelson CM. Snail1, Snail2, and E47 promote mammary epithelial branching morphogenesis. EMBO J. 2011;30(13):2662–2674. | |
Gras B, Jacqueroud L, Wierinckx A, et al. Snail family members unequally trigger EMT and thereby differ in their ability to promote the neoplastic transformation of mammary epithelial cells. PLoS One. 2014;9(3):e92254. | |
Zhang A, Wang Q, Han Z, et al. Reduced expression of Snail decreases breast cancer cell motility by downregulating the expression and inhibiting the activity of RhoA GTPase. Oncol Lett. 2013;6(2):339–346. | |
Okubo T, Truong TK, Yu B, et al. Down-regulation of promoter 1.3 activity of the human aromatase gene in breast tissue by zinc-finger protein, Snail (SnaH). Cancer Res. 2001;61(4):1338–1346. | |
Martin TA, Goyal A, Watkins G, Jiang WG. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol. 2005;12(6):488–496. | |
Muenst S, Däster S, Obermann EC, et al. Nuclear expression of snail is an independent negative prognostic factor in human breast cancer. Dis Markers. 2013;35(5):337–344. | |
Hill L, Browne G, Tulchinsky E. ZEB/miR-200 feedback loop: at the crossroads of signal transduction in cancer. Int J Cancer. 2013;132(4):745–754. | |
Wang G, Guo X, Hong W, et al. Critical regulation of miR-200/ZEB2 pathway in Oct4/Sox2-induced mesenchymal-to-epithelial transition and induced pluripotent stem cell generation. Proc Natl Acad Sci U S A. 2013;110(8):2858–2863. | |
Korpal M, Ell BJ, Buffa FM, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2012;17(9):1101–1108. | |
Kasimir-Bauer S, Hoffmann O, Wallwiener D, Kimmig R, Fehm T. Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 2012;14:R15. | |
van Nes JG, de Kruijf EM, Putter H, et al. Co-expression of SNAIL and TWIST determines prognosis in estrogen receptor-positive early breast cancer patients. Breast Cancer Res Treat. 2012;133(1):49–59. | |
Cubillo E, Diaz-Lopez A, Cuevas EP, et al. E47 and Id1 interplay in epithelial-mesenchymal transition. PLoS One. 2013;8(3):e59948. | |
Perk J, Gil-Bazo I, Chin Y, et al. Reassessment of id1 protein expression in human mammary, prostate, and bladder cancers using a monospecific rabbit monoclonal anti-id1 antibody. Cancer Res. 2006;66(22):10870–10877. | |
Nassour M, Idoux-Gillet Y, Selmi A, et al. Slug controls stem/progenitor cell growth dynamics during mammary gland morphogenesis. PLoS One. 2012;7(12):e53498. | |
Fang X, Cai Y, Liu J, et al. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and stem-like cell self-renewal. Oncogene. 2011;30(47):4707–4720. | |
Vesuna F, Lisok A, Kimble B, Raman V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia. 2009;11(12):1318–1328. | |
Yang MH, Hsu DS, Wang HW, et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010; 12(10):982–992. | |
Wang D, Lu P, Zhang H, et al. Oct-4 and Nanog promote the epithelial-mesenchymal transition of breast cancer stem cells and are associated with poor prognosis in breast cancer patients. Oncotarget. 2014; 5(21):10803–10815. | |
Li R, Liang J, Ni S, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7(1):51–63. | |
Yin L, Castagnino P, Assoian RK. ABCG2 expression and side population abundance regulated by a transforming growth factor beta-directed epithelial-mesenchymal transition. Cancer Res. 2008; 68(3):800–807. | |
Mallini P, Lennard T, Kirby J, Meeson A. Epithelial-to-mesenchymal transition: what is the impact on breast cancer stem cells and drug resistance. Cancer Treat Rev. 2014;40:341–348. | |
Tang B, Yoo N, Vu M, et al. Transforming growth factor-beta can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res. 2007;67(18):8643–8652. | |
Yori JL, Johnson E, Zhou G, Jain MK, Keri RA. Kruppel-like factor 4 inhibits epithelial-to-mesenchymal transition through regulation of E-cadherin gene expression. J Biol Chem. 2010;285(22):16854–16863. | |
Koopmansch B, Berx G, Foidart JM, Gilles C, Winkler R. Interplay between KLF4 and ZEB2/SIP1 in the regulation of E-cadherin expression. Biochem Biophys Res Commun. 2013;431(4):652–657. | |
Akaogi K, Nakajima Y, Ito I, et al. KLF4 suppresses estrogen- dependent breast cancer growth by inhibiting the transcriptional activity of ERalpha. Oncogene. 2009;28(32):2894–2902. | |
Hu D, Zhou Z, Davidson NE, Huang Y, Wan Y. Novel insight into KLF4 proteolytic regulation in estrogen receptor signaling and breast carcinogenesis. J Biol Chem. 2012;287(17):13584–13597. | |
Liu Y, Li H, Feng J, et al. Lin28 induces epithelial-to-mesenchymal transition and stemness via downregulation of let-7a in breast cancer cells. PLoS One. 2013;8(12):e83083. | |
