")
Back to Journals » International Journal of Nanomedicine » Volume 12
The long-term fate of mesenchymal stem cells labeled with magnetic resonance imaging-visible polymersomes in cerebral ischemia
Authors Duan X, Lu L, Wang Y, Zhang F, Mao J, Cao M, Lin B, Zhang X, Shuai X , Shen J
Received 19 July 2017
Accepted for publication 16 August 2017
Published 8 September 2017 Volume 2017:12 Pages 6705—6719
DOI https://doi.org/10.2147/IJN.S146742
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dongwoo Khang
Xiaohui Duan, 1,* Liejing Lu, 1,* Yong Wang, 2 Fang Zhang, 1 Jiaji Mao, 1 Minghui Cao, 1 Bingling Lin, 1 Xiang Zhang, 1 Xintao Shuai, 2,3 Jun Shen 1
1Department of Radiology, Sun Yat-Sen Memorial Hospital, 2PCFM Lab of Ministry of Education, School of Materials Science and Engineering, 3BME Center, Zhongshan School of Medicine, Sun Yat-Sen University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Understanding the long-term fate and potential mechanisms of mesenchymal stem cells (MSCs) after transplantation is essential for improving functional benefits of stem cell-based stroke treatment. Magnetic resonance imaging (MRI) is considered an attractive and clinically translatable tool for longitudinal tracking of stem cells, but certain controversies have arisen in this regard. In this study, we used SPION-loaded cationic polymersomes to label green fluorescent protein (GFP)-expressing MSCs to determine whether MRI can accurately reflect survival, long-term fate, and potential mechanisms of MSCs in ischemic stroke therapy. Our results showed that MSCs could improve the functional outcome and reduce the infarct volume of stroke in the brain. In vivo MRI can verify the biodistribution and migration of grafted cells when pre-labeled with SPION-loaded polymersome. The dynamic change of low signal volume on MRI can reflect the tendency of cell survival and apoptosis, but may overestimate long-term survival owing to the presence of iron-laden macrophages around cell graft. Only a small fraction of grafted cells survived up to 8 weeks after transplantation. A minority of these surviving cells were differentiated into astrocytes, but not into neurons. MSCs might exert their therapeutic effect via secreting paracrine factors rather than directing cell replacement through differentiation into neuronal and/or glial phenotypes.
Keywords: mesenchymal stem cells, magnetic resonance imaging, superparamagnetic iron oxide nanoparticles, polymersome, ischemic stroke, green fluorescence protein
Erratum for this paper has been published
Introduction
Stroke is the most common cause of adult-acquired long-term disability and a major cause of death worldwide.1 Transplantation of mesenchymal stem cells (MSCs) has emerged as a novel and promising candidate strategy for the treatment of stroke owing to their multipotential, immunomodulatory, and immunodepressive properties, as well as the ease to isolate and expand them.2,3 Numerous animal studies and preliminary clinical trials have investigated the performance of MSCs in stroke therapy.2–5 Despite the success of MSCs in improving animal stroke outcomes,2,3 recent clinical trials have indicated that the therapeutic benefit of MSCs remained limited in the treatment of stroke patients.4,5 This unexpected, suboptimal therapeutic outcome might be associated with the poor survival, retention, and engraftment of transplanted stem cells.6–8 Therefore, knowledge of the long-term fate of stem cells including survival, biodistribution, migration, and differentiation after transplantation is essential for improving functional benefits of MSC-based stroke treatment. Monitoring of the fate of therapeutic cells is typically achieved by postmortem histological analysis at predetermined time points after cell transplantation – which is laborious and invasive and, thus, cannot elucidate the long-term retention of transplanted cells in the same living organism.
Cellular magnetic resonance imaging (MRI) is considered an attractive and clinically translatable tool for longitudinal tracking of stem cells because of its noninvasiveness, deep tissue penetration, lack of irradiation, superior spatial resolution, as well as the capacity for high sensitivity, low toxicity, and biocompatibility necessarily associated with cellular labeling.9 At present, in vivo real-time tracking of stem cells has been well established in ischemic stroke, which is mainly based on MRI of superparamagnetic iron oxide nanoparticles (SPION)-labeled stem cells.10–12 However, SPION-based MRI does not provide reliable information on long-term cell viability and might overestimate the survival of SPION-labeled therapeutic MSCs in myocardial infarction.13,14 The ability of SPION-based MRI to reliably evaluate long-term cell engraftment and survival remains controversial. Further studies have shown that long-lasting hypo-signal intensity on MRI mainly originated from macrophages that engulfed SPION,15,16 or from extracellular iron particles that persisted in the interstitial space after the grafted labeled cells died.17 However, almost all of these investigations mainly focused on cell fate in myocardial infarction. The long-term fate of transplanted stem cells and their relationship with dynamic signal changes on MRI in ischemic stroke remain unclear and need to be elucidated.
