")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
The Microbiome in Comedonal Contents of Inflammatory Acne Vulgaris is Composed of an Overgrowth of Cutibacterium Spp. and Other Cutaneous Microorganisms
Authors Akaza N , Takasaki K, Nishiyama E, Usui A, Miura S, Yokoi A, Futamura K , Suzuki K , Yashiro Y, Yagami A
Received 30 June 2022
Accepted for publication 25 August 2022
Published 21 September 2022 Volume 2022:15 Pages 2003—2012
DOI https://doi.org/10.2147/CCID.S379609
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Narifumi Akaza,1 Kazuto Takasaki,2 Eri Nishiyama,2 Atsuko Usui,2 Shiori Miura,1 Aya Yokoi,3,4 Kyoko Futamura,4 Kayoko Suzuki,4 Youichi Yashiro,1 Akiko Yagami4
1Research Laboratories, Nippon Menard Cosmetic Co., Ltd., Nagoya, Japan; 2Fasmac Co., Ltd., Kanagawa, Japan; 3Nihombashi Irodori Dermatology Clinic, Tokyo, Japan; 4Department of Allergology, Fujita Health University School of Medicine, Nagoya, Japan
Correspondence: Narifumi Akaza, Research Laboratories, Nippon Menard Cosmetic Co., Ltd., 2-7 Torimi-cho, Nishi-ku, Nagoya, Aichi, 451-0071, Japan, Tel +81 52 531 6263, Fax +81 52 531 6277, Email [email protected]
Background: Acne vulgaris (acne) and cutaneous resident microorganisms are considered to be closely related. However, the bacterial and fungal microbiota in the comedonal contents of inflammatory acne lesions have not yet been investigated in detail.
Purpose: To clarify the relationship between cutaneous microorganisms and acne, we examined the microbiome in the comedonal contents of inflammatory acne and on the facial skin of patients with acne using 16s rRNA and ITS gene sequencing with a next-generation sequencer (NGS).
Patients and Methods: Twenty-two untreated Japanese acne outpatients were examined. The comedonal contents of inflammatory acne lesions on the face were collected using a comedo extractor. Skin surface samples from facial skin were collected using the swab method.
Results: The results obtained revealed that the predominant bacteria in the comedonal contents of inflammatory acne were Cutibacterium spp. (more prominent in areas with large amounts of sebum), while those on the skin surface were Staphylococcus spp. Malassezia spp., particularly Malassezia restricta, were the predominant fungi in both the comedonal contents of inflammatory acne and on the skin surface. The bacterial microbiome in comedonal contents exhibited stronger metabolic activity, including the production of enzymes related to acne, than that on the skin surface.
Conclusion: These results indicate that acne is an inflammatory disease involving the overgrowth of Cutibacterium acnes and other cutaneous resident microorganisms, including Malassezia spp.
Keywords: acne vulgaris, Cutibacterium, Staphylococcus, Malassezia, microbiome, microbiota, next generation sequencing
Introduction
Acne vulgaris (acne) is a common, chronic, and inflammatory skin disorder of the pilosebaceous apparatus. Four factors contribute to acne: the hypersecretion of sebum, the abnormal proliferation and differentiation of keratinocytes in hair follicles, bacterial colonization, and host inflammatory responses.1 Among these factors, the predominant cutaneous bacteria, Cutibacterium acnes (formerly known as Propionibacterium acnes) is considered to mediate inflammatory responses and lead to sub-clinical and inflammatory acne lesions. Several mechanisms of acne pathogenesis involving C. acnes have been proposed, including the induction of inflammation,2,3 comedone formation,4 and enhancements in sebaceous gland activity.5 Lipase (EC 3.1.1.3), protease (EC 3.4), phosphatase (EC 3.1.3), hyaluronate lyase (EC 4.2.2.1), endoglycoceramidase (EC 3.2.1.123), and neuraminidase (EC 3.2.1.18) produced by C. acnes impair hair follicles, sebaceous glands, and the dermal extracellular matrix, ultimately aggravating inflammation.1,6 Moreover, free fatty acids produced by lipase contribute to the formation of comedones.7
Inflammatory comedonal contents include many Staphylococcus and Malassezia spp. as well as C. acnes.8 Malassezia spp., which are lipophilic fungi, promote the secretion of pro-inflammatory cytokines from keratinocytes.9 Moreover, the lipase activity of these fungi is approximately 100-fold stronger than that of C. acnes.10 These findings indicate that not only C. acnes, but also cutaneous resident microorganisms, including Malassezia spp., are involved in acne pathogenesis.
