Back to Journals » Cancer Management and Research » Volume 11
The molecular mechanisms underlying BCR/ABL degradation in chronic myeloid leukemia cells promoted by Beclin1-mediated autophagy
Authors Huang X, Li Y, Shou L, Li L, Chen Z, Ye X, Qian W
Received 22 January 2019
Accepted for publication 16 May 2019
Published 6 June 2019 Volume 2019:11 Pages 5197—5208
DOI https://doi.org/10.2147/CMAR.S202442
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Xianbo Huang,1,* Ying Li,1,* Lihong Shou,2 Li Li,1 Zhenzhen Chen,3 Xiujin Ye,1 Wenbin Qian1,4
1Department of Hematology, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou 310003, People’s Republic of China; 2Department of Hematology, The Central Hospital of Huzhou City, Huzhou 313000, People’s Republic of China; 3Department of Hematology, Hangzhou First People’s Hospital, Hangzhou 310003, People’s Republic of China; 4Malignant Lymphoma Diagnosis and Therapy Center, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou 310003, People’s Republic of China
*These authors contributed equally to this work
Background: The development of drug resistance and the persistence of leukemia stem cells are major obstacles for the use of tyrosine kinase inhibitors (TKIs) in the treatment of chronic myeloid leukemia (CML). The induction of autophagic death in tumor cells represents a new route for leukemia treatment. Our previous study showed that infection of CML cells with oncolytic viruses carrying the autophagy gene Beclin1 downregulated BCR/ABL protein expression and significantly increased the killing effect of the oncolytic viruses on CML cells via autophagy activation. However, the specific molecular mechanisms underlying the regulation of BCR/ABL and Beclin1-dependent CML cell killing remain unclear.
Methods: A physical interaction between BCR/ABL and Beclin1 was characterized via GST-pulldown, co-IP and dual-luciferase reporter assays. Cell proliferation was examined via CCK-8 and clone formation assays. The expression levels of the related proteins were measured via Western blotting. Autophagosomes were observed under transmission electron microscopy. Lentiviral vectors carrying Atg7/UVRAG shRNA or the Beclin1 gene were used to modulate the expression levels of the indicated genes. Immunofluorescence were performed to examine colocalization of BCR/ABL and p62/SQSTM1. CD34+,CD38−, cells were isolated from bone marrow samples from CML patients via fluorescence-activated cell sorting.
Results: In this study, we observed that Beclin1 directly interacts with BCR/ABL. Beclin1 inhibited the activity of the BCR/ABL promoter to downregulate the level of BCR/ABL protein and to promote the downstream colocalization of p62/SQSTM1 and BCR/ABL to autolysosomes for degradation via activation of the autophagy signaling pathway. In CML cell lines, primary cells and CD34+,CD38−, leukemia stem cells, Beclin1 overexpression significantly inhibited cell growth and proliferation and induced autophagy.
Conclusion: Taken together, our results suggest that autophagy induction via Beclin1 overexpression might offer new approaches for treating TKI-resistant CML and for promoting the clearance of leukemia stem cells, both of which have important clinical implications.
Keywords: CML, autophagy, BCR/ABL, Beclin1, leukemia stem cell
Introduction
Chronic myeloid leukemia (CML) is a clonal proliferative neoplasm derived from pluripotent hematopoietic stem cells. This cancer is characterized by aberrant expression of the BCR/ABL oncogene, which is created via the chromosomal translocation t(9;22)(q34;q11). The BCR/ABL fusion protein possesses constitutive tyrosine kinase activity and causes uncontrolled myeloid cell proliferation.1 The advent of tyrosine kinase inhibitors (TKIs), eg, imatinib, greatly improved the prognosis and survival of CML patients.2 However, TKI resistance, which mainly results from BCR/ABL point mutations, gene amplification, or BCR/ABL-independent mechanisms, is now a major clinical problem for CML treatment.3 Although second-generation TKIs, such as nilotinib and dasatinib, have overcome most of the challenges stemming from drug resistance conferred by BCR/ABL mutations, the treatment of CML patients harboring the BCR/ABL T315I mutation is still ineffective.4 More importantly, TKIs cannot clear leukemia stem cells (LSCs) from CML patients.5 Therefore, studying and elucidating new targets and strategies for CML treatment still have important implications.
Autophagy is a lysosomal degradation pathway that is essential for survival, differentiation, development, and homeostasis. This process principally plays an adaptive role to protect organisms against diverse pathologies; therefore, abnormal autophagy is closely associated with the development and progression of many diseases, including tumors.6 Autophagy is a complex process that plays dual roles in cancer, as it can have both tumor suppressive and oncogenic effects under different conditions.7 On the one hand, autophagy is a pro-survival mechanism when organisms experience environmental stress, and it is also associated with drug-resistance in tumor cells (ie, protective autophagy).7,8 For example, as imatinib can induce protective autophagy in CML cells, the application of autophagy inhibitors can significantly enhance its antitumor effects.9,10 On the other hand, autophagy is also a type of antitumor mechanism. Previous studies have shown that deficiencies or low expression levels of autophagy-related genes, such as Beclin1, UVRAG, and Atg7, promote the development and progression of many tumors, including myeloid leukemia, and such genetic dysregulation is associated with poor prognosis.11–15 Recent research has shown that inducing autophagy may overcome drug resistance and exert an antileukemia effect,16,17 indicating that autophagy stimulation might offer a new treatment strategy for CML patients.
