Back to Journals » International Journal of Nanomedicine » Volume 12
The mechanisms of graphene-based materials-induced programmed cell death: a review of apoptosis, autophagy, and programmed necrosis
Authors Ou L, Lin S, Song B, Liu J, Lai R, Shao L ![]()
Received 26 April 2017
Accepted for publication 13 July 2017
Published 7 September 2017 Volume 2017:12 Pages 6633—6646
DOI https://doi.org/10.2147/IJN.S140526
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Lingling Ou,1,2 Shaoqiang Lin,2 Bin Song,1 Jia Liu,1 Renfa Lai,2 Longquan Shao1
1Department of Stomatology, Nanfang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 2Department of Stomatology, the First Affiliated Hospital of Jinan University, Guangzhou, People’s Republic of China
Abstract: Graphene-based materials (GBMs) are widely used in many fields, including biomedicine. To date, much attention had been paid to the potential unexpected toxic effects of GBMs. Here, we review the recent literature regarding the impact of GBMs on programmed cell death (PCD). Apoptosis, autophagy, and programmed necrosis are three major PCDs. Mechanistic studies demonstrated that the mitochondrial pathways and MAPKs (JNK, ERK, and p38)- and TGF-β-related signaling pathways are implicated in GBMs-induced apoptosis. Autophagy, unlike apoptosis and necroptosis which are already clear cell death types, plays a vital pro-survival role in cell homeostasis, so its role in cell death should be carefully considered. However, GBMs always induce unrestrained autophagy accelerating cell death. GBMs trigger autophagy through inducing autophagosome accumulation and lysosome impairment. Mitochondrial dysfunction, ER stress, TLRs signaling pathways, and p38 MAPK and NF-κB pathways participate in GBMs-induced autophagy. Programmed necrosis can be activated by RIP kinases, PARP, and TLR-4 signaling in macrophages after GBMs exposure. Though apoptosis, autophagy, and necroptosis are distinguished by some characteristics, their numerous signaling pathways comprise an interconnected network and correlate with each other, such as the TLRs, p53 signaling pathways, and the Beclin-1 and Bcl-2 interaction. A better understanding of the mechanisms of PCD induced by GBMs may allow for a thorough study of the toxicology of GBMs and a more precise determination of the consequences of human exposure to GBMs. These determinations will also benefit safety assessments of the biomedical and therapeutic applications of GBMs.
Keywords: graphene based materials, cell toxicity, programmed cell death, mechanisms
Introduction
Graphene materials have various applications and are widely used in the biomedical field in tumor therapy,1–3 diagnostics,4,5 bio-imaging,6–8 and drug delivery.9,10 The widespread use of graphene materials has prompted concerns about the possible human health and environmental safety impacts of these materials. GBMs present in biomedical and non-biomedical products have shown potential toxicity to human beings, animals, and cells.11–13 Several reviews have summarized that GBMs resulted in various degrees of cell death.14,15 For example, GO and rGO treatments resulted in dose-dependent cell death in A549 cells and HUVEC cells, and the functionalization decreased cytotoxicity and cell death.16,17 Cell death is a signal that reflects the toxicity of GBMs,18–20 and this review focuses on the mechanisms of GBMs-induced PCD.
A systematic classification of cell death was recently presented in a set of recommendations by the Nomenclature Committee on Cell Death.21 PCD is proposed to be death of a cell in any pathological format, when mediated by an intracellular program. PCD balances cell death with survival of normal cells when the equilibrium becomes disturbed. It is suggested that PCD is mediated by specific cellular mechanisms and that several signal pathways are activated and involved in these processes.22 Several PCD modalities, including apoptosis, autophagy, and programmed necrosis (also called necroptosis), have been investigated in the studies of the toxicity of nanomaterials.23,24 In particular, apoptosis, autophagy, and necroptosis are usually involved in GBMs toxicology.25–27 Many studies have reported that the different physicochemical properties of GBMs result in various cellular toxicities, and GBMs induce cell death in dose- or time-dependent manner.28 However, the detailed mechanism of PCD induced by GBMs has not been well elucidated. Since many other reviews had illustrated that the different physicochemical properties of GBMs largely impacted on the cell toxicity,29–31 we concentrate on summarizing the pathways of PCD induced by GBMs in this review, and leave aside the effect of administration route, administration dose, and the physicochemical properties of GBMs.
Notably, apoptosis, autophagy, and necroptosis can be distinguished by their unique morphological differences and physiological processes; however, the underlying signaling pathways are intricate and delicate.32,33 Evidence has illustrated that apoptosis and necroptosis are two manners of cell death, whereas autophagy can play either pro-survival or pro-death roles. Under some conditions, apoptosis, autophagy, and necroptosis pathways are somehow cross-linked with each other. Herein, we review some studies on GBMs-induced PCD and summarize part of the signaling pathways, and analyze some differences and correlation between PCDs.
GBMs induce apoptosis in cells
Apoptosis is one of the most important toxic features described by studies of GBMs toxicity. GBMs induce apoptosis both in vivo and in vitro, and the extent of apoptosis differs depending on the varying properties of GBMs, including surface area, layer number, lateral dimension, functional groups, and surface chemistry. Recently, hydrophilic GO showed cellular uptake resulting in apoptosis following cell morphological changes from an elongated to a spherical morphology; conversely, hydrophobic rGO was found to be mostly adsorbed at cell surface without internalization at a dose of 20 mg/L.34–36 Graphene layers (10 μg/mL) induced apoptosis by activating caspase-3 in a time-dependent manner.37 GO nanoribbons (100 μg/mL) and GO polyethylenimine (1.6 μg/mL) triggered severe apoptosis by translocating to the cell nucleus.38,39 However, polyvinyl pyrrolidone-coated GO and nano-GO coated with polyethylene glycol triggered weaker apoptosis compared to GO in a dose-dependent manner.40,41
GBMs-induced apoptosis somehow promoted diseases. The cell cycle arrest and DNA fragmentation caused by GO could induce germline apoptosis, affecting gonad development at the concentration of 10 mg/L.42 GQDs may translocate to the brain and localize to the nucleus of the diencephalon, causing Parkinson’s disease-like symptoms by inducing cell apoptosis and senescence.43 Long-term and chronic exposure to GBMs may result in some diseases or do harm to the health.
However, numerous studies have demonstrated the therapeutic value of GBMs in cancer cells. GBMs, similarly to several anticancer compounds, exert their inhibitory effect on tumors by arresting the cell cycle at a specific checkpoint, activating apoptosis, or a combined effect of both processes.44 In vivo, GO and rGO injections decrease the mass and volume of tumors, and in vitro, GO and rGO (50 μg/mL) treatments induce apoptosis by the upregulation of caspase-3 and membrane leakage.45,46 GQDs (100 μg/mL) induced apoptosis of U251 human glioma cells displaying morphological and biochemical characteristics, such as phosphatidylserine externalization, caspase activation, and DNA fragmentation.47 Similarly, PEGylated silver nanoparticles coated with GQDs exerted a distinct antitumor effect at the level of 100 μg/mL by triggering apoptosis only in cancer cells without impacting the viability of normal cells.48 Consequently, understanding of how to improve the antitumor effects and decrease the toxicity of normal cells may allow good use of GBMs in biomedicine.