MSCs might exert therapeutic effects in experimental stroke models via multiple mechanisms, such as differentiation into cell types relevant to repair, modulation of the immune system, promotion of angiogenesis and neurogenesis, or secretion of neuroprotective and neurotrophic factors.18,19 Initially, it was assumed that transdifferentiation of MSCs into neuronal and glial cells to replace damaged cells might play a pivotal role in stroke recovery.18,20 Nonetheless, growing evidence suggested that the enhanced function recovery of MSCs might not be mainly associated with the transdifferentiation of MSCs.20 Since MSCs can secrete a vast array of cytokines, chemokines, angiogenic factors, and growth factors, paracrine action might play an essential role in brain regeneration in MSCs therapy.19,20 Therefore, it needs further clarification whether it is with a definite single or integrated multiple mechanisms that MSCs function in stroke.
In this study, we used SPION-loaded cationic polymersomes to label green fluorescent protein (GFP)-MSCs (GFP-MSCs). Cationic polymersomes can be used to deliver SPION into MSCs in an efficient and rapid way.21 GFP has been widely used as an excellent fluorescent marker, which can be used to verify the presence of living stem cells and eliminate the interference from hemorrhaged and dead cells.22 After GFP-MSCs were transplanted in a rat model of ischemic stroke, serial MRI was performed to track the long-term distribution and migration of grafted cells. GFP fluorescence was used as image labels to detect the survival and differentiation of grafted cells, and then to verify the findings of MRI (Figure 1). The aim of this study is to determine whether an MRI can accurately reflect the survival of MSCs and to investigate the long-term fate and potential mechanisms of MSCs in ischemic stroke therapy.
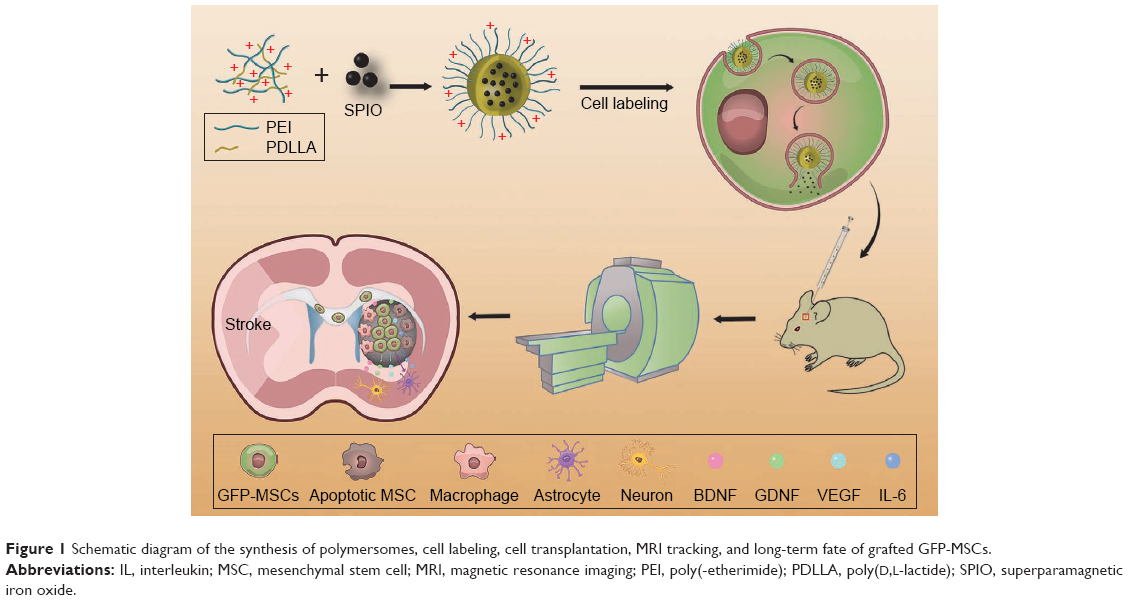 | Figure 1 Schematic diagram of the synthesis of polymersomes, cell labeling, cell transplantation, MRI tracking, and long-term fate of grafted GFP-MSCs. |
Materials and methods
Animals
Animal experiments were approved by the Institutional Animal Care Committee of Sun Yat-Sen University, and all procedures were in accordance with the guidelines for the care and use of laboratory animals and the ethics review process of Sun Yat-Sen University. Fifty-four adult male SD rats, weighing 250–280 g, were used to establish animal models of acute ischemic cerebral injury. The animals were purchased from the Animal Experiment Center of Sun Yat-Sen University and were housed in a standard animal facility with standard food and ad libitum water.