Analyses of the microbiome of acne patients have been performed using culture-independent technologies, namely, DNA sequencing. Bek-Thomsen et al demonstrated that the predominant bacteria in pilosebaceous units was Cutibacterium spp.11 Fitz-Gibbon et al compared the skin microbiome between acne patients and healthy individuals by sampling pilosebaceous units on their noses, and showed that while the relative abundance of C. acnes was similar, strain population structures significantly differed between the two cohorts.12 Li et al analyzed the skin microbiomes of acne patients and healthy controls using skin swabs, and found that the skin microbiome of acne patients was significantly different from that in healthy controls.13 Dreno et al also examined the skin microbiome of acne patients using skin swabs, and reported that the predominant bacteria were Staphylococcus spp.14 The bacteria as well as fungi and C. acnes phages in samples of pilosebaceous units on the nose of acne patients have also been investigated.15 However, previous findings on acne patients were obtained using samples from the skin surface and pilosebaceous units (not all were inflamed); therefore, the comedonal contents of inflammatory acne lesions remain unclear. Furthermore, the fungal microbiome has not been examined in detail.
The aim of the present study was to elucidate the relationship between the cutaneous bacterial and fungal microbiomes and acne using a comprehensive analysis involving 16s rRNA and ITS gene sequencing with a next-generation sequencer (NGS) of the facial comedonal contents of inflammatory acne and the facial skin surface of patients with acne in order to clarify the relationship between cutaneous microorganisms and acne.
Material and Methods
Subjects
Twelve female acne outpatients (aged 36 ± 10 years [range, 17–50]), and 10 male acne outpatients (aged 28 ± 12 years [range, 13–47]) not previously treated with topical or systemic steroids, antibiotics, and/or antifungal agents, retinoids, anti-inflammatory agents, and/or any chemical peeling agents within the past 6 months at Fujita Health University Bantane Hospital (Nagoya, Japan) between February 2018 and March 2018 were examined. Information on patients is shown in Supplementary Table 1. The severity of acne was in accordance with the Japanese Acne Study Group criteria.16
The study procedures were conducted in accordance with the Declaration of Helsinki. The study protocol and ethical approval were granted by the Fujita Health University Ethics Committee (approval no. HM17-275). Informed consent was obtained from all subjects prior to the initiation of the study. For subjects under the age of 18, we obtained informed consent from their parents. All subjects signed a consent form for the publication of the case details and images. Data confidentiality and participants’ privacy were strictly maintained, and the participants were informed that they had the right to withdraw from the study at any time.
Sample Collection
The comedonal contents in 26 inflammatory acne lesions (papules or pustules) were collected using a comedo extractor from the following areas of facial skin: the forehead, nose, and chin, defined as the T-zone (areas of high sebum secretion), and the cheek, defined as the U-zone (an area of low sebum secretion).17 All comedonal contents were sealed immediately after being placed into a sampling tube. Skin surface samples were also obtained by the swab method using cotton-tipped swabs (1A1504, Japan Cotton Buds, Tokyo, Japan) and TE buffer (pH 8.0) (Fujifilm, Osaka, Japan) containing 2% polysorbate 80 (Merck, Darmstadt, Germany) from a 5-cm2 area of the forehead (T-zone) and cheek (U-zone) without using any cosmetics. Whenever possible, samples were taken from the skin without using any cosmetics.18 Samples were stored at −20°C.
DNA Extraction
Comedonal samples were placed in TE buffer containing 2% polysorbate 80 (pH 7.0) and crushed using a sonication device (Bioruptor, Cosmo Bio, Tokyo, Japan). After the addition of 0.4 mg/mL of crude achromopeptidase (Fujifilm) to comedonal and skin surface samples, they were treated at 55°C for 10 minutes. The DNA of each sample was extracted by a treatment with 5% DNA Extraction Reagent (GenCheck, Fasmac, Kanagawa, Japan) at 100°C for 10 minutes.
Next Generation Sequencing
The V1-2 region of the 16S rRNA gene for the bacterial community analysis was amplified using 27 F (AGRGTTYGATYMTGGCTCAG) and 337R (CTGCTGCCTYCCGTA) primers, while the ITS-1 region for fungi was amplified using ITS1-F (CTTGGTCATTTAGAGGAAGTAA) and ITS1-R (GTTCAAAGAYTCGATGATTCAC) primers. PCR amplification was performed using 2 × KAPA HiFi Hot Start ReadyMix (KAPA Biosystems, Wilmington, MA) according to the manufacturer’s instructions and the library preparation protocol from Illumina Inc. (https://jp.support.illumina.com/downloads/16s_metagenomic_sequencing_library_preparation.html). Libraries were quantified using the Qubit fluorometer with the Qubit dsDNA HS assay kit (Thermo Fisher Scientific, MA, USA) and the library size was assessed using the Agilent BioAnalyzer with high-sensitivity DNA chips (Agilent Technologies, CA, USA). Libraries were pooled at equimolar concentrations in preparation for sequencing.