Liao B, Bao X, Liu L, et al. MicroRNA cluster 302–367 enhances somatic cell reprogramming by accelerating a mesenchymal-to-epithelial transition. J Biol Chem. 2011;286(19):17359–17364. | |
Subramanyam D, Lamouille S, Judson RL, et al. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat Biotechnol. 2011;29(5):443–448. | |
Mani SA, Yang J, Brooks M, et al. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A. 2007;104(24):10069–10074. | |
Hollier BG, Tinnirello AA, Werden SJ, et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013;73(6):1981–1992. | |
Chimge NO, Baniwal SK, Little GH, et al. Regulation of breast cancer metastasis by Runx2 and estrogen signaling: the role of SNAI2. Breast Cancer Res. 2011;13:R127. | |
Hong SH, Nah HY, Lee YJ, et al. Expression of estrogen receptor-alpha and -beta, glucocorticoid receptor, and progesterone receptor genes in human embryonic stem cells and embryoid bodies. Mol Cells. 2004;18(3):320–325. | |
Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci U S A. 2008;105(15):5774–5779. | |
Kabos P, Haughian JM, Wang X, et al. Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res Treat. 2011; 128(1):45–55. | |
Li X, Chen S, Sun T, et al. The transcriptional regulation of SOX2 on FOXA1 gene and its application in diagnosis of human breast and lung cancers. Clin Lab. 2014;60(6):909–918. | |
Bernardo GM, Keri RA. FOXA1: a transcription factor with parallel functions in development and cancer. Biosci Rep. 2012;32(2):113–130. | |
Liu YN, Lee WW, Wang CY, Chao TH, Chen Y, Chen JH. Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene. 2005;24(56):8277–8290. | |
Jung JW, Park SB, Lee SJ, Seo MS, Trosko JE, Kang KS. Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS One. 2011;6(11):e28068. | |
Simões BM, Piva M, Iriondo O, et al. Effects of estrogen on the proportion of stem cells in the breast. Breast Cancer Res Treat. 2011; 129(1):23–35. | |
Fillmore CM, Gupta PB, Rudnick JA, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A. 2010;107(50):21737–21742. | |
Hiscox S, Jiang WG, Obermeier K, et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Cancer. 2006;118(2):290–301. | |
Asselin-Labat ML, Vaillant F, Sheridan JM, et al. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010; 465(7299):798–802. | |
Joshi PA, Jackson HW, Beristain AG, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465(7299):803–807. | |
Al Saleh S, Al Mulla F, Luqmani YA. Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS One. 2011;6(6):e20610. | |
Arteaga CL, Tandon AK, Von Hoff DD, Osborne CK. Transforming growth factor beta: potential autocrine growth inhibitor of estrogen receptor-negative human breast cancer cells. Cancer Res. 1988;48:3898–3904. | |
Shipitsin M, Campbell LL, Argani P, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. | |
Vesuna F, Lisok A, Kimble B, et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-α. Oncogene. 2012;31(27):32223–3234. | |
Lapidus RG, Ferguson AT, Ottaviano YL, et al. Methylation of estrogen receptor gene 5′ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin Cancer Res. 1996;2(5):805–810. | |
Sun Y, Wang Y, Fan C, et al. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol Cancer. 2014;13:137. | |
Soares R, Balogh G, Guo S, Gärtner F, Russo J, Schmitt F. Evidence for the notch signaling pathway on the role of estrogen in angiogenesis. Mol Endocrinol. 2004;18:2333–2343. | |
D’Angelo RC, Ouzounova M, Davis A, et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol Cancer Ther. 2015;14(3):779–787. | |
Wang H, Liu H, Li X, et al. Estrogen receptor α-coupled Bmi1 regulation pathway in breast cancer and its clinical implications. BMC Cancer. 2014;14:122. | |
Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137(4):647–658. | |
Godar S, Ince TA, Bell GW, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134(1):62–73. | |
Hermeking H. MicroRNAs in thep53 network: micromanagement of tumour suppression. Nat Rev Cancer. 2012;12(9):613–626. | |
Rieber M, Strasberg-Rieber M. p53 inactivation decreases dependence on estrogen/ERK signalling for proliferation but promotes EMT and susceptibility to 3-bromopyruvate in ERα+ breast cancer MCF-7 cells. Biochem Pharmacol. 2014;88(2):169–177. | |
Zhang Y, Yan W, Chen X. Mutant p53 disrupts MCF-10A cell polarity in three-dimensional culture via epithelial-to-mesenchymal transitions. J Biol Chem. 2011;286(18):16218–16228. | |