Beclin1 is a very important initiator and regulatory factor of autophagy.18 Beclin1 expression is generally downregulated in leukemia cells;19,20 however, the induction of high Beclin1 expression can exert an antitumor effect via autophagy activation.21,22 Our previous work showed that treating CML cells with chimeric oncolytic adenoviruses carrying Beclin1 induced autophagy, inhibited BCR/ABL expression, and significantly enhanced the killing effect of the oncolytic viruses in leukemia cells.23 The present study aimed to further investigate and elucidate the mechanisms underlying this BCR/ABL downregulation and the killing of leukemia cells, especially LSCs, via Beclin1-mediated autophagy.
Materials and methods
Cell lines
The human K562 and HEK 293T cell lines were obtained from the China Center for Type Culture Collection (CCTCC, Wuhan, China). The murine leukemic 32D cells carrying the T315I-mutant BCR/ABL (32Dp210-T315I) allele were kindly provided by Professor L. Qiu (Harbin Institute of Hematology and Oncology, Harbin, China). The cells were grown in RPMI-1640 medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS) and were maintained at 37 °C in a humidified atmosphere with 5% CO2. The Ethics Committee of the First Affiliated Hospital of Zhejiang University (Hangzhou, China) approved the human cell lines used in the current study.
Primary cells
Bone marrow (BM) samples were obtained from patients during CML blast crisis (CML-BC, including those carrying the T315I mutation) prior to the initiation of chemotherapy. Mononuclear cells (MNCs) were isolated from the BM samples via Ficoll-Paque (Haoyang Biotech, Tianjin, China) density gradient centrifugation according to the manufacturer’s instructions. CD34+CD38− cells were isolated from the MNCs via fluorescence-activated cell-sorting (FACS), as previously described.24 The CD34-FITC and CD38-PE antibodies were purchased from BD Biosciences (San Jose, CA, USA). The CD34+CD38−-enriched populations were usually greater than 95% pure. CD34+CD38− primary cells were incubated in Iscove’s modified Dulbecco’s medium (IMDM; Gibco) supplemented with 10% FBS and containing stem cell factor (SCF; 100 ng/ml), interleukin (IL)-3 (20 ng/ml), IL-6 (20 ng/ml), and granulocyte-macrophage colony-stimulating factor (GM-CSF; 100 ng/ml) in a humidified atmosphere with 5% CO2, as previously described.25 Cytokines were obtained from PeproTech (Rocky Hill, NJ, USA). The Ethics Committee of the First Affiliated Hospital of Zhejiang University approved the use of human specimens in the current study, and written informed consent was obtained from all patients.
GST pull-down assay
The plasmid encoding the GST-tagged isoform of BCR/ABL (pLEF GST- BCR/ABL, p210) was obtained from Addgene (plasmid #38158; Cambridge, MA, USA). The GST-tagged BCR/ABL recombinant protein was expressed in Escherichia coli and purified using Glutathione-Sepharose 4B beads (GE Healthcare, Uppsala, Sweden). The Flag-Beclin1 plasmid was kindly provided by Prof. Jie Jin from Zhejiang University. The Beclin1 mutants (Beclin1ΔN50, Beclin1ΔN100, Beclin1ΔN300, Beclin1ΔC50, Beclin1ΔC100, Beclin1ΔC150, and Beclin1ΔC200) were generated using a PCR-based mutagenesis method. Equal amounts of GST or the GST fusion proteins bound to Glutathione-Sepharose beads were incubated with lysates from HEK 293T cells that were previously transfected with plasmids encoding either Flag-tagged wild type Beclin1 or the mutated Beclin1 constructs using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). The beads were washed three times, and the bound proteins were then analyzed via 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After washing, the protein bands were visualized via autoradiography.
Coimmunoprecipitation (Co-IP) assay
The plasmid encoding HA-tagged full-length p210 BCR/ABL was kindly provided by Prof. Rongzhen Xu from Zhejiang University. The plasmids encoding Flag-Beclin1 and the mutated constructs are described above. K562 cells were electrotransfected with the plasmids carrying the HA- and Flag-tagged constructs using the Gene Pulser Xcell electroporation system (Bio-Rad, Hercules, CA, USA). After incubation for 24 h, the cells were washed with phosphate-buffered saline (PBS) and then lysed in CHAPS lysis buffer (20 mM Tris-HCl, pH 7.5, 137 mM NaCl, 2 mM EDTA, 10% glycerol, and 2% CHAPS) containing protease inhibitors. The total cell lysate (5 mg of protein) was precleared with 20 µl of protein A/G-Plus Agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 3 h; then, the supernatants were collected via centrifugation at 3,000 rpm for 3 min at 4 °C. The supernatants were incubated with the indicated antibodies overnight at 4 C with gentle agitation. After three washes with CHAPS buffer, the immunocomplexes were mixed with 2× SDS sample buffer, boiled for 5 min, and then subjected to Western blot analysis.
Stable Beclin1 overexpression
The recombinant lentiviral vector carrying Beclin1 (LV-Beclin1) and the negative control (LV-NC) vector were purchased from Hanheng Biotech (Shanghai, China). Cells were transfected with the lentiviruses at a multiplicity of infection (MOI) of 100, cultured at 37 °C for 48 h, and then selected with puromycin (Gibco). Cellular Beclin1 expression was evaluated via Western blot analysis.
Dual luciferase reporter assay
The pGL3-BCR/ABL promoter constructs were prepared as described in the previous report by Marega et al26 K562 cells overexpressing Beclin1 (K562-Beclin1) and the negative control cells (K562-NC) were transfected with the pGL3-BCR/ABL promoter construct via electroporation for 24 h. Luciferase activity was measured using a Dual-Luciferase Assay Kit (Promega Corporation, Madison, WI, USA) according to the manufacturer’s instructions.