Sequencing was performed using the Illumina MiSeq sequencer (Illumina, San Diego, CA, USA) with V3 reagents to generate 2 × 300-bp paired-end reads. Initial sequence data processing was performed using MiSeq Reporter software version 1.3.17.0 (Illumina, CA, U.S.A) to de-multiplex samples and remove adapters and primer sequences, and sequence data were then exported in the FASTQ format. Sequencing was performed within FASMAC Co., Ltd.
Data Analysis
Sequence data were processed in QIIME2 (version 2021.2.0),19 a comprehensive bioinformatics platform for the microbiome analysis. DADA2 was used to process the sequence variants selected with a Phred-like quality score of less than 20 as calculated by the Illumina MiSeq sequencer. Sequence variants were taxonomically assigned at the genus level or lower taxonomic ranks using the data sets of Silva version 132 (https://www.arb-silva.de/) for the 16S rRNA region and UNITE version 8.2 (https://unite.ut.ee/) for the ITS region. q2-gcn-norm plugin for QIIME2 was used for normalizing sequences by 16S rRNA gene copy number (GCN) based on rrnDB database (https://rrndb.umms.med.umich.edu/). We only included data on taxa (genera and species) represented by a frequency of ≥1% of reads in at least one individual. All other taxa were pooled into a separate group called “Other bacteria” or “Other fungi”.
Xanthomonadaceae derived from achromopeptidase in DNA extraction were excluded from data in advance and also from the analysis. Moreover, Moraxellaceae, Oxalobacteraceae, Planococcaceae, and Comamonadaceae were excluded because these microorganisms were considered to be derived from the cotton swabs used for sample collection based on their abundance in control samples with cotton swabs only. The number of DNA reads used for the analysis is shown in Supplementary Figure 1.
Statistical Analysis
All statistical analyses were conducted in QIIME2’s diversity analyses were available through the q2-diversity plugin, which supports computing alpha and beta diversity metrics, applying related statistical tests, and generating interactive visualizations. Alpha diversity was assessed by Shannon’s Index, Pielou’s Evenness Index, Faith’s Phylogenetic Diversity, and observed OTUs. Alpha diversity indices were calculated from the Kruskal–Wallis test with QIIME2 for non–normally distributed variables. Data normalization was performed to normalize the varying sequencing depth among samples, referring to the minimum frequency calculated automatically. Since data did not have a normal distribution, continuous variables were summarized in terms of medians and interquartile ranges (IQRs). The beta diversity of weighted UniFrac values was also calculated using QIIME2 and analyzed by a permutational multivariate analysis of variance (PERMANOVA) with 999 permutations. A principal coordinates analysis (PCoA) was performed to obtain principal coordinates and visualize complex multidimensional data. Data for all alpha and beta diversity metrics were downloaded from outputs generated with the “QIIME2 view” online software (https://view.qiime2.org/). The PICRUSt2 plugin was used for integration between the output files of the default QIIME2 pipelines and PICRUSt220 for metagenome predictions, such as EC, KO, and MetaCyc pathway. Multiple run data contained the same mock sequences and combined for one analysis, referring to them. Sequencing data are available on the DDBJ Read Archive (ID: DRA013024). The Mann–Whitney U-test was employed for statistical analyses of the skin surface of the forehead and comedonal contents of the T-zone, and also of the skin surface of the cheek and comedonal contents of the U-zone using R version 4.1.1 software (R Core Team, 2021). All tests used p = 0.05 as a threshold for significance.
Results
The genus classifications of 16s rRNA in comedonal contents and on the skin surface are shown in Figure 1. The major bacterial genera in comedonal contents and on the skin surface were Cutibacterium, Staphylococcus, Corynebacterium, and Streptococcus spp. The predominant bacteria genus on the skin surface was Staphylococcus spp., whereas Cutibacterium spp. were predominant in comedonal contents. Comedonal contents on the T-zone had a greater ratio of Cutibacterium spp. than those on the U-zone and a lower ratio of Staphylococcus spp. The predominant Cutibacterium species was C. acnes, and the predominant Staphylococcus species was S. epidermidis in comedonal contents and on the skin surface of acne patients (Supplementary Table 2). The alpha diversity of 16s rRNA is shown in Figure 2. In an analysis using Faith’s phylogenetic diversity and the observed OTUs, diversities were higher in comedonal contents than on the skin surface, while no significant differences were observed by the Shannon index and Pielou’s evenness index. Alpha diversity values varied more widely in comedonal contents than on the skin surface in every analysis. The beta diversity (weighted UniFrac values) of 16s rRNA is shown in Figure 3. Significant differences were observed between the bacterial microbiome of comedonal contents and that on the skin surface. The functional prediction of 16s rRNA is shown in Figure 4 and Supplementary Figure 2. The bacterial microbiome in comedonal contents exhibited stronger metabolic activity than that on the skin surface (Supplementary Figure 2). In the results obtained on enzymes involved in acne, namely, lipase (EC 3.1.1.3), phosphatase (EC 3.1.3), neuraminidase (EC 3.2.1.18), endoglycoceramidase (EC 3.2.1.123), protease (EC 3.4), and hyaluronate lyase (EC 4.2.2.1), metabolic activity in the bacterial microbiome in comedonal contents was stronger than that on the skin surface for every enzyme (Figure 4).