Cationic polymersomes and cell labeling
Cationic polymersomes were synthesized from diblock copolymers of poly(-etherimide) and poly(D,L-lactide) (PEI-PDLLA), and SPION-loaded cationic polymersomes were prepared and characterized as described in our previous study.21 SD rat bone marrow-derived GFP-MSCs were purchased from Cyagen Bioscience Technology Co. (Guangzhou, People’s Republic of China) and cultured in a standard culture medium consisting of Dulbecco’s Modified Eagle’s Medium (DMEM, GiBco, NY, USA) containing 10% fetal bovine serum (FBS, GiBco, NY, USA), penicillin, streptomycin, and glutamine (Sigma, St Louis, MO, USA) at 37°C in a humidified 5% CO2 atmosphere. GFP-MSCs of three to five passages were used in the following experiments. For cell labeling, GFP-MSCs were incubated with SPION-loaded cationic polymersomes with an iron concentration of 15.0 μg Fe/mL and an incubation time of 1.5 h, as previously described.21 Labeling efficacy was assessed by Prussian blue staining, transmission election microscopy (TEM), and in vitro MRI. Twenty-four hours after labeling, labeling cytotoxicity was assessed by detecting cell viability, apoptosis rate, and the intracellular reactive oxygen species (ROS) level, according to previously reported methods.21
Animal model and cell transplantation
A left cerebral acute ischemic infarct was established in 54 adult rats by using direct intraluminal suture occlusion of the middle cerebral artery (MCAO), as previously described.21,23 Two days after MCAO, animals were randomly selected to receive stereotactic injection of 5×105 GFP-MSCs pre-labeled with SPION-loaded polymersomes, unlabeled GFP-MSCs, or phosphate-buffered saline (PBS; n=18 in each group) into the right striatum (stereotaxic coordinates: 3.0 mm lateral to bregma, 0.5 mm rostral to bregma, and 6.0 mm deep from the pial surface) using a 26 s gauge needle attached to a 25-μL Hamilton syringe mounted on a microinjector. Before the injection, GFP-MSCs were suspended in 3 μL PBS, and cell viability was determined to be greater than 90%. The cell suspension was injected at a constant rate of 0.3 μL/min. After injection, the needle was left in place for an additional 15 min before retraction.
MRI
In vitro MRI was performed on a clinical 1.5-T system (Intera; Philips Medical Systems, Best, the Netherlands) with an 11-cm circular coil. For in vitro MRI, 5×105 labeled and unlabeled GFP-MSCs were harvested and suspended in 200 μL 2% agarose gel (Invitrogen, Merelbeke, Belgium), and then transferred into 96-well plates. The imaging sequences included fast spin echo (FSE) T1-weighted images (T1WI) and T2-weighted images (T2WI), fast field echo (FFE) T2*-weighted images (T2*WI), and T2-mapping. T2-mapping was acquired by using single-section multi-spin-echo sequences. The detailed acquisition parameters and derivation of T2 relaxation times from T2-mapping were described previously.21,23
In vivo MRI was performed on a clinical 3.0-T system (Achieva; Philips Medical Systems) with a 50×50 mm 4-channel phased array rat coil (Shanghai Chenguang Medical Technologies, People’s Republic of China) at 1, 2, 3, 4, 6, and 8 weeks after transplantation. The acquisition sequences included axial and coronal FSE T2WI (TR/TE =800/60 ms; NSA =2) and FFE T2*WI (TR/TE =500/18 ms; Flip angel =20°; NSA =3). Other acquisition parameters were field of view (FOV) =60 mm, matrix =256×256, section thickness/gap =1.0/0.0 mm.
The low signal volume and signal–contrast ratio of transplanted cells on T2WI and T2*WI at each time point were measured and calculated using ImageJ (NIH, Bethesda, MD, USA). For each slice, the hypointense region was manually outlined by using the method of region of interest (ROI). Slices of hypointense areas were then summated to generate low signal volume. The signal–contrast ratio at the injection site was expressed as a percentage and calculated as follows: signal–contrast ratio = (SINC − SIHA)/SINC ×100%. Where SINC represents the signal intensity of the normal cerebrum, and SIHA represents the signal intensity of the hypointense area.
