Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
The Mediation of Circulating Inflammatory Proteins in the Causal Pathway from Immune Cells to COPD
Received 8 October 2024
Accepted for publication 3 February 2025
Published 7 February 2025 Volume 2025:20 Pages 245—257
DOI https://doi.org/10.2147/COPD.S495073
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Min Zhang
Kunrong Yan, Yingjian Wang, Peng Xin
Department of Anaesthesia, Chaoyang Second Hospital, Chaoyang, Liaoning, People’s Republic of China
Correspondence: Peng Xin, Email [email protected]
Objective: Observational studies have indicated that immune cells and circulating inflammatory proteins may play a dual role in the progression of COPD; however, the precise mechanisms remain uncertain. The objective of this study was to ascertain the causal relationship between immune cells and COPD and to quantify the potential role of circulating inflammatory proteins as mediators.
Methods: A two-sample Mendelian randomisation analysis was conducted involving 731 immune cells, 91 inflammatory proteins and COPD, utilising summary-level data from genome-wide association studies. The causal relationships between immune cells, inflammatory proteins and COPD were sequentially analysed by multivariate Mendelian randomisation and validated using Bayesian weighted Mendelian randomisation. Subsequently, sensitivity analyses were conducted, employing Cochran’s Q test to assess heterogeneity, MR-PRESSO and MR-Egger tests to assess pleiotropy, and reverse MR and Steiger directionality tests to rule out reverse causality. Lastly, a two-step approach was employed to ascertain the proportion of inflammatory proteins that mediate immune cell-mediated effects in COPD.
Results: The combination of the inverse variance weighting method and the Bayesian weighting algorithm identified 30 immune cells that were found to be causally associated with COPD, as well as eight inflammatory proteins that were associated with COPD. By two-step analysis, six inflammatory proteins were found to mediate the effects of eight immune cell phenotypes on COPD, with CXCL10 having the highest percentage of mediation at 14.49%, followed by IL20RA at 11.47%.
Conclusion: This study provides a comprehensive investigation of the causal relationship between immune cells and COPD, as well as an estimation of the proportion of the effect of inflammatory proteins as mediators. These findings facilitate the identification of individuals at high risk of COPD and offer novel insights for early prevention and clinical intervention.
Keywords: Mendelian randomization, Bayesian weighted Mendelian randomization, immune cells, circulating inflammatory proteins, COPD, mediation analysis
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a complex lung disease that is primarily caused by damage to the airways (eg, bronchitis and bronchiectasis) or alveoli (eg, emphysema). The disease is typified by chronic respiratory symptoms and a progressive decline in lung function.1 Today COPD is a major public health problem worldwide, accounting for over 50% of all cases of chronic respiratory disease and ranking as the third leading cause of mortality worldwide.2,3 As the population ages, the morbidity and mortality rates of COPD continue to increase, with significant social and economic consequences.4,5
In terms of pathophysiology, immune cells play a multifaceted role in maintaining homeostasis in the body and promoting injury repair. The lungs may be a pivotal site for the host’s innate and adaptive immune responses in their interactions with diverse microbial pathogens.6 However, it is postulated that aberrant activation of the immune system, particularly the proliferation and activation of neutrophils, macrophages and T cells, is a principal factor in the perpetuation of chronic inflammation and the deterioration of lung function.7 These immune cells not only contribute directly to lung tissue damage but also serve to further exacerbate the lesions by releasing a range of inflammatory mediators.
Observational studies have demonstrated a notable elevation in the number of immune cells within the lung tissue of patients diagnosed with COPD in comparison to healthy controls.8,9 Additionally, these studies have revealed a substantial enhancement in immune cell responses.10 In particular, it has been demonstrated that CD68+ myeloid antigen-presenting cells, CD4+ T cells, and CD8+ T cells undergo proliferation in the lungs of COPD patients, resulting in sustained inflammation.11,12 However, certain immune cells may exert a protective effect against the progression of COPD, including CD4+ T cells and resting natural killer cells. Therefore, although immune cells play a significant role in the inflammatory microenvironment of the lung, the precise causal relationship between immune cells and COPD, as well as the underlying mechanisms, remain unclear.
When external stimuli such as smoking or pollutants enter the lungs, immune cells are activated and release a number of inflammatory mediators, including cytokines and chemokines. These mediators not only attract more immune cells to the damaged tissue, but also cause them to secrete large amounts of inflammatory proteins. Relevant studies have shown that C-reactive protein (CRP) is associated with increased acute exacerbations of COPD13 and similarly IL-1β, IL-17A, TNF-α and IL-6 are significantly correlated with the severity of emphysema or persistent airway inflammation and severe exacerbations.14–16 These inflammatory proteins act as amplifiers in the inflammatory response, further activating and recruiting more immune cells, creating a self-reinforcing cycle of inflammation. This positive feedback mechanism results in the maintenance and exacerbation of a chronic inflammatory state in COPD patients, ultimately leading to irreversible lung tissue damage and progressive loss of lung function. Thus, the interaction between immune cells and inflammatory proteins is an important basis for the pathophysiology of COPD. An in-depth understanding of this interaction will not only help to elucidate the pathogenesis of COPD, but may also provide new biomarkers and targets for the diagnosis and treatment of the disease.
Mendelian randomisation (MR) analysis, which uses single nucleotide polymorphisms (SNPs) from genome-wide association studies (GWAS) as instrumental variables (IVs), is a robust tool in epidemiological studies. The core idea is to use genetic variants identified at conception as a tool to assess causal relationships between risk factors and specific diseases.17–19 As confounders are an important source of confounding in causal inference in epidemiological studies, this method can provide more reliable causal inference by avoiding confounding and reverse causation in traditional observational studies. Therefore, our aim was to use MR to determine the causal relationship between immune cells and COPD and to assess the extent to which circulating inflammatory proteins influence the association between the two.
Materials and Methods
Study Design
To assess the causal relationship between exposure factors (immune cells and circulating inflammatory proteins) and outcome (COPD) in MR analyses, SNPs that are strongly correlated with these exposures are usually selected as IVs. Effective MR analyses require that IVs satisfy the following three basic assumptions (Figure 1a), 1 Strong association assumption: The IVs are strongly correlated with the exposure factors, ensuring that these genetic variants can explain the variability in exposure.2 Independence assumption: The IVs are not associated with confounding factors that affect exposure and outcome, to avoid bias.3 Exclusivity assumption: IVs only influence outcomes through exposure factors, and there are no other ways of directly influencing outcomes.20
 |
Figure 1 Schematic diagrams depicting the experimental design. (a) Three major assumptions to be followed in MR analysis; (b) Schematic of the methodology for a two-step MR analysis; (c) Overall technical line of this study. |
In causal analyses, the effects of exposure factors on outcomes can be divided into total and indirect effects. The former refers to the direct effect of exposure factors on the outcome, whereas the latter affects the outcome indirectly through mediating factors. Therefore, we used two-step MR (TSMR) and multivariate MR (MVMR) methods with immune cells as exposure factors, COPD as outcome factors, and selected circulating inflammatory proteins as potential mediators to elucidate the possible mediating role of inflammatory proteins in the causal pathway between immune cells and COPD (Figure 1b).