Since apoptosis induced by GBMs emerges in many situations, we summarize some information to describe GBMs-induced apoptosis, such as the physiochemical properties and functionalization of GBMs, the exposed cells, the exposed dose and time, and so on. The information is collectively presented in Table 1.
 | Table 1 GBMs induced apoptosis |
The signaling pathways involved in GBM-induced apoptosis
Apoptosis, or type I PCD, is a gene-controlled cell death process maintaining cell homeostasis and is characterized by specific morphological changes such as cell shrinkage, nuclear condensation and fragmentation, and the appearance of apoptotic bodies.49,50 Apoptosis is divided into extrinsic (death receptor pathway) and intrinsic (mitochondrial pathway) pathways.51 The extrinsic pathway is mediated by the TNF receptor superfamily, and the binding ligands are Fas ligand, TNF-R, or TRAIL.52 The Fas/Fas-L complex recruits the FADD and pro-caspase-8 to form the DISC. This protein complex serves as a platform for caspase activation, and the autocatalytic activation of caspase-8 at the DISC leads to the proteolysis of pro-caspase-3 into active caspase-3, triggering the apoptotic process.53 The intrinsic pathway is under the control of mitochondrial proenzymes with a variety of functions, including the dissipation of the mitochondrial transmembrane potential and the release of proapoptotic proteins into the cytosol, as well as a second, mitochondria-derived activator of caspases. After being triggered by extracellular stimuli or intracellular signals, internal cytochrome c is released into the cytosol from the permeable mitochondrial membranes and recruits Apaf-1 and pro-caspase-9 to the apoptosome, which activates the downstream caspase-9/3 signaling cascade and consequently results in apoptosis.54,55
Apoptosis is a complex process that can be activated through several signaling pathways.56 Recently, much attention has been paid to clarifying the intricate mechanism of apoptosis in GBMs-induced toxicity. GBMs-induced apoptosis usually has the fundamental features of caspase activation, DNA fragmentation, increased oxidative stress, and calcium efflux.57,58
Mechanistic studies demonstrated that mitochondria are implicated in apoptotic cell death, which act as major control points responsible for regulating apoptosis. It is well accepted that mitochondria participate in the intrinsic pathway of apoptosis and that they release soluble proteins from the intermembrane space to the cytosol to initiate caspase activation.59,60 For example, caspase-3, one of the most important executioner caspases, and PARP are activated by mitochondrial pathways that initiate pristine graphene-mediated apoptosis.61,62 Exposure to graphene increases intracellular ROS generation, including mitochondria-produced superoxide, which subsequently results in the loss of the MMP.62,63 Disruptions of the MMP may lead to broken electron transfer chain, decline in ATP synthesis, and release of proapoptotic molecules from the mitochondria to cytoplasm. Similarly, GO and rGO treatments also cause mitochondrial damage and accelerate apoptosis by generating oxidative stress.64,65 Exposure to few-layer graphene caused elevation of cytoplasmic Ca2+ concentration and mitochondrial membrane depolarization, and eventually, the abnormal opening of the mitochondrial permeability transition pore led to more severe mitochondrial damage and the initiation of apoptosis.57 Notably, TLRs also contribute to the loss of MMP, consequently inducing apoptosis after exposure to rGO.66
In addition to mitochondrial pathways, the MAPKs (JNK, ERK, and p38)- and TGF-β-related signaling pathways are also involved in GBMs-induced apoptosis.62 The MAPK (JNK, ERK, and p38) signaling pathway involves a family of serine/threonine protein kinases that regulates cell proliferation, cell survival, and apoptosis.67,68 JNK activates the Bcl-2 protein family proapoptotic members, Bim and Bax, eliciting MOMP during GBM-induced apoptosis.69,70 Subsequently, mitochondrial proapoptotic factors activate caspase-3, an important effector caspase involved in apoptosis.71 The p38 protein is activated by ROS and can induce apoptosis following the stimulation of TNF-α, the recruitment of caspase-8 and caspase-3, and the activation of the downstream effector protein PARP. In addition, TGF-β participating in cell apoptosis has been shown in several cell lines;72–74 however, the TGF-β-related signaling pathways have not been well studied in GBM-induced apoptosis. The proteins downstream of TGF-β, Smad proteins, activate proapoptotic factors and relocate from the mitochondria to the cytosol to regulate cell apoptosis. These pathways may be a good research direction for further study of GBMs-induced apoptosis. GBMs-induced apoptosis pathways are important parts of GBMs-induced toxicity, and some of them are delineated in Figure 1.
 | Figure 1 The signaling pathways involved in GBM-induced apoptosis. The schematic diagram delineates the extrinsic (death receptor pathway) and intrinsic (mitochondrial pathway) pathways of apoptosis. GBMs may induce apoptosis through mitochondrial pathways and the MAPKs- and TGF-β-related signaling pathways. Mitochondria act as major control points involving in regulation of apoptosis. Caspases (caspase-3, caspase-8), PARP, and Bcl-2 protein family proapoptotic members (Bim and Bax) are activated and mitochondrial outer membrane permeabilization and the loss of mitochondrial membrane permeabilization occur, and are involved in the pathways during GBM-induced apoptosis. |
GBMs induce autophagy in cells
In the current toxicology study of GBMs, there is no detailed research on distinguishing between autophagy, autophagic cell death, and cell death caused by autophagy. Rather, we just discuss autophagy triggered or inhibited by GBMs in the following text. GO triggers autophagy in macrophages in a concentration-dependent manner. Furthermore, GO can induce autophagy in various cell lines, including SNU-449, Mahlavu, A549, HEK293, and RAW 264.7 macrophages, at a concentration of 100 μg/mL by stimulating the TLR signaling cascades.75 GO exposure to the human-hamster hybrid (AL) cells significantly elicited genuine autophagy by increasing the level of LC3-II and the autophagic flux even at a low dose (1 μg/mL) or an early time point (4 h).76 GO was phagocytosed by murine colon carcinoma (CT26) cells and simultaneously triggered autophagy by TLR-4/9 signaling cascades at a dose of 50 μg/mL.77 It was also suggested that GO was a potent autophagy inducer in CT26 colon cancer cells, and thus in chemotherapy, GO combined with the chemotherapy drug cisplatin (CDDP) (GO/CDDP) diverted the autophagic flux and improved antitumor effects at a dose of 50 μg/mL.78 GNPs promoted autophagy by increasing the conversion from LC3B-I to LC3B-II and the level of p62 protein at 24 h after exposure in a dose-dependent manner in BEAS-2B cells.79 Similarly, GQDs also significantly dose-dependently increased ROS generation and induced autophagy with an increase in the expression of LC3-II/I and Beclin-1 in macrophages or U251 human glioma cells.26,47 Since autophagy may eliminate GBM particles or provide energy through cleaning the remnant organelle in cells, it may become a primary index to evaluate the toxicity of GBMs in occupational exposure or biomedical application. A summary of GBMs’ physicochemical properties, exposed cells, toxicokinetics, and the autophagy results from the literature reviewed here is listed in Table 2.
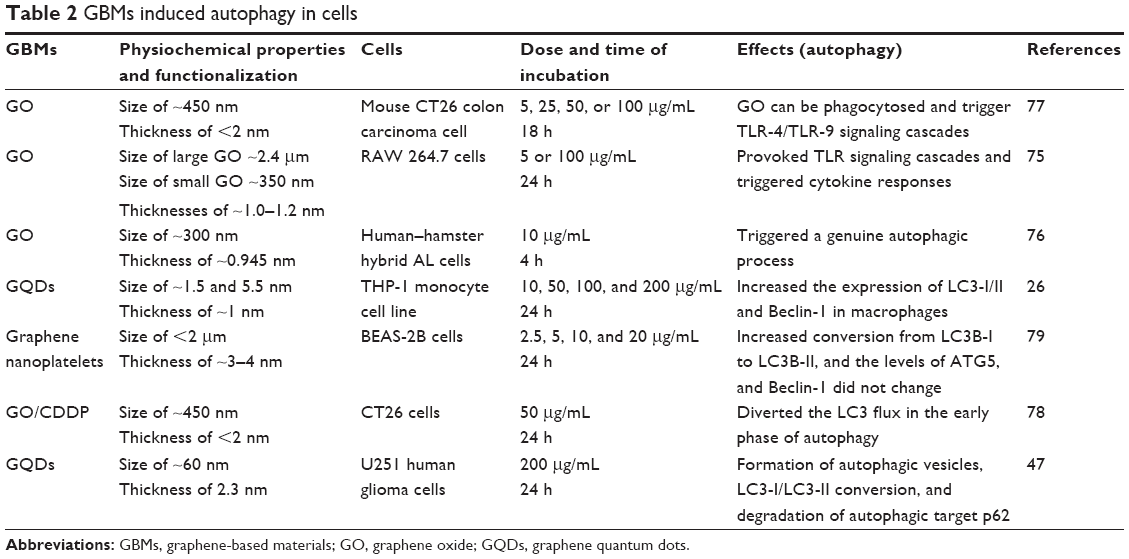 | Table 2 GBMs induced autophagy in cells |
The signaling pathways involved in GBMs-induced autophagy
Autophagic cell death is sometimes called type II PCD, and some experts had come up with an opposite opinion that autophagy might not cause cell death, and autophagy is different from autophagic cell death. They said that autophagic cell death is the cell death caused by autophagy rather than the cell death that autophagy participates in. Moreover, they pointed out that if the inhibitor of autophagy cannot prevent cell death, it cannot be called autophagic cell death.80 Until now, the underlying mechanism of autophagy is far from clear. In addition, in the study of toxicity induced by GBMs, there are no detailed debates about whether autophagy can be called type II PCD. Furthermore in most situations, autophagy is taken as one kind of cell death or the main reason of cell death. GBMs always induced unrestrained autophagy, which would accelerate rather than prevent cell death.