Therapeutic effect
To observe the therapeutic effect of grafted cells, a modified neurological severity score (mNSS) was used to grade the neurological function recovery,24 and the infarct volume was measured on T2WI. All animals (n=6 for each group) underwent behavioral tests to obtain mNSS before MCAO and after 1, 2, 3, 4, 6, and 8 weeks after MRI examinations. mNSS is the composite of the motor (muscle status, abnormal movement), sensory (visual, tactile, and proprioceptive), and reflex tests. The higher the score, the more severe the injury. The infarct volume of the rat brain was measured using ImageJ at 1, 2, 3, 4, 6, and 8 weeks after cell transplantation. The hyperintense region of cerebral infarct in each slice and in the contralateral hemisphere were manually outlined. Slice hyperintense areas were then summated to generate infarct volume as a percentage of contralateral intact brain volume.21,23
Histology
Immediately after MRI, three rats in each group were randomly sacrificed by anesthetic overdose, then transcardially perfused with saline and, further, with 4% paraformaldehyde in PBS at 2, 4, and 8 weeks after cells transplantation. Brains were harvested and post-fixed for 1–2 h and then cryoprotected in 20% sucrose solution. Conventional paraffin-embedding was performed, and contiguous 12-μm thickness sections were cut in the coronal plane.
Immunohistochemistrical staining for the macrophage marker CD11b and Prussian blue staining were performed to identify the distribution of iron particles in grafted cells and their relationship with macrophages. In brief, sections were stained with Prussian blue staining and eosin. Then, the same slides were co-stained with antibodies against CD11b (1:200; Abcam, Cambridge, UK). To determine the survival, differentiation, phagocytosis, and migration of transplanted cells, immunofluorescence staining for glial fibrillary acidic protein (GFAP, specific for astrocytes), neuronal nuclear antigen (NeuN, specific for neurons), and CD11b (specific for activated microglia) was undertaken. In brief, after blocking by goat or donkey serum for 30 min at room temperature, sections were incubated with primary antibodies against GFAP (1:200, Abcam), NeuN (1:200, Abcam) and CD11b (1:200, Abcam) at 4°C overnight, and then with Alexa Fluor 647-conjugated secondary antibodies (1:200, Invitrogen) at room temperature in the dark for 2 h. Thereafter, the sections were washed three times with PBS, followed by incubation with DAPI for 10 min at room temperature to label the nuclei. After a final PBS wash, the sections were examined under a confocal microscope (LSM 780; Carl Zeiss). Transplanted cells were identified by the fluorescent GFP label under confocal microscopy.
The survival, differentiation, and phagocytosis of grafted cells were quantified semiquantitatively according to a previously described method.21,25,26 In brief, the total numbers of positive cells in the random, predefined rectangular fields (0.23×0.23 mm) were counted (×400 magnification) at the level of implantation. The fields were positioned in the injection site or corpus callosum by referring to positive areas determined by Prussian blue staining on an adjacent coronal section. The total numbers of GFP-MSCs co-localizing with other markers (GFAP, NeuN, and CD11b), as detected by double staining, were counted. Five fields of each section were randomly selected. The total number of GFP-positive survived cells was counted and the percentage of GFP-positive cells was calculated per field (0.23×0.23 mm2) in the injection site by using the following formula: (GFP-positive cells/total DAPI-stained nuclei) ×100%. Five fields of each section were randomly selected.25
TUNEL analysis
The TUNEL assay was applied to identify apoptosis of grafted MSCs at 1, 2, and 3 weeks after transplantation. TUNEL staining was performed by using an in situ apoptosis detection kit (Life Technologies Corporation, USA) according to the manufacturer’s instructions. After TUNEL staining, the sections were examined under a confocal microscope (LSM 780; Carl Zeiss). The total number of TUNEL-positive GFP-MSCs was counted and the percentage of TUNEL-positive cells was calculated per field (0.23×0.23 mm2) in the injection site by using the following formula: (TUNEL-positive nuclei/total DAPI-stained nuclei) ×100%. Five fields of each section were randomly selected.25,26
Enzyme-linked immunosorbent assay (ELISA)
ELISA assay was conducted to determine the neurotrophic factors and cytokine released from grafted MSCs at 2, 4, and 8 weeks after cell transplantation. After lethal pentobarbital injection, rat brains were rapidly removed and the ipsilateral cerebral hemisphere into which cell injection was done was dissected on ice immediately. Then, the samples were homogenized in 1 g/mL homogenate buffer, and centrifuged with a speed of 10,000 rpm for 10 minutes at 4°C. Supernatants were collected for secretion measurement. The concentrations of BDNF, GDNF, VEGF, and IL-6 were determined with ELISA kit (Boster, Wuhan, People’s Republic of China) according to the manufacturer’s instructions. All samples were analyzed in triplicate.
