Back to Archived Journals » Research and Reports in Biology » Volume 6
The maternal immune system during pregnancy and its influence on fetal development
Authors Morelli S, Mandal M, Goldsmith LT, Kashani BN, Ponzio NM
Received 9 January 2015
Accepted for publication 1 May 2015
Published 1 October 2015 Volume 2015:6 Pages 171—189
DOI https://doi.org/10.2147/RRB.S80652
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Zvi Kelman
Video abstract presented by Sara S Morelli.
Views: 28518
Sara S Morelli,1 Mili Mandal,2 Laura T Goldsmith,1 Banafsheh N Kashani,1 Nicholas M Ponzio3,4
1Department of Obstetrics, Gynecology and Women's Health, New Jersey Medical School, Rutgers University, Newark, 2Department of Pharmacology and Toxicology, Ernest Mario School of Pharmacy, Rutgers University, Piscataway, 3Department of Pathology and Laboratory Medicine, New Jersey Medical School, Rutgers University, Newark, 4Graduate School of Biomedical Sciences, Rutgers University, Newark, NJ, USA
Abstract: The maternal immune system plays a critical role in the establishment, maintenance, and completion of a healthy pregnancy. However, the specific mechanisms utilized to achieve these goals are not well understood. Various cells and molecules of the immune system are key players in the development and function of the placenta and the fetus. Effector cells of the immune system act to promote and yet limit placental development. The T helper 1 (Th1)/T helper 2 (Th2) immune shift during pregnancy is well established. A fine balance between proinflammatory and anti-inflammatory influences is required. We herein review the evidence regarding maternal tolerance of fetal tissues and the underlying cell-mediated immune and humoral (hormones and cytokines) mechanisms. We also note the many unanswered questions in our understanding of these mechanisms. In addition, we summarize the clinical manifestations of an altered maternal immune system during pregnancy related to susceptibility to common viral, bacterial, and parasitic infections, as well as to autoimmune diseases.
Keywords: maternal–fetal interface, immune system, fetal tolerance, lymphocyte subsets, decidua, pregnancy
Introduction
The relationship between mother and fetus has fascinated immunologists for decades. Survival of the semiallogeneic fetus was used by Billingham et al1 in 1953 as an example of immune tolerance to the fetus by the maternal immune system. Numerous hypotheses related to placental protection of the fetus, including expression (or lack of expression) of histocompatibility antigens on fetal tissues, maternal immune tolerance to fetal antigens, and inhibition and/or regulation of maternal antifetal immune responses have been put forth to explain the survival of the “immunogenic” fetus. Yet, the mechanisms still remain to be totally clarified.
Part of the difficulty in studying these mechanisms is due to the variation among species in which such investigations are conducted. Mice are used for many of these investigations because of their short gestational time, relatively lower cost, well-defined genetics (including mutant, transgenic, and knockout strains), and availability of a wide spectrum of antibodies and reagents to perform immunologic and molecular studies. However, differences in the reproductive system in general, and the feto–materno–placental unit in particular, as well as differences in the development and function of immune elements, often preclude direct extension of results observed in mice to humans. In contrast, studies designed to investigate such questions in humans are unethical, and studies incorporating nonhuman primates for these investigations raise similar moral issues and are also prohibitively expensive.
Therefore, our review is not designed to address all the unanswered questions surrounding the significance of the maternal immune system during pregnancy and its influence on fetal development. Rather, our goals are to identify the gaps in the knowledge and understanding about the topic from the published literature about various species and to acknowledge contexts wherein differences preclude a direct comparison with humans. Notwithstanding these differences however, investigations conducted in other species, such as rodents, do serve to identify possible strategies to address some of these unanswered questions.
Additionally, we take an interdisciplinary approach as coauthors who bring clinical and basic science perspectives and expertise in reproductive and immunological disciplines. Thus, we address topics related to definition of the maternal–fetal interface, as well as the significance of maternal immune responses in regulating key early events during both pregnancy (eg, implantation, angiogenesis, and vascular remodeling) and in development of the fetal immune system. We then review current understanding about maternal tolerance of fetal tissues and the underlying cellular and humoral immune mechanisms. Finally, we examine clinical manifestations of an altered maternal immune system during pregnancy related to susceptibility to certain viral, bacterial, and parasitic infections, as well as to autoimmune disorders.
Description and definition of the maternal–fetal interface
Maternal: decidua
In women, invasion by the trophoblast is extensive, encompassing the endometrium as well as the inner third of the myometrium.2 To accommodate this, a pronounced remodeling process must occur, involving multiple cellular compartments of the uterus in preparation for implantation and establishment and support of pregnancy. This process, decidualization, occurs in humans on a cyclic basis beginning in the midluteal phase of the menstrual cycle, independently of pregnancy. In contrast, in rodents and most other species, decidualization requires the presence of a blastocyst. The term maternal decidua thus refers to the uterine mucosal layer (endometrium) after it has undergone decidualization, the requisite and complex differentiation process involving the multiple cellular compartments of the endometrium in preparation for embryo implantation.
The parenchymal cellular compartments of the maternal decidua include the glandular epithelial compartment, the luminal epithelial compartment, the endothelium of the spiral arteries, and the decidualized stromal cells, all of which undergo dramatic transformation in preparation for pregnancy. The glandular epithelium acquires increased secretory activity under the influence of maternal progesterone.3 Dramatic remodeling of spiral arteries occurs during decidualization, discussed in greater detail later in this review. The endometrial stromal fibroblasts that undergo dramatic morphologic and biochemical differentiation in preparation for implantation and support of pregnancy become known as decidual cells, or decidualized stromal cells. Decidualized stromal cells no longer have the characteristic spindle shape of the endometrial stromal fibroblast and, instead, have acquired an epithelioid phenotype, characterized by progressive cell enlargement, rounding of the nucleus, and expansion of the rough endoplasmic reticulum and Golgi complex, all consistent with the transformation into a secretory cell.3 Major secretory products of decidualized stromal cells include prolactin and insulin-like growth-factor-binding protein-1, the hallmark proteins widely used as phenotypic markers of decidualization.4 These cells also secrete a number of cytokines and growth factors (eg, interleukin [IL]-11, epidermal growth factor [EGF], heparin-binding EGF-like growth factor), which further regulate the process of decidualization in an autocrine and/or paracrine manner.5
In addition to the parenchymal cellular compartments making up the maternal decidua, various populations of immune cells exist in the human endometrium throughout the menstrual cycle. In early pregnancy, leukocytes are abundant, comprising 30%–40% of all human decidual stromal compartment cells.6 The basalis layer of the human endometrium contains lymphoid aggregates composed of T-cells and a small number of B-cells. In the functionalis layer of the proliferative phase, few uterine natural killer (uNK) cells, T-cells, and macrophages are scattered throughout the stromal compartment.7 Although the numbers of T-cells and macrophages remain largely unchanged throughout the luteal phase and during the process of decidualization,7 there is a dramatic increase in the number of uNK cells postovulation, playing a critical role in preparation of the endometrium for pregnancy. With regard to decidual immune cell populations during early pregnancy, studies using flow cytometry and immunostaining of human tissues demonstrate that the majority of first-trimester human decidual leukocytes are uNK cells (~70%), followed by macrophages (~20%).8 T-cells make up approximately 10%–20% of decidual leukocytes, and dendritic cells (DCs) and B-cells are rare.8 As in humans, uNK cells are the predominant leukocyte population in the decidua of the rhesus macaque and the mouse, but studies to determine relative numbers of other leukocyte populations in murine decidua are lacking. The functions of each immune cell type at the maternal–fetal interface are discussed in more detail in this review, with a particular focus on uNK cells.