Autophagy is characterized by the formation of autophagosomes, with double membrane-bound structures wrapping organelles and cytoplasmic macromolecules that subsequently fuse with lysosomes to form autolysosomes, thereby degrading the contents of the encapsulated vacuole.81,82 mTOR is the checkpoint at the initiation step of autophagy. Autophagy is initiated by negatively regulating the PI3K/Akt pathway and mTOR activity; however, Bcl-2 can bind to Beclin-1 and reverse this outcome. Moreover, the activated AMPK can induce autophagy through a phosphorylation event that stimulates TSC1/TSC2 activity, and the TSC1/TSC2 complex can negatively regulate mTORC1. In contrast, the phosphorylation and inactivation of TSC1/TSC2 allow for mTOR activation via the PI3K/Akt signaling pathway inhibiting autophagy.83–86 Many signaling pathways of autophagy have been verified in diseases, but those pathways need to be further studied in biocompatibility and toxicology of GBMs.
The classic process of autophagy is autophagosome accumulation and LC3-I/LC3-II conversion. Nascent LC3 is modified at its c-terminal domains by Atg4 and becomes LC3-I, which subsequently couples with phosphatidylethanolamine to become LC3-II. LC3-II then translocates to the autophagosome membrane,87–89 and the short hairpin RNA against LC3-II can block the autophagic response.47 GO induced autophagosome accumulation and lysosome impairment disrupting intracellular homeostasis and promoted cell death after GBMs treatment in primary murine peritoneal macrophages and other cell lines.90,91 GO overload on lysosomal vesicles caused lysosomal membrane destabilization and significant lysosome fusion, which consequently resulted in the blockade of autophagosomes and autophagosome accumulation.81 It was elicited that lysosomal membrane permeabilization, lysosomal membrane rupture, and cytoskeleton disruption might be involved in lysosomal membrane destabilization, eventually resulting in autophagosome accumulation.92 GO also leads to a decrease of p62, a protein that directly binds to LC3 and selectively incorporates into autophagosomes.93 Moreover, p62 is preferentially degraded by autolysosomes; this degradation process is inversely correlated with autophagosomal maturation and degradation.94 However, a study reported that GO inhibited the maturation of autolysosomes and prevented the fusion between autophagosomes and lysosomes, thereby reducing the integrity of autophagic flux.95 GBMs led to different consequences of autophagy depending on the various concentrations, exposure times, and particle sizes.
Mitochondrial dysfunction and ER stress play vital roles in GBMs-induced autophagy. GBMs have been shown to induce mitochondrial damage and provoke autophagy in a human bronchial epithelial cell line (BEAS-2B cells).79 ROS and NO generation after GNPs treatment resulted in decreased numbers of mitochondria, ER, and Golgi apparatus, and mitochondrial fission-initiated mitochondrial autophagy.96 GNPs dramatically increased the levels of p62 and Bcl-2 proteins to induce autophagy.79 The adaptor protein p62 can bind to ubiquitinated mitochondrial proteins and LC3 on autophagosomes, recruiting autophagic membranes for mitochondrial clearance.97 Furthermore, Bcl-2 and Bcl-xL are antiapoptotic proteins that bind to Beclin-1, and the dissociation of the Bcl-2–Beclin-1 complex is essential for the initiation of autophagy.98 Moreover, the dissociation of the Bcl-2–Beclin-1 complex allows Beclin-1 to be activated by AMBRA1, a protein that may reside on mitochondria and bind to Bcl-2 until it is released by autophagic stimuli.99 Autophagy selectively removed mitochondria and adjusted the number of mitochondria to supply energy, adapting to the cell survival environment after GBMs exposure.
Different TLRs such as TLR-2, TLR-3, TLR-4, and TLR-7 are proposed as autophagy inductors,100–102 and some TLR signaling pathways participate in GO-induced autophagy. GO-activated autophagy is mediated through TLR-4/9 signaling pathways associated with the downstream signaling proteins MyD88, TRAF6, and NF-κB.75 GO binds to TLRs activating MyD88 or TRIF, and recruiting the downstream proteins TRAF6 and Beclin-1. Beclin-1 can interact with TRAF6 to accelerate its activation and the formation of autophagosomes.101,103 Similarly, GO can be phagocytosed by CT26 colon cancer cells, triggering autophagy by both TLR-4 and TLR-9 signaling cascades. TLRs transduce signals first by MyD88, followed by TRAF6, NF-κB, and IRF7, eventually resulting in various cellular responses including secretion of cytokines TNF-α, IL-1β, and IFNs.77 Except for TLR-4/9, the other TLRs have not been well studied in GBM-induced autophagy. It can be forecasted that the other TLRs may attract a lot of attention in the future.
Other signaling pathways are also involved in GBMs-induced autophagy. The p38 MAPK and NF-κB pathways participate in GQD-induced autophagy in THP-1 macrophages. GQDs significantly increase the phosphorylation of p38 MAPK and p65 and promote NF-κB translocation to the nucleus.26 The overexpression or activation of p38α disrupts the p38–mAtg9 interaction and thereby influences LC3 lipidation and p62 degradation, thus controlling the induction of autophagy.104 Moreover, mTORC1 is a major checkpoint in autophagy, and the activation or inhibition of mTOR pathway has been studied in other diseases or nanomaterials that may be investigated in future studies of GBM-induced PCD. Some pathways of GBM-induced autophagy are demonstrated in Figure 2.