 |
Figure 1 Genus classification of 16s rRNA clones in comedonal contents of inflammatory acne and on the skin surface of acne patients. (A) Individual distribution of the main bacterial genus arranged in descending order of Cutibacterium spp.; (B) boxplots of Cutibacterium and Staphylococcus spp. are shown. f, Female; m, male; T, T-zone; U, U-zone. Only bacteria with an average detection rate of 1% or higher are listed, while those with an average detection rate of less than 1% are classified as others. The Mann–Whitney U-test was employed for a statistical analysis of the skin surface of the forehead and cheek, the comedonal contents of the T-zone and U-zone, the skin surface of the forehead and the comedonal contents of T-zone, and the skin surface of cheek and the comedonal contents of the U-zone. Significant differences were observed between Cutibacterium and Staphylococcus spp. in the comedonal contents of the T-zone and on the skin surface of the forehead (P < 0.05). Significant differences were also noted between Cutibacterium and Staphylococcus spp. in the comedonal contents of the T- and U-zones (P < 0.05). No significant differences were found in any bacteria on the skin surface (forehead and cheek) or in comedonal contents (T- and U-zones) between females and males. |
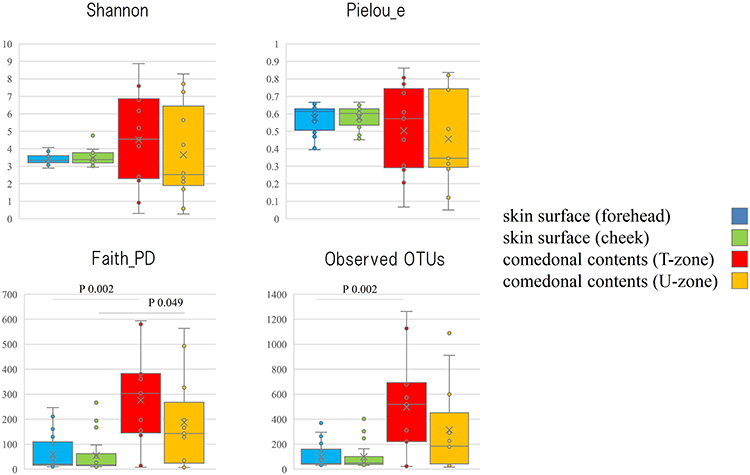 |
Figure 2 Alpha diversity of 16s rRNA clones in comedonal contents of inflammatory acne and on the skin surface of acne patients. The Mann–Whitney U-test was employed for a statistical analysis of the skin surface of the forehead and cheek, the comedonal contents of the T-zone and U-zone, the skin surface of the forehead and the comedonal contents of the T-zone, and the skin surface of the cheek and the comedonal contents of the U-zone. In the analysis using Faith’s phylogenetic diversity and the observed OTUs, diversities were higher in the comedonal contents of the T- and U-zones than on the skin surface of the forehead and cheek (P < 0.05); however, no significant differences were observed by the Shannon index or Pielou’s evenness index. No significant differences were found in either index on the skin surface (forehead and cheek) or in comedonal contents (T- and U-zones) between females and males. |
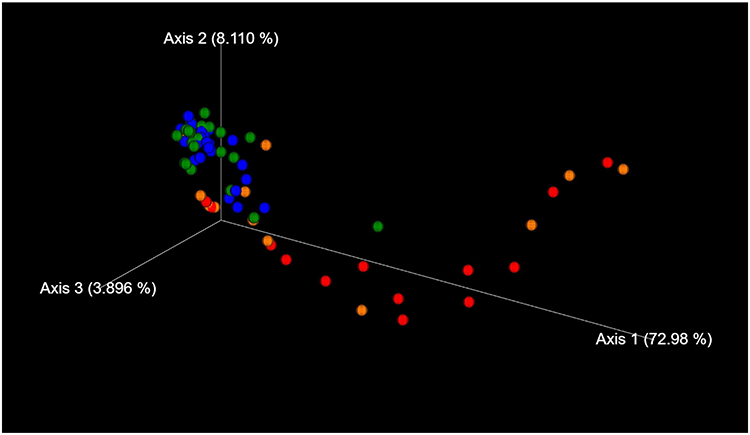 |
Figure 3 Beta diversity (weighted UniFrac values) of 16s rRNA clones in comedonal contents of inflammatory acne and on the skin surface of acne patients. Blue, skin surface (forehead); green, skin surface (cheek); red, comedonal contents of the T-zone; orange, comedonal contents of the U-zone. Significant differences were observed between the bacterial microbiome of the comedonal contents and that on the skin surface by PERMANOVA. P value; the skin surface of the forehead versus the comedonal contents of the T-zone, <0.001; the skin surface of the cheek versus the comedonal contents of the U-zone, 0.006. |
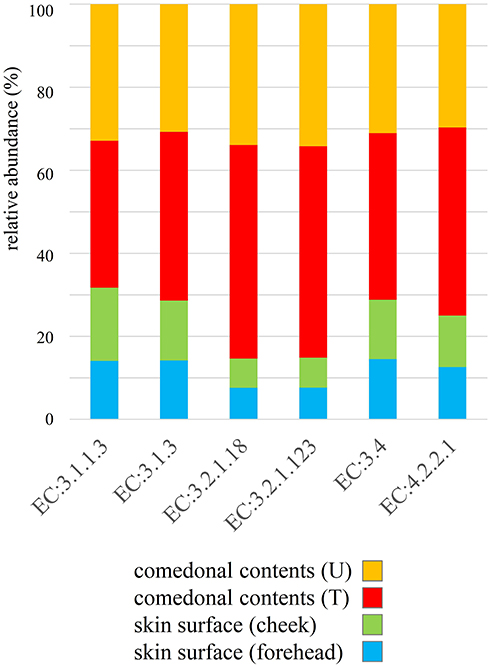 |
Figure 4 Functional prediction of 16s rRNA clones in comedonal contents of inflammatory acne and on the skin surface of acne patients. Data are arranged in descending order of comedonal contents. f, Female; m, male; T, T-zone; U, U-zone. The median value of each group was used as data, and ratios are shown in the graph. Enzymes related to acne; lipase, EC 3.1.1.3; phosphatase, EC 3.1.3; neuraminidase, EC 3.2.1.18; endoglycoceramidase, EC 3.2.1.123; protease, EC 3.4; hyaluronate lyase, EC 4.2.2.1. |