Specifically, we examined the causal effects between immune cells and COPD, inflammatory proteins and COPD, and immune cells and inflammatory proteins. Using the coefficient product method, we can calculate the indirect effect of exposure factors on the outcome and thus clarify the specific role of inflammatory proteins in this causal chain. The overall study design is shown in Figure 1c.
Data Sources
The immunocytogenetic data used in this study were derived from a 2020 GWAS analysis of 3,757 European individuals focusing on 272 factors associated with blood immune cells.21 This analysis included 731 immune traits, including absolute cell counts (n=118), relative cell counts (n=192), mean fluorescence intensities reflecting surface antigen levels (n=389), and morphological parameters (n=32). These characteristics were further classified into seven cell groups: monocytes (n=43), B cells (n=190), CD cells (n=64), myeloid cells (n=64), TBNK cells (n=124), T cells in maturation (n=79) and Treg cells (n=167). Full data are available in the GWAS Catalogue database (login IDs GCST90001391 to GCST90002121) (https://www.ebi.ac.uk/gwas/).
Genetic data on circulating inflammatory proteins were derived from a genome-wide Protein Quantity Trait Locus (pQTL) study using the Olink Target platform on 91 proteins in the plasma of 14,824 participants, representing 97.4% of the European population.22 The full data are freely available in the GWAS Catalogue database (login IDs GCST90274758 to GCST90274848).
The summary statistics for COPD are derived from the FinnGen database Consortium version R11, which includes 394,244 European individuals (of which 21,617 are cases and 372,627 are controls). The database contains genetic information on more than 400,000 Finns and aims to provide insight into the genetic basis of various diseases through GWAS and other genetic data. It should be noted that these GWAS data come from different consortia or organisations, and theoretically there is no overlap of samples.
Instrumental Variable Selection
In this study, we initially screened SNPs associated with immune cells, inflammatory proteins, and COPD by genome-wide association analyses as IVs for MR analyses. To ensure that the IVs met the first assumption of MR (ie, that the IVs were strongly associated with the exposure variables), we employed a P < 5×10−8 threshold for screening SNPs. However, the number of instrumental variables obtained under this rigorous screening criterion was more limited. To ensure the inclusion of a sufficient number of positive SNPs and to enhance the statistical efficacy of the analyses, the screening criterion of P value was relaxed to P < 1×10−5. This allowed for the capture of a greater number of genetic markers associated with the exposure variables.23 However, genetic variants with similar genomic locations are more likely to be co-inherited, resulting in a higher than random probability that they occur on the same chromosome. Therefore, we used the clump_data() function in the R package TwoSampleMR to constrain the linkage disequilibrium condition (set kilobase pairs (kb) = 10,000, r2 = 0.001) to ensure that the selected SNPs are independent.
To further ensure the accuracy of the data, potential duplicates or palindromes were removed using the harmonise_data() function with the parameter set to ‘action=2’. The F-test value was then calculated for each SNP using the formula F = (N-K-1)R2/K(1-R2) to assess the strength of the correlation between loci and exposure factors. According to the criteria, if the F value was less than 10, the SNP was considered a weak instrumental variable and was excluded. In the formula, R2 is the proportion of variance explained by the IVs, N is the sample size, and K is the number of SNPs.24 Through this rigorous screening and validation process, we ensured that the selected IVs had sufficient statistical validity and independence for subsequent Mendelian randomisation analyses.
Statistical Analysis
We analysed potential associations between immune cells, inflammatory proteins and COPD on a pair-by-pair basis using the MVMR method. The specific analyses were performed in the R 4.4.1 environment using the R package TwoSampleMR. Five different methods were used to assess causality, including inverse variance weighting (IVW), MR-Egger regression, weighted median, weighted mode and simple mode methods. Among these, IVW, as the main method, takes into account the heterogeneity of variance-specific causal estimates and combines ratio estimates through meta-analysis to produce unbiased results, assuming that all selected instrumental variables are valid.25 In addition, we introduced the BWMR_updated.R function, which implements a Bayesian weighting algorithm, to test whether the positive results revealed by the IVW method are equally statistically significant in a Bayesian framework. The Bayesian weighting algorithm provides additional evidence for causal inference by comparing a priori distributions with data-driven posterior distributions.26 The results were adjusted using the Bonferroni correction method to avoid the accumulation of false positives due to multiple testing. Specifically, the corrected significance threshold was set at P < 0.05/N, where N represents the number of causal analyses performed. For P-values falling between 0.05 and the corrected threshold, although they did not meet the strict significance criteria, they still suggest potential causal associations. To better interpret causality, we converted the derived β-values to odds ratios (OR) and computed 95% confidence intervals (CI).
In order to verify the accuracy of causality and to rule out critical factors that may affect the causal hypothesis, a number of quality control measures were applied to assess the sensitivity, heterogeneity and multiplicity of the study results. Firstly, the p-value of the Cochran’s Q test was calculated using the IVW method to assess the effect of heterogeneity; secondly, the MR-PRESSO global test and the MR-Egger intercept were used to correct for horizontal polytropy and to exclude outliers. If the P value was greater than 0.05, heterogeneity and pleiotropy were considered to have no significant effect on the study results. In addition, we performed reverse MR analyses and Steiger directionality tests to ensure a positive causal relationship between exposure and outcome. For the reverse MR analysis, a P value greater than 0.05 ruled out the effect of reverse causality; for the Steiger directionality test, a P value less than 0.05 indicated that the positive association of causality was reliable.27
To assess the mediating effect of inflammatory proteins, we calculated the total effect (β_all) between immune cells and COPD using the IVW method. The mediating effect (β1β2) was then obtained by calculating the product of the beta value of immune cells for inflammatory proteins (β1) and the beta value of inflammatory proteins for COPD (β2). The mediation ratio is then expressed as β1β2/β_all to measure the mediating role of inflammatory proteins in the causal pathway between immune cells and COPD.28
Results
Selection of IVs
Based on the preset screening criteria (P < 1×10−5, kb = 10,000, r2 = 0.001), 731 immune cells had significant IVs ranging from 3–759, 91 inflammatory proteins had significant IVs ranging from 6–54, and 140 significant IVs were used for COPD in reverse MR. Detailed information on SNPs, effector alleles, allele frequencies and beta values for each IV is stored in Supplementary Table 1.The smallest F value for these SNPs was 19.508, indicating the lowest probability of weak instrumental bias.
Causal Effects of Immune Cells on COPD
In the MVMR analysis, IVW was used as the primary analytical tool, suggesting that there were 54 immune cells with a possible causal relationship to COPD (Figure 2a; Supplementary Table 2). The 54 immune cells were further verified by BWMR analysis, which retained 43 immune cells (Figure 2b). To exclude the influence of possible confounders, we performed a sensitivity analysis of the 43 causal associations (Table 1) and found that 12 immune cells, including HLA DR++ monocyte %leukocyte, CD27 on CD24+ CD27+ B cell, and HLA DR on CD14+ monocyte, were heterogeneous or pleiotropic (P < 0.05), suggesting that exposure variables may affect outcomes in different ways through multiple biological pathways or in different settings. After their exclusion, there remained 30 immune cells with significant causal effects on COPD (Figure 2c): 4 lymphocyte count, 4 leukocyte count, 4 myeloid white cell count, and 18 blood protein measurements.