Weisz L, Damalas A, Liontos M, et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007;67(6):2396–2401. | |
Schramek D, Leibbrandt A, Sigl V, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 2010;468(7320):98–102. | |
Liu M, Sakamaki T, Casimiro MC, et al. The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. 2010;70(24):10464–10473. | |
Tsubaki M, Komai M, Fujimoto S, et al. Activation of NF-κB by the RANKL/RANK system up-regulates snail and twist expressions and induces epithelial-to-mesenchymal transition in mammary tumor cell lines. J Exp Clin Cancer Res. 2013;32:62. | |
Bertheau P, Lehmann-Che J, Varna M, et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 2013;22 Suppl 2:S27–S29. | |
Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16(11):528–535. | |
Mimoto R, Taira N, Takahashi H, et al. DYRK2 controls the epithelial-mesenchymal transition in breast cancer by degrading Snail. Cancer Lett. 2013;339(2):214–225. | |
Liu T, Sun B, Zhao X, et al. OCT4 expression and vasculogenic mimicry formation positively correlate with poor prognosis in human breast cancer. Int J Mol Sci. 2014;15(11):19634–19649. | |
Piva M, Domenici G, Iriondo O, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6(1):66–79. | |
Finicelli M, Benedetti G, Squillaro T, et al. Expression of stemness genes in primary breast cancer tissues: the role of SOX2 as a prognostic marker for detection of early recurrence. Oncotarget. 2014; 5(20):9678–9688. | |
Huang YH, Luo MH, Ni YB, et al. Increased SOX2 expression in less differentiated breast carcinomas and their lymph node metastases. Histopathology. 2014;64(4):494–503. | |
Nagata T, Shimada Y, Sekine S, et al. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer. 2014; 21(1):96–101. | |
Pandya AY, Talley LI, Frost AR, et al. Nuclear localization of KLF4 is associated with an aggressive phenotype in early-stage breast cancer. Clin Cancer Res. 2004;10(8):2709–2719. | |
Pietiläinen T, Lipponen P, Aaltomaa S, Eskelinen M, Kosma VM, Syrjänen K. Expression of c-myc proteins in breast cancer as related to established prognostic factors and survival. Anticancer Res. 1995;15(3):959–964. | |
Dueck AC, Reinholz MM, Geiger XJ, et al. Impact of c-MYC protein expression on outcome of patients with early-stage HER2+ breast cancer treated with adjuvant trastuzumab NCCTG (alliance) N9831. Clin Cancer Res. 2013;19(20):5798–5807. | |
Ren J, Jin F, Yu Z, et al. MYC overexpression and poor prognosis in sporadic breast cancer with BRCA1 deficiency. Tumour Biol. 2013; 34(6):3945–3958. | |
Schlotter CM, Vogt U, Bosse U, Mersch B, Wassmann K. C-myc, not HER-2/neu, can predict recurrence and mortality of patients with node-negative breast cancer. Breast Cancer Res. 2003;5(2):R30–R36. | |
Rodriguez-Pinilla SM, Jones RL, Lambros MB, et al. MYC amplification in breast cancer: a chromogenic in situ hybridisation study. J Clin Pathol. 2007;60(9):1017–1023. | |
Feng C, Neumeister V, Ma W, et al. Lin28 regulates HER2 and promotes malignancy through multiple mechanisms. Cell Cycle. 2012; 11(13):2486–2494. | |
Xie R, Wang Y, Nie W, et al. Lin28B expression correlates with aggressive clinicopathological characteristics in breast invasive ductal carcinoma. Cancer Biother Radiopharm. 2014;29(5):215–220. | |
Moody SE, Perez D, Pan TC, et al. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8(3):197–209. | |
Guo W, Keckesova Z, Donaher JL, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148(5):1015–1028. | |
Riaz M, Sieuwerts AM, Look MP, et al. High TWIST1 mRNA expression is associated with poor prognosis in lymph node-negative and estrogen receptor-positive human breast cancer and is co-expressed with stromal as well as ECM related genes. Breast Cancer Res. 2012;14:R123. | |
Markiewicz A, Ahrends T, Welnicka-Jaskiewicz M, et al. Expression of epithelial to mesenchymal transition-related markers in lymph node metastases as a surrogate for primary tumor metastatic potential in breast cancer. J Transl Med. 2012;10:226. | |
Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22(5–6):396–403. | |
May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011;13:202. | |
Papadaki MA, Kallergi G, Zafeiriou Z, et al. Co-expression of putative stemness and epithelial-to-mesenchymal transition markers on single circulating tumour cells from patients with early and metastatic breast cancer. BMC Cancer. 2014;14:651. | |
Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One. 2013;8(9):e74250. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.