The species classifications of ITS in comedonal contents and on the skin surface are shown in Figure 5. The major genus in comedonal contents and on the skin surface was Malassezia spp., particularly M. restricta, followed by M. globosa. Other than Malassezia spp., Candida parapsilosis, Aureobasidium pullulans, and Cladophialophora boppii were detected in some samples. The fungal microbiome did not significantly differ between comedonal contents and the skin surface. The alpha and beta diversities of ITS are shown in Figure 6. In the analysis using Pielou’s evenness index, diversities in comedonal contents were higher in the T-zone than in the U-zone, while no significant differences were observed by Shannon index’s, Pielou’s evenness index, or the observed OTUs. The beta diversity (weighted UniFrac values) of ITS is shown in Figure 7. There were no significant differences between the fungal microbiome of comedonal contents and that on the skin surface.
 |
Figure 5 Species classifications of ITS clones in comedonal contents of inflammatory acne and on the skin surface of acne patients. The individual distribution of the main fungal species arranged in descending order of M. restricta is shown. f, Female; m, male; T, T-zone; U, U-zone. Only fungi with an average detection rate of 1% or higher are listed, and fungi with an average detection rate of less than 1% are classified as others. The Mann–Whitney U-test was employed for a statistical analysis of the skin surface of the forehead and cheek, the comedonal contents of the T-zone and U-zone, the skin surface of the forehead and the comedonal contents of the T-zone, and the skin surface of the cheek and the comedonal contents of the U-zone. No significant differences were observed between comedonal contents and the skin surface. Furthermore, no significant differences were noted in any fungus on the skin surface (forehead and cheek) or in comedonal contents (T- and U-zones) between females and males. |
 |
Figure 6 Alpha diversity of ITS clones in comedonal contents of inflammatory acne and on the skin surface of acne patients. f, Female; m, male; T, T-zone; U, U-zone. The Mann–Whitney U-test was employed for a statistical analysis of the skin surface of the forehead and cheek, the comedonal contents of the T-zone and U-zone, the skin surface of the forehead and the comedonal contents of the T-zone, and the skin surface of the cheek and the comedonal contents of the U-zone. In an analysis using Pielou’s evenness index, diversities in comedonal contents were higher in the T-zone than in the U-zone, while no significant differences were observed by Shannon’s index, Pielou’s evenness index, or the observed OTUs (P < 0.05). |
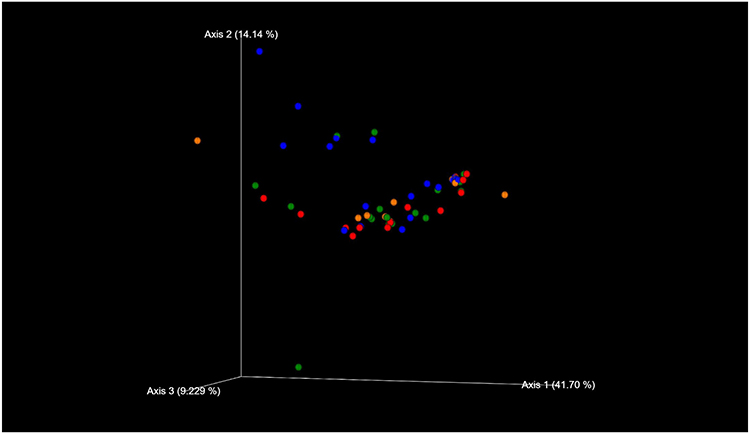 |
Figure 7 Beta diversity (weighted UniFrac values) of ITS clones in comedonal contents of inflammatory acne and on the skin surface of acne patients. Blue, skin surface (forehead); green, skin surface (cheek); red, comedonal contents of the T-zone; orange, comedonal contents of the U-zone. There were no significant differences between the fungal microbiome of the comedonal contents and that on the skin surface by PERMANOVA (P > 0.05). |
Discussion
Akaza et al previously investigated the microbiome in the comedonal contents of inflammatory acne and on the skin surface of patients with acne using real-time PCR.8 However, this method cannot detect all microorganisms in samples because of the necessity for targeting; Akaza et al previously targeted Cutibacterium, Staphylococcus, and Malassezia spp. Therefore, to comprehensively analyze the microbiota in the comedonal contents of inflammatory acne and on the skin surface of patients with acne, we herein performed an analysis using the NGS technique. The results obtained confirmed that the predominant bacteria in the comedonal contents of inflammatory acne and on the skin surface were Cutibacterium and Staphylococcus spp., while the predominant fungi were Malassezia spp. (Figures 1 and 5). A sex difference was previously reported in the cutaneous microbiome of healthy subjects in a quantitative study using real-time PCR,21 however, the results obtained in the present study showed no significant sex differences in the microbiome in the comedonal contents of inflammatory acne or on the skin surface of patients with acne. The amount of sebum is generally lower in healthy females than in healthy males; however, it is similar in females and males with acne.22 Therefore, we consider that there is no significant difference in the microbiome on the skin surface between females and males. Moreover, it was also clarified that there is no significant difference in the microbiome on the skin surface between forehead and cheek.