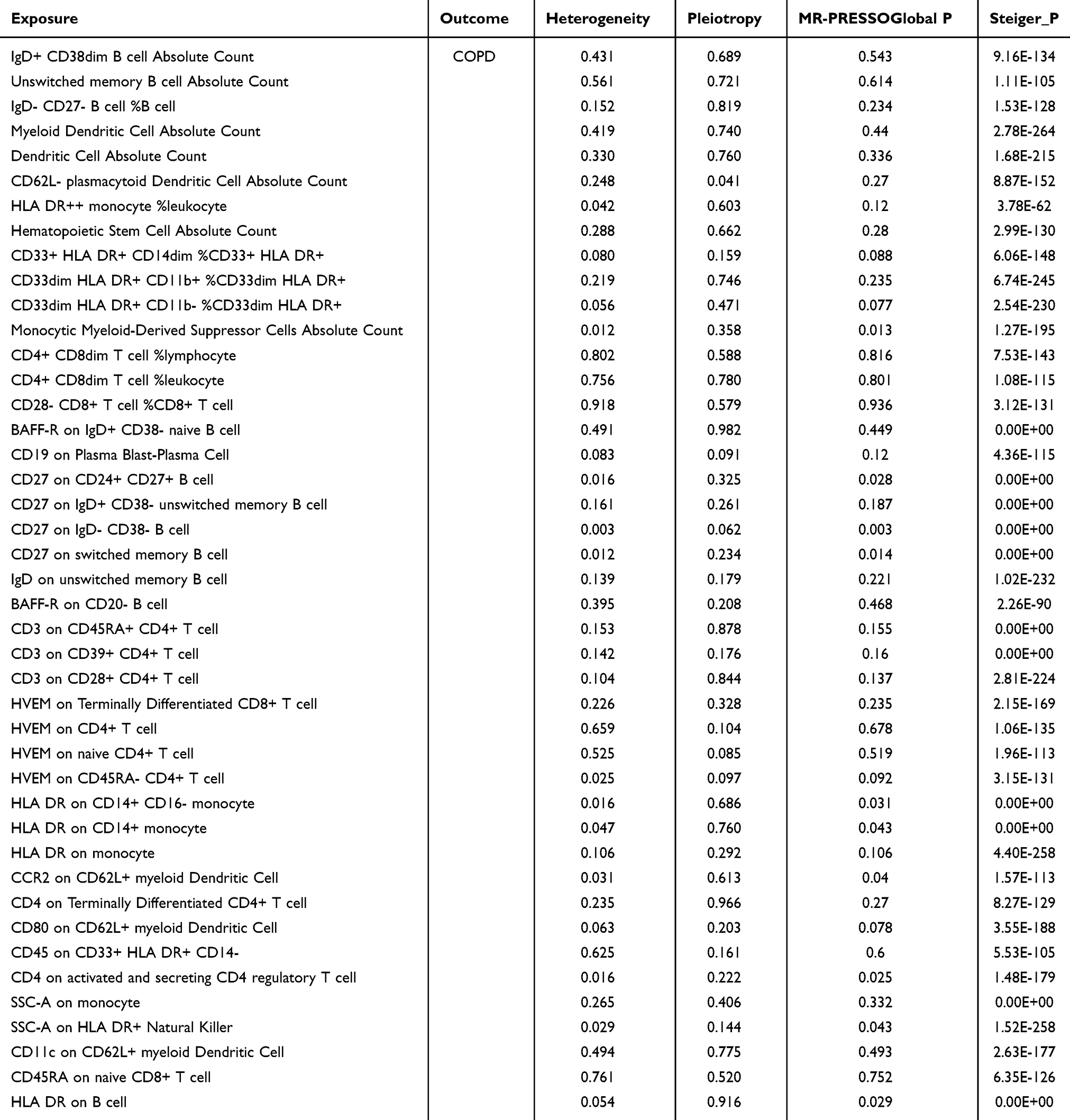 |
Table 1 Sensitivity Tests of the Causal Association Between 43 Immune Cells and COPD |
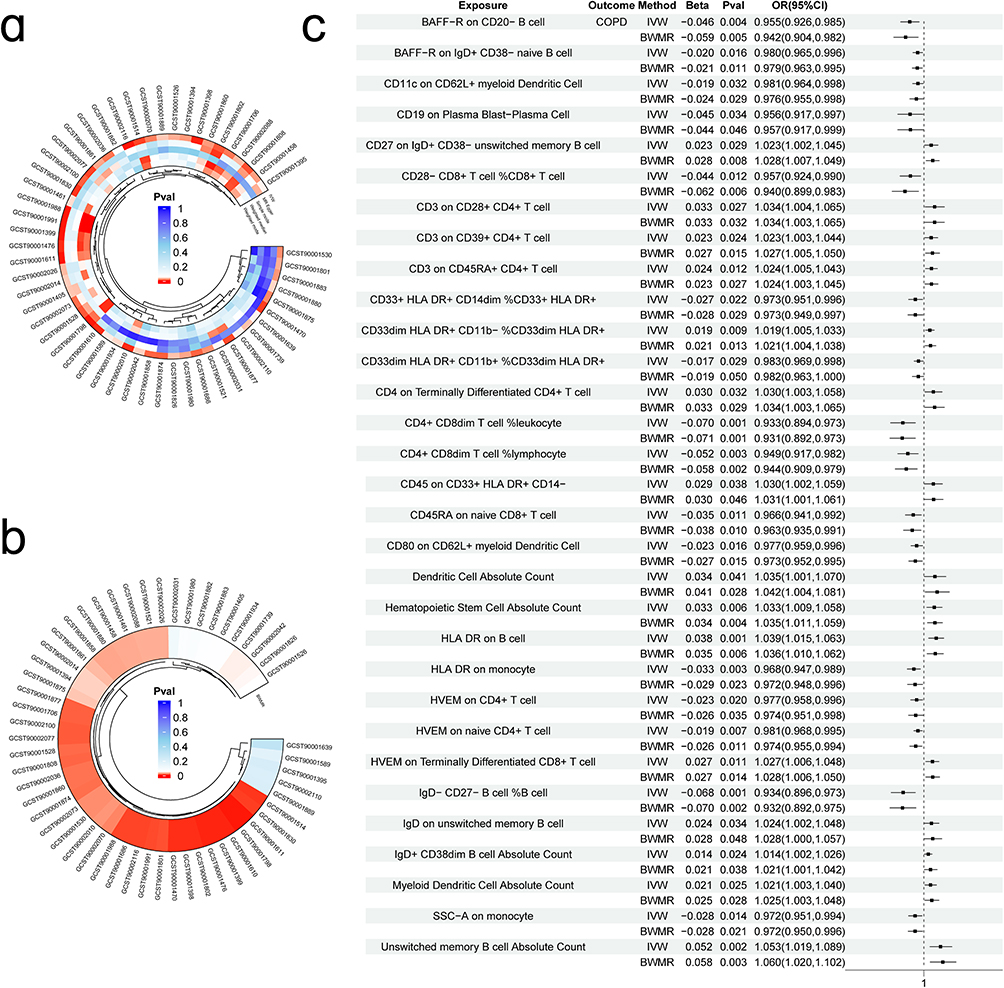 |
Figure 2 Immune cells with a causal link to COPD. (a) Circle plot visualising significant causal relationships between immune cells and COPD; (b) BWMR validation of significant causal relationships between immune cells and COPD; (c) Forest plot of the results of 30 immune cells analysed using two MR methods (OR greater than 1 indicates a positive correlation, while less than 1 indicates a negative correlation). |
Among them, 14 immune cells, such as CD3 on CD45RA+ CD4+ T cells, CD3 on CD39+ CD4+ T cells and absolute number of dendritic cells, showed a potential positive association (OR > 1), whereas IgD- CD27- B cells %B cells, CD4+ CD8dim T cells %lymphocytes, BAFF-R on CD20- B cells and 16 other immune cells negatively modulated the risk of developing COPD (OR < 1); Furthermore, the directions of the β-estimates reflecting these causal associations were consistent in MVMR and BWMR methods (Figure 2c), increasing the robustness of our findings. Meanwhile, reverse MR analysis showed (Supplementary Figure 1) that genetic susceptibility to COPD had no effect on any immune cell trait (P > 0.05), and the Steiger test (Table 1) further confirmed the reverse causal association (P < 0.05).