Cell viability assay
Cell viability was measured with the Cell Counting Kit-8 (Dojindo, Tokyo, Japan). K562-NC and K562-Beclin1 cells were incubated for 24 h with 3-methyladenine (3-MA; Selleck Chemicals, Houston, TX, USA) or epoxomicin (Epo; Merck Millipore, Billerica, MA, USA) and plated in 96-well plates (5×104 cells/ml). 32Dp210-T315I-Beclin1 cells were also plated in 96-well plates (5×104 cells/ml) and incubated for 0, 24, 48 and 72 h. The absorbance at 450 nm was measured with a microplate spectrophotometer (Bio-Rad).
Western blot analysis
Total proteins were extracted from the collected cells. Equal amounts of protein were separated via SDS-PAGE and then transferred onto polyvinylidene fluoride (PVDF) membranes (Merck Millipore). The membranes were subsequently blocked for 2 h in Tris-buffered saline containing 0.1% Tween and 5% nonfat dry milk and then incubated with primary antibodies overnight at 4 °C. After incubation with horseradish peroxidase-conjugated secondary antibodies (1:5000; Multi Sciences Biotech, Hangzhou, China), the PVDF membranes were visualized using chemiluminescence (ECL; Biological Industries, Beit Ahemeq, Israel). The specific Caspase-3 inhibitor Z-DEVD-FMK (DEVD) and the tyrosine kinase inhibitor imatinib were obtained from Selleck Chemicals. The primary antibodies used were as follows: anti-Beclin1, anti-cleaved PARP, anti-cleaved Caspase-3, anti-Atg5, anti-Atg7, anti-UVRAG, anti-p-BCR/ABL (Tyr177), anti-Flag, anti-HA, and anti-GAPDH, purchased from Cell Signaling Technology (Beverly, MA, USA), and anti-BCR/ABL, anti-p62/SQSTM1, and anti-LC3 I/II, purchased from Abcam (Cambridge, UK).
Transmission electron microscopy
Cultured cells were washed twice in PBS and fixed overnight in 2.5% glutaraldehyde in PBS (pH 7.4). The samples were treated with 1.0% osmium tetroxide, dehydrated via washes with a series of solutions with graduated ethanol concentrations (50%, 70%, 90%, and 100%), and then embedded in durcupan resin. Ultrathin sections (65 nm) were cut with an ultramicrotome, poststained with 1% uranyl acetate and 0.1% lead citrate, and examined with a TECNAI 10 electron microscope (Philips Electronic Instruments, Mahwah, NJ, USA) at 60 kV.
Gene knockdown via short hairpin RNA
Recombinant lentiviral vectors carrying specific short hairpin RNAs (shRNAs) against Atg7 and UVRAG and a negative control shRNA (scramble) were purchased from Hanheng Biotechnology. The cells were infected with the lentivirus for 48 h and then selected with puromycin. Cell lines with stable gene knockdown were collected for further experiments.
Immunofluorescence studies
K562-NC and K562-Beclin1 cells were incubated with or without CA074 (Calbiochem, Darmstadt, Germany) for 24 h and then fixed with 4% formaldehyde. After permeabilization with 0.3% Triton X-100 and incubation with goat serum (Dawen Biotech, Hangzhou, China), the cells were incubated with the indicated primary antibodies overnight at 4 °C. Next, the cells were incubated with Alexa Fluor 488 (green) or 647 (red) dye-labeled secondary antibodies (Abcam). After washing with PBS, the nuclei were counterstained with DAPI (Southern Biotech, AL, USA). The fluorescence was observed under a fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Colony-forming cell (CFC) assay
CML cells infected with LV-Beclin1 or LV-NC were plated in complete methylcellulose medium (MethoCult H4434; StemCell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. The cells were cultured in a humidified incubator at 37 °C with 5% CO2 for 14 days. Granulocyte-macrophage colony-forming units (GM-CFUs) were counted under a light microscope (Olympus, Tokyo, Japan) and recorded for statistical analysis.
Statistical analysis
The experimental results are presented as the mean ± standard deviation. The differences between groups were evaluated via analysis of variance (ANOVA) and Student’s t-test. P<0.05 was considered significant.