Cutibacterium spp. were the predominant bacteria in the comedonal contents of inflammatory acne (Figure 1). This result suggests that Cutibacterium spp. are the bacteria most closely involved in acne inflammation. Differences in the α-diversity (Faith’s phylogenetic diversity and the observed OTUs) and β-diversity of 16s rRNA between the comedonal contents of inflammatory acne and on the facial skin surface suggest that the microbiota in the former differs from that on the latter (Figures 2 and 3). Furthermore, the bacterial microbiome of comedonal contents exhibited stronger metabolic activity than that on the skin surface in most metabolic pathways, including enzymes related to acne in functional predictions (Figure 4 and Supplementary Figure 2). The microbiome in the comedonal contents of inflammatory acne appears to be in a state in which the proportion of Cutibacterium spp. is elevated and metabolic activity is stronger than that on the skin surface.
Cutibacterium spp. were previously reported to be the predominant bacteria on the facial skin surface of healthy subjects.23 However, in the present study, Staphylococcus spp. were the predominant bacteria on the facial skin surface of patients with acne (Figure 1). Dreno et al also showed that Staphylococcus spp. were more abundant than Cutibacterium spp., and were also more abundant on the surface of comedones, papules, and pustules than on non-lesional skin.14 Bek-Thomsen et al identified Staphylococcus spp. as the predominant bacteria on the facial skin surface of patients with acne.11 In a comparison of the microbiome on the cheeks of healthy subjects and patients with acne, Staphylococcus spp. were more abundant in acne patients than in healthy subjects.13 Therefore, Staphylococcus spp., not Cutibacterium spp., are regarded as the predominant bacteria on the facial skin surface of patients with acne. C. acnes and Staphylococcus epidermidis were previously reported to be in a competitive relationship.24,25 Furthermore, S. epidermidis has been suggested to play a beneficial role in acne by limiting P. acnes (C. acnes) overcolonisation and inflammation.26 Our hypothesis may appear to be inconsistent with these findings. However, we suspect that the imbalance in the skin microbiota caused by acne may be responsible. The reasons for Staphylococcus spp. being the predominant bacteria on the skin surface of patients with acne, which differs from healthy subjects, warrant further study.
No significant differences were observed between the bacterial microbiota in the T- and U-zones on the facial skin surface (Figure 1). On the other hand, the population of Cutibacterium spp. in the comedonal contents of inflammatory acne in the T-zone was greater than that on the facial skin surface in the T-zone and in the comedonal contents of inflammatory acne in the U-zone. The T-zone is an area of high sebum secretion.17 The number of Cutibacterium spp. is generally considered to correlate with the amount of sebum secreted on the skin.22 However, the present results indicate that sebum secretion affects the population of Cutibacterium spp. in the microbiome rather than their numbers. Moreover, the involvement of Cutibacterium spp. in acne is more prominent in areas with larger amounts of sebum.