Causal Effects of Circulating Inflammatory Proteins on COPD
Consistent with the methods described above, we identified eight inflammatory proteins with causal effects on COPD using MVMR and BWMR approaches (Figure 3a and b; Supplementary Table 3). Specifically, we found that increased expression of adenosine deaminase, CUB domain-containing protein 1(CSMD1) and interleukin-33(IL-33) was negatively associated with the risk of developing COPD. Conversely, five inflammatory proteins, including C-X-C motif chemokine 10 (CXCL10), interleukin-20 receptor subunit alpha (IL20RA) and STAM-binding protein (STAMBP), were found to positively regulate COPD, indicating that higher expression levels of these proteins were associated with an increased risk of developing the disease (Figure 3c). Sensitivity analysis (Table 2) indicated that the causal effects of these eight proteins on COPD did not show significant heterogeneity or horizontal pleiotropy (P > 0.05). Furthermore, reverse MR analysis and the Steiger test confirmed that the causal relationships were not affected by reverse causality (Figure 3d).
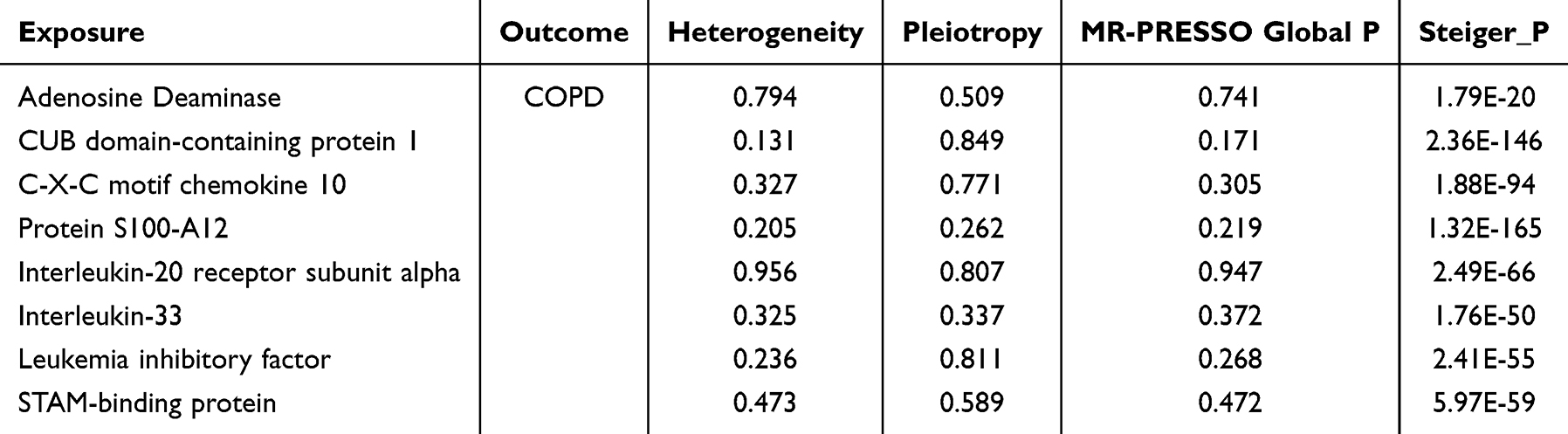 |
Table 2 Sensitivity Tests of the Causal Association Between 8 Circulating Inflammatory Proteins and COPD |
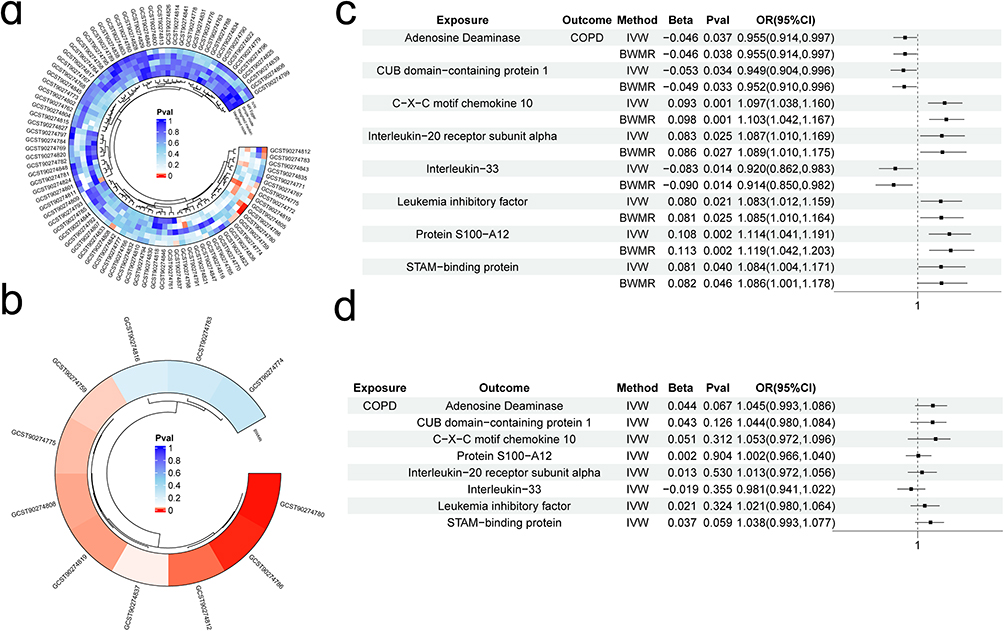 |
Figure 3 Circulating inflammatory proteins with a causal relationship to COPD.(a)Inflammatory proteins identified by the MVMR method as having a causal relationship with COPD; (b) BWMR validation of significant causal relationships between inflammatory proteins and COPD; (c) Forest plot of the results for eight proteins analyzed using two MR methods; (d) Reverse causality analysis. |
Mediated Mendelian Randomization Analysis
Based on the immune cells and circulating inflammatory proteins previously identified as having significant causal relationships with COPD, we performed further analysis using the MVMR method, treating immune cells as exposures and inflammatory proteins as outcomes, to establish their associations. As a result, we identified 16 causal relationships between 10 immune cells and 7 proteins, as shown in Figure 4. Notably, our results suggest that several immune cells may have a causal relationship with the same inflammatory protein. For example, CD3 on CD45RA+ CD4+ T cells was negatively associated with leukaemia inhibitory factor, whereas absolute dendritic cell count and HLA-DR on monocytes positively regulated leukaemia inhibitory factor expression. In addition, we performed a simple sensitivity analysis of the MR results to further confirm their robustness. As shown in Table 3, HLA-DR on monocytes showed heterogeneity in its causal relationship with CUB domain-containing protein 1, indicating the influence of confounding factors, leading to its exclusion. The remaining results showed no significant heterogeneity, pleiotropy or reverse causality.