Fetal: placenta, fetal membranes (amnion and chorion)
Structurally, the interface between the uterine mucosa and the extraembryonic tissues is commonly referred to as the maternal–fetal interface. This is represented in Figure 1, which depicts the maternal immune cells and the fetal trophoblast.9
 | Figure 1 Schematic depiction of the human maternal–fetal interface including maternal immune cells such as uterine natural killer (uNK) cells, macrophages, (the predominant immune cell types) and T helper (Th) cells, T-cytotoxic (Tc) cells, dendritic cells, as well as invading trophoblast cells. |
Extraembryonic cells in direct contact with maternal cells are the trophoblast cells, derived from the trophectoderm layer surrounding the blastocyst. In women, invasion by the trophoblast into maternal spiral arteries substantially increases uterine blood flow, puts maternal blood in direct contact with fetal trophoblast cells, and ensures sufficient delivery of maternal nutrients and oxygen to the placenta.10 However, the maternal and fetal circulations do not mix. After attachment of the blastocyst to the endometrial luminal epithelium, trophoblast cells invade the decidua as depicted in Figure 1. The trophoblast, composed of an inner cell layer (cytotrophoblast) and outer cell layer (syncytiotrophoblast), does not give rise to the fetus itself, but rather to the placenta and fetal membranes (amnion and chorion). As the blastocyst and surrounding trophoblast invade the decidua, one pole of the blastocyst remains oriented toward the endometrial lumen, and the other remains buried in the decidua, which will develop into the anchoring cytotrophoblasts and villous trophoblasts, contributing to formation of the placenta, chorion, and amnion. Of note are the species differences in the degree of invasion by trophoblast cells, which have been documented in detail elsewhere.11 In distinct contrast to the process in women, trophoblast invasion is minimal in rodents.11,12
Significance of maternal immune responses during pregnancy
Immune cell subtypes and their functional significance
Immune cells accumulating in the human endometrium at the time of decidualization play critical and diverse roles at the maternal–fetal interface, including functions in implantation, placental development, and immunity against infectious diseases. Of all decidual leukocyte populations, the most abundant are the phenotypically unique uNK cells. These cells dramatically increase in number in the human endometrium 3–5 days postovulation, accounting for 25%–40% of endometrial leukocytes prior to implantation and accounting for ~70% of decidual leukocytes in the first trimester.7,8 It is critical to note that uNK cells are both phenotypically and functionally distinct from peripheral NK cells. Phenotypically, they are identified by expression of the NK cell marker CD56, expressed at high concentrations (CD56bright), but they lack expression of CD16, found on most peripheral NK cells (CD56dimCD16+).7 In terms of function, peripheral CD56dimCD16+ NK cells are highly cytotoxic, mediating both natural and antibody-dependent killing, whereas uNK cells are only weakly cytotoxic and do not normally kill trophoblast cells.13 In addition, uNK cells are a potent source of immunoregulatory cytokines,14 matrix metalloproteinases (MMPs),15 and angiogenic factors.16 These various factors mediate extracellular matrix remodeling, trophoblast invasion, and angiogenesis, which are key processes in placentation and establishment of early pregnancy at the maternal-fetal interface.17
In addition to uNK cells, decidual macrophages are relatively abundant, comprising ~20% of the human decidual leukocyte population in the first trimester.8 In normal pregnancy, most of the macrophages at the maternal–fetal interface are of the M2 (immunomodulatory) phenotype.18 Present in decidua prior to the presence of extravillous trophoblast,19 macrophages play a role in early spiral artery remodeling by producing factors associated with tissue remodeling (MMP-9) and angiogenesis (vascular endothelial growth factor [VEGF]).18 Apoptosis is an important event during spiral artery remodeling and trophoblast invasion, and decidual macrophages phagocytose apoptotic cells in remodeled vascular wall and apoptotic trophoblast cells, thereby preventing the release of proinflammatory substances from the apoptotic cells into the decidua.20 First-trimester decidual macrophages may also be responsible for inhibition of human uNK cell–mediated lysis of invasive cytotrophoblast, mediated by decidual secretion of transforming growth factor-beta-1 (TGF-²1), as demonstrated in human in vitro studies.21 In distinct contrast to human uNK cells, which peak in number at 20 weeks gestation and are nearly absent in the decidua at term,12 decidual macrophages are present throughout pregnancy, but the precise role of decidual macrophages at the end of pregnancy remains unknown.18
T-cells are also fairly abundant in human decidua, comprising ~10%–20% of the human decidual leukocyte population,22,23 of which 30%–45% are CD4+ T-cells and 45%–75% are CD8+ T-cells.23 The main function of T-cells in the decidua, particularly of CD4+ T-regulatory (Treg) cells, is generally thought to be the promotion of tolerance to the fetus24 (discussed in detail later in this review). However, because a variety of different T-cell subsets are present, the complex interactions of T-cells in the decidua have not been completely defined.25 Human in vitro studies of CD8+ T-cells isolated from first-trimester decidua demonstrate that these cells exhibit cytotoxic activity as well as cytokine production (predominantly interferon-gamma [IFN-γ] and IL-8).26 Since decidual CD8+ T-cell supernatants increase the in vitro invasive capacity of extravillous trophoblast cells, secreted products of CD8+ T-cells may play a role in regulation of trophoblast invasion, but precise mediators have not yet been identified.26
DCs, which are antigen-presenting cells that play a critical role in regulation of the adaptive immune response, make up a very small portion of human decidual leukocytes. However, no single specific marker for DCs exists and their phenotypic definition is therefore controversial, thereby limiting the existing studies of decidual DCs.27 Using lineage-negative and human leukocyte antigen-DR-positive (HLA-DR+) status as a combination marker for DCs, Gardner and Moffett28 demonstrated that decidual DCs comprised ~1% of first-trimester human decidual leukocytes. Due to the rarity of this cell population, functional studies of human decidual DCs are scarce. Human in vitro studies have demonstrated that decidual DCs, isolated from early-pregnancy decidua, are more likely than peripheral DCs to prime naïve CD4+ T-cells into a Th2 phenotype, suggesting a potential role for decidual DCs in averting Th1-mediated rejection of the fetus.29 Decidual DCs also appear to regulate uNK cell function, since coculture of decidual DCs with uNK cells stimulated uNK cell proliferation and activation.30 In vivo functional studies of decidual DCs exist only in mice and are more definitive. Decidual DC–depleted mice exhibit severely impaired implantation, impaired decidual proliferation and differentiation, impaired angiogenesis, impaired differentiation of uNK cells, and resorption of embryos.31,32 Therefore, at least in mice, decidual DCs play an important role in decidualization and establishment and maintenance of early pregnancy.
Mechanisms by which immune cells (focus: uNK cells) regulate key early events in establishment of pregnancy: implantation, angiogenesis, and vascular remodeling
uNK cells regulate trophoblast invasion
Studies performed by Hanna et al33 provided strong evidence that human uNK cells play a role in regulation of trophoblast invasion. These investigators demonstrated that uNK cells isolated from first-trimester human decidua express the chemokines IL-8 and IFN-inducible protein (IP)-10, and that purified human invasive trophoblasts express the chemokine receptors for these ligands: CXCR1 (IL-8 receptor) and CXCR3 (IP-10 receptor). The ability of uNK cells, but not peripheral blood NK cells, to induce trophoblast migration in an in vitro trophoblast migration assay was significantly reduced in the presence of neutralizing antibodies to IL-8 and IP-10. These investigators subsequently performed in vivo studies in which NK cell subsets embedded in Matrigel were injected into the subcutaneous tissues of nude mice, and human trophoblast cells were injected around the Matrigel plug. These in vivo experiments further demonstrated that uterine, but not peripheral, NK cells promoted trophoblast invasion, and that migration of trophoblast cells into the Matrigel plug was significantly reduced in the presence of IL-8- and IP-10-neutralizing antibodies. Overall, these studies demonstrated the ability of uNK cells to positively regulate invasion of trophoblast, mediated by the uNK-derived cytokines IL-8 and IP-10.33 However, trophoblast invasiveness into maternal decidua must be tightly regulated. The balance of factors involved in regulation of invasion is not yet precisely determined. Excessive invasion predisposes to placenta accreta, a potentially life-threatening obstetrical condition in which the placenta attaches abnormally to the uterine myometrium.34 Interestingly, human uNK cells also have the ability to inhibit trophoblast invasion, as demonstrated by Lash et al35 using in vitro Matrigel invasion assays. These investigators demonstrated that human uNK cells isolated from early human pregnancy decidua are a source of IFN-γ, which inhibits trophoblast invasion by increasing apoptosis of extravillous trophoblast cells and decreasing trophoblast secretion of MMP-2.35 Thus, the fine balance required to avoid either underinvasion or overinvasion of trophoblast in early human pregnancy is regulated, at least in part, by the various cytokines derived from human uNK cells present in decidua.
Role of uNK cells in angiogenesis and vascular remodeling in early pregnancy
In humans, extensive vascular remodeling must occur to allow for placentation and establishment of early pregnancy, as well as to support the demands of a growing fetus. The decidual spiral arteries must be transformed into larger-diameter vessels with low resistance and high flow, capable of transporting nutrients and oxygen to the fetus.22 In addition, the endothelium of these vessels is replaced by extravillous trophoblast cells that have migrated from the placenta, allowing for diversion of blood flow into the space surrounding the placental villous tree and thereby permitting nutrient and gas exchange between mother and fetus.36 Not only is adequate vascular remodeling critical for the establishment of a normal pregnancy, but abnormalities in these early events are associated with later complications of pregnancy such as preeclampsia and intrauterine growth restriction, which can have a major impact on fetal and neonatal health.34
A critical role for uNK cells in vascular remodeling has been demonstrated in both murine in vivo and human in vitro studies. However, it is important to note significant differences among species in terms of strategies to increase blood flow to the site of maternal–placental exchange. In humans, extensive invasion and destruction of preexisting arteries by trophoblast occurs. In nonhuman primates such as rhesus macaques, trophoblastic invasion and modification of uterine arteries occurs, but unlike in humans, invasion of decidual stroma by trophoblast in the rhesus monkey occurs only to a minimal extent.12 In mice, the extent to which the trophoblast invades both the decidual stroma and uterine arteries is even more limited.12 Rodent models thus have limited value in advancing our understanding of mechanisms of vascular remodeling that facilitate human pregnancy. Nevertheless, there are in vivo studies performed in mice that cannot be performed in humans, and the availability of nonhuman primates for such in vivo studies in early pregnancy is limited. Therefore, much of the existing data on uNK cell functions in vascular remodeling are derived from murine studies.
Multiple murine in vivo studies demonstrate that uNK cells play a critical role in the remodeling of endometrial spiral arteries both prior to and during pregnancy. The earliest studies demonstrating a critical role for uNK cells in vascular remodeling in pregnancy were those conducted by Guimond et al,37 who demonstrated several reproductive abnormalities in the Tgµ26 mouse strain, which is deficient in NK cells. Multiple vascular abnormalities associated with implantation sites, including thickening of the media and adventitia, endothelial damage, reduction in placental size, and onset of fetal loss at Day 10 of gestation, were demonstrated in NK-cell-deficient mice. Subsequent studies from the same laboratory38 demonstrated that bone marrow transplantation from severe combined immunodeficient mice (which lack T- and B- lymphocytes but not NK cells) to NK-cell-deficient mice led to restoration of the uNK cell population in recipients, reduced anomalies in decidual blood vessels, increased placental size, and restored fetal viability. Overall, these studies provide strong support for a critical role of murine uNK cells in decidualization, placentation, and the appropriate vascularization of implantation sites.
The role of murine uNK cells in vascular remodeling and decidualization appears to be mediated via IFN-γ, since transgenic mice that lack IFN-γ or its receptor fail to initiate modification of decidual arteries and exhibit necrosis of decidual cells, and treatment of NK-deficient mice with recombinant IFN-γ rescues decidual morphology and initiates decidual vessel modification.39,40 However, whether human uNK cells regulate decidual vascular remodeling via IFN-γ is yet to be definitively determined. The data regarding IFN-γ expression by human uNK cells are conflicting, likely due to differences in methodology between studies and the status of cytokine stimulation of the uNK cells being studied. Evidence for production of IFN-γ in unstimulated human uNK cells is limited, but after exposure to stimulatory cytokines such as IL-2, IL-12, or IL-15, human uNK cells isolated from first-trimester decidua exhibit significantly increased IFN-γ secretion.41,42 In addition, because IFN-γ is rapidly secreted once produced, and expression of IFN-γ mRNA and protein by human uNK cells rapidly decreases after 24–48 hours in culture,35 conflicting data regarding IFN-γ expression by human uNK cells may be attributable to length of time in culture before measurement. In a nonhuman primate model of early pregnancy, the major population of CD56bright uNK cells isolated from early-pregnancy rhesus monkey decidua is not a source of IFN-γ.43 Therefore, while compelling evidence exists to support the role of IFN-γ in decidual vascular remodeling in rodents, whether uNK cell-derived IFN-γ plays an equally important role in vascular remodeling in humans and in nonhuman primates remains unclear.