Regarding fungi, Malassezia spp., particularly M. restricta, were predominant in the comedonal contents of inflammatory acne and on the skin surface of patients with acne (Figure 5). Previous findings showed that Malassezia spp., particularly M. restricta, were predominant on the skin surface of healthy subjects which is consistent with the present results.27 Furthermore, our findings are in accordance with previous studies that demonstrated that M. globosa, M. dermatis, and M. sympodialis are also found on the skin surface. Moreover, in contrast to bacteria, no significant difference was observed in the α-diversity or β-diversity of ITS between the comedonal contents of inflammatory acne and the facial skin surface (Figures 6 and 7). Based on these results, the fungal microbiome appears to be homeostatic, in contrast to the bacterial microbiome.
In the present study, we attempted to functionally predict the microbiome based on NGS data (Figures 4 and Supplementary Figure 2). The results obtained confirmed that the greater proportion of Cutibacterium spp. in the microbiome exhibited stronger metabolic activity, including the production of enzymes related to acne, namely, lipase (EC 3.1.1.3), phosphatase (EC 3.1.3), neuraminidase (EC 3.2.1.18), endoglycoceramidase (EC 3.2.1.123), protease (EC 3.4), and hyaluronate lyase (EC 4.2.2.1). These enzymes are involved in inflammation and the formation of comedones.1,6,7 These results suggest that Cutibacterium spp. affect the metabolic activity of the microbiome on the skin, and are the microorganisms most closely involved in acne. The present results showing the presence of various microorganisms in the comedonal contents of acne indicate that acne is an inflammatory disease involving not only Cutibacterium spp., but also various cutaneous microorganisms. However, the suppression of Cutibacterium spp. is generally one of the most effective methods for attenuating the inflammation of acne. On the other hand, as shown in Figure 1, Cutibacterium spp. was less abundant in some acne samples, and, thus, caution is needed for the prevention and treatment of acne.
The present study mainly examined patients with mild acne who account for the majority of acne patients. The relationship between the severity of acne and the skin microbiome warrants further study.
Conclusion
The present study confirmed that the predominant bacteria in the comedonal contents of inflammatory acne were Cutibacterium spp. (more prominent in areas with large amounts of sebum), while those on the skin surface were Staphylococcus spp. Malassezia spp., particularly M. restricta, are the predominant fungi in the comedonal contents of inflammatory acne and on the skin surface. The bacterial microbiome in comedonal contents exhibited stronger metabolic activity, including the production of enzymes related to acne, than that on the skin surface. These results suggest that the microbiota in the comedonal contents of inflammatory acne is a state in which the number of Cutibacterium spp., which exhibit strong metabolic activity, is increased based on the cutaneous microbiome (the microbiome on the skin surface). In other words, acne is an inflammatory disease involving the overgrowth of C. acnes and other cutaneous resident microorganisms, including Malassezia spp. However, cutaneous resident microorganisms also inhabit healthy skin without acne. Therefore, cutaneous resident microorganisms are not the only cause of acne, and cutaneous immune deficiency targeted towards some of these microorganisms in the host may be needed.
Consent Statement
The authors certify that they have obtained all appropriate patient consent forms. The patients or their parents (for subjects under the age of 18) signed a consent form for the publication of the case details and images.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Xu H, Li H. Acne, the skin microbiome, and antibiotic treatment. Am J Clin Dermatol. 2019;20:335–344. doi:10.1007/s40257-018-00417-3
2. Akaza N, Akamatsu H, Kishi M, et al. Effects of Propionibacterium acnes on various mRNA expression levels in normal human epidermal keratinocytes in vitro. J Dermatol. 2009;36:213–223. doi:10.1111/j.1346-8138.2009.00626.x
3. Ingham E, Holland KT, Gowland G, Cunliffe WJ. Partial purification and characterization of lipase (EC 3.1.1.3) from Propionibacterium acnes. J Gen Microbiol. 1981;124:393–401. doi:10.1099/00221287-124-2-393
4. Jarrousse V, Castex-Rizzi N, Khammari A, Charveron M, Dreno B. Modulation of integrins and filaggrin expression by Propionibacterium acnes extracts on keratinocytes. Arch Dermatol Res. 2007;299:441–447. doi:10.1007/s00403-007-0774-5