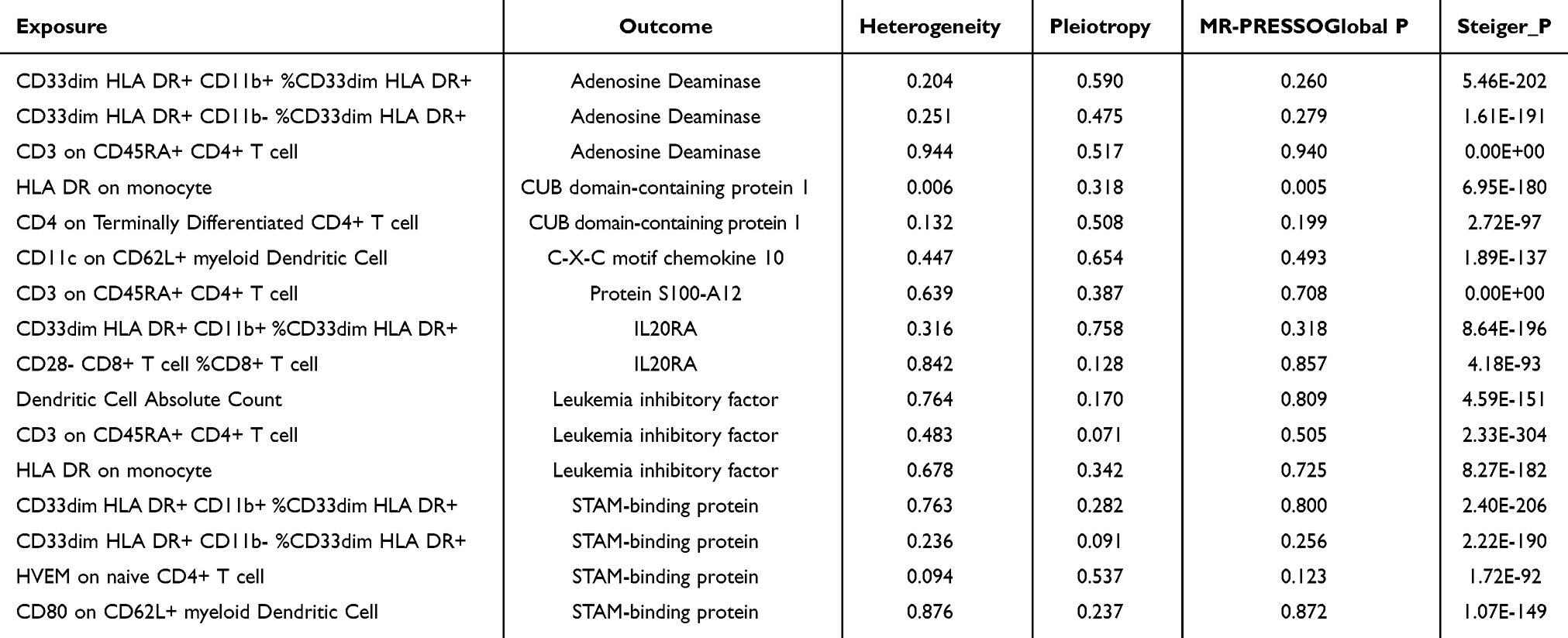 |
Table 3 Sensitivity Analysis of the Causal Relationships Between Immune Cells and Inflammatory Proteins |
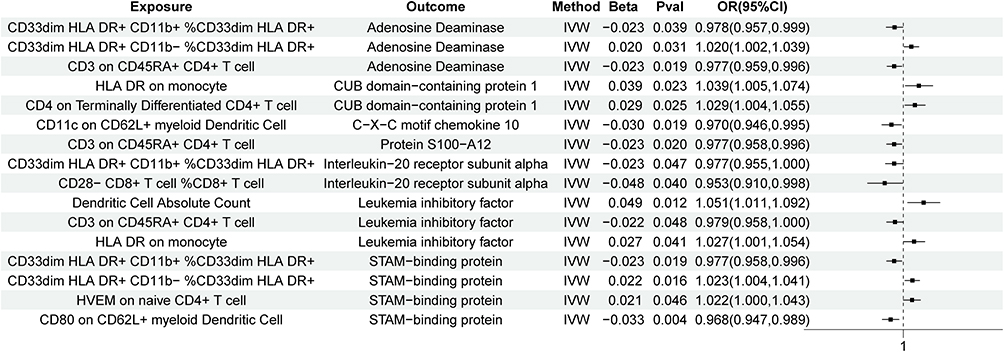 |
Figure 4 The relationship between immune cells and inflammatory proteins that have significant associations with COPD. |
Potential Mediation Effect Calculation
In mediation analysis, a necessary condition for an inflammatory protein to act as a mediator is that the indirect effect of an immune cell on COPD through the inflammatory protein must be consistent in direction with the overall effect of the immune cell on COPD. Therefore, after organising the relationships between the 10 immune cells, 7 inflammatory proteins and COPD, we calculated β_all (the effect of immune cells on COPD), β1 (the effect of immune cells on inflammatory proteins) and β2 (the effect of inflammatory proteins on COPD). After determining the indirect effect (β1β2), we calculated the percentage of the mediation effect by dividing the indirect effect by the total effect. The results of the analysis are shown in Table 4, where we found that six inflammatory proteins mediated the relationship between eight immune cell phenotypes and COPD. Among them, CXCL10 showed the highest mediation effect, accounting for 14.49% of the total effect in the inhibition of COPD progression by CD11c on CD62L+ myeloid dendritic cells, followed by IL20RA with a mediation proportion of 11.47% and leukaemia inhibitory factor with 11.45%, among others.