Results
Beclin1 and the BCR/ABL fusion protein directly interact
First, bioinformatics predictions of the interaction between Beclin1 and BCR/ABL were performed using the STRING database (
 | Figure 1 The interaction between BCR/ABL and Beclin1. (A) Prediction of the interaction between Beclin1 and BCR/ABL using bioinformatics analysis. (B) Schematic structures of the Beclin1 mutant constructs, which were generated using a PCR-based mutagenesis method. (C) The BCR/ABL-binding motifs in Beclin1 were evaluated using Flag-Beclin1 and the deletion mutant constructs, which were cotransfected into HEK 293T cells with GST or GST-tagged BCR/ABL. The interactions between the proteins were determined via GST pull-down assays. (D) K562 cells electrotransfected with the indicated plasmids were subjected to immunoprecipitation with an anti- Flag antibody. The immunoprecipitated complexes were separated via SDS-PAGE and probed with an anti- Flag antibody to detect Flag-tagged Beclin1 and the mutant constructs and an anti-HA antibody to detect HA-tagged BCR/ABL. (E) K562-Beclin1 or K562-NC cells were electrotransfected with the pRL-TK and pGL3-BCR/ABL promoter constructs. The dual-luciferase reporter assay revealed the binding of Beclin1 to the BCR/ABL promoter in K562 cells. The results are presented as the mean ± standard deviation (SD); **P<0.01. Data are representative of three independent experiments. Abbreviation: NC, negative control. |
Beclin1 overexpression downregulated BCR/ABL expression in CML cells, inhibited cell growth and proliferation, and induced autophagy
To examine whether Beclin1 overexpression in CML cells inhibits BCR/ABL expression and induces autophagic death, K562 cells were transfected with a lentiviral vector carrying the Beclin1 gene to robustly increase the Beclin1 protein level (Figure 2A). Western blot analysis suggested that the BCR/ABL level in Beclin1-overexpressing cells was significantly decreased (Figure 2A). Apoptosis of CML cells can be accompanied by caspase-dependent BCR/ABL degradation.27 Our results show that Beclin1 overexpression did not induce the cleavage and activation of caspase-3 and PARP, which lie downstream in the apoptosis pathway, and the caspase-3 inhibitor DEVD did not reverse BCR/ABL downregulation (Figure 2A). These results suggest that the BCR/ABL downregulation mediated by Beclin1 overexpression is not associated with cell apoptosis. Based on the results of CCK-8 detection assays, it was clear that Beclin1 overexpression inhibited K562 cell growth and had a synergistic effect with the proteasome inhibitor Epo (which can be used as an autophagy activator28,29), whereas the autophagy inhibitor 3-MA30 reversed, at least partially, the inhibitory effect of Beclin1 overexpression on cell growth. These results suggest that the mechanism underlying the inhibition of K562 cell survival by Beclin1 overexpression is closely associated with autophagy (Figure 2B). We also used TEM to show that Beclin1 overexpression promoted autophagosome formation in cells and that it had a synergistic effect with Epo that could be partially reversed by 3-MA (Figure 2C). Western blot analysis showed that Beclin1 overexpression induced the expression of the autophagy-related genes Atg5, Atg7, and UVRAG and promoted an increase in the conversion of LC3-I into LC3-II downstream in the autophagy pathway, while downregulating the p62/SQSTM1 level (Figure 2D), suggesting the occurrence of autophagy. These phenomena were enhanced by Epo and partially reversed by 3-MA (Figure 2D), and these observations are consistent with those of the TEM and CCK-8 experiments. However, neither 3-MA nor Epo had significant effects on the BCR/ABL downregulation induced by Beclin1 overexpression, and these observations require further study.
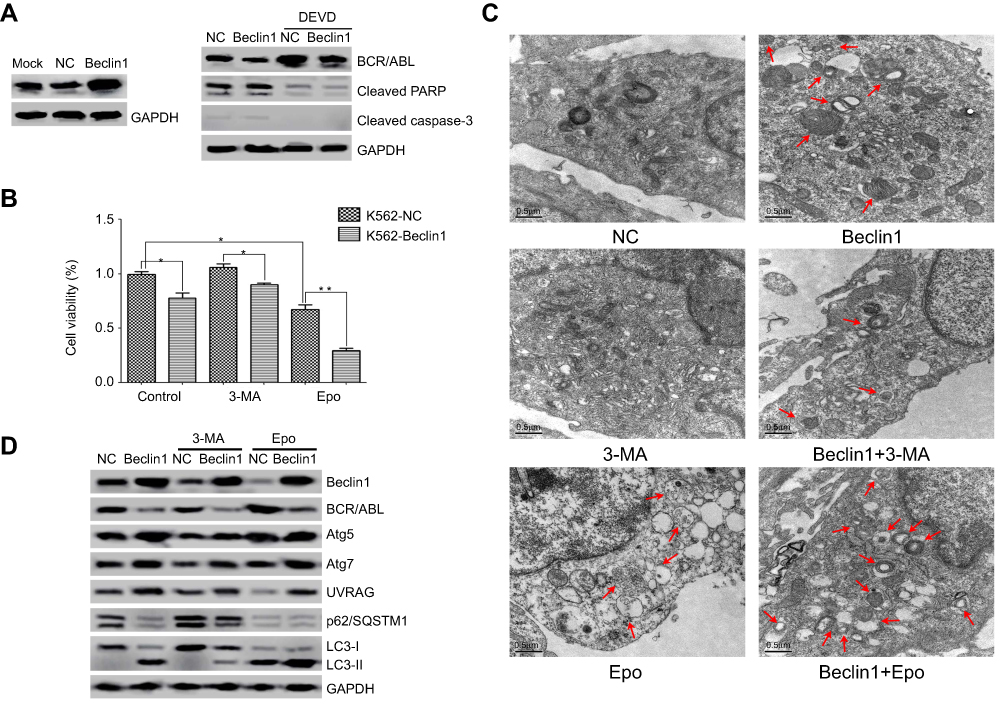 | Figure 2 Beclin1 overexpression in K562 cells induces BCR/ABL downregulation and promotes cell death via autophagic stimulation. (A) K562-NC or K562-Beclin1 cells were subjected to Western blot analysis to assess the levels of BCR/ABL, cleaved caspase-3 and cleaved PARP. The specific caspase-3 inhibitor DEVD was used to inhibit caspase-dependent apoptosis. (B) The indicated cells were cultured with or without the autophagy inhibitor 3-MA (5 mM) or the proteasomal inhibitor Epo (500 nM) for 24 h. Cell viability was measured using the CCK-8 assay. Data are presented as the mean ± SD of three independent experiments; *P<0.05, **P<0.01. (C) Representative TEM photomicrographs of K562-Beclin1 and control K562 cells treated with 3-MA (5 mM) or Epo (500 nM) for 24 h and untreated cells. Arrows, autophagic vacuoles. Scale bars, 0.5 μm. (D) Western blot analysis of the levels of BCR/ABL and other autophagy-related proteins in K562-Beclin1 and K562-NC cells. Cells were pretreated in the absence or presence of 3-MA (5 mM) or Epo (500 nM) for 24 h. The results are representative of three independent experiments. Abbreviations: NC, negative control; 3-MA, 3-methyladenine; Epo, epoxomicin; DEVD, Z-DEVD-FMK. |
Beclin1 overexpression induced autophagy-dependent Atg7 and UVRAG expression and the colocalization of BCR/ABL and p62/SQSTM1 to autolysosomes for degradation
Atg7 is an important regulatory factor of autophagy, and it forms protein complexes with Beclin1 to activate the classical autophagy signaling pathway.31 Furthermore, as a Beclin1-binding protein, UVRAG mediates the activation of the Beclin1-PI(3)KC3 complex to promote autophagy and to further inhibit tumor cell growth and proliferation.12 Our previous results confirmed that the autophagy induced by Beclin1 overexpression was accompanied by upregulation of the Atg7 and UVRAG levels. Next, the effects of Atg7 and UVRAG downregulation were investigated. Hanheng Biotechnology synthesized three specific shRNAs targeting Atg7 or UVRAG and constructed lentiviral vectors for infecting K562 cells. The shRNAs with the highest interference efficiency were selected for subsequent experiments (Figure 3A). The downregulation of either Atg7 or UVRAG significantly reversed the LC3-II upregulation mediated by Beclin1 overexpression, suggesting that autophagy was inhibited. Moreover, these results show that the silencing of Atg7 or UVRAG expression partially reversed Beclin1-mediated BCR/ABL downregulation, suggesting the influence of some autophagy-related factors (Figure 3B).