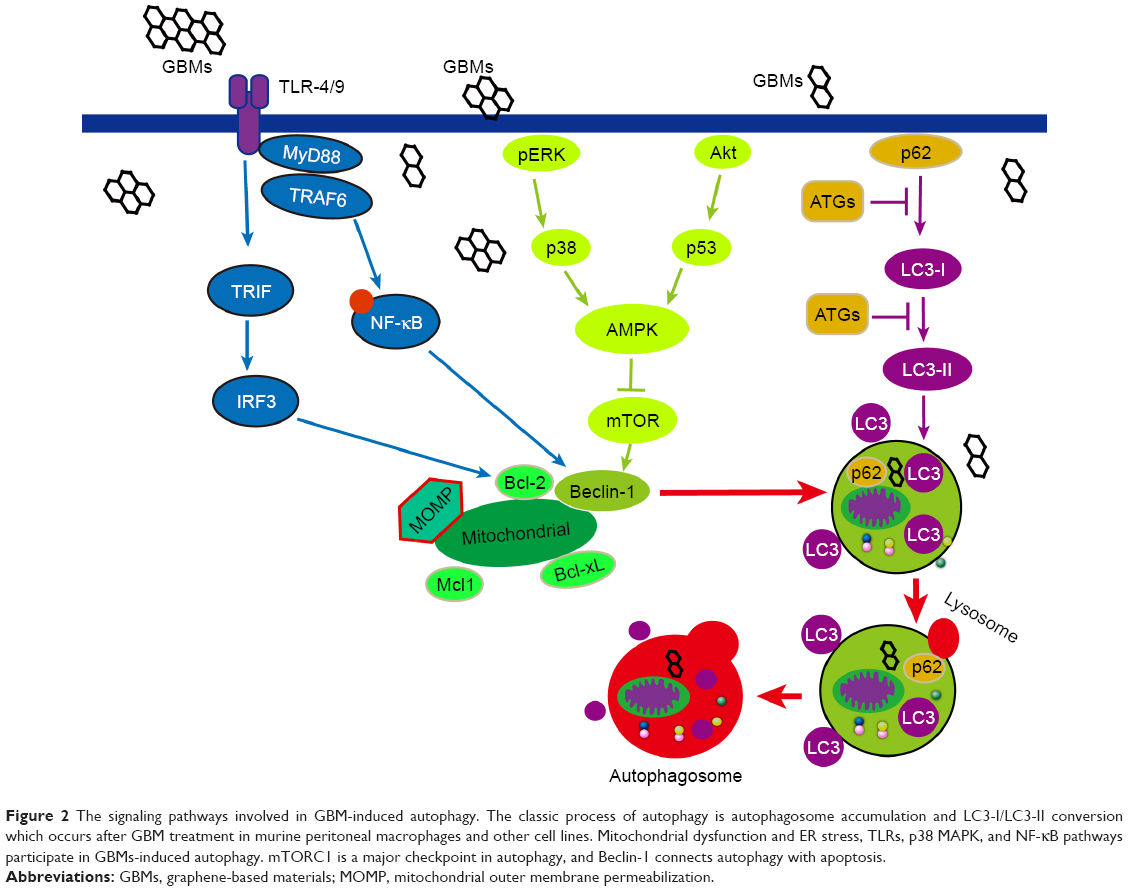 | Figure 2 The signaling pathways involved in GBM-induced autophagy. The classic process of autophagy is autophagosome accumulation and LC3-I/LC3-II conversion which occurs after GBM treatment in murine peritoneal macrophages and other cell lines. Mitochondrial dysfunction and ER stress, TLRs, p38 MAPK, and NF-κB pathways participate in GBMs-induced autophagy. mTORC1 is a major checkpoint in autophagy, and Beclin-1 connects autophagy with apoptosis. |
GBMs induce necroptosis and relative pathways involved
There are fewer studies of GBM-induced necroptosis compared to apoptosis and autophagy, and the mechanisms of necroptosis are not well studied. With the discovery of key regulators of necrotic death, such as RIP kinases and PARP, the concept of programmed necrosis has recently gained ground. RIP3 phosphorylation forms a tight RIP1–RIP3 complex, which is often needed to initiate the necrotic programme.105 The results of previous studies have demonstrated that GO/CDDP potentiates CT26 cell death through programmed necrosis when the cells are exposed to a concentration of 50 mg/mL for 24 h. GO/CDDP induces programmed necrosis by increasing the levels of RIP1, RIP3, and HMGB1, a nuclear protein that is released into cytosol and then released from cells after the initiation of necrosis.78
Programmed necrosis, type III PCD, is a crucial type of cell death for the preservation of tissue homoeostasis, the elimination of damaged cells, and immune responses.106,107 Signs of programmed necrosis include cell swelling, organelle dysfunction, and cell lysis.108,109 Although necrosis has always been considered an accidental form of cell death, the concept of programmed necrosis has been universally recognized since the discovery of key mediators of programmed necrosis, such as RIP kinases and PARP.53 The death receptor (TNFR1) and TLRs can induce RIPK1- and RIPK3-dependent necrosis in the presence of FADD, caspase-8, TRADD, and TIR-related adaptor proteins. Furthermore, calcium-activated calpain can invoke JNK resulting in necroptosis.110,111
Programmed necrosis can also be activated by TLR-4 signaling in macrophages. GO (40 μg/mL)-induced programmed necrosis in macrophages occurs through a TLR-4-dependent signaling pathway and is partially TNF-dependent; this latter process also involves RIP-dependent and RIP-independent signaling pathways.27 GO disrupts the integrity of the cytoskeleton of macrophages and promotes ROS generation, which is critical to TLR-4-mediated macrophagic necrosis. Blocking TLR-4 signaling with a selective inhibitor or using TLR-4-deficient BMDMs leads to resistance against GO-triggered necrosis. Moreover, GO increases TNF-α secretion upon activation of the TLR-4 pathway, and TNF-α triggers programmed necrosis via the activation of the kinases RIP1 and RIP3 or other signaling pathways. The study of the mechanism underlying GO-mediated programmed necrosis is just a good beginning, the outcome the relative mechanism is far from clear and complete. Other programmed necrosis pathways involved in the diseases have not been discussed in the study of GBMs. Some of the pathways of GO-triggered programmed necrosis are displayed in Figure 3.
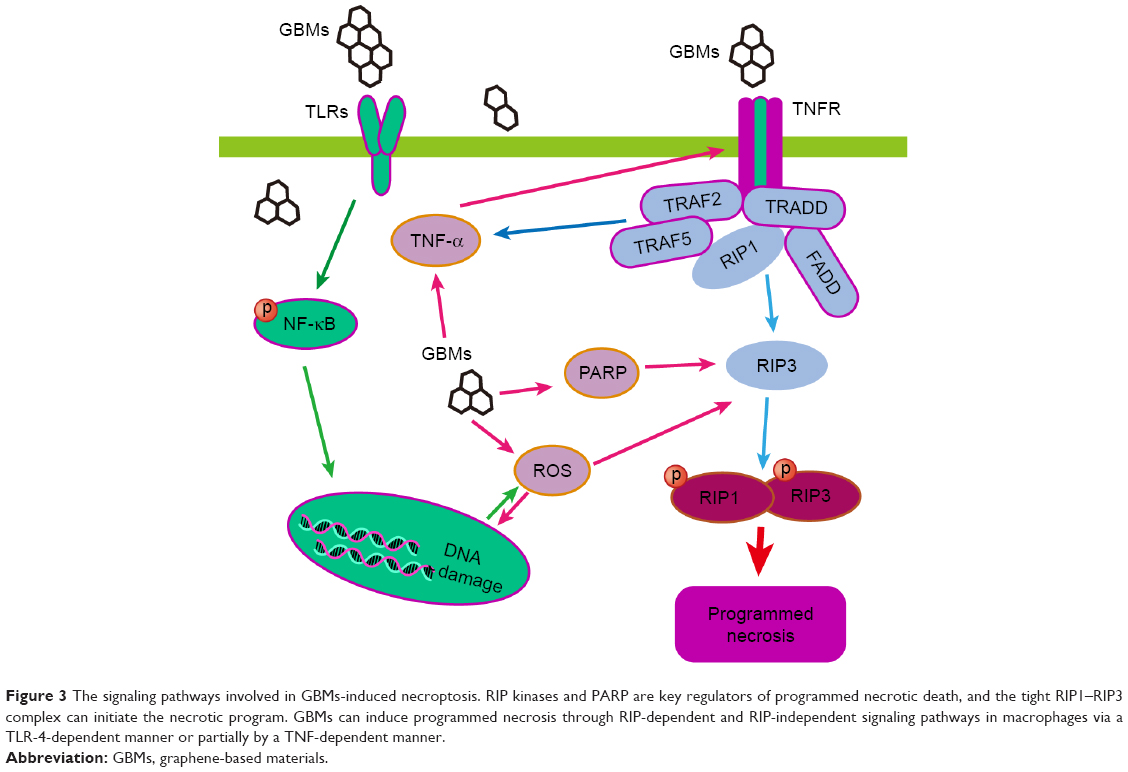 | Figure 3 The signaling pathways involved in GBMs-induced necroptosis. RIP kinases and PARP are key regulators of programmed necrotic death, and the tight RIP1–RIP3 complex can initiate the necrotic program. GBMs can induce programmed necrosis through RIP-dependent and RIP-independent signaling pathways in macrophages via a TLR-4-dependent manner or partially by a TNF-dependent manner. |
Some differences and relationships of GBMs-induced PCDs
Differences in PCD
From the discussion above, we find that we cannot sum up the law that GBMs induced what kinds of PCDs under certain circumstance. Different modes of PCDs appear in the same cells, which may be related to the cell phenotype, the physicochemical property and exposure time of GBMs, etc.112 Often, several kinds of PCDs coexist in the same cell; however, we have not yet studied which kinds of PCDs come first in research of GBMs, nor do we try to understand how GBMs induce several PCDs and maintain the balance. All of these queries may be answered in the future.