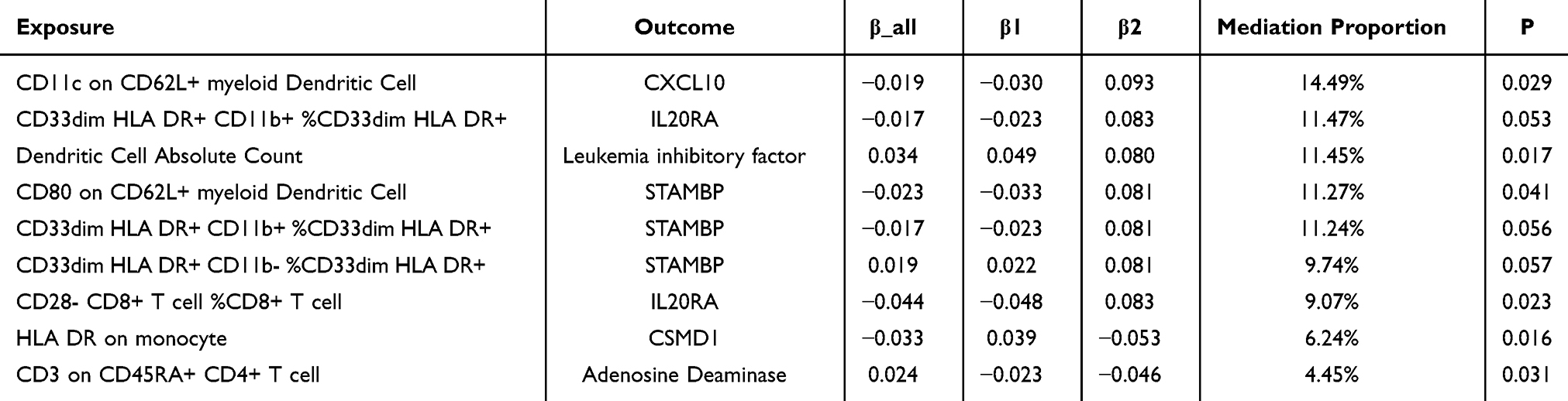 |
Table 4 Mediation Proportion of Inflammatory Proteins in the Causal Effect of Immune Cells on COPD |
Results
Traditionally, the role of the immune system in the development of COPD has been recognised as a critical factor influenced by both genetic and environmental factors. Numerous observational studies have shown that immune cells are closely associated with the development and progression of COPD.29 Immune cells play a key role in the pathogenesis of COPD by initiating and maintaining chronic airway inflammation,30 damaging lung tissue and regulating the intensity and duration of immune responses. However, causal inferences based on observational studies are subject to uncertainty due to potential confounding factors. Therefore, this study identified potential causal effects between 30 types of immune cells and COPD using Mendelian randomisation and a large number of publicly available summary statistics.
Our study is the first to confirm a causal relationship between immune cell phenotypes and COPD, which is consistent with current research. Our results show that 16 types of immune cells are risk factors for the development of COPD, including four types of B cells, five types of T cells and three types of myeloid dendritic cells. Studies have shown that B cells and their products are significantly increased in the blood and lungs of COPD patients, which is associated with inflammation and autoimmune mechanisms.31,32 The increase in memory B cells is associated with impaired lung function and small airway dysfunction, and the presence of CD24 on memory B cells is associated with an increased risk of COPD.33 Regarding T cells, Treg cell function is reduced in COPD patients, while CD4+ T cells (especially the Th1 subtype) and CD8+ T cells are increased,34,35 which are associated with lung inflammation and airway damage. CD4+CD25- Foxp3+ T cells exacerbate lung inflammation by promoting the generation of Th17 effector cells, while an increase in CD45RA+CD8+ T cells is associated with tissue damage.36,37 Among myeloid cells, CD14+ CD16+ monocytes, a subset expressing CD14 and CD16, regulate inflammatory responses and tissue repair and are closely linked to the pathogenesis of COPD,38–40 serving as key drivers of lung inflammation and tissue remodelling.41 In addition, dendritic cells (DCs) in COPD show upregulation of co-stimulatory molecules such as CD40, CD86 and CD80, which enhance the stimulation of downstream T cells, with mDC2 being particularly prominent in the induction of helper T cells.42
Many systemic manifestations of COPD are thought to be mediated by elevated levels of inflammatory proteins such as IL-6, TNF-α and CRP. Numerous studies have confirmed that CRP is a common biomarker of systemic inflammation in COPD patients, with plasma CRP levels being higher in COPD patients than in healthy individuals.43,44 Interestingly, CRP is regulated by IL-6, as this cytokine can initiate an acute phase response by inducing the production of acute phase proteins, including CRP, in hepatocytes. In contrast to these studies, our research has identified several other inflammatory proteins associated with COPD. Among these, increased expression of five inflammatory proteins, including CXCL10, STAMBP and IL20RA, is associated with an increased risk of COPD, while adenosine deaminase, CSMD1 and IL33 are protective against COPD. In support of our findings, related studies, such as that by Qiuping Li45 et al, have shown that Icariin inhibits the chemotactic enhancement of CD8+ T cells by targeting the CXCL10/CXCR3 axis, suggesting that CD8+ T cells may be a novel therapeutic target for reducing lung tissue damage and improving lung function in COPD. In addition, Kittipong Maneechotesuwan46 et al found that simvastatin could reverse the inhibition of adenosine deaminase activity by IL-13, thereby preventing the progression of COPD, which is consistent with our conclusion that adenosine deaminase plays a protective role in COPD.
Our mediation analysis identified the role of six inflammatory proteins, with CXCL10 mediating 14.49% of the protective effect of CD11c on CD62L+ myeloid dendritic cells against COPD. Specifically, CD11c on CD62L+ myeloid dendritic cells represent a specialised subset of dendritic cells that play a critical role in the immune system, particularly in inflammation and immune responses. CD11c is an integrin alpha chain protein that is widely expressed on immune cells such as dendritic cells and macrophages and is commonly used as a marker for myeloid dendritic cells.47 It is involved in cell adhesion and signalling and is a key molecule in the antigen presentation process. CD62L is a cell adhesion molecule primarily expressed on lymphocytes and some myeloid cells, and contributes to cell homing to lymphoid organs and rolling migration at sites of inflammation,48 thereby regulating the distribution and activation state of immune cells. CXCL10, on the other hand, is a chemokine with significant immunomodulatory functions produced by various cell types, including endothelial cells, macrophages, fibroblasts and T cells. It plays an important role in inflammation and immune responses.49 Studies have shown that several chemokines (CCL2, CCL3, CCL5, CX3CL1, CXCL8, CXCL9, CXCL10, CXCL11 and CXCL12) and inflammatory mediators (MMP-9, MMP-12, IL-18 and neutrophil counts) are significantly increased in the serum of COPD patients compared to healthy controls.50 In our study, CD11c on CD62L+ myeloid dendritic cells was negatively correlated with both COPD and CXCL10, suggesting that the increased expression of these immune cells may contribute to their protective effect against COPD, possibly through suppression of CXCL10 expression.
Second, the proportion of COPD suppression mediated by IL10RA through CD33dim HLA-DR+ CD11b+ cells is 11.47%. CD33dim HLA-DR+ CD11b+ represent a specific subset of immune cells, typically identified by flow cytometry as monocytes with low CD33 expression and high HLA-DR and CD11b expression. Studies have shown that the increase in these cells is closely associated with the persistence of inflammation and immune responses. They may influence the severity of COPD by secreting inflammatory mediators and activating immune pathways such as the NF-κB and MAPK pathways.51–53 In particular, these cells can secrete pro-inflammatory cytokines such as IL-6 and TNF-α, which exacerbate airway inflammation and tissue damage.54 IL20RA plays a critical mediating role in this process. IL20RA not only regulates the function of these immune cells, but also influences their sensitivity to inflammatory responses. In COPD patients, upregulation of IL20RA expression in these immune cell populations may enhance their responsiveness to inflammation and further contribute to COPD progression. Although current research on the relationship between CD33dim HLA-DR+ CD11b+ cells and COPD is limited, these findings provide a strong direction for future studies.