Rather, the finding that human uNK cells isolated from first-trimester decidua are a potent source of the angiogenic factors angiopoietin (Ang)1, Ang2, VEGF, and PLGF16,33 supports an important role for these cells in the vascular remodeling required for successful human pregnancy. Functional studies by Hanna et al33 demonstrated that human uNK cells isolated from first-trimester decidua are potent secretors of angiogenic factors such as VEGF and placental growth factor (PLGF). Supernatants derived from human uterine (but not peripheral) NK cells promoted in vitro angiogenesis, as demonstrated by an increased ability of human umbilical vascular endothelial cells to form network-like structures, a process inhibited in the presence of VEGF- and PLGF-neutralizing proteins. In addition, these investigators33 demonstrated the in vivo ability of human uNK cells to promote angiogenesis and growth of human trophoblast choriocarcinoma (JEG-3) tumor cells when injected subcutaneously into nude mice. In vivo angiogenic properties of uNK cells were inhibited in the presence of a VEGF- and PLGF-neutralizing protein. These studies provide strong evidence that the angiogenic properties of human uNK cells are mediated, at least in part, by their secretion of VEGF and PLGF.
Influence of maternal immune response on development of the fetal immune system
Compelling clinical data demonstrate that children of mothers exposed to certain infectious organisms during pregnancy have significantly higher frequencies of neurological disorders,44–53 including schizophrenia and autism spectrum disorders. In such scenarios, the etiology of these disorders has been linked to activation of the maternal inflammatory/immune responses (reviewed by Jonakait54 and Patterson55). Rodent studies in which the maternal immune system is activated during pregnancy replicate these clinical findings and provide validated mouse models of these disorders.46,47,51,56–66 Thus, maternal immune stimulation during pregnancy acts as an environmental risk factor that affects development of the brain and the immune system in the offspring.
The underlying mechanisms of these phenomena have been studied primarily in prenatal rodent models, in which pregnant dams are injected with either infectious pathogens or synthetic agents that mimic viral or bacterial infections (namely, lipopolysaccharides and polyinosinic:polycytidylic acid [poly(I:C)]). Offspring of such immunostimulated pregnant dams exhibit immune dysregulation and behavioral abnormalities, as well as chemical and structural anomalies of the brain, which are similar to those seen in individuals with schizophrenia and autism spectrum disorders.63,67–72
There is a transient increase of cytokines (IL-1, IL-6, IL-12, tumor necrosis factor-alpha [TNF-α], granulocyte-macrophage colony stimulating factor) in the blood and amniotic fluid of immunostimulated pregnant dams,73,74 which appears to influence development of the fetal immune system, a concept known as “fetal programming”.75–79 Mandal et al73,74,80 have also shown that offspring of immunostimulated pregnant dams exhibit accelerated development and heightened responsiveness of Th1, Th17, and cytotoxic effector T-cell subsets, indicating a proinflammatory phenotype in these offspring.
We hypothesized that in utero exposure of the fetus to cytokines elicited by maternal immune stimulation acts as a “first hit” to influence fetal programming of the immune system, which persists postnatally and into adulthood. Such alterations of normal fetal programming results in development of a “proinflammatory” phenotype, and upon subsequent postnatal exposure to an immune stimulus (ie, second hit), the offspring of the immunostimulated pregnant dams exhibit exacerbated responses in comparison to offspring of phosphate-buffered saline (PBS)-injected dams. Such a scenario is also consistent with the “multiple hit” concept of mental disorders.81,82 In the context of neurodevelopmental disorders, this would mean that abnormalities of behavior and immune dysregulation observed in some affected children could reflect such altered fetal programming that is manifested postnatally upon encounter with a second hit (eg, infection) to their immune system. We tested this hypothesis in adult offspring of immunostimulated pregnant dams using well-documented in vivo experimental models that involve activation of the innate and/or adaptive immune systems. In each of these models, the adult offspring of immunostimulated dams mounted a more robust inflammatory response than adult offspring of control dams injected with PBS.73,83 Thus, offspring from immunostimulated dams exhibit behavioral anomalies reminiscent of those seen in individuals with some neurodevelopmental disorders, such as schizophrenia and autism. In addition to their behavioral abnormalities, our studies show that as a result of in utero exposure to products of maternal immune stimulation, these adult offspring also exhibit a “proinflammatory” phenotype that confers a vulnerability to develop immune-mediated pathology after birth and into adulthood.73,74,80
In this regard, the results obtained from our investigations in mouse models have provided the scientific rationale for an ongoing translational research project to determine whether similar molecular pathogenic mechanisms are involved in a cohort of autistic children who also exhibit diagnostic evidence of immune dysregulation.84 Using DNA obtained from the Autism Genetic Resource Exchange database, we initiated a study to determine whether polymorphisms in selected maternal cytokine genes occurred more frequently in mothers of these autistic children. Our results show that mothers of autistic children in this cohort have significantly higher frequencies of proinflammatory cytokine gene polymorphisms, thereby conferring the genetic capability to respond more vigorously to immune stimulation by producing the types and amounts of cytokines that promote inflammatory reactions. Moreover, analysis of preliminary data from the offspring indicates that the autistic children of these mothers inherit the maternal genotype. Thus, results obtained from our investigation of the experimental prenatal mouse model of maternal immune stimulation during pregnancy73 appear to have biological relevance to humans.
Maternal–fetal tolerance
Billingham et al1 in 1953 were the first to propose the concept of immune tolerance during pregnancy. They hypothesized that the semiallogeneic fetus is able to survive due to regulation of the immunologic interactions between mother and fetus. Such regulation can be caused by a lack of fetal antigen expression and/or functional suppression of maternal immune response.1
HLAs that are expressed in the fetal membranes are tolerogenic rather than immunogenic,85 and expression of major histocompatibility complex (MHC) proteins at the maternal–fetal interface is tightly regulated during pregnancy.86 The MHC class I genes are subdivided into classes Ia and Ib. The MHC class Ia is further subdivided into HLA-A, B, and C and class Ib is subdivided into HLA-E, F, and G. HLA class II (HLA-D) genes are not translated in human trophoblast cells.87 Human trophoblast cells express one MHC class Ia (HLA-C) and all MHC class Ib molecules. In human placenta, fetal trophoblast cells do not express MHC class Ia (HLA-A and B) molecules that are responsible for the rejection of allografts in humans.88,89 Interactions between HLA-C and decidual NK cells may also cause infiltration of trophoblast into maternal tissue. Pregnancies with mismatched fetal HLA-C exhibit a greater number of activated T-cells and functional Tregs in decidual tissues compared to HLA-C-matched pregnancies.90 This suggests that in uncomplicated pregnancies, decidual T-cells recognize fetal HLA-C at the maternal–fetal interface but are prevented from inducing a destructive immune response.91
Regarding pregnancy, one of the most important questions is how the fetal–placental unit escapes maternal rejection. Although there is a continuous interaction between the fetus and maternal cells throughout pregnancy, the fetus acts as a privileged site that is protected from immune rejection.91 Expression of MHC molecules on trophoblast cells is repressed in most of the species as a strategy to avoid recognition and destruction by the maternal immune cells.92 Peripheral blood lymphocytes from pregnant mares demonstrate reduced capacity to develop into effector cytotoxic T lymphocytes.93 This reduction in T-cell-mediated alloreactivity returns to normal after termination of pregnancy and is not observed in nonpregnant mares. In addition, extracts from Day 80 placentas from mares have been shown to inhibit proliferation of maternal lymphocytes, and coculture of trophoblast cells with maternal lymphocytes caused reduction in proliferation and cytokine production.94
Cell-mediated immunity: mechanisms promoting maternal–fetal tolerance
The Th1–Th2 shift in pregnancy
Pregnancy is a complex immunological state, wherein the mother must tolerate the “foreign” fetus, and thus requires a degree of immunosuppression. On the other hand, the mother must maintain sufficient immune function to fight off infection. One mechanism that plays a role in maintenance of successful pregnancy is a switch from the Th1 cytokine profile to the Th2 profile. This switch is more prominent at the maternal–fetal interface. Th2 cells accumulate in decidua, and uterine DCs can drive naïve T-cells to become Th2 cells.95,96 Therefore, the switch to a Th2 phenotype is due to both migration of Th2 cells and induction of Th2 cells at the maternal–fetal interface, but there is little change in the systemic immune system.96 The hypothesis of Th2 predominance and downregulation of Th1 response during pregnancy was proposed by Wegmann et al,97 which is supported by both murine and human studies. In mice, the proinflammatory cytokines IFN-γ and TNF-α, or stimulation of toll-like receptors, induce miscarriage, which can be reversed by inhibitors of Th1 cytokines or by administration of anti-inflammatory IL-10 (Th2 cytokine).98 However, IFN-γ also plays an important role in vascular remodeling in early murine pregnancy. Therefore, Th1-type immunity appears to be controlled to avoid overstimulation during pregnancy. Progesterone, estradiol, prostaglandin D2 (PGD2), and leukemic inhibitory factor generated during pregnancy promote the Th2 profile and are, in part, responsible for the Th2 bias associated with normal pregnancy.96 However, transgenic Th2 cytokine single-knockout mice such as IL-4−/−, IL-10−/−99 and mice with single, double, triple, and quadruple gene deletions of IL-4, IL-5, IL-9, and IL-13 have normal pregnancies, suggesting that a predominant Th2-type immunity might not be essential for successful pregnancy.100
An increase of Th2 cytokines IL-4, IL-10, and monocyte-colony stimulating factor in the peripheral blood and the maternal–fetal interface is associated with successful pregnancy. Trophoblast, decidua, and amnion contribute to the Th2 cytokine-biased environment by production of IL-13, IL-10, IL-4, and IL-6.101–103 Human placental cytotrophoblasts have been shown to produce the immunosuppressive cytokine IL-10.101 In addition, macrophages and Tregs present within decidua during pregnancy also produce IL-10 and are involved in maintenance of immune tolerance toward allogeneic fetal antigens.91 The placenta also produces PGD2, which can act as a chemoattractant for Th2 cells to the maternal–fetal interface via the Th2 receptor CRTH2 (a chemoattractant receptor-homologous molecule expressed on Th2 cells). Women suffering recurrent pregnancy loss have reduced expression of CRTH2+ cells than women undergoing elective termination of pregnancy.104 Anti-inflammatory cytokines IL-4 and IL-10 inhibit Th1 cells and macrophages, which in turn prevent fetal allograft rejection. In addition, these cytokines also inhibit TNF-α, cyclooxygenase-2 (COX-2), and prostaglandin E2 in amnion-derived cells, which prevent the onset of labor.24,105–107
Labor is often associated with a proinflammatory state with reversal back to Th1 rather than Th2. Studies indicate increases in Th1 proinflammatory cytokines and reduction in Th2 cytokines in women who are in active labor. Fetal membranes, myometrium, amnion, amniotic fluid, and decidua produce proinflammatory cytokines IL-1² and TNF-α at term and can induce nuclear factor kappa B. This transcription factor regulates the expression of labor-associated genes such as COX-2, IL-8, and MMP-9 and triggers a cascade of labor-inducing events. Despite the proinflammatory nature of Th1 cytokines, they are essential for successful pregnancy, contributing to timely labor.108–110
Role of Tregs in pregnancy
CD4+CD25+ Tregs are a subpopulation of T-cells responsible for the maintenance of immunological self-tolerance by suppressing self-reactive lymphocytes in a cell contact-dependent manner by production of TGF-² and IL-10.111,112 Tregs express transcription factor forkhead box transcription factor (FoxP3), which acts as a major regulator in their development and function.113 There are two main Treg subsets: naturally occurring or thymic Tregs (tTregs) and induced or extrathymic/peripheral Tregs (pTregs). tTregs are CD4+CD25+Foxp3+ and express cytotoxic T lymphocyte-associated antigen 4. pTregs develop from naïve T-cells after exposure to antigens in the periphery and exposure to either IL-10 or TGF-² and can be either Foxp3- or Foxp3+.114,115 Owing to their immunosuppressive function, Tregs also play a key role during pregnancy by maintaining maternal–fetal tolerance.