Statistical analysis
Data are presented as mean ± SD. Statistical comparisons were conducted by using two independent samples Student’s t-test, one-way analysis of variance (ANOVA), or two-way repeated measures ANOVA. Multiple comparisons were analyzed using the Student–Newman–Keuls test. Statistical analysis was conducted by using SPSS 15.0 for Windows (SPSS Inc., Chicago, IL, USA). p-value <0.05 was considered statistically significant.
Results
Cell-labeling efficacy and cytotoxicity
On in vitro MRI, GFP-MSCs labeled with SPION-loaded polymersomes showed an obvious hypointense signal on T1WI, T2WI, and T2*WI, and had a decreased T2 value compared with unlabeled GFP-MSCs (Figure 2A). Prussian blue staining revealed that almost 100% of the labeled GFP-MSCs were positively stained, with abundant blue-stained particles inside cells (Figure 2B). TEM showed that dense iron particles were present in the cytosol (Figure 2D). No such blue-stained particles and iron particles were seen in unlabeled cells (Figure 2C and E). Cell viability, apoptosis rate, and intracellular ROS level in labeled cells were not significantly different from that in unlabeled cells (p>0.05; Figure 3). These indicated no noticeable detrimental influence from polymersomes-based labeling of cells.
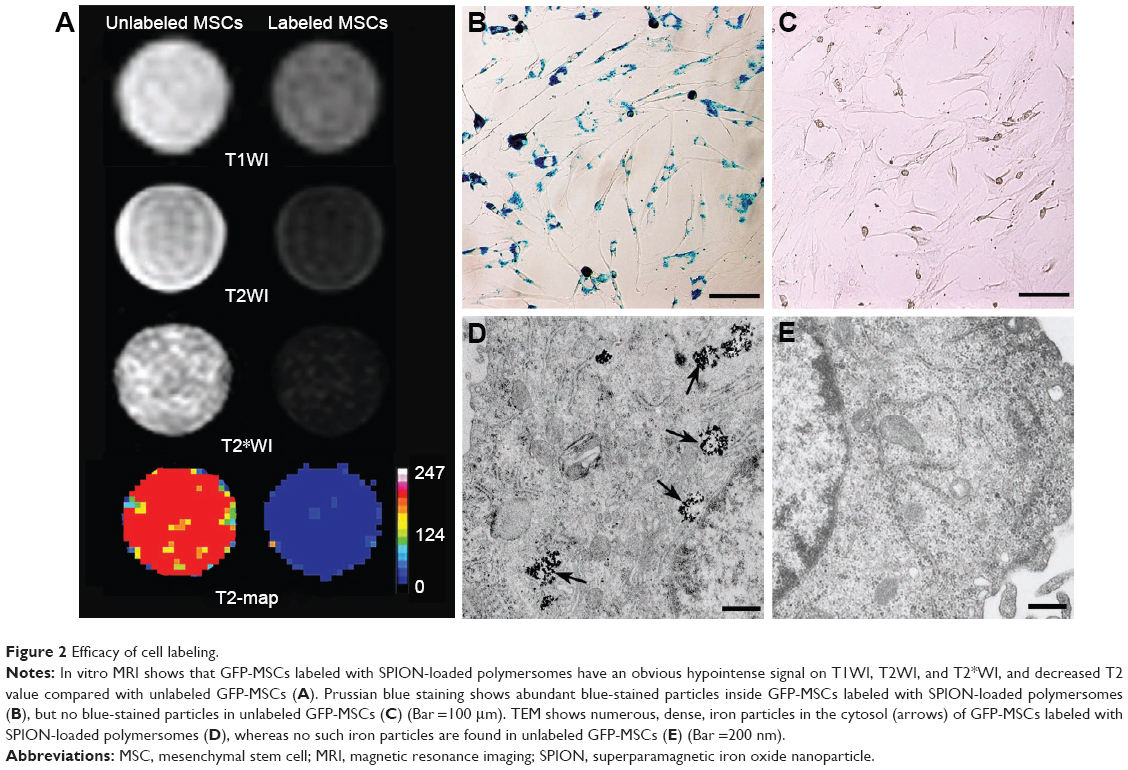 | Figure 2 Efficacy of cell labeling. |
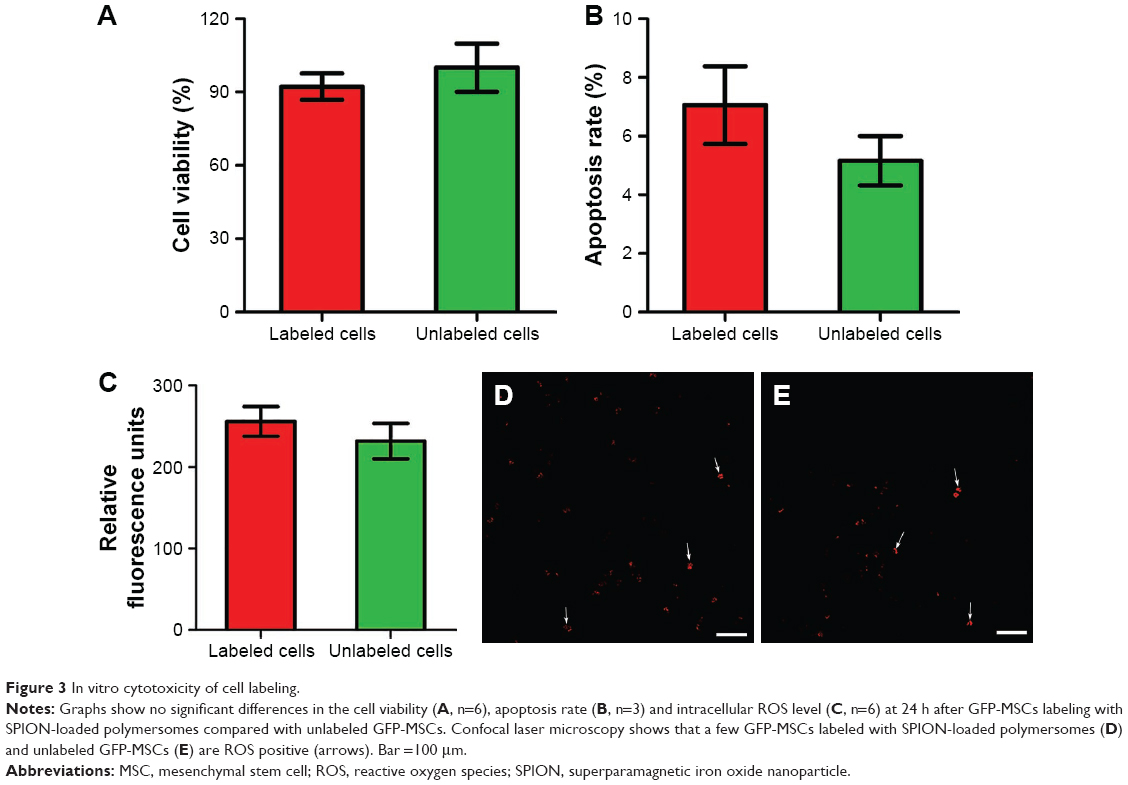 | Figure 3 In vitro cytotoxicity of cell labeling. |
Therapeutic effect
The infarcted brain in animals grafted with polymersomes-labeled GFP-MSCs, unlabeled GFP-MSCs, and PBS appeared as hyperintense signals on T2WI (Figure 4A–C). The infarct volume in three groups showed a slow decline from Week 1 to Week 8 after transplantation. There were significantly lower infarct volumes in animals grafted with labeled and unlabeled GFP-MSCs than that in animals treated with PBS at 4, 6, and 8 weeks after transplantation (p<0.05), whereas no significant difference was found between animals grafted with labeled and unlabeled GFP-MSCs at each time point (p>0.05; Figure 4D).