 | Figure 3 Beclin1 overexpression-induced autophagy requires Atg7 and UVRAG in K562 cells and promotes BCR/ABL degradation via an interaction with p62/SQSTM1. (A) The knockdown efficiency of Atg7 or UVRAG by lentiviral vectors in K562 cells was determined via Western blotting. (B) The BCR/ABL and LC3-I/II expression levels in Atg7- and UVRAG-silenced K562 cells were assessed using Western blotting. (C) Fluorescence images showing p62/SQSTM1 (green) and BCR/ABL (red) in the indicated cells (K562-NC, K562-Beclin1, and K562-Beclin1 cells treated with 10 μM CA074 for 24 h). The nuclei were stained with DAPI (blue). The merged images show the colocalization of p62/SQSTM1 and BCR/ABL (yellow). All experiments were repeated at least three times. Abbreviations: NC, negative control; shRNA, short hairpin RNA. |
Arsenic trioxide (As2O3) induces autophagy in CML cells. As2O3 also promotes the formation of a protein complex between p62/SQSTM1 and BCR/ABL that leads to their colocalization in autolysosomes for degradation via the cathepsin B protease.32 Immunofluorescence experiments showed that Beclin1 overexpression caused perinuclear localization of BCR/ABL in K562 cells, resulting in an obvious colocalization phenomenon with p62/SQSTM1. Further experiments showed that the cathepsin B inhibitor CA074 significantly inhibited BCR/ABL and p62/SQSTM1 colocalization (Figure 3C). Therefore, we speculate that with the help of p62/SQSTM1, BCR/ABL enters autolysosomes for cathepsin B-dependent degradation, which might be an important mechanism underlying the BCR/ABL downregulation induced by Beclin1 overexpression.
Beclin1 overexpression downregulated BCR/ABL, inhibited growth and induced autophagy in TKI resistant CML cells carrying the T315I mutation
To test the cytotoxic effects of imatinib on CML cells harboring the T315I mutation, 32D murine myeloid cells stably expressing T315I-mutant BCR/ABL (32Dp210-T315I) were treated with different imatinib concentrations for 24 h. The Western blot results showed that the expression and activation levels of BCR/ABL did not change significantly, indicating that the 32Dp210-T315I cells were resistant to TKI treatment (Figure 4A). Next, we investigate the effects of Beclin1 overexpression in 32Dp210-T315I cells. The cells were transfected with the lentiviral vector carrying Beclin1 gene. Western blot analysis suggested that Beclin1 overexpression resulted in a decreased level of BCR/ABL expression and activation and facilitated an increase in the conversion of downstream LC3-I into LC3-II (Figure 4B). Bone marrow mononuclear cells (BMMCs) carrying the T315I mutation were isolated from CML-BC patients and also infected with the Beclin1-overexpressing lentiviruses, and CFC assays were then performed. The results showed that the number of GM-CFUs in the Beclin1-overexpressing primary cells was significantly lower compared with that of the control group, indicating that Beclin1-mediated autophagy can overcome TKI-resistance and exert a significant growth inhibition in CML (Figure 4C).