Several studies have confirmed an increase in Tregs during pregnancy in blood, lymph nodes, and thymus, followed by decrease from midgestation onward until they reach nonpregnant levels at term or shortly thereafter. They play a critical role in embryo implantation and in the maintenance of the maternal immune tolerance against semiallogeneic fetal antigens.116,117 Evidence suggests that Tregs during pregnancy are specific to paternal alloantigens, which protects the fetus from rejection by the mother’s immune system.118 Expansion of Tregs in decidua from normal pregnant women suppresses maternal Th1/Th17 activity on the semiallogeneic fetus.119
Murine experiments have shown increased levels of Tregs in both syngeneic and allogeneic matings, suggesting alloantigen-independent Treg expansion.120 Treg expansion appears to be regulated by estradiol. This is supported by in vitro studies, which show that physiological levels of estradiol not only expand Tregs but also stimulate conversion of CD4+CD25- T-cells into CD4+CD25+ T-cells.121 On the other hand, Zhao et al122 observed no increase in Tregs in ovariectomized mice. Moreover, they detected higher number of Tregs in pregnant mice from allogeneic versus syngeneic matings, suggesting an involvement of paternal antigens in Treg expansion.122 Recently, Robertson et al123 showed that seminal fluid can drive Treg expansion. Therefore, both antigen-dependent and antigen-independent mechanisms are likely to be involved in Treg expansion.
Tregs express various chemokine receptors whose ligands are expressed at the maternal–fetal interface, which might contribute to chemokine-mediated migration of Tregs to the decidua.120 Furthermore, other immune cells produce large amounts of CCL17, CCL4, and CCL1,124–126 which might attract Tregs specifically expressing CCR4 and CCR8.127,128 Besides chemokine-mediated migration of Tregs, integrins, similar to CD62L, seem to play an important role in Treg migration, as neutralizing CD62L-specific antibody blocks expansion of Tregs in draining lymph nodes and results in allograft rejection. Schumacher et al129 have shown the importance of human chorionic gonadotropin as one of the main attractants of Tregs to the maternal–fetal interface.
Aluvihare et al117 first noted that Tregs increased in all lymphoid organs in allogeneic matings of C57BL/6 female mice with CBA males. They also adoptively transferred lymphocytes from BALB/c females, either allopregnant from C57BL/6 males or synpregnant from BALB/c males, into T-cell-deficient BALB/c females, which were then mated with C57BL/6 males. Pregnancy proceeded normally when whole lymphocyte populations were transferred. In contrast, lymphocytes depleted of Tregs resulted in fetal resorptions, and there was a massive infiltration of T-cells into the implantation sites.117 Zenclussen130 and Zenclussen et al131 have shown complete prevention of abortion in the CBA × DBA/2J model of naturally occurring spontaneous abortions by transferring Tregs from alloimmunized mice, and they also reported that no abortions occurred in the CBA × BALB/c and CBA × CBA control matings. Finally, Chen et al116 demonstrated that stimulation of Tregs, either directly by low dose of IL-2 or indirectly by Fms-related tyrosine kinase 3 ligand, led to normal pregnancy rates in CBA × DBA/2J abortion-prone mice. The results of these experiments all demonstrate that in allogenic matings, Tregs are necessary for prevention of a maternal immune response against the fetus.
Clinical manifestations of an altered immune system in pregnancy
The notion of pregnancy as an altered state of immune suppression is well documented.132–136 Pregnancy is a time period that poses a risk of increased susceptibility to infectious diseases, and the maternal immune system is solely responsible for defending against infectious microorganisms and protecting the fetus because both the fetal and the placental responses are limited.132,136 The Th1/Th2 immune shifts in pregnancy are well established and have provided a platform to further study the immune system.136 This has led to refining our understanding about the immune system and the development of a new paradigm regarding pregnancy and immune function. This newer theory proposes that the immune system during pregnancy is a functional and active system, wherein not only a maternal immune response exists but also a fetal–placental immune response, which in combination is powerful in defending both the mother and the fetus.133,136 With this notion, the immune system is not suppressed, but rather in a modulated state, and therefore, this explains why pregnant women have differential responses to various pathogens.133 During this altered response, signals are generated in the placenta, which modulate the maternal immune system to behave uniquely to different microorganisms.133 Although these old and new paradigms surrounding the immunology of pregnancy differ, it is clear that the immune system’s goal in pregnancy is to ensure that a pregnancy progresses successfully, while still providing protection for both mother and fetus from external pathogens.
Endocrine regulation of immune cells
Hormone concentrations vary with the initiation of pregnancy, and there are specific fluctuations in hormone levels throughout each trimester of pregnancy. In general, pregnancy hormones are thought to suppress maternal alloresponses, while promoting pathways of tolerance.137 Hormonal shifts are thought to reduce the number of DCs and monocytes, decrease macrophage activity, while blocking NK cells, T-cells, and B-cells.137 Each of the major pregnancy-associated hormones is thought to directly and indirectly affect the function of the major immune cells and thus impacts the immune milieu during pregnancy. These alterations are discussed in Table 1.
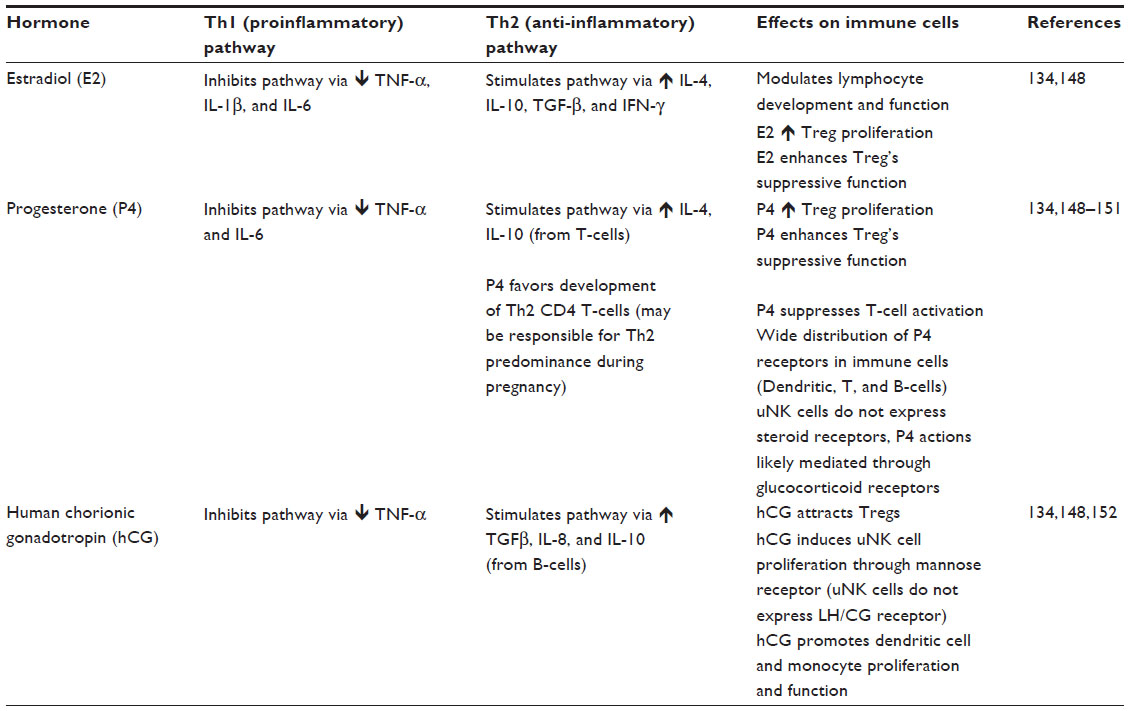 | Table 1 Endocrine regulation of immune cells and immune function |
 , decreased;
, decreased; 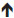 , increased.