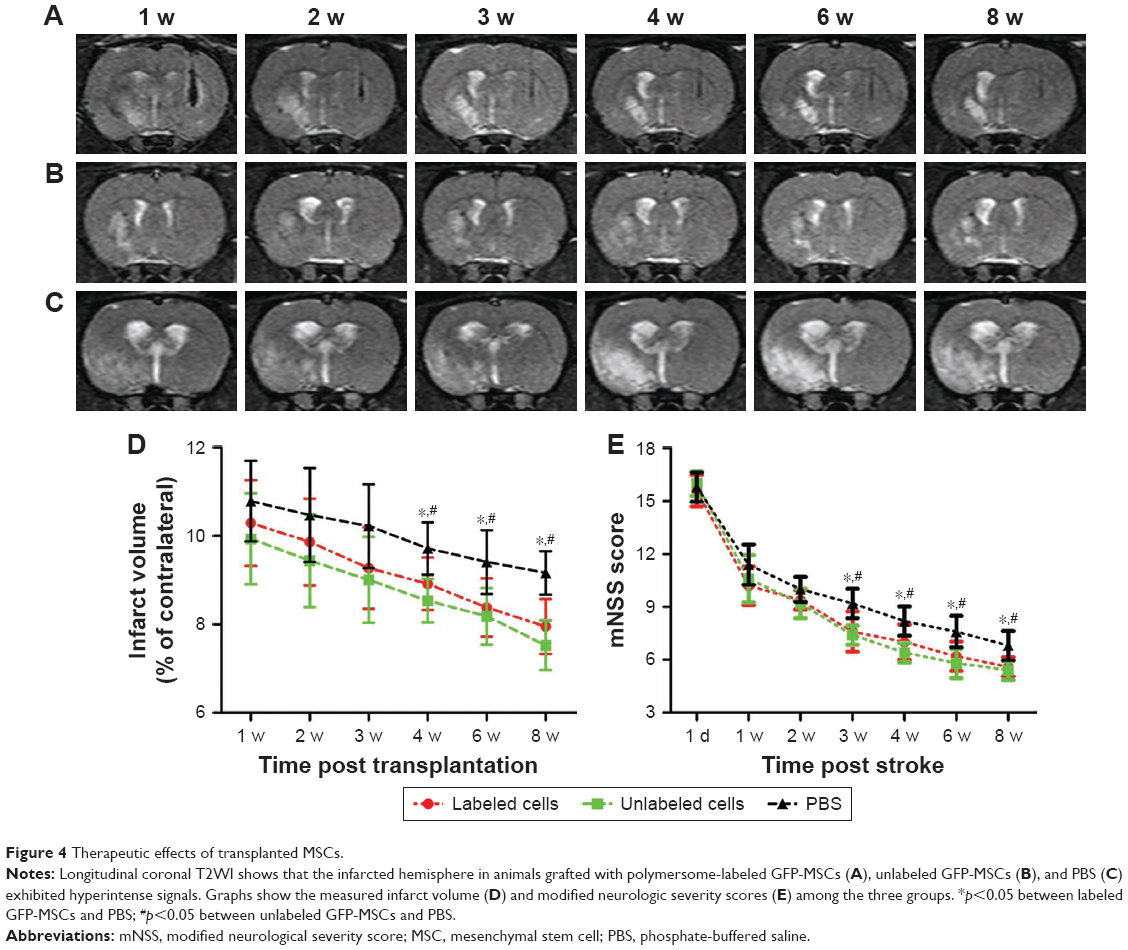 | Figure 4 Therapeutic effects of transplanted MSCs. |
Neurological functions in three groups were gradually recovered over time. mNSS was significantly lower in animals treated with labeled MSCs or unlabeled GFP-MSCs than that in animals treated with PBS at 3, 4, 6, and 8 weeks after MCAO (p<0.05). No significant difference was found between animals grafted with labeled and unlabeled GFP-MSCs at each time point (Figure 4E).
In vivo MRI and cell apoptosis
After transplantation, the grafted GFP-MSCs pre-labeled with SPION-loaded polymersomes manifested as strong hypointense areas in the left striatum on T2WI (Figure 4A) and T2*WI (Figure 5A). These hypointense signals were markedly reduced in size at 2 weeks, then gradually reduced in size but remained until 8 weeks after transplantation on T2WI and T2*WI. These hypointense areas in the right striatum were much smaller on T2WI, compared with T2*WI. A spotty slightly hypointense signal was found in the ipsilateral corpus callosum on T2*WI at 2 weeks after transplantation in animals grafted with polymersome-labeled GFP-MSCs (Figure 5A). No such hypointense signals were found in the striatum and corpus callosum in animals grafted with unlabeled GFP-MSCs and PBS (Figure 5B and C).