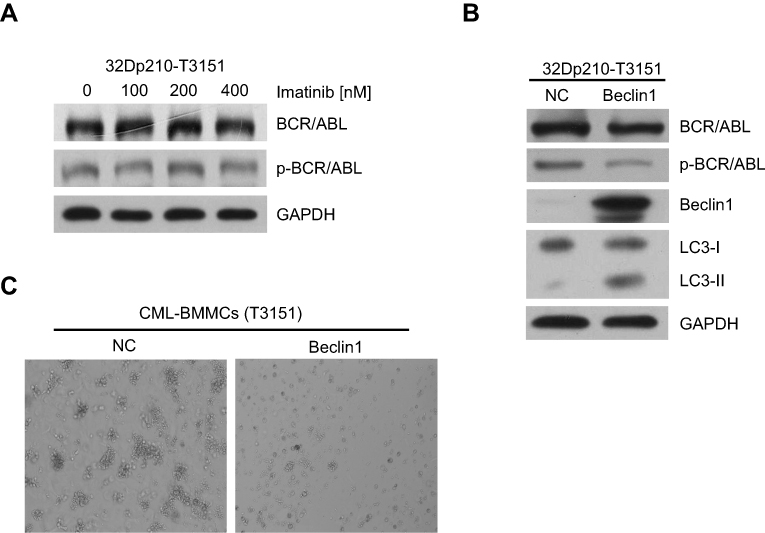 | Figure 4 Beclin1 overexpression in T315I mutant CML cells induces BCR/ABL downregulation and promotes autophagic cell death. (A) 32Dp210-T315I cells were cultured with different imatinib concentrations for 24 h, and the expression and activation of BCR/ABL protein were assessed via Western blotting. (B) Western blot analysis of BCR/ABL, p-BCR/ABL, Beclin1 and LC3-I/II levels in Beclin1-overexpressing or negative control 32Dp210-T315I cells. (C) CFC assays were performed with BMMCs derived from CML-BC patients with T315I mutant BCR/ABL. The primary cells were infected with LV-Beclin1 or LV-NC. After 14 days of culture in methylcellulose medium, GM-CFUs were observed. All experiments were repeated at least three times. Abbreviations: BMMCs, bone marrow mononuclear cells; NC, negative control; GM-CFUs, granulocyte-macrophage colony-forming units. |
Beclin1 overexpression downregulated BCR/ABL expression in CD34+CD38− CML stem cells and induced autophagy
To study the effects of Beclin1 overexpression in LSCs from CML patients, BM CD34+CD38− cells were isolated from patients with CML-BC using Ficoll density gradient centrifugation and FACS. After cell identification, the purity was 96.37±1.6%, indicating that the cells could be used for subsequent experiments (Figure 5A). The CD34+CD38− cells were infected with Beclin1-overexpressing lentivirus, and the effect of high Beclin1 expression on the colony formation ability of the LSCs was investigated via CFC assays. The number of GM-CFUs in the Beclin1-overexpressing CD34+CD38− group was significantly lower than that in the control group, suggesting that Beclin1 overexpression exerts significant growth inhibition in LSCs in CML (Figure 5B). Next, we performed Western blots to analyze the effects of Beclin1 overexpression on the levels of BCR/ABL and autophagy pathway proteins in CD34+CD38− cells. As shown in Figure 5C, Beclin1 overexpression significantly inhibited BCR/ABL protein expression in LSCs. Furthermore, we observed higher levels of the autophagy-related proteins Atg5, Atg7, and UVRAG, a significant increase in downstream LC3-II levels, and a reduction in the p62/SQSTM1 level. These results suggest that Beclin1 overexpression induces autophagy in CD34+CD38− CML stem cells.
 | Figure 5 Beclin1 overexpression downregulates BCR/ABL expression and induces autophagy in CML-LSCs. (A) Representative dot plots showing the purity of the CD34+CD38− LSCs collected via FACS from CML primary cells. (B) The CFC assay was performed with CML-LSCs infected with LV-Beclin1 or LV-NC. After 14 days of culture in methylcellulose medium, GM-CFUs were observed and counted (*P<0.05). (C) Western blot analysis of the expression levels of BCR/ABL and other autophagy-related proteins in CML-LSCs with and without Beclin1 overexpression. Three biological replicates were performed for each experiment. Abbreviations: NC, negative control; FITC, fluorescein isothiocyanate; PE, phycoerythrin; GM-CFUs, granulocyte-macrophage colony-forming units. |
Discussion
The BCR/ABL fusion protein is the most critical disease-causing factor of CML. The inhibition or clearance of BCR/ABL is considered the most effective method for CML treatment.1 Although TKIs have achieved great success in CML treatment, increasing TKI resistance and the persistence of LSCs remain major challenges.2,3,5 Many recent studies have indicated that inhibition of BCR/ABL gene transcription and induction of BCR/ABL protein degradation may be new routes for CML treatment.33–35 Some drugs, such as As2O3 or imatinib, promote autolysosomal BCR/ABL degradation by inducing autophagy.10,32,36 Currently, the specific mechanisms underlying autophagy-dependent BCR/ABL degradation in CML cells are not clear. Our previous study confirmed that oncolytic adenoviruses carrying the Beclin1 gene had an enhanced killing effect against CML cells that was accompanied by BCR/ABL downregulation.23 Beclin1 is a tumor suppressor and a critical factor for autophagy regulation.6 In this study, we showed that Beclin1 directly interacts with BCR/ABL and inhibits its expression at the transcriptional level by inhibiting its promoter activity. These findings led us to further investigate the relationship between autophagy and CML treatment. In cancer, the role of autophagy is complex and clearly depends on tumor stage, type and the driving oncogene.37 In general, autophagy is thought to promote longevity and suppress tumorigenesis in healthy individuals, mainly via its function of protecting cells from genotoxic stress.37 However, a number of studies have described tumor-promoting roles for autophagy after tumor formation as well as during metastasis, where it supports cancer cell survival.38–40 The autophagy signaling pathway is normally defective or inhibited in leukemia cells, which can lead to poor prognosis.19,20 These observations suggest that the induction of autophagy may serve as a new route for CML treatment.