, increased.Evidence of altered immune function in pregnancy: effects of infectious organisms on pregnancy
The alterations in the immune system during pregnancy are well established, and subsequently, these changes result in increased susceptibility to certain viral, bacterial, and parasitic infections.132 This increased susceptibility is believed to result from the suppression of cell-mediated immunity, as pregnancy promotes a shift away from the Th1 to the Th2 immune environment.132,134 Additionally, infection with certain pathogens has been documented to result in severe symptoms in pregnant patients because of these immune changes.133,138 However, it is important to note that, in certain infectious diseases among gravid patients, the morbidity and mortality vary between developed and nondeveloped countries. For example, pregnant women with varicella in the US or Canada fare better than those diagnosed in underdeveloped countries, where resources are limited.139 Thus, some bias may result when evaluating the severity of disease states in pregnant women depending on geographical distribution.
Table 2 summarizes the more commonly recognized and studied pathogens related to pregnancy. As seen in Table 2, infectious diseases during pregnancy are associated with not only maternal risks but fetal risks as well. These fetal effects result from infections that cross the placenta, which can cause miscarriage, congenital anomalies, or even fetal death.133 As a result, the American Congress of Obstetricians and Gynecologists and the US Centers for Disease Control and Prevention recommend that all women be vaccinated for influenza and tetanus, diphtheria, and pertussis (Tdap) during pregnancy.140–142 Both these vaccines appear to be safe when administered during pregnancy, with few maternal and fetal adverse events.142,143 In contrast, live vaccines, such as measles–mumps–rubella (MMR) and varicella, are not recommended during pregnancy due to the theoretical risks to the fetus.141,142
 | Table 2 Common infectious organisms in pregnancy |
The risk of infection during pregnancy is a serious matter, not only for concerns of maternal well-being but also the potential fetal risks, which may have long-term consequences. Animal studies have elucidated that the placenta may trigger fetal inflammatory response syndrome (FIRS), which is the diagnosis of a placental infection without the growth of an organism, from the microbiology standpoint.133,136 FIRS is serious and results in increased circulating levels of cytokines, such as IL-1, IL-6, IL-8, and TNF-α.133 These inflammatory shifts have been demonstrated to increase the risk of fetal abnormalities, such as ventriculomegaly or hemorrhages. Furthermore, human studies have demonstrated an association between FIRS and the development of autism, schizophrenia, neurosensorial deficits, and psychosis.133,136 These observations further validate the experimental mouse models described earlier in which immunostimulation induces high levels of proinflammatory cytokines in blood and amniotic fluid of pregnant dams, which are likely involved in the etiology of neurodevelopmental disorders exhibited in their offspring.63,72–74 In contrast, bacterial infections that reach the decidua trigger a proinflammatory response that leads to the development of intrauterine infections.144 This is through the activation of pattern recognition receptors (PRRs) and increased secretion of cytokines, such as IL-1 and TNF-α.145 Combined, these contribute to poor pregnancy outcomes, disruption in fetal development, or preterm births with resultant low-birth-weight infants.146,147 Thus, it is important to recognize that pregnancy can cause increased disease susceptibility, which not only affects maternal morbidity but contributes to detrimental long-term fetal and neonatal outcomes.
Evidence of altered immune function in pregnancy: effects of pregnancy on autoimmune disease
As discussed, pregnancy confers a shift from Th1- to Th2-mediated immunity, and this shift affects disease status in women with known autoimmune diseases. In general, the hormonal milieu induced by pregnancy shifts the cytokine profile away from cell-mediated immunity (Th1 type of immunity) and, therefore, improves inflammatory-type autoimmune diseases.132 In contrast, autoimmune diseases that are humorally (or antibody) mediated are exacerbated, as pregnancy favors increased Th2-related activities, as well as a Th2 cytokine profile.132,148 For details, please view Table 3.
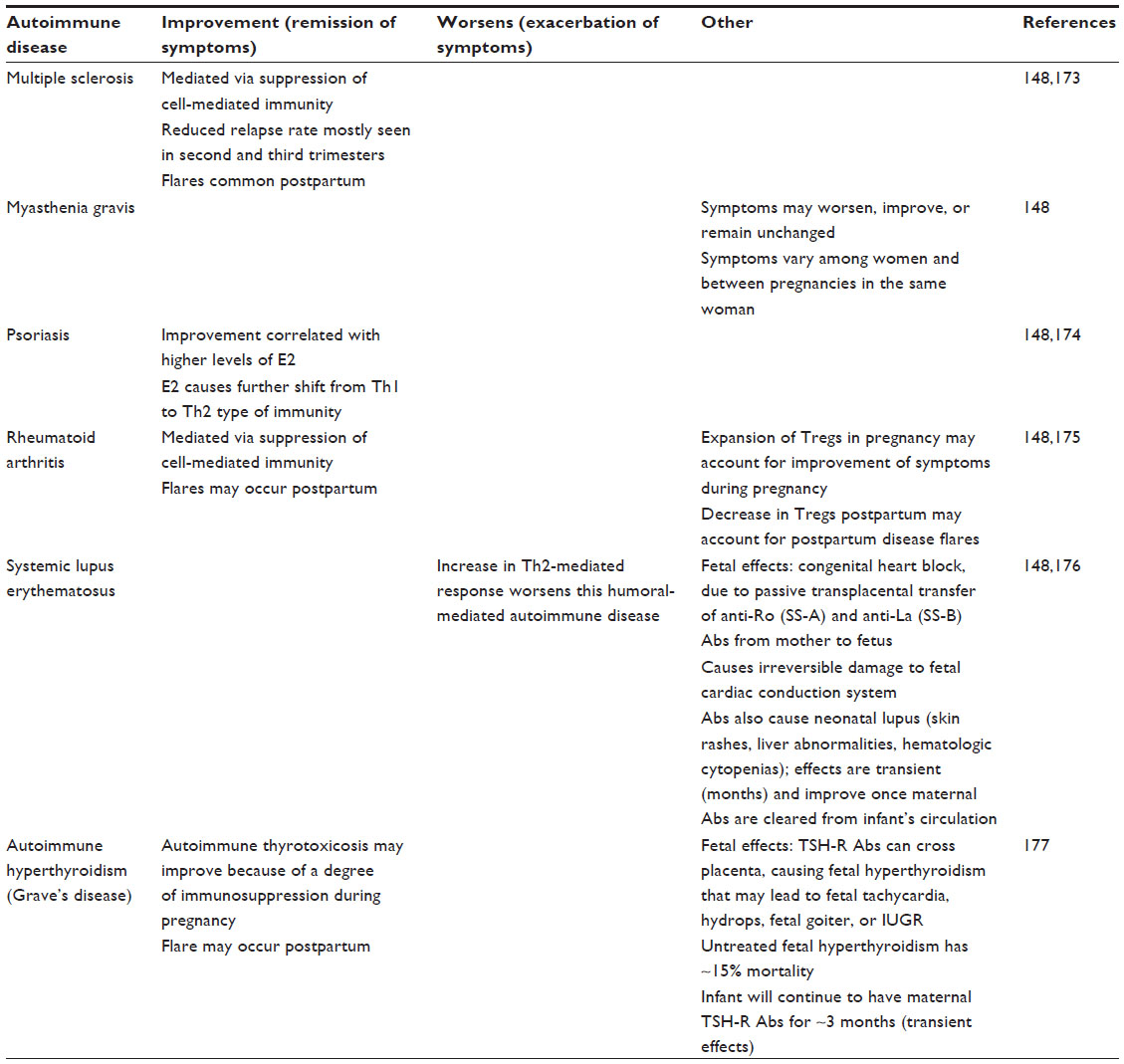 | Table 3 Autoimmune disease in pregnancy |
Conclusion and future outlook
Pregnancy in women is a dynamic state, with different mechanisms used during different trimesters to enable and ensure successful establishment, maintenance, and timely termination of the pregnancy. Mechanisms operative in early pregnancy to establish the pregnancy may differ from those needed to maintain the pregnancy and from those required to ensure successful and timely labor and delivery. Recent data challenge the notion that pregnancy is simply an immunosuppressed state protecting the allogeneic fetus from attack by the maternal immune system. The evidence suggests that rather, pregnancy may be a state of upregulated innate immune response and decreased cell-mediated response. Unique decidual lymphoid cell populations actively contribute to placental development and to tolerance of the fetus. Although substantial progress in the understanding of the function of immune cells during pregnancy, especially early pregnancy, has been achieved, many unanswered questions regarding regulation of their proliferation and function by endocrine and other factors still remain. The published results from human studies and animal models clearly indicate that a fine balance between proinflammatory and anti-inflammatory influences is critical for successful pregnancy. Thus, the future challenge for translational research in reproductive immunology will be to define more completely those factors that favor optimal immunological environments that promote fetal health and development at specific stages of pregnancy, so that evidence-based regulatory therapeutic strategies can then be designed.
Acknowledgments
The authors thank Yingting Zhang for her assistance with the manuscript. Mili Mandal is currently affiliated with Oncology, R&D, GlaxoSmithKline, Collegeville, PA, US.
Disclosure
The authors report no conflicts of interest in this work.