This study uses Mendelian randomization to assess the causal relationship between immune cells and COPD, minimizing reverse causality and controlling for potential confounding factors. By utilizing existing large-scale GWAS data, the study avoids the need for long-term follow-up, saving time and resources. While the mediating role of inflammatory proteins is relatively small, our findings highlight their role in the relationship between immune cells and COPD and identify immune cells and proteins causally associated with COPD, providing valuable insights for future research. Although the mediating effect of inflammatory proteins between immune cells and COPD is limited, their significance in immune responses and disease progression should not be overlooked. Further exploration of the role of these inflammatory proteins provides potential therapeutic targets for clinical practice, which could aid in the early diagnosis and personalized treatment of COPD. In the future, a better understanding of the causal relationship between immune cells and COPD, combined with the mediating role of inflammatory proteins, may help develop novel immunomodulatory therapies, improving the prognosis and quality of life for COPD patients.
However, there are several notable limitations to this study. First, our analysis is limited to individuals of European descent, which may limit the generalisability of our findings to other ethnic groups. The inclusion of more diverse populations is essential for a comprehensive understanding of the global epidemiology of COPD, and future studies should aim to include these diverse groups.Second, to include more genetic variants associated with the phenotype, we relaxed the threshold for selecting instrumental variables, which may increase the risk of violating the strong instrument assumption of the MR design. Nevertheless, we addressed this by excluding weak instrumental variables through F-statistic screening (F > 10).Finally, when determining mediation effects, most proteins had low mediation proportions, suggesting the potential presence of other unknown mediators. In addition, our results are theoretical and need to be validated by clinical or animal studies. Further research, including cell and animal studies, is needed to elucidate these mechanisms.
Conclusion
This study provides a comprehensive assessment of the causal relationships between immune cell phenotypes, circulating inflammatory proteins and COPD. We identified eight immune cells with a causal association with COPD, mediated by six different inflammatory proteins. Our findings highlight the importance of the potential mechanisms linking immune cells, inflammatory proteins and COPD. This suggests that future interventions could focus on modulating immune cell levels by targeting the inflammatory microenvironment, thereby improving COPD prevention and treatment strategies. In addition, these findings may help to identify populations at risk of COPD.
Data Sharing Statement
The original data used in the study are included in the article and its supplementary materials. For further inquiries, please contact the corresponding author directly.
Ethics Approval and Consent to Participate
This study complies with the conditions for exemption from review as stated in the “Ethical Review Measures for Life Sciences and Medical Research Involving Humans.” Since the data do not involve identifiable personal information, the study avoids direct human subject research and meets the requirements for ethical exemption.
Acknowledgments
We thank all the participants and investigators involved in the GWAS, as well as all the authors for their contributions to this article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare no competing interests in this work.
References
1. Global Initiative for Chronic Obstructive Lung Disease (Gold). Global strategy for the diagnosis, management and prevention of chronic obstructive lung disease (2024 report). Available from: https://Goldcopd.Org/.
2. Soriano JB, Kendrick PJ, Paulson KR, GBD Chronic Respiratory Disease Collaborators. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet Respir Med. 2020;8(6):585–596. doi:10.1016/S2213-2600(20)30105-3
3. WHO. Global health estimates: leading causes of death. cause-specific mortality 2000–2019. Available from: https://Www.Who.Int/Data/Gho/Data/Themes/Mortality-and-Global-Health-Estimates/Ghe-Leading-Causes-of-Death.
4. Meghji J, Mortimer K, Agusti A, et al. Improving lung health in low-income and middle-income countries: from challenges to solutions. Lancet. 2021;397(10277):928–940. doi:10.1016/S0140-6736(21)00458-X
5. Adeloye D, Song P, Zhu Y, Campbell H, Sheikh A, Rudan I. Global, regional, and national prevalence of, and risk factors for, chronic obstructive pulmonary disease (COPD) in 2019: a systematic review and modelling analysis. Lancet Respir Med. 2022;10(5):447–458. doi:10.1016/S2213-2600(21)00511-7
6. Planer JD, Morrisey EE. After the storm: regeneration, repair, and reestablishment of homeostasis between the alveolar epithelium and innate immune system following viral lung injury. Annu Rev Pathol. 2023;18(1):337–359. doi:10.1146/annurev-pathmechdis-031621-024344
7. Bracke KR, Polverino F. Blunted adaptive immune responses and acute exacerbations of COPD: breaking the code. Eur Respir J. 2023;62(2):2301030. doi:10.1183/13993003.01030-2023
8. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
9. Kalathil SG, Lugade AA, Pradhan V, et al. T-regulatory cells and programmed death 1+ T cells contribute to effector T-cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(1):40–50. doi:10.1164/rccm.201312-2293OC
10. Polverino F, Cosio BG, Pons J, et al. B cell-activating factor. an orchestrator of lymphoid follicles in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192(6):695–705. doi:10.1164/rccm.201501-0107OC
11. Villaseñor-Altamirano AB, Jain D, Jeong Y, et al. Activation of CD8+ T cells in chronic obstructive pulmonary disease lung. Am J Respir Crit Care Med. 2023;208(11):1177–1195. doi:10.1164/rccm.202305-0924OC
12. de Fays C, Geudens V, Gyselinck I, et al. Mucosal immune alterations at the early onset of tissue destruction in chronic obstructive pulmonary disease. Front Immunol. 2023;14:1275845. doi:10.3389/fimmu.2023.1275845
13. Butler CC, Gillespie D, White P, et al. C-reactive protein testing to guide antibiotic prescribing for COPD exacerbations. N Engl J Med. 2019;381(2):111–120. doi:10.1056/NEJMoa1803185
14. Serum IL-1β and IL-17 levels in patients with COPD: associations with clinical parameters - PubMed [Internet]. [cited August 31, 2024]. Available from: https://pubmed.ncbi.nlm.nih.gov/28490868/.