PCDs are distinguished by some characteristics. Apoptosis is morphologically characterized by cell shrinkage, chromatin aggregation, DNA fragmentation, and apoptotic body formation. In apoptosis, the early degradation of cytoskeleton occurs, but organelles degradation is preserved until the later stages. By contrast, autophagy occurs in the absence of chromatin condensation with massive autophagic vacuolization in the cytoplasm.81,113 Thus, the early disruption of organelles occurs, but the cytoskeleton is preserved until the late stage in the process of autophagy. Necroptosis is another morphologically and mechanistically distinct modality of cell death, characterized by mitochondrial lysis and lysosomal membrane rupture. In contrast to apoptosis and autophagy, necroptosis is characterized by a significant inflammatory response.114 Furthermore, unlike apoptosis which occurs in a caspase-dependent manner, necroptosis is caspase-independent. The phosphorylation and formation of RIP1 and RIP3 which only occur in necroptosis are specific indicators of necroptosis.105,115
Several cross-linked pathways in PCD
Although apoptosis, autophagy, and necroptosis have different characteristics, their signaling pathways have certain correlations with each other. For instance, the suppression of apoptosis can trigger autophagy,116 while the absence of the caspase signaling pathway can trigger programmed necrosis; however, a specific inhibitor of programmed necrosis, Nec-1, can revert the death pathway to apoptosis.53 From studies of other diseases, we conclude that the same signaling pathway plays different roles in different PCD processes and that different signaling pathways cross-link with each other to activate the same downstream effector molecules.
Biologists have investigated how apoptosis, autophagy, and necroptosis are interconnected.53 Apoptosis and autophagy are two primary PCD mechanisms that interact with each other and often occur in the same cells.117 Apoptosis and autophagy can share key molecular regulators, such as p53, Akt, JNK, and Beclin-1.118 Apoptosis can suppress autophagy partly via the caspase kinase-mediated cleavage of essential autophagy proteins.119 Autophagy stimulates apoptosis by depleting endogenous inhibitors or attenuates apoptosis by selectively reducing proapoptotic proteins.120 Apoptosis, autophagy, and necroptosis are initiated by a combination of related death receptors and ligands. For example, apoptosis and necrosis appear to be regulated by different forms of caspase-8: a fully processed, heterotetrameric form of caspase-8 induces apoptosis; however, caspase-8 mutants and caspase-8 inhibition activate necroptosis in some cell lines.106,121 Autophagy can trigger or block necroptosis in response to starvation and antigen stimulation in several cell lines.21,122–124
Oxidative stress is involved in the toxic effects of GBMs. The generation of ROS is considered the primary cause of cell death.31,34 The increased ROS generation leads to apoptosis and autophagy with an increase in the expression levels of apoptotic and autophagic effectors, such as caspase-3, caspase-9, Beclin-1, Bax, Bad, and LC3-I/II, after exposure to GBMs.26,27 ROS induces the loss of the mitochondrial membrane that potentially initiates both apoptosis and necrosis.125,126
Numerous signaling pathways comprise an interconnected network of apoptosis, autophagy, and necroptosis, such as the TLRs, p53 signaling pathways, and the Beclin-1 and Bcl-2 interaction. It is known that apoptosis can be induced via TLR-2, TLR-6, and TLR-9 pathways accompanied by the downstream proteins NIK and IκB kinase in GBMs-mediated toxicity.66,127–129 Interestingly, autophagy is elicited through the TLR-4 and TLR-9 pathways accompanied by the downstream adaptor proteins, MyD88, TRIF, and TRAF6, which were activated in RAW 264.7 macrophage cells and CT26 colon cancer cells after exposure to GO.75,77 Moreover, programmed necrosis can also be triggered via the TLR-4 pathway, so TLR-4-deficient BMDMs or a selective TLR-4 inhibitor CLI-095 constrains the effects of GO triggering programmed necrosis.27 p53, a vital regulator of apoptosis, can modulate the MOMP and thereby govern the Bcl-2 family to trigger apoptosis.130 In addition, p53 negatively controls autophagy through the AMPK/mTOR-dependent pathway by inhibiting mTOR via the activation of AMPK or the transactivation of DRAM.131,132 p53 is also involved in the regulation of necrosis.133–136 The functional and structural interactions between Beclin-1 and the antiapoptotic proteins Bcl-2 and Bcl-xL participate to some degree in the cross-linking of apoptosis, autophagy, and necroptosis.98,106,137,138
Collectively, studies have indicated that apoptosis, autophagy, and necroptosis are independent but interconnected by similar pathways and share initiator or effector molecules. A part of the interrelationship of apoptosis, autophagy, and programmed necrosis is shown in Figure 4.
 | Figure 4 The interrelationships between programmed cell death. The schematic diagram depicts that apoptosis, autophagy, and necroptosis are independent but interconnected. They are often mediated by similar pathways, such as the TLR signaling pathway, the Beclin-1 and Bcl-2 interaction, and the p53 signaling pathway, and share initiator or effector molecules, comprising a complicated and delicate network. |
Summary
After summarizing and analyzing hundreds of articles on the toxic study of GBMs, we found that one or two or more kinds of PCDs occur simultaneously or successively in the same cells, and different types of cell deaths emerge simultaneously or successively depending on the physicochemical property, exposure dose, and exposure time of GBMs (these factors are not the focus of this review). The molecular mechanisms of PCD are multifaceted, complex, and interconnected, and the mechanism of PCD induced by GBMs is far from clear and complete. Understanding the mechanisms of PCD and relationships between apoptosis, autophagy, and programmed necrosis may benefit to figure out further the mechanism of GBMs-induced toxicity. The cognition of autophagy and apoptosis was limited by the experimental conditions and experimental methods years ago. The experimental techniques are improving over time, and our cognition of PCD is improving. Some original conclusions may need to be revised, redefined, and changed. Studies of GBMs-induced necroptosis are limited, and hence, more concerns are needed.
The role of autophagy in cell death should be carefully considered. Some researchers believed that autophagy may more likely be a kind of regulation mechanism, related to apoptosis or necrosis pathway components. The instinct of autophagy to remove the damaged proteins and organelles is of great significance for cell survival. Autophagy may make the last desperate efforts for survival in the dying cells.139 Some studies have suggested that autophagy occurs ahead of apoptosis and necroptosis, and autophagy can promote or inhibit apoptosis/necroptosis. The ultimate goal of autophagy restraining apoptosis and necrosis may be to maintain cell survival. Once it cannot maintain survival, autophagy must promote cell death in a reasonable way to help maintain local homeostasis.140,141 From the study on GBMs toxicity, we can identify that the duty of autophagy is to degrade the intracellular proteins and abnormal cellular components, which may ultimately lead to cell death. However, there is no enough evidence to prove that autophagic cell death occurs through autophagosome accumulation, and hence, more work is needed to distinguish the autophagic cell death induced by GBMs.