References
Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature. 1953;172(4379):603–606. | |
Brosens JJ, Pijnenborg R, Brosens IA. The myometrial junctional zone spiral arteries in normal and abnormal pregnancies: a review of the literature. Am J Obstet Gynecol. 2002;187(5):1416–1423. | |
Gellersen B, Brosens IA, Brosens JJ. Decidualization of the human endometrium: mechanisms, functions, and clinical perspectives. Semin Reprod Med. 2007;25(6):445–453. | |
Gellersen B, Brosens J. Cyclic AMP and progesterone receptor cross-talk in human endometrium: a decidualizing affair. J Endocrinol. 2003;178(3):357–372. | |
Dimitriadis E, White CA, Jones RL, Salamonsen LA. Cytokines, chemokines and growth factors in endometrium related to implantation. Hum Reprod Update. 2005;11(6):613–630. | |
Lash GE, Robson SC, Bulmer JN. Review: functional role of uterine natural killer (uNK) cells in human early pregnancy decidua. Placenta. 2010;31(Suppl):S87–S92. | |
King A. Uterine leukocytes and decidualization. Hum Reprod Update. 2000;6(1):28–36. | |
Bulmer JN, Morrison L, Longfellow M, Ritson A, Pace D. Granulated lymphocytes in human endometrium: histochemical and immunohistochemical studies. Hum Reprod. 1991;6(6):791–798. | |
Weiss G, Goldsmith LT, Taylor RN, Bellet D, Taylor HS. Inflammation in reproductive disorders. Reprod Sci. 2009;16(2):216–229. | |
Moffett A, Colucci F. Uterine NK cells: active regulators at the maternal-fetal interface. J Clin Invest. 2014;124(5):1872–1879. | |
Moffett A, Loke C. Immunology of placentation in eutherian mammals. Nat Rev Immunol. 2006;6(8):584–594. | |
Moffett-King A. Natural killer cells and pregnancy. Nat Rev Immunol. 2002;2(9):656–663. | |
King A, Birkby C, Loke YW. Early human decidual cells exhibit NK activity against the K562 cell line but not against first trimester trophoblast. Cell Immunol. 1989;118(2):337–344. | |
Koopman LA, Kopcow HD, Rybalov B, et al. Human decidual natural killer cells are a unique NK cell subset with immunomodulatory potential. J Exp Med. 2003;198(8):1201–1212. | |
Naruse K, Lash GE, Innes BA, et al. Localization of matrix metalloproteinase (MMP)-2, MMP-9 and tissue inhibitors for MMPs (TIMPs) in uterine natural killer cells in early human pregnancy. Hum Reprod. 2009;24(3):553–561. | |
Lash GE, Schiessl B, Kirkley M, et al. Expression of angiogenic growth factors by uterine natural killer cells during early pregnancy. J Leukoc Biol. 2006;80(3):572–580. | |
Yagel S. The developmental role of natural killer cells at the fetal-maternal interface. Am J Obstet Gynecol. 2009;201(4):344–350. | |
Faas MM, Spaans F, De Vos P. Monocytes and macrophages in pregnancy and pre-eclampsia. Front Immunol. 2014;5:298. | |
Smith SD, Dunk CE, Aplin JD, Harris LK, Jones RL. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am J Pathol. 2009;174(5):1959–1971. | |
Abrahams VM, Kim YM, Straszewski SL, Romero R, Mor G. Macrophages and apoptotic cell clearance during pregnancy. Am J Reprod Immunol. 2004;51(4):275–282. | |
Co EC, Gormley M, Kapidzic M, et al. Maternal decidual macrophages inhibit NK cell killing of invasive cytotrophoblasts during human pregnancy. Biol Reprod. 2013;88(6):155. | |
Wallace AE, Fraser R, Cartwright JE. Extravillous trophoblast and decidual natural killer cells: a remodelling partnership. Hum Reprod Update. 2012;18(4):458–471. | |
Nancy P, Erlebacher A. T cell behavior at the maternal-fetal interface. Int J Dev Biol. 2014;58(2–4):189–198. | |
Piccinni MP. T cell tolerance towards the fetal allograft. J Reprod Immunol. 2010;85(1):71–75. | |
Bulmer JN, Williams PJ, Lash GE. Immune cells in the placental bed. Int J Dev Biol. 2010;54(2–3):281–294. | |
Scaife PJ, Bulmer JN, Robson SC, Innes BA, Searle RF. Effector activity of decidual CD8+ T lymphocytes in early human pregnancy. Biol Reprod. 2006;75(4):562–567. | |
Hsu P, Nanan RK. Innate and adaptive immune interactions at the fetal-maternal interface in healthy human pregnancy and pre-eclampsia. Front Immunol. 2014;5:125. | |
Gardner L, Moffett A. Dendritic cells in the human decidua. Biol Reprod. 2003;69(4):1438–1446. | |
Miyazaki S, Tsuda H, Sakai M, et al. Predominance of Th2-promoting dendritic cells in early human pregnancy decidua. J Leukoc Biol. 2003;74(4):514–522. | |
Laskarin G, Redzovic A, Rubesa Z, et al. Decidual natural killer cell tuning by autologous dendritic cells. Am J Reprod Immunol. 2008;59(5):433–445. | |
Plaks V, Birnberg T, Berkutzki T, et al. Uterine DCs are crucial for decidua formation during embryo implantation in mice. J Clin Invest. 2008;118(12):3954–3965. | |
Krey G, Frank P, Shaikly V, et al. In vivo dendritic cell depletion reduces breeding efficiency, affecting implantation and early placental development in mice. J Mol Med (Berl). 2008;86(9):999–1011. | |
Hanna J, Goldman-Wohl D, Hamani Y, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. 2006;12(9):1065–1074. | |
Norwitz ER. Defective implantation and placentation: laying the blueprint for pregnancy complications. Reprod Biomed Online. 2007;14 Spec No 1:101–109. | |
Lash GE, Otun HA, Innes BA, et al. Interferon-gamma inhibits extravillous trophoblast cell invasion by a mechanism that involves both changes in apoptosis and protease levels. FASEB J. 2006;20(14):2512–2518. | |
Erlebacher A. Immunology of the maternal-fetal interface. Annu Rev Immunol. 2013;31:387–411. | |
Guimond MJ, Luross JA, Wang B, Terhorst C, Danial S, Croy BA. Absence of natural killer cells during murine pregnancy is associated with reproductive compromise in TgE26 mice. Biol Reprod. 1997;56(1):169–179. | |
Guimond MJ, Wang B, Croy BA. Engraftment of bone marrow from severe combined immunodeficient (SCID) mice reverses the reproductive deficits in natural killer cell-deficient tg epsilon 26 mice. J Exp Med. 1998;187(2):217–223. | |
Ashkar AA, Di Santo JP, Croy BA. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J Exp Med. 2000;192(2):259–270. | |
Monk JM, Leonard S, McBey BA, Croy BA. Induction of murine spiral artery modification by recombinant human interferon-gamma. Placenta. 2005;26(10):835–838. | |
Manaster I, Gazit R, Goldman-Wohl D, et al. Notch activation enhances IFNgamma secretion by human peripheral blood and decidual NK cells. J Reprod Immunol. 2010;84(1):1–7. | |
Vigano P, Gaffuri B, Somigliana E, Infantino M, Vignali M, Di Blasio AM. Interleukin-10 is produced by human uterine natural killer cells but does not affect their production of interferon-gamma. Mol Hum Reprod. 2001;7(10):971–977. | |
Dambaeva SV, Durning M, Rozner AE, Golos TG. Immunophenotype and cytokine profiles of rhesus monkey CD56bright and CD56dim decidual natural killer cells. Biol Reprod. 2012;86(1):1–10. | |
Croonenberghs J, Bosmans E, Deboutte D, Kenis G, Maes M. Activation of the inflammatory response system in autism. Neuropsychobiology. 2002;45:1–6. | |
Deykin E, MacMahon B. Viral exposure and autism. Am J Epidemiol. 1979;109:628–638. | |
Hagberg H, Mallard C. Effect of inflammation on central nervous system development and vulnerability: review. Curr Opin Neurol. 2005;18: 117–123. | |
Hornig M, Weissenbock H, Horscroft N, Lipkin WI. An infection-based model of neurodevelopmental damage. Proc Natl Acad Sci. 1999;96: 12101–12107. | |
Lipkin W, Hornig M. Microbiology and immunology of autism spectrum disorders. Novartis Found Symp. 2003;251:129–143. | |
Malek-Ahmadi P. Cytokines and etiopathogenesis of pervasive developmental disorders. Med Hypotheses. 2001;56(3):321–324. | |
Pardo C, Eberhart C. The neurobiology of autism. Brain Pathol. 2007;17:434–447. | |
Patterson P. Maternal infection: window on neuroimmune interactions in fetal brain development and mental illness. Curr Opin Neurobiol. 2002;12:115–118. | |
Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. 2006;32(2):200–202. | |
Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167(3):261–280. | |
Jonakait G. The effects of maternal inflammation on neuronal development: possible mechanisms. Int J Dev Neurosci. 2007;25:415–425. | |
Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17:389–394. | |
Bell M, Hallenbeck J. Effects of intrauterine inflammation on developing rat brain. J Neurosci Res. 2002;70:570–579. | |
Carvey P, Chang Q, Lipton J, Ling Z. Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long-term losses of dopamine neurons in offspring: a potential, new model of Parkinson’s disease. Front Biosci. 2003;8:s826–s837. | |
Fatemi SH, Earle J, Kanodia R, et al. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Clin Mol Neurobiol. 2002;22: 25–33. | |
Hornig M, Solbrig M, Horscroft N, Weissenbock H, Lipkin W. Borna disease virus infection of adult and neonatal rats: models for neuropsychiatric disease. Curr Top Microbiol Immunol. 2001;253:157–177. | |
Pletnikov MV, Jones ML, Rubin SA, Moran TH, Carbone KM. Rat model of autism spectrum disorders. Genetic background effects on Borna disease virus-induced development brain damage. Ann NY Acad Sci. 2001;939:318–319. | |
Pletnikov M, Rubin S, Schwartz G, Moran T, Carbone K. Persistent neonatal Borna disease virus (BDV) infection of teh brain causes chronic emotional abnormalities in adult rats. Physiol Behav. 1999;66: 823–831. | |
Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23(1):297–302. | |
Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27(40):10695–10702. | |