Infection of a CML cell line with lentiviruses carrying the Beclin1 gene significantly inhibited cell growth and proliferation, successfully induced autophagy, and downregulated BCR/ABL protein expression. We further investigated the relationship between autophagy and BCR/ABL. The results indicated that at the initial stage of autophagy, Beclin1 interacts with UVRAG to form protein complexes that promote autophagy development.41 Furthermore, the Atg7 protein is necessary during autophagic vacuole formation.42,43 Our study confirmed that Beclin1-induced autophagy was accompanied by upregulation of the Atg7 and UVRAG levels. Silencing of Atg7 and UVRAG protein expression could significantly reverse the increased autophagy. These results further confirm that the development of autophagy requires the involvement of autophagy-related proteins, such as Beclin1, UVRAG, and Atg7. We also showed that the silencing of Atg7 and UVRAG in Beclin1-overexpressing K562 cells partially reversed BCR/ABL downregulation. Studies by other groups have also indicated that Atg7 silencing reverses autophagy-dependent BCR/ABL degradation.32,36 The downstream autophagy factors p62/SQSTM1 and LC3 interact with each other to participate in autophagosome formation, and the latter fuses with lysosomes to form autolysosomes, resulting in the degradation of the autophagic contents.44 Goussetis et al confirmed that treatment of CML with arsenic induced p62/SQSTM1-assisted BCR/ABL localization to autolysosomes for cathepsin B-dependent degradation.32 Our study also showed that Beclin1 promoted the colocalization of p62/SQSTM1 and BCR/ABL in K562 cells, which was reversed by treatment with the cathepsin B inhibitor CA074. The data from the T315I-mutant CML cell line 32Dp210-T315I further suggest that Beclin1 overexpression can overcome TKI-resistance and that it exerts its anti-CML effect via autophagy induction and the promotion of BCR/ABL inactivation and degradation. In summary, our study indicates that Beclin1 inhibits BCR/ABL expression at the transcriptional level and induces autophagy-dependent BCR/ABL degradation.
The persistence of LSCs, which cannot be cleared by TKIs, remains a challenge in CML treatment.2,3,5 As2O3 exerts its CML-LSC killing effect by activating autophagy.32 Our experiments showed that autophagy induced by Beclin1 overexpression could also decrease the BCR/ABL level in CML-LSCs and that it significantly inhibited cell growth. Taken together, these results suggest that autophagy induction could serve as a new route to promote CML-LCS clearance.
Conclusion
In summary, defective or abnormal autophagy is closely associated with the development and progression of leukemia and drug resistance. Our study showed that Beclin1-mediated autophagy activation in CML cell lines and primary cells inhibited BCR/ABL gene expression, promoted autophagy-dependent degradation of the BCR/ABL protein, and killed tumor cells. Our study suggests that autophagy activation may be a new strategy for treating TKI-resistant CML and for promoting LSC clearance, both of which have important clinical implications.
Ethics approval and informed consent
The Ethics Committee of the First Affiliated Hospital of Zhejiang University (Hangzhou, China) approved the use of human cell lines and specimens in the current study. Written informed consent was obtained from all patients. All methods and experimental protocols were approved by the First Affiliated Hospital of Zhejiang University.
Acknowledgments
This research was supported by funds from the Natural Science Foundation of Zhejiang Province (No. LQ19H080005 and No. LY19H080005), the Science Technology Department of Zhejiang Province (No. 2016C33137 and No. 2018C03016-1), and the Health Department of Zhejiang Province (No. 2015KYA210).
Author contributions
WQ and XY designed the study; XH and YL conducted the study; LS, LL and ZC contributed new analytical tools and reagents; XH and LS analyzed the data and XH drafted the manuscript. Critical revision of the manuscript was done by XY and WQ. All authors contributed to the data analysis and drafting and revision of the article and gave their final approval of the version to be published. All of the authors agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mughal TI, Radich JP, Deininger MW, et al. Chronic myeloid leukemia: reminiscences and dreams. Haematologica. 2016;101(5):541–558. doi:10.3324/haematol.2015.139337
2. Wei G, Rafiyath S, Liu D. First-line treatment for chronic myeloid leukemia: dasatinib, nilotinib, or imatinib. J Hematol Oncol. 2010;3:47. doi:10.1186/1756-8722-3-50
3. Yang K, Fu LW. Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: a review. Crit Rev Oncol Hematol. 2015;93(3):277–292. doi:10.1016/j.critrevonc.2014.11.001
4. Soverini S, Rosti G, Iacobucci I, Baccarani M, Martinelli G. Choosing the best second-line tyrosine kinase inhibitor in imatinib-resistant chronic myeloid leukemia patients harboring Bcr-Abl kinase domain mutations: how reliable is the IC50? Oncologist. 2011;16(6):868–876. doi:10.1634/theoncologist.2010-0388
5. Chomel JC, Bonnet ML, Sorel N, et al. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood. 2011;118(13):3657–3660. doi:10.1182/blood-2011-02-335497
6. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018
7. Kumar A, Singh UK, Chaudhary A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med Chem. 2015;7(12):1535–1542. doi:10.4155/fmc.15.88
8. Lin YX, Wang Y, An HW, et al. Peptide-based autophagic gene and cisplatin co-delivery systems enable improved chemotherapy resistance. Nano Lett. 2019;19:2968–2978. [Epub ahead of print]. doi:10.1021/acs.nanolett.9b00083
9. Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119(5):1109–1123. doi:10.1172/JCI35660