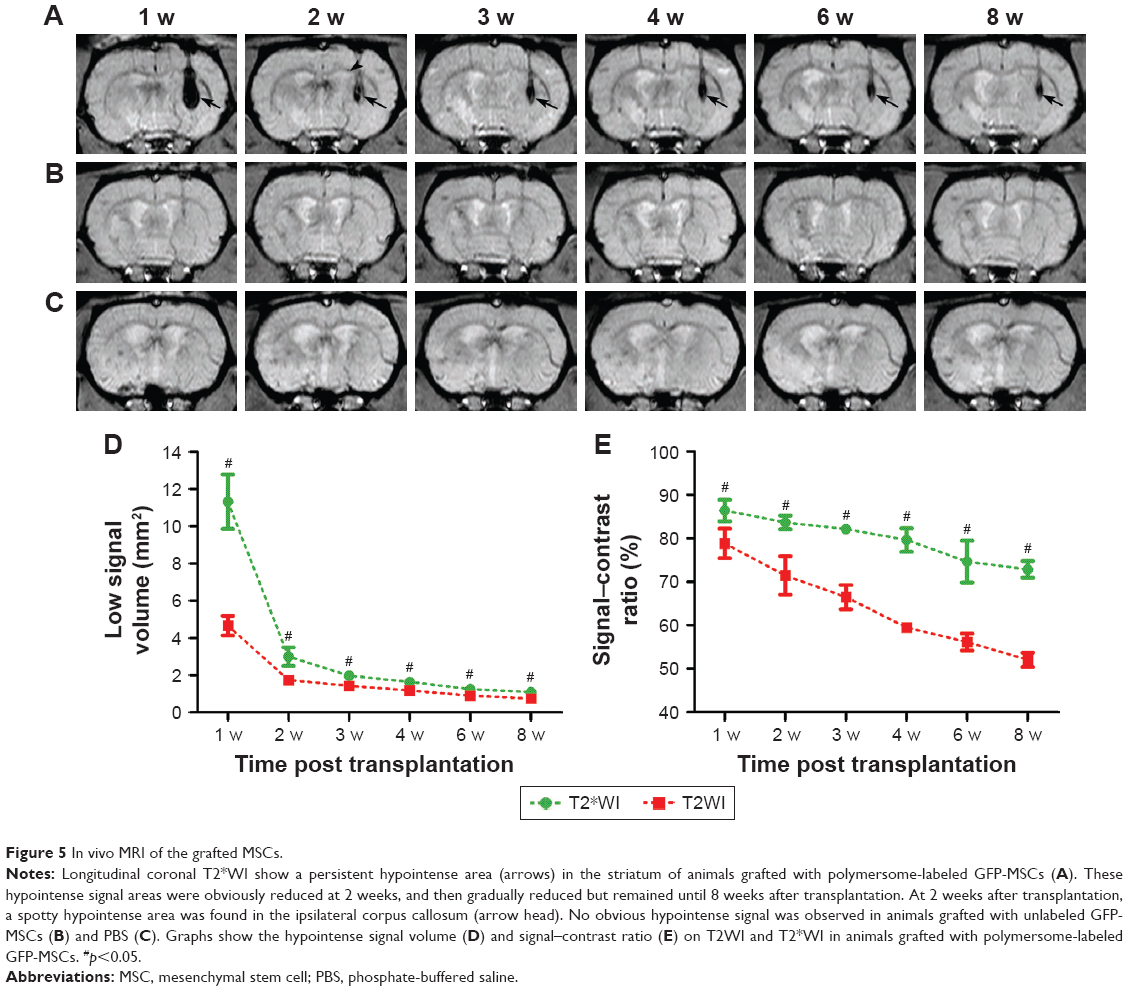 | Figure 5 In vivo MRI of the grafted MSCs. |
The dynamic change of hypointense signal volume and signal–contrast ratio on T2WI and T2*WI over time were semiquantitatively calculated. A rapid and significant decrease of the hypointense signal volume was found at 2 weeks, followed by a slight and gradual decrease at 3, 4, 6, and 8 weeks after cell transplantation (p<0.05; Figure 5D). A gradual and moderate decrease was found in the signal–contrast ratio on T2WI and T2*WI at 2, 3, 4, 6, and 8 weeks (Figure 5E). Additionally, the low signal volume and signal–contrast ratio on T2WI were significantly smaller than those on T2*WI at each time point (p<0.05).
To determine the reason of the rapid decrease of low signal volume at 2 weeks after cells transplantation, the survival and apoptosis of GFP-MSCs were assessed at 1, 2, and 3 weeks after transplantation. Histology showed that numerous GFP-MSCs were found at the implantation site in labeled and unlabeled GFP-MSCs groups 1 week after transplantation, indicating successful cell engraftment and robust survival (Figure 6). The number of GFP-MSCs decreased markedly at 2 weeks after transplantation. TUNEL assay confirmed that the total number of apoptotic GFP-MSCs peaked at 1 week after transplantation (Figure 6). There were no statistical differences in the survival and apoptosis of GFP-MSCs between the labeled and unlabeled GFP-MSCs groups at each time point (p>0.05). In addition, the percentage of TUNEL-positive cells in animals grafted with labeled and unlabeled cells was significant higher than that in animals grafted with PBS at each time point (p<0.05), whereas no significant difference was found between animals grafted with labeled and unlabeled cells at each time point (p>0.05; Figure 6).