Weissenbock H, Hornig M, Hickey W, Lipkin W. Microglial activation and neuronal apoptosis in Bornavirus infected neonatal Lewis rats. Brain Pathol. 2000;10:260–272. | |
Lancaster K, Dietz D, Moran T, Pletnikov M. Abnormal social behaviors in young and adult rats neonatally infected with Borna disease virus. Behav Brain Res. 2007;176:141–148. | |
Rousset CI, Chalon S, Cantagrel S, et al. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr Res. 2006;59:428–433. | |
Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8. | |
Conroy SM, Nguyen V, Quina LA, et al. Interleukin-6 produces neuronal loss in developing cerebellar granule neuron cultures. J Neuroimmunol. 2004;155:43–54. | |
Gilmore J, Fredrik J, Vadlamudi S, Lauder J. Prenatal infection and risk for schizophrenia: IL-1beta, IL-6, and TNFalpha inhibit cortical neuron dendrite development. Neuropsychopharmacology. 2004;29: 1221–1229. | |
Nawa H, Takei N. Recent progress in animal modeling of immune inflammatory processes in schizophrenia: implication of specific cytokines. Neurosci Res. 2006;56:2–13. | |
Samuelsson A, Jennische E, Hansson H, Holmang A. Prenatal exposure to Interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Amer J Physiol Regul Integr Comp Phyusiol. 2006;290: R1345–R1356. | |
Ponzio NM, Servatius R, Beck K, Marzouk A, Kreider T. Cytokine levels during pregnancy influence immunological profiles and neurobehavioral patterns of the offspring. Ann N Y Acad Sci. 2007;1107:118–128. | |
Mandal M, Donnelly R, Elkabes S, et al. Maternal immune stimulation during pregnancy shapes the immunological phenotype of offspring. Brain Behav Immun. 2013;33:33–45. | |
Mandal M, Marzouk AC, Donnelly R, Ponzio NM. Maternal immune stimulation during pregnancy affects adaptive immunity in offspring to promote development of TH17 cells. Brain Behav Immun. 2011;25(5):863–871. | |
Barker DJ. Intrauterine programming of adult disease. Mol Med Today. 1995;1(9):418–423. | |
Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: critical role for the immune system. Front Behav Neurosci. 2009;3:1–14. | |
Barrett EG. Maternal influence in the transmission of asthma susceptibility. Pulm Pharmacol Ther. 2008;21(3):474–484. | |
Bellinger DL, Lubahn C, Lorton D. Maternal and early life stress effects on immune function: relevance to immunotoxicology. J Immunotoxicol. 2008;5(4):419–444. | |
Conrad ML, Ferstl R, Teich R, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J Exp Med. 2009;206(13):2869–2877. | |
Mandal M, Marzouk AC, Donnelly R, Ponzio NM. Preferential development of Th17 cells in offspring of immunostimulated pregnant mice. J Reprod Immunol. 2010;87(1–2):97–100. | |
Keshavan MS. Development, disease and degeneration in schizophrenia: a unitary pathophysiological model. J Psychiatr Res. 1999;33(6):513–521. | |
Keshavan MS, Hogarty GE. Brain maturational processes and delayed onset in schizophrenia. Dev Psychopathol. 1999;11(3):525–543. | |
Ponzio NM, Mandal M, Elkabes S, et al. Pro-inflammatory phenotype induced by maternal immune stimulation during pregnancy. In: Fitzgerald M, editor. Recent Advances in Autism Spectrum Disorders. Vol 1. Rijeka: In Tech; 2013:113–141. | |
Ramanathan M, Ponzio N, Limson F, Shah S, Fernandes H. Maternal cytokine regulation in the pathogenesis of autism. In: Program and abstracts of the 9th Annual International Meeting for Autism Research; May 20–22, 2010; Philadelphia, PA. Abstract 136.101. | |
Blaschitz A, Hutter H, Dohr GHLA. Class I protein expression in the human placenta. Early Pregnancy. 2001;5(1):67–69. | |
Braud VM, Allan DS, McMichael AJ. Functions of nonclassical MHC and non-MHC-encoded class I molecules. Curr Opin Immunol. 1999;11(1):100–108. | |
Hunt JS, Andrews GK, Wood GW. Normal trophoblasts resist induction of class I HLA. J Immunol. 1987;138(8):2481–2487. | |
Hunt JS, Orr HT. HLA and maternal-fetal recognition. FASEB J. 1992;6(6):2344–2348. | |
Claas FH, Gijbels Y, van der Velden-de Munck J, van Rood JJ. Induction of B cell unresponsiveness to noninherited maternal HLA antigens during fetal life. Science. 1988;241(4874):1815–1817. | |
Tilburgs T, Scherjon SA, van der Mast BJ, et al. Fetal-maternal HLA-C mismatch is associated with decidual T cell activation and induction of functional T regulatory cells. J Reprod Immunol. 2009;82(2):148–157. | |
Chen SJ, Liu YL, Sytwu HK. Immunologic regulation in pregnancy: from mechanism to therapeutic strategy for immunomodulation. Clin Dev Immunol. 2012;2012:258391. | |
Noronha LE, Antczak DF. Maternal immune responses to trophoblast: the contribution of the horse to pregnancy immunology. Am J Reprod Immunol. 2010;64(4):231–244. | |
Baker JM, Bamford AI, Antczak DF. Modulation of allospecific CTL responses during pregnancy in equids: an immunological barrier to interspecies matings? J Immunol. 1999;162(8):4496–4501. | |
Flaminio MJ, Antczak DF. Inhibition of lymphocyte proliferation and activation: a mechanism used by equine invasive trophoblast to escape the maternal immune response. Placenta. 2005;26(2–3):148–159. | |
Thellin O, Coumans B, Zorzi W, Igout A, Heinen E. Tolerance to the foeto-placental ‘graft’: ten ways to support a child for nine months. Curr Opin Immunol. 2000;12(6):731–737. | |
Sykes L, MacIntyre DA, Yap XJ, Teoh TG, Bennett PR. The Th1:th2 dichotomy of pregnancy and preterm labour. Mediators Inflamm. 2012;2012:967629. | |
Wegmann TG, Lin H, Guilbert L, Mosmann TR. Bidirectional cytokine interactions in the maternal-fetal relationship: is successful pregnancy a TH2 phenomenon? Immunol Today. 1993;14(7):353–356. | |
Chaouat G, Assal Meliani A, Martal J, et al. IL-10 prevents naturally occurring fetal loss in the CBA × DBA/2 mating combination, and local defect in IL-10 production in this abortion-prone combination is corrected by in vivo injection of IFN-tau. J Immunol. 1995;154(9):4261–4268. | |
Svensson L, Arvola M, Sallstrom MA, Holmdahl R, Mattsson R. The Th2 cytokines IL-4 and IL-10 are not crucial for the completion of allogeneic pregnancy in mice. J Reprod Immunol. 2001; 51(1):3–7. | |
Fallon PG, Jolin HE, Smith P, et al. IL-4 induces characteristic Th2 responses even in the combined absence of IL-5, IL-9, and IL-13. Immunity. 2002;17(1):7–17. | |
Roth I, Corry DB, Locksley RM, Abrams JS, Litton MJ, Fisher SJ. Human placental cytotrophoblasts produce the immunosuppressive cytokine interleukin 10. J Exp Med. 1996;184(2):539–548. | |
Jones CA, Finlay-Jones JJ, Hart PH. Type-1 and type-2 cytokines in human late-gestation decidual tissue. Biol Reprod. 1997;57(2):303–311. | |
Bennett WA, Lagoo-Deenadayalan S, Brackin MN, Hale E, Cowan BD. Cytokine expression by models of human trophoblast as assessed by a semiquantitative reverse transcription-polymerase chain reaction technique. Am J Reprod Immunol. 1996;36(5):285–294. | |
Michimata T, Sakai M, Miyazaki S, et al. Decrease of T-helper 2 and T-cytotoxic 2 cells at implantation sites occurs in unexplained recurrent spontaneous abortion with normal chromosomal content. Hum Reprod. 2003;18(7):1523–1528. | |
Gilmour JS, Hansen WR, Miller HC, Keelan JA, Sato TA, Mitchell MD. Effects of interleukin-4 on the expression and activity of prostaglandin endoperoxide H synthase-2 in amnion-derived WISH cells. J Mol Endocrinol. 1998;21(3):317–325. | |
Keelan JA, Sato TA, Mitchell MD. Comparative studies on the effects of interleukin-4 and interleukin-13 on cytokine and prostaglandin E2 production by amnion-derived WISH cells. Am J Reprod Immunol. 1998;40(5):332–338. | |
Goodwin VJ, Sato TA, Mitchell MD, Keelan JA. Anti-inflammatory effects of interleukin-4, interleukin-10, and transforming growth factor-beta on human placental cells in vitro. Am J Reprod Immunol. 1998;40(5):319–325. | |
Keelan JA, Marvin KW, Sato TA, Coleman M, McCowan LM, Mitchell MD. Cytokine abundance in placental tissues: evidence of inflammatory activation in gestational membranes with term and preterm parturition. Am J Obstet Gynecol. 1999;181(6):1530–1536. | |
Lindstrom TM, Bennett PR. The role of nuclear factor kappa B in human labour. Reproduction. 2005;130(5):569–581. | |
Elliott CL, Loudon JA, Brown N, Slater DM, Bennett PR, Sullivan MH. IL-1beta and IL-8 in human fetal membranes: changes with gestational age, labor, and culture conditions. Am J Reprod Immunol. 2001;46(4):260–267. | |
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995; 155(3):1151–1164. | |
Kingsley CI, Karim M, Bushell AR, Wood KJ. CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J Immunol. 2002;168(3):1080–1086. | |
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–1061. | |
Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. | |
Apostolou I, Sarukhan A, Klein L, von Boehmer H. Origin of regulatory T cells with known specificity for antigen. Nat Immunol. 2002;3(8):756–763. | |
Chen T, Darrasse-Jèze G, Bergot AS, et al. Self-specific memory regulatory T cells protect embryos at implantation in mice. J Immunol. 2013;191(5):2273–2281. | |
Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol. 2004;5(3):266–271. | |
Moldenhauer LM, Diener KR, Thring DM, Brown MP, Hayball JD, Robertson SA. Cross-presentation of male seminal fluid antigens elicits T cell activation to initiate the female immune response to pregnancy. J Immunol. 2009;182(12):8080–8093. | |
Saito S, Nakashima A, Shima T, Ito M. Th1/Th2/Th17 and regulatory T-cell paradigm in pregnancy. Am J Reprod Immunol. 2010;63(6):601–610. | |