10. Crowley LC, Elzinga BM, O’Sullivan GC, McKenna SL. Autophagy induction by Bcr-Abl-expressing cells facilitates their recovery from a targeted or nontargeted treatment. Am J Hematol. 2011;86(1):38–47. doi:10.1002/ajh.21914
11. Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820. doi:10.1172/JCI20039
12. Liang C, Feng P, Ku B, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8(7):688–699. doi:10.1038/ncb1426
13. Watson AS, Mortensen M, Simon AK. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle. 2011;10(11):1719–1725. doi:10.4161/cc.10.11.15673
14. Ramsingh G, Jacoby MA, Shao J, et al. Acquired copy number alterations of miRNA genes in acute myeloid leukemia are uncommon. Blood. 2013;122(15):44–51. doi:10.1182/blood-2013-03-488007
15. Zare-Abdollahi D, Safari S, Movafagh A, et al. Expression analysis of BECN1 in acute myeloid leukemia: association with distinct cytogenetic and molecular abnormalities. Int J Lab Hematol. 2016;38(2):125–132. doi:10.1111/ijlh.12454
16. Laane E, Tamm KP, Buentke E, et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ. 2009;16(7):1018–1029. doi:10.1038/cdd.2009.46
17. Bonapace L, Bornhauser BC, Schmitz M, et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest. 2010;120(4):1310–1323. doi:10.1172/JCI39987
18. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. doi:10.1038/cdd.2010.191
19. Liang PQ, Miao M, Liu ZG, et al. Expression of autophagy genes in acute myeloid leukemia: associations with clinical characteristics and prognosis. Neoplasma. 2018;65(5):807–814. doi:10.4149/neo_2018_171028N691
20. Radwan SM, Hamdy NM, Hegab HM, El-Mesallamy HO. Beclin-1 and hypoxia-inducible factor-1α genes expression: potential biomarkers in acute leukemia patients. Cancer Biomark. 2016;16(4):619–626. doi:10.3233/CBM-160603
21. Cicchini M, Chakrabarti R, Kongara S, et al. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy. 2014;10(11):2036–2052. doi:10.4161/auto.34398
22. Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42(1):23–35. doi:10.1016/j.molcel.2011.02.009
23. Tong Y, You L, Liu H, et al. Potent antitumor activity of oncolytic adenovirus expressing Beclin-1 via induction of autophagic cell death in leukemia. Oncotarget. 2013;4(6):860–874. doi:10.18632/oncotarget.1018
24. Graham SM, Jørgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–325.
25. Jin Y, Lu Z, Ding K, et al. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res. 2010;70(6):2516–2527. doi:10.1158/0008-5472.CAN-09-3950
26. Marega M, Piazza RG, Pirola A, et al. BCR and BCR-ABL regulation during myeloid differentiation in healthy donors and in chronic phase/blast crisis CML patients. Leukemia. 2010;24(8):1445–1449. doi:10.1038/leu.2010.101
27. Shi X, Chen X, Li X, et al. Gambogic acid induces apoptosis in imatinib-resistant chronic myeloid leukemia cells via inducing proteasome inhibition and caspase-dependent Bcr-Abl downregulation. Clin Cancer Res. 2014;20(1):151–163. doi:10.1158/1078-0432.CCR-13-1063
28. Tang B, Cai J, Sun L, et al. Proteasome inhibitors activate autophagy involving inhibition of PI3K-Akt-mTOR pathway as an anti-oxidation defense in human RPE cells. PLoS One. 2014;9(7):e103364. doi:10.1371/journal.pone.0103364
29. Sha Z, Schnell HM, Ruoff K, Goldberg A. Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy activation. J Cell Biol. 2018;217(5):1757–1776. doi:10.1083/jcb.201708168
30. Morselli E, Galluzzi L, Kepp O, et al. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta. 2009;1793(9):1524–1532. doi:10.1016/j.bbamcr.2009.01.006
31. Jin S, Wei J, You L, Liu H, Qian W. Autophagy regulation and its dual role in blood cancers: a novel target for therapeutic development. Oncol Rep. 2018;39(6):2473–2481. doi:10.3892/or.2018.6370
32. Goussetis DJ, Gounaris E, Wu EJ, et al. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012;120(17):3555–3562. doi:10.1182/blood-2012-01-402578
33. Shi X, Jin Y, Cheng C, et al. Triptolide inhibits Bcr-Abl transcription and induces apoptosis in STI571-resistant chronic myelogenous leukemia cells harboring T315I mutation. Clin Cancer Res. 2009;15(5):1686–1697. doi:10.1158/1078-0432.CCR-08-2141
34. Nimmanapalli R, Bali P, O’Bryan E, et al. Arsenic trioxide inhibits translation of mRNA of bcr-abl, resulting in attenuation of Bcr-Abl levels and apoptosis of human leukemia cells. Cancer Res. 2003;63(22):7950–7958.
35. Mao JH, Sun XY, Liu JX, et al. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. Proc Natl Acad Sci U S A. 2010;107(50):21683–21688. doi:10.1073/pnas.1016311108
36. Elzinga BM, Nyhan MJ, Crowley LC, O’Donovan TR, Cahill MR, McKenna SL. Induction of autophagy by Imatinib sequesters Bcr-Abl in autophagosomes and down-regulates Bcr-Abl protein. Am J Hematol. 2013;88(6):455–462. doi:10.1002/ajh.23428
37. Chen HY, White E. Role of autophagy in cancer prevention. Cancer Prev Res (Phila). 2011;4(7):973–983. doi:10.1158/1940-6207.CAPR-10-0387
38. Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi:10.1016/j.ccr.2006.06.001
39. Strohecker AM, White E. Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy. 2014;10(2):384–385. doi:10.4161/auto.27320
40. Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85(6):830–838. doi:10.1124/mol.114.091850
41. Wu S, He Y, Qiu X, et al. Targeting the potent Beclin 1-UVRAG coiled-coil interaction with designed peptides enhances autophagy and endolysosomal trafficking. Proc Natl Acad Sci U S A. 2018;115(25):E5669–E5678. doi:10.1073/pnas.1721173115
42. Yu L, Alva A, Su H, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304(5676):1500–1502. doi:10.1126/science.1096645
43. Karvela M, Baquero P, Kuntz EM, et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy. 2016;12(6):936–948. doi:10.1080/15548627.2016.1162359
44. Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi:10.1016/S0076-6879(08)03612-4
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.