15. Bradford E, Jacobson S, Varasteh J, et al. The value of blood cytokines and chemokines in assessing COPD. Respir Res. 2017;18(1):180. doi:10.1186/s12931-017-0662-2
16. Bai Y, Zhou Q, Fang Q, Song L, Chen K. Inflammatory cytokines and t-lymphocyte subsets in serum and sputum in patients with bronchial asthma and chronic obstructive pulmonary disease. Med Sci Monit. 2019;25:2206–2210. doi:10.12659/MSM.913703
17. Beeghly-Fadiel A, Khankari NK, Delahanty RJ, et al. A Mendelian randomization analysis of circulating lipid traits and breast cancer risk. Int J Epidemiol. 2020;49(4):1117–1131. doi:10.1093/ije/dyz242
18. Titova OE, Michaëlsson K, Larsson SC. Sleep duration and stroke: prospective cohort study and Mendelian randomization analysis. Stroke. 2020;51(11):3279–3285. doi:10.1161/STROKEAHA.120.029902
19. Ahmed M, Mulugeta A, Lee SH, Mäkinen VP, Boyle T, Hyppönen E. Adiposity and cancer: a Mendelian randomization analysis in the UK biobank. Int J Obes Lond. 2021;45(12):2657–2665. doi:10.1038/s41366-021-00942-y
20. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. 2017;318(19):1925–1926. doi:10.1001/jama.2017.17219
21. Orrù V, Steri M, Sidore C, et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat Genet. 2020;52(10):1036–1045. doi:10.1038/s41588-020-0684-4
22. Zhao JH, Stacey D, Eriksson N, et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat Immunol. 2023;24(9):1540–1551. doi:10.1038/s41590-023-01588-w
23. Wang Q, Dai H, Hou T, et al. Dissecting causal relationships between gut microbiota, blood metabolites, and stroke: a Mendelian randomization study. J Stroke. 2023;25(3):350–360. doi:10.5853/jos.2023.00381
24. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45(6):1961–1974. doi:10.1093/ije/dyw220
25. Bowden J, Del Greco MF, Minelli C, et al. Improving the accuracy of two-sample summary-data Mendelian randomization: moving beyond the NOME assumption. Int J Epidemiol. 2019;48(3):728–742. doi:10.1093/ije/dyy258
26. Zhao J, Ming J, Hu X, Chen G, Liu J, Yang C. Bayesian weighted Mendelian randomization for causal inference based on summary statistics. Bioinformatics. 2020;36(5):1501–1508. doi:10.1093/bioinformatics/btz749
27. Zhong Y, Wang F, Meng X, Zhou L. The associations between gut microbiota and inflammatory skin diseases: a bi-directional two-sample Mendelian randomization study. Front Immunol. 2024;15:1297240. doi:10.3389/fimmu.2024.1297240
28. Carter AR, Sanderson E, Hammerton G, et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur J Epidemiol. 2021;36(5):465–478. doi:10.1007/s10654-021-00757-1
29. Bhat TA, Panzica L, Kalathil SG, Thanavala Y. Immune dysfunction in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2015;12(Suppl 2):S169–175. doi:10.1513/AnnalsATS.201503-126AW
30. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026. doi:10.1016/S0140-6736(11)60988-4
31. Faner R, Cruz T, Casserras T, et al. Network analysis of lung transcriptomics reveals a distinct B-cell signature in emphysema. Am J Respir Crit Care Med. 2016;193(11):1242–1253. doi:10.1164/rccm.201507-1311OC
32. Spatial transcriptomics resolve an emphysema-specific lymphoid follicle b cell signature in chronic obstructive pulmonary disease - PubMed [Internet]. [cited August 31, 2024]. Available from: https://pubmed.ncbi.nlm.nih.gov/37934672/.
33. Habener A, Grychtol R, Gaedcke S, et al. IgA+ memory B-cells are significantly increased in patients with asthma and small airway dysfunction. Eur Respir J. 2022;60(5):2102130. doi:10.1183/13993003.02130-2021
34. Xue W, Ma J, Li Y, Xie C. Role of CD4 + T and CD8 + T lymphocytes-mediated cellular immunity in pathogenesis of chronic obstructive pulmonary disease. J Immunol Res. 2022;2022:1429213. doi:10.1155/2022/1429213
35. Albertson TE, Chenoweth JA, Pearson SJ, Murin S. The pharmacological management of asthma-chronic obstructive pulmonary disease overlap syndrome (ACOS). Expert Opin Pharmacother. 2020;21(2):213–231. doi:10.1080/14656566.2019.1701656
36. Generation and immune regulation of CD4+CD25-Foxp3+ T cells in chronic obstructive pulmonary disease – PubMed [Internet]. [cited August 31, 2024]. Available from: https://pubmed.ncbi.nlm.nih.gov/30842769/.
37. Foo SY, Zhang V, Lalwani A, et al. Regulatory T cells prevent inducible BALT formation by dampening neutrophilic inflammation. J Immunol. 2015;194(9):4567–4576. doi:10.4049/jimmunol.1400909
38. Lee Y, Song J, Jeong Y, Choi E, Ahn C, Jang W. Meta-analysis of single-cell RNA-sequencing data for depicting the transcriptomic landscape of chronic obstructive pulmonary disease. Comput Biol Med. 2023;167:107685. doi:10.1016/j.compbiomed.2023.107685
39. Huang W, Luo T, Lan M, et al. Identification and characterization of a ceRNA regulatory network involving LINC00482 and PRRC2B in peripheral blood mononuclear cells: implications for COPD pathogenesis and diagnosis. Int J Chron Obstruct Pulmon Dis. 2024;19:419–430. doi:10.2147/COPD.S437046
40. Single-cell sequencing of lung macrophages and monocytes reveals novel therapeutic targets in COPD – PubMed [Internet]. [cited August 31, 2024]. Available from: https://pubmed.ncbi.nlm.nih.gov/38132091/.
41. Wohnhaas CT, Baßler K, Watson CK, et al. Monocyte-derived alveolar macrophages are key drivers of smoke-induced lung inflammation and tissue remodeling. Front Immunol. 2024;15:1325090. doi:10.3389/fimmu.2024.1325090
42. Lung dendritic cell expression of maturation molecules increases with worsening chronic obstructive pulmonary disease – PubMed [Internet]. [cited August 31, 2024]. Available from: https://pubmed.ncbi.nlm.nih.gov/19729666/.
43. Yende S, Waterer GW, Tolley EA, et al. Inflammatory markers are associated with ventilatory limitation and muscle dysfunction in obstructive lung disease in well functioning elderly subjects. Thorax. 2006;61(1):10–16. doi:10.1136/thx.2004.034181
44. Broekhuizen R, Wouters EFM, Creutzberg EC, Amwj S. Raised CRP levels mark metabolic and functional impairment in advanced COPD. Thorax. 2006;61(1):17–22. doi:10.1136/thx.2005.041996
45. Li Q, Sun J, Cao Y, et al. Icaritin inhibited cigarette smoke extract-induced CD8+ T cell chemotaxis enhancement by targeting the CXCL10/CXCR3 axis and TGF-β/Smad2 signaling. Phytomedicine. 2022;96:153907. doi:10.1016/j.phymed.2021.153907
46. Maneechotesuwan K, Kasetsinsombat K, Wongkajornsilp A, Barnes PJ. Simvastatin up-regulates adenosine deaminase and suppresses osteopontin expression in COPD patients through an IL-13-dependent mechanism. Respir Res. 2016;17(1):104. doi:10.1186/s12931-016-0424-6
47. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449(7161):419–426. doi:10.1038/nature06175
48. Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22(1):129–156. doi:10.1146/annurev.immunol.21.090501.080131
49. Gangur V, Birmingham NP, Thanesvorakul S. Chemokines in health and disease. Vet Immunol Immunopathol. 2002;86(3–4):127–136. doi:10.1016/S0165-2427(02)00018-1
50. Hao W, Li M, Pang Y, Du W, Huang X. Increased chemokines levels in patients with chronic obstructive pulmonary disease: correlation with quantitative computed tomography metrics. Br J Radiol. 2021;94(1118):20201030. doi:10.1259/bjr.20201030
51. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–964. doi:10.1038/nri1733
52. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi:10.1101/cshperspect.a001651
53. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18. doi:10.1038/sj.cr.7290105
54. Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. 2000;117(4):1162–1172. doi:10.1378/chest.117.4.1162
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Exploring the Causal Relationship Between Frailty and Chronic Obstructive Pulmonary Disease: Insights From Bidirectional Mendelian Randomization and Mediation Analysis
Cheng Z, Wu J, Xu C, Yan X
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:193-205
Published Date: 25 January 2025