Leber A, Teles A, Zenclussen AC. Regulatory T cells and their role in pregnancy. Am J Reprod Immunol. 2010;63(6):445–459. | |
Tai P, Wang J, Jin H, et al. Induction of regulatory T cells by physiological level estrogen. J Cell Physiol. 2008;214(2):456–464. | |
Zhao JX, Zeng YY, Liu Y. Fetal alloantigen is responsible for the expansion of the CD4(+)CD25(+) regulatory T cell pool during pregnancy. J Reprod Immunol. 2007;75(2):71–81. | |
Robertson SA, Guerin LR, Bromfield JJ, Branson KM, Ahlstrom AC, Care AS. Seminal fluid drives expansion of the CD4+CD25+ T regulatory cell pool and induces tolerance to paternal alloantigens in mice. Biol Reprod. 2009;80(5):1036–1045. | |
Bystry RS, Aluvihare V, Welch KA, Kallikourdis M, Betz AG. B cells and professional APCs recruit regulatory T cells via CCL4. Nat Immunol. 2001;2(12):1126–1132. | |
Schaniel C, Pardali E, Sallusto F, et al. Activated murine B lymphocytes and dendritic cells produce a novel CC chemokine which acts selectively on activated T cells. J Exp Med. 1998;188(3):451–463. | |
Tang HL, Cyster JG. Chemokine Up-regulation and activated T cell attraction by maturing dendritic cells. Science. 1999;284(5415):819–822. | |
Iellem A, Mariani M, Lang R, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2001;194(6):847–853. | |
Colantonio L, Iellem A, Sinigaglia F, D’Ambrosio D. Skin-homing CLA+ T cells and regulatory CD25+ T cells represent major subsets of human peripheral blood memory T cells migrating in response to CCL1/I-309. Eur J Immunol. 2002;32(12):3506–3514. | |
Schumacher A, Brachwitz N, Sohr S, et al. Human chorionic gonadotropin attracts regulatory T cells into the fetal-maternal interface during early human pregnancy. J Immunol. 2009;182(9):5488–5497. | |
Zenclussen AC. CD4(+)CD25+ T regulatory cells in murine pregnancy. J Reprod Immunol. 2005;65(2):101–110. | |
Zenclussen AC, Gerlof K, Zenclussen ML, et al. Abnormal T-cell reactivity against paternal antigens in spontaneous abortion: adoptive transfer of pregnancy-induced CD4+CD25+ T regulatory cells prevents fetal rejection in a murine abortion model. Am J Pathol. 2005;166(3):811–822. | |
Jamieson DJ, Theiler RN, Rasmussen SA. Emerging infections and pregnancy. Emerg Infect Dis. 2006;12(11):1638–1643. | |
Mor G, Cardenas I. The immune system in pregnancy: a unique complexity. Am J Reprod Immunol. 2010;63(6):425–433. | |
Muzzio D, Zygmunt M, Jensen F. The role of pregnancy-associated hormones in the development and function of regulatory B cells. Front Endocrinol (Lausanne). 2014;5:39. | |
Polese B, Gridelet V, Araklioti E, Martens H, Perrier d’Hauterive S, Geenen V. The endocrine milieu and CD4 T-lymphocyte polarization during pregnancy. Front Endocrinol (Lausanne). 2014;5:106. | |
Racicot K, Kwon JY, Aldo P, Silasi M, Mor G. Understanding the complexity of the immune system during pregnancy. Am J Reprod Immunol. 2014;72(2):107–116. | |
Schumacher A, Costa SD, Zenclussen AC. Endocrine factors modulating immune responses in pregnancy. Front Immunol. 2014;5:196. | |
Schminkey DL, Groer M. Imitating a stress response: a new hypothesis about the innate immune system’s role in pregnancy. Med Hypotheses. 2014;82(6):721–729. | |
Zhang HJ, Patenaude V, Abenhaim HA. Maternal outcomes in pregnancies affected by varicella zoster virus infections: population-based study on 7.7 million pregnancy admissions. J Obstet Gynaecol Res. 2014;41(1):62–68. | |
American College of Obstetricians and Gynecologists. Update on immunization and pregnancy: tetanus, diptheria, and pertussis vaccination. ACOG Committee Opinion no 566. Obstet Gynecol. 2013;121(6):1411–1414. | |
American College of Obstetricians and Gynecologists (ACOG) Immunization Expert Work Group. Immunizations and routine obstetric-gynecologic care. Washington, DC: ACOG; 2013. | |
Centers for Disease Control and Prevention (CDC). Guidelines for Vaccinating Pregnant Women. 2013. Advisory Committee on Immunization Practices (ACIP) recommended immunization schedule for persons aged 0 through 18 Years. Atlanta, GA: CDC; 2013. | |
Munoz FM, Weisman LE, Read JS, et al. Assessment of safety in newborns of mothers participating in clinical trials of vaccines administered during pregnancy. Clin Infect Dis. 2014;59(Suppl 7):S415–S427. | |
Kraus TA, Engel SM, Sperling RS, et al. Characterizing the pregnancy immune phenotype: results of the viral immunity and pregnancy (VIP) study. J Clin Immunol. 2012;32(2):300–311. | |
Raj RS, Bonney EA, Phillippe M. Influenza, immune system, and pregnancy. Reprod Sci. 2014;21(12):1434–1451. | |
Brabin BJ, Romagosa C, Abdelgalil S, et al. The sick placenta-the role of malaria. Placenta. 2004;25(5):359–378. | |
Rogerson SJ, Hviid L, Duffy PE, Leke RFG, Taylor DW. Malaria in pregnancy: pathogenesis and immunity. Lancet Infect Dis. 2007;7(2):105–117. | |
Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol. 2014;35(3):347–369. | |
Mjosberg J, Berg G, Jenmalm MC, Ernerudh J. FOXP3+ regulatory T cells and T helper 1, T helper 2, and T helper 17 cells in human early pregnancy decidua. Biol Reprod. 2010;82(4):698–705. | |
Lee JH, Ulrich B, Cho J, Park J, Kim CH. Progesterone promotes differentiation of human cord blood fetal T cells into T regulatory cells but suppresses their differentiation into Th17 cells. J Immunol. 2011;187(4):1778–1787. | |
Henderson TA, Saunders PT, Moffett-King A, Groome NP, Critchley HO. Steroid receptor expression in uterine natural killer cells. J Clin Endocrinol Metab. 2003;88(1):440–449. | |
Kosaka K, Fujiwara H, Tatsumi K, et al. Human chorionic gonadotropin (HCG) activates monocytes to produce interleukin-8 via a different pathway from luteinizing hormone/HCG receptor system. J Clin Endocrinol Metab. 2002;87(11):5199–5208. | |
Siewiera J, El Costa H, Tabiasco J, et al. Human cytomegalovirus infection elicits new decidual natural killer cell effector functions. PLoS Pathog. 2013;9(4):e1003257. | |
Gabriel G, Arck PC. Sex, immunity and influenza. J Infect Dis. 2014; 209(Suppl 3):S93–S99. | |
Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS. Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry. 2013;70(7):677–685. | |
Brown AS, Begg MD, Gravenstein S, et al. Serologic evidence of prenatal influenza in the etiology of schizophrenia. JAMA Psychiatry. 2004;61(8):774–780. | |
Lamont RF, Sobel JD, Carrington D, et al. Varicella-zoster virus (chickenpox) infection in pregnancy. BJOG. 2011;118(10):1155–1162. | |
Mandelbrot L. Fetal varicella – diagnosis, management, and outcome. Prenat Diagn. 2012;32(6):511–518. | |
Baud D, Greub G. Intracellular bacteria and adverse pregnancy outcomes. Clin Microbiol Infect. 2011;17(9):1312–1322. | |
Williams D, Dunn S, Richardson A, Frank JF, Smith MA. Time course of fetal tissue invasion by Listeria monocytogenes following an oral inoculation in pregnant guinea pigs. J Food Prot. 2011;74(2):248–253. | |
Poulsen KP, Czuprynski CJ. Pathogenesis of listeriosis during pregnancy. Anim Health Res Rev. 2013;14(1):30–39. | |
Lamont RF, Sobel J, Mazaki-Tovi S, et al. Listeriosis in human pregnancy: a systematic review. J Perinat Med. 2011;39(3):227–236. | |
Rowe JH, Ertelt JM, Xin L, Way SS. Listeria monocytogenes cytoplasmic entry induces fetal wastage by disrupting maternal Foxp3+ regulatory T cell-sustained fetal tolerance. PLoS Pathog. 2012;8(8):e1002873. | |
Nguyen HT, Pandolfini C, Chiodini P, Bonati M. Tuberculosis care for pregnant women: a systematic review. BMC Infect Dis. 2014;14(617):1–10. | |
Sugarman J, Colvin C, Moran AC, Oxlade O. Tuberculosis in pregnancy: an estimate of the global burden of disease. Lancet Glob Health. 2014;2(12):e710–e716. | |
Bates M, Ahmed Y, Kapata N, Maeurer M, Mwaba P, Zumla A. Perspectives on tuberculosis in pregnancy. Int J Infect Dis. 2015;32:124–127. | |
Dewan P, Gomber S, Das S. Congenital tuberculosis: a rare manifestation of a common disease. Paediatr Int Child Health. 2014;34(1):60–62. | |
Molina RL, Diouf K, Nour NM. Tuberculosis and the obstetrician-gynecologist: a global perspective. Rev Obstet Gynecol. 2013;6(3–4):174–181. | |
Beeson JG, Duffy PE. The immunology and pathogenesis of malaria during pregnancy. Curr Top Microbiol Immunol. 2005;297: 187–227. | |
Adams Waldorf KM, McAdams RM. Influence of infection during pregnancy on fetal development. Reproduction. 2013;146(5):R151–R162. | |
Li XL, Wei HX, Zhang H, Peng HJ, Lindsay DS. A meta analysis on risks of adverse pregnancy outcomes in Toxoplasma gondii infection. PLoS One. 2014;9(5):e97775. | |
Han M, Jiang Y, Lao K, et al. sHLA-G involved in the apoptosis of decidual natural killer cells following Toxoplasma gondii infection. Inflammation. 2014;37(5):1718–1727. | |
Miller DH, Fazekas F, Montalban X, Reingold SC, Trojano M. Pregnancy, sex and hormonal factors in multiple sclerosis. Mult Scler. 2014;20(5):527–536. | |
Murase JE, Chan KK, Garite TJ, Cooper DM, Weinstein GD. Hormonal effect on psoriasis in pregnancy and post partum. Arch Dermatol. 2005;141(5):601–606. | |
Amin S, Peterson EJ, Reed AM, Mueller DL. Pregnancy and rheumatoid arthritis: insights into the immunology of fetal tolerance and control of autoimmunity. Curr Rheumatol Rep. 2011;13(5):449–455. | |
Friedman DM, Duncanson LJ, Glickstein J, Buyon JP. A review of congenital heart block. Images Paediatr Cardiol. 2003;5(3):36–48. | |
Frise CJ, Williamson C. Endocrine disease in pregnancy. Clin Med J R Coll Phys Lond. 2013;13(2):176–181. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.