To clarify the mechanisms of GBM-induced PCD is beyond the scope of this review. However, the insights regarding GBM-induced PCD could be harnessed for more reliable assessments of the health effects of GBMs. The dose and time of occupational exposure of GBMs is huge, and hence, relative studies are not enough. Moreover, since GBMs can induce PCD in cancer cells, the application of GBMs in therapy will benefit to curing tumors/cancers combined with their powerful drug delivery function. However, we need to pay more attention to this study approach.
Abbreviations
BMDMs, bone marrow-derived macrophages; GBMs, graphene-based materials; GNPs, graphene nanoplatelets; GO, graphene oxide; GQDs, graphene quantum dots; MMP, mitochondrial membrane potential; MOMP, mitochondrial outer membrane permeabilization; PCD, programmed cell death; rGO, reduced graphene oxide.
Acknowledgment
This review was supported by the National Natural Science Foundation of China (81550011, 51172283, 81400557), Natural Science Foundation of Guangdong Province (2015A030313299), Guangdong Provincial Medical Research Foundation (A2016360), and Science and Technology Planning Project of Guangdong Province (2016A010103017).
Disclosure
The authors report no conflicts of interest in this work.
References
Yang K, Feng L, Liu Z. Stimuli responsive drug delivery systems based on nano-graphene for cancer therapy. Adv Drug Deliv Rev. 2016;105(Pt B):228–241. | ||
Hu D, Zhang J, Gao G, Sheng Z, Cui H, Cai L. Indocyanine green-loaded polydopamine-reduced graphene oxide nanocomposites with amplifying photoacoustic and photothermal effects for cancer theranostics. Theranostics. 2016;6(7):1043–1052. | ||
Battogtokh G, Ko YT. Graphene oxide-incorporated pH-responsive folate-albumin-photosensitizer nanocomplex as image-guided dual therapeutics. J Control Release. 2016;234:10–20. | ||
Teixeira SR, Lloyd C, Yao S, et al. Polyaniline-graphene based α-amylase biosensor with a linear dynamic range in excess of 6 orders of magnitude. Biosens Bioelectron. 2016;85:395–402. | ||
Shirai A, Henares TG, Sueyoshi K, Endo T, Hisamoto H. Fast and single-step immunoassay based on fluorescence quenching within a square glass capillary immobilizing graphene oxide-antibody conjugate and fluorescently labelled antibody. Analyst. 2016;141(11):3389–3394. | ||
Lin J, Chen X, Huang P. Graphene-based nanomaterials for bioimaging. Adv Drug Deliv Rev. 2016;105(Pt B):242–254. | ||
Weng X, Neethirajan S. A microfluidic biosensor using graphene oxide and aptamer-functionalized quantum dots for peanut allergen detection. Biosens Bioelectron. 2016;85:649–656. | ||
Ali Tahir A, Ullah H, Sudhagar P, Asri Mat Teridi M, Devadoss A, Sundaram S. The application of graphene and its derivatives to energy conversion, storage, and environmental and biosensing devices. Chem Rec. 2016;16(3):1591–1634. | ||
McCallion C, Burthem J, Rees-Unwin K, Golovanov A, Pluen A. Graphene in therapeutics delivery: problems, solutions and future opportunities. Eur J Pharm Biopharm. 2016;104:235–250. | ||
Liu H, Li T, Liu Y, Qin G, Wang X, Chen T. Glucose-reduced graphene oxide with excellent biocompatibility and photothermal efficiency as well as drug loading. Nanoscale Res Lett. 2016;11(1):211. | ||
Sanchez VC, Jachak A, Hurt RH, Kane AB. Biological interactions of graphene-family nanomaterials: an interdisciplinary review. Chem Res Toxicol. 2012;25(1):15–34. | ||
Gurunathan S, Kim JH. Synthesis, toxicity, biocompatibility, and biomedical applications of graphene and graphene-related materials. Int J Nanomedicine. 2016;11:1927–1945. | ||
Orecchioni M, Ménard-Moyon C, Delogu LG, Bianco A. Graphene and the immune system: challenges and potentiality. Adv Drug Deliv Rev. 2016;105(Pt B):163–175. | ||
Lalwani G, D’Agati M, Khan AM, Sitharaman B. Toxicology of graphene-based nanomaterials. Adv Drug Deliv Rev. 2016;105(Pt B):109–144. | ||
Zhang B, Wei P, Zhou Z, Wei T. Interactions of graphene with mammalian cells: molecular mechanisms and biomedical insights. Adv Drug Deliv Rev. 2016;105(Pt B):145–162. | ||
Horváth L, Magrez A, Burghard M, Kern K, Forró L, Schwaller B. Evaluation of the toxicity of graphene derivatives on cells of the lung luminal surface. Carbon. 2013;64(11):45–60. | ||
Chong Y, Ma Y, Shen H, et al. The in vitro and in vivo toxicity of graphene quantum dots. Biomaterials. 2014;35(19):5041–5048. | ||
Singh Z. Applications and toxicity of graphene family nanomaterials and their composites. Nanotechnol Sci Appl. 2016;9:15–28. | ||
Patlolla AK, Randolph J, Kumari SA, Tchounwou PB. Toxicity evaluation of graphene oxide in kidneys of Sprague-Dawley rats. Int J Environ Res Public Health. 2016;13(4):380. | ||
Lewinski N, Colvin V, Drezek R. Cytotoxicity of nanoparticles. Small. 2008;4(1):26–49. | ||
Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19(1):107–120. | ||
Andón FT, Fadeel B. Programmed cell death: molecular mechanisms and implications for safety assessment of nanomaterials. Acc Chem Res. 2013;46(3):733–742. | ||
Liu J, Feng X, Wei L, Chen L, Song B, Shao L. The toxicology of ion-shedding zinc oxide nanoparticles. Crit Rev Toxicol. 2016;46(4):348–384. | ||
Stern ST, Johnson DN. Role for nanomaterial-autophagy interaction in neurodegenerative disease. Autophagy. 2008;4(8):1097–1100. | ||
Bhattacharya S, Mishra S, Gupta P, et al. Liquid phase collagen modified graphene that induces apoptosis. RSC Adv. 2015;5:44447–44457. | ||
Qin Y, Zhou ZW, Pan ST, et al. Graphene quantum dots induce apoptosis, autophagy, and inflammatory response via p38 mitogen-activated protein kinase and nuclear factor-kappaB mediated signaling pathways in activated THP-1 macrophages. Toxicology. 2015;327:62–76. | ||
Qu G, Liu S, Zhang S, et al. Graphene oxide induces toll-like receptor 4 (TLR4)-dependent necrosis in macrophages. ACS Nano. 2013;7(7):5732–5745. | ||
Ou L, Song B, Liang H, et al. Toxicity of graphene-family nanoparticles: a general review of the origins and mechanisms. Part Fibre Toxicol. 2016;13(1):57. | ||
Yang K, Li Y, Tan X, Peng R, Liu Z. Behavior and toxicity of graphene and its functionalized derivatives in biological systems. Small. 2013;9(9–10):1492–1503. | ||
Seabra AB, Paula AJ, de Lima R, Alves OL, Durán N. Nanotoxicity of graphene and graphene oxide. Chem Res Toxicol. 2014;27(2):159–168. | ||
Jarosz A, Skoda M, Dudek I, Szukiewicz D. Oxidative stress and mitochondrial activation as the main mechanisms underlying graphene toxicity against human cancer cells. Oxid Med Cell Longev. 2016;2016:5851035. | ||
Bialik S, Zalckvar E, Ber Y, Rubinstein AD, Kimchi A. Systems biology analysis of programmed cell death. Trends Biochem Sci. 2010;35(10):556–564. | ||
Tan ML, Ooi JP, Ismail N, Moad AI, Muhammad TS. Programmed cell death pathways and current antitumor targets. Pharm Res. 2009;26(7):1547–1560. | ||
Chatterjee N, Eom HJ, Choi J. A systems toxicology approach to the surface functionality control of graphene-cell interactions. Biomaterials. 2014;35(4):1109–1127. | ||
Zhang W, Yan L, Li M, et al. Deciphering the underlying mechanisms of oxidation-state dependent cytotoxicity of graphene oxide on mammalian cells. Toxicol Lett. 2015;237(2):61–71. | ||
Hatamie S, Akhavan O, Sadrnezhaad SK, Ahadian MM, Shirolkar MM, Wang HQ. Curcumin-reduced graphene oxide sheets and their effects on human breast cancer cells. Mater Sci Eng C Mater Biol Appl. 2015;55:482–489. | ||