5. Iinuma K, Sato T, Akimoto N, et al. Involvement of Propionibacterium acnes in the augmentation of lipogenesis in hamster sebaceous glands in vivo and in vitro. J Invest Dermatol. 2009;129:2113–2119. doi:10.1038/jid.2009.46
6. Lee YB, Byun BJ, Kim HS. Potential role of the microbiome in acne: a comprehensive review. J Clin Med. 2019;8:987. doi:10.3390/jcm8070987
7. Katsuta Y, Iida T, Hasegawa K, Inomata S, Denda M. Function of oleic acid on epidermal barrier and calcium influx into keratinocytes is associated with N-methyl D-aspartate-type glutamate receptors. Br J Dermatol. 2009;160:69–74. doi:10.1111/j.1365-2133.2008.08860.x
8. Akaza N, Akamatsu H, Numata S, et al. Microorganisms inhabiting follicular contents of facial acne are not only Propionibacterium but also Malassezia spp. J Dermatol. 2016;43:906–911. doi:10.1111/1346-8138.13245
9. Akaza N, Akamatsu H, Takeoka S, Mizutani H, Nakata S, Matsunaga K. Increased hydrophobicity in Malassezia species correlates with increased proinflammatory cytokine expression in human keratinocytes. Med Mycol. 2012;50:802–810. doi:10.3109/13693786.2012.678019
10. Akaza N, Akamatsu H, Takeoka S, et al. Malassezia globosa tends to grow actively in summer conditions more than other cutaneous Malassezia species. J Dermatol. 2012;39:613–616. doi:10.1111/j.1346-8138.2011.01477.x
11. Bek-Thomsen M, Lomholt HB, Kilian M. Acne is not associated with yet-uncultured bacteria. J Clin Microbiol. 2008;46:3355–3360. doi:10.1128/JCM.00799-08
12. Fitz-Gibbon S, Tomida S, Chiu B-H, et al. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J Invest Dermatol. 2013;133:2152–2160. doi:10.1038/jid.2013.21
13. Li CX, You ZX, Lin YX, Liu HY, Su J. Skin microbiome differences relate to the grade of acne vulgaris. J Dermatol. 2019;46:787–790. doi:10.1111/1346-8138.14952
14. Dreno B, Martin R, Moyal D, Henley JB, Khammari A, Seité S. Skin microbiome and acne vulgaris: Staphylococcus, a new actor in acne. Exp Dermatol. 2017;26:798–803. doi:10.1111/exd.13296
15. Barnard E, Shi B, Kang D, Craft N, Li H. The balance of metagenomic elements shapes the skin microbiome in acne and health. Sci Rep. 2016;6:39491. doi:10.1038/srep39491
16. Hayashi N, Akamatsu H, Kawashima M; Acne Study Group. Establishment of grading criteria for acne severity. J Dermatol. 2008;35:255–260. doi:10.1111/j.1346-8138.2008.00462.x
17. Youn SW, Park ES, Lee DH, Huh CH, Park KC. Does facial sebum excretion really affect the development of acne? Br J Dermatol. 2005;153:919–924. doi:10.1111/j.1365-2133.2005.06794.x
18. Numata S, Akamatsu H, Akaza N, et al. Quantitative effect of face washing on cutaneous resident microbiota in female subjects who wear make-up. J Dermatol. 2012;39:1100–1101. doi:10.1111/j.1346-8138.2012.01644.x
19. Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi:10.1038/s41587-019-0209-9
20. Douglas GM, Maffei VJ, Zaneveld JR, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38:685–688. doi:10.1038/s41587-020-0548-6
21. Akaza N, Akamatsu H, Sasaki Y, et al. Cutaneous Malassezia microbiota of healthy subjects differ by sex, body part and season. J Dermatol. 2010;37:786–792. doi:10.1111/j.1346-8138.2010.00913.x
22. Akaza N, Akamatsu H, Numata S, et al. Fatty acid compositions of triglycerides and free fatty acids in sebum depend on amount of triglycerides, and do not differ in presence or absence of acne vulgaris. J Dermatol. 2014;41:1069–1076. doi:10.1111/1346-8138.12699
23. Grice EA, Kong HH, Conlan S, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi:10.1126/science.1171700
24. Wang Y, Kuo S, Shu M, et al. Staphylococcus epidermidis in the human skin microbiome mediates fermentation to inhibit the growth of Propionibacterium acnes: implications of probiotics in acne vulgaris. Appl Microbiol Biotechnol. 2014;98:411–424. doi:10.1007/s00253-013-5394-8
25. Xia X, Li Z, Liu K, Wu Y, Jiang D, Lai Y. Staphylococcal LTA-induced miR-143 inhibits Propionibacterium acnes-mediated inflammatory response in skin. J Invest Dermatol. 2016;136:621–630. doi:10.1016/j.jid.2015.12.024
26. Claudel J-P, Auffret N, Leccia M-T, Poli F, Corvec S, Dréno B. Staphylococcus epidermidis: a potential new player in the physiopathology of acne? Dermatology. 2019;235:287–294. doi:10.1159/000499858
27. Zhang E, Tanaka T, Tajima M, Tsuboi R, Nishikawa A, Sugita T. Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiol Immunol. 2011;55:625–632. doi:10.1111/j.1348-0421.2011.00364.x
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.