Zhang Y, Ali SF, Dervishi E, et al. Cytotoxicity effects of graphene and single-wall carbon nanotubes in neural phaeochromocytoma-derived PC12 cells. ACS Nano. 2010;4(6):3181–3186. | ||
Mbeh DA, Akhavan O, Javanbakht T, Mahmoudi M, Yahia LH. Cytotoxicity of protein corona-graphene oxide nanoribbons on human epithelial cells. Appl Surf Sci. 2014;320:596–601. | ||
Ding Z, Zhang Z, Ma H, Chen Y. In vitro hemocompatibility and toxic mechanism of graphene oxide on human peripheral blood T lymphocytes and serum albumin. ACS Appl Mater Interfaces. 2014;6(22):19797–19807. | ||
Orecchioni M, Bedognetti D, Sgarrella F, Marincola FM, Bianco A, Delogu LG. Impact of carbon nanotubes and graphene on immune cells. J Transl Med. 2014;12:138. | ||
Jeong J, Cho HJ, Choi M, Wang SL, Chung BH, Lee JS. In vivo toxicity assessment of angiogenesis and the live distribution of nano-graphene oxide and its PEGylated derivatives using the developing zebrafish embryo. Carbon. 2015;93:431–440. | ||
Zhao Y, Wu Q, Wang D. An epigenetic signal encoded protection mechanism is activated by graphene oxide to inhibit its induced reproductive toxicity in Caenorhabditis elegans. Biomaterials. 2016;79:15–24. | ||
Ren C, Hu X, Li X, Zhou Q. Ultra-trace graphene oxide in a water environment triggers Parkinson’s disease-like symptoms and metabolic disturbance in zebrafish larvae. Biomaterials. 2016;93:83–94. | ||
Chan KT, Meng FY, Li Q, et al. Cucurbitacin B induces apoptosis and S phase cell cycle arrest in BEL-7402 human hepatocellular carcinoma cells and is effective via oral administration. Cancer Lett. 2010;294(1):118–124. | ||
Gurunathan S, Han JW, Kim ES, Park JH, Kim JH. Reduction of graphene oxide by resveratrol: a novel and simple biological method for the synthesis of an effective anticancer nanotherapeutic molecule. Int J Nanomedicine. 2015;10:2951–2969. | ||
Jaworski S, Sawosz E, Kutwin M, et al. In vitro and in vivo effects of graphene oxide and reduced graphene oxide on glioblastoma. Int J Nanomedicine. 2015;10:1585–1596. | ||
Markovic ZM, Ristic BZ, Arsikin KM, et al. Graphene quantum dots as autophagy-inducing photodynamic agents. Biomaterials. 2012;33(29):7084–7092. | ||
Habiba K, Encarnacion-Rosado J, Garcia-Pabon K, et al. Improving cytotoxicity against cancer cells by chemo-photodynamic combined modalities using silver-graphene quantum dots nanocomposites. Int J Nanomedicine. 2016;11:107–119. | ||
Nishida K, Yamaguchi O, Otsu K. Crosstalk between autophagy and apoptosis in heart disease. Circ Res. 2008;103(4):343–351. | ||
Saptarshi SR, Duschl A, Lopata AL. Biological reactivity of zinc oxide nanoparticles with mammalian test systems: an overview. Nanomedicine (Lond). 2015;10(13):2075–2092. | ||
Eum KH, Lee M. Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem. 2011;348(1–2):61–68. | ||
Meier P, Vousden KH. Lucifer’s labyrinth – ten years of path finding in cell death. Mol Cell. 2007;28(5):746–754. | ||
Ouyang L, Shi Z, Zhao S, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45(6):487–498. | ||
Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta. 2012;1824(1):22–33. | ||
Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55(3):178–194. | ||
Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998;281(5384):1860–1863. | ||
Sasidharan A, Swaroop S, Chandran P, Nair S, Koyakutty M. Cellular and molecular mechanistic insight into the DNA-damaging potential of few-layer graphene in human primary endothelial cells. Nanomedicine. 2016;12(5):1347–1355. | ||
Choi YJ, Kim E, Han J, Kim JH, Gurunathan S. A novel biomolecule-mediated reduction of graphene oxide: a multifunctional anti-cancer agent. Molecules. 2016;21(3):375. | ||
Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. | ||
Vaux DL. Apoptogenic factors released from mitochondria. Biochim Biophys Acta. 2011;1813(4):546–550. | ||
Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46(8):497–510. | ||
Li Y, Liu Y, Fu Y, et al. The triggering of apoptosis in macrophages by pristine graphene through the MAPK and TGF-beta signaling pathways. Biomaterials. 2012;33(2):402–411. | ||
Markovic ZM, Harhaji-Trajkovic LM, Todorovic-Markovic BM, et al. In vitro comparison of the photothermal anticancer activity of graphene nanoparticles and carbon nanotubes. Biomaterials. 2011;32(4):1121–1129. | ||
Pinto AM, Gonçalves IC, Magalhães FD. Graphene-based materials biocompatibility: a review. Colloids Surf B Biointerfaces. 2013;111:188–202. | ||
Russier J, Treossi E, Scarsi A, et al. Evidencing the mask effect of graphene oxide: a comparative study on primary human and murine phagocytic cells. Nanoscale. 2013;5(22):11234–11247. | ||
Reshma SC, Syama S, Mohanan PV. Nano-biointeractions of PEGylated and bare reduced graphene oxide on lung alveolar epithelial cells: a comparative in vitro study. Colloids Surf B Biointerfaces. 2016;140:104–116. | ||
Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17(1–4):287–296. | ||
Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. | ||
Zhou S, Wang Y, Zhu JJ. Simultaneous detection of tumor cell apoptosis regulators Bcl-2 and Bax through a dual-signal-marked electrochemical immunosensor. ACS Appl Mater Interfaces. 2016;8(12):7674–7682. | ||
Dudek I, Skoda M, Jarosz A, Szukiewicz D. The molecular influence of graphene and graphene oxide on the immune system under in vitro and in vivo conditions. Arch Immunol Ther Exp (Warsz). 2016;64(3):195–215. | ||
Ramesh S, Wildey G, Howe PH. Transforming growth factor beta (TGFbeta)-induced apoptosis: the rise & fall of Bim. Cell Cycle. 2009;8(1):11–17. | ||
Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial–mesenchymal transition. Mol Biol Cell. 2012;23(5):781–791. | ||
Franco DL, Mainez J, Vega S, et al. Snail1 suppresses TGF-beta-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J Cell Sci. 2010;123(Pt 20):3467–3477. | ||
Zhu B, Zhai J. Prohibitin regulates TGF-beta induced apoptosis as a downstream effector of Smad-dependent and -independent signaling. Prostate. 2010;70(1):17–26. | ||
Chen GY, Yang HJ, Lu CH, et al. Simultaneous induction of autophagy and toll-like receptor signaling pathways by graphene oxide. Biomaterials. 2012;33(27):6559–6569. | ||
Liu Y, Wang X, Wang J, et al. Graphene oxide attenuates the cytotoxicity and mutagenicity of PCB 52 via activation of genuine autophagy. Environ Sci Technol. 2016;50(6):3154–3164. | ||
Chen GY, Chen CL, Tuan HY, et al. Graphene oxide triggers toll-like receptors/autophagy responses in vitro and inhibits tumor growth in vivo. Adv Healthc Mater. 2014;3(9):1486–1495. | ||
Chen GY, Meng CL, Lin KC, et al. Graphene oxide as a chemosensitizer: diverted autophagic flux, enhanced nuclear import, elevated necrosis and improved antitumor effects. Biomaterials. 2015;40:12–22. | ||
Park EJ, Lee GH, Han BS, et al. Toxic response of graphene nanoplatelets in vivo and in vitro. Arch Toxicol. 2015;89(9):1557–1568. | ||
Shimizu S, Yoshida T, Tsujioka M, Arakawa S. Autophagic cell death and cancer. Int J Mol Sci. 2014;15(2):3145–3153. | ||
Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9(3):217–222. | ||
Lin C, Tsai SC, Tseng MT, et al. AKT serine/threonine protein kinase modulates baicalin-triggered autophagy in human bladder cancer T24 cells. Int J Oncol. 2013;42(3):993–1000. | ||
Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18(23):2893–2904. | ||
Arsham AM, Neufeld TP. Thinking globally and acting locally with TOR. Curr Opin Cell Biol. 2007;18(6):589–597. | ||
Budanov A, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134(3):451–460. | ||
Chan EY, Longatti A, Mcknight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29(1):157–171. | ||
Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. | ||
Patel AS, Lin L, Geyer A, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One. 2012;7(7):L56–L69. | ||
Ma X, Wu Y, Jin S, et al. Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment. ACS Nano. 2011;5(11):8629–8639. | ||
Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283(33):22847–22857. | ||
Wan B, Wang ZX, Lv QY, et al. Single-walled carbon nanotubes and graphene oxides induce autophagosome accumulation and lysosome impairment in primarily cultured murine peritoneal macrophages. Toxicol Lett. 2013;221(2):118–127. | ||
Johnson-Lyles DN, Peifley K, Lockett S, et al. Fullerenol cytotoxicity in kidney cells is associated with cytoskeleton disruption, autophagic vacuole accumulation, and mitochondrial dysfunction. Toxicol Appl Pharmacol. 2010;248(3):249–258. | ||
Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10(3):431–441. | ||
Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. | ||
Rubinsztein DC, Cuervo AM, Ravikumar B, et al. In search of an “autophagomometer”. Autophagy. 2009;5(5):585–589. | ||
Roy A, Kolattukudy PE. Monocyte chemotactic protein-induced protein (MCPIP) promotes inflammatory angiogenesis via sequential induction of oxidative stress, endoplasmic reticulum stress and autophagy. Cell Signal. 2012;24(11):2123–2131. | ||
Thomas RL, Gustafsson AB. Mitochondrial autophagy – an essential quality control mechanism for myocardial homeostasis. Circ J. 2013;77(10):2449–2454. | ||
Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. | ||
Strappazzon F, Vietri-Rudan M, Campello S, et al. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 2011;30(7):1195–1208. | ||
Anand PK, Tait SW, Lamkanfi M, et al. TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation. J Biol Chem. 2011;286(50):42981–42991. | ||
Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3(123):ra42. | ||
Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27(7):1110–1121. | ||
Xu C, Liu J, Hsu LC, Luo Y, Xiang R, Chuang TH. Functional interaction of heat shock protein 90 and Beclin 1 modulates Toll-like receptor-mediated autophagy. FASEB J. 2011;25(8):2700–2710. | ||
Webber JL. Regulation of autophagy by p38alpha MAPK. Autophagy. 2010;6(2):292–293. | ||
Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. | ||
Han J, Zhong CQ, Zhang DW. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol. 2011;12(12):1143–1149. | ||
Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2013;9(3):112–119. | ||
Wu W, Liu P, Li J. Necroptosis: an emerging form of programmed cell death. Crit Rev Oncol Hematol. 2012;82(3):249–258. | ||
Mccall K. Genetic control of necrosis – another type of programmed cell death. Curr Opin Cell Biol. 2010;22(6):882–888. | ||
Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135(7):1161–1163. | ||
Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32(1):37–43. | ||
Patschan S, Goligorsky MS. Autophagy: the missing link between non-enzymatically glycated proteins inducing apoptosis and premature senescence of endothelial cells? Autophagy. 2008;4(4):521–523. | ||
Li JJ, Hartono D, Ong CN, Bay BH, Yung LY. Autophagy and oxidative stress associated with gold nanoparticles. Biomaterials. 2010;31(23):5996–6003. | ||
Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11. | ||
He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–1111. | ||
Amelio I, Melino G, Knight RA. Cell death pathology: cross-talk with autophagy and its clinical implications. Biochem Biophys Res Commun. 2011;414(2):277–281. | ||
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752. | ||
Fimia GM, Piacentini M. Regulation of autophagy in mammals and its interplay with apoptosis. Cell Mol Life Sci. 2010;67(10):1581–1588. | ||
Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. | ||
Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. | ||
Peter ME. Programmed cell death: apoptosis meets necrosis. Nature. 2011;471(7338):310–312. | ||
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833(12):3448–3459. | ||
Bonapace L, Bornhauser BC, Schmitz M, et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest. 2010;120(4):1310–1323. | ||
Farkas T, Daugaard M, Jäättelä M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J Biol Chem. 2011;286(45):38904–38912. | ||
Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115(10):2679–2688. | ||
Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304(3):463–470. | ||
Kim AS, Chae CH, Kim J, Choi JY, Kim SG, Băciu̧t G. Silver nanoparticles induce apoptosis through the Toll-like receptor 2 pathway. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113(6):789–798. | ||
Chakraborty DC, Mukherjee G, Banerjee P, Banerjee KK, Biswas T. Hemolysin induces Toll-like receptor (TLR) independent apoptosis but multiple TLR associated parallel activation of macrophages. J Biol Chem. 2011;286(40):34542–34551. | ||
Lim EJ, Park DW, Lee JG, et al. Toll-like receptor 9-mediated inhibition of apoptosis occurs through suppression of FoxO3a activity and induction of FLIP expression. Exp Mol Med. 2010;42(10):712–720. | ||
Vaseva A, Marchenko N, Ji K, Tsirka S, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149(7):1536–1548. | ||
Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102(23):8204–8209. | ||
Tasdemir E, Maiuri MC, Galluzzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–687. | ||
Baumann K. Cell death: multitasking p53 promotes necrosis. Nat Rev Mol Cell Biol. 2012;13(8):480–481. | ||
Greay SJ, Ireland DJ, Kissick HT, et al. Induction of necrosis and cell cycle arrest in murine cancer cell lines by Melaleuca alternifolia (tea tree) oil and terpinen-4-ol. Cancer Chemother Pharmacol. 2010;65(5):877–888. | ||
Palanisamy AP, Cheng G, Sutter AG, et al. Mitochondrial uncoupling protein 2 induces cell cycle arrest and necrotic cell death. Metab Syndr Relat Disord. 2014;12(2):132–142. | ||
Franke S, Sommer M, Rüster C, et al. Advanced glycation end products induce cell cycle arrest and proinflammatory changes in osteoarthritic fibroblast-like synovial cells. Arthritis Res Ther. 2009;11(5):R136. | ||
Ono K, Wang X, Kim SO, Armstrong LC, Bornstein P, Han J. Metaxin deficiency alters mitochondrial membrane permeability and leads to resistance to TNF-induced cell killing. Protein Cell. 2010;1(2):161–173. | ||
Borkan SC. The role of BCL-2 family members in acute kidney injury. Semin Nephrol. 2016;36(3):237–250. | ||
Colell A, Ricci JE, Tait S, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129(5):983–997. | ||
Djavaheri-Mergny M, Amelotti M, Mathieu J, et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281(41):30373–30382. | ||
Sasnauskiene A, Kadziauskas J, Vezelyte N, Jonusiene V, Kirveliene V. Damage targeted to the mitochondrial interior induces autophagy, cell cycle arrest and, only at high doses, apoptosis. Autophagy. 2009;5(5):743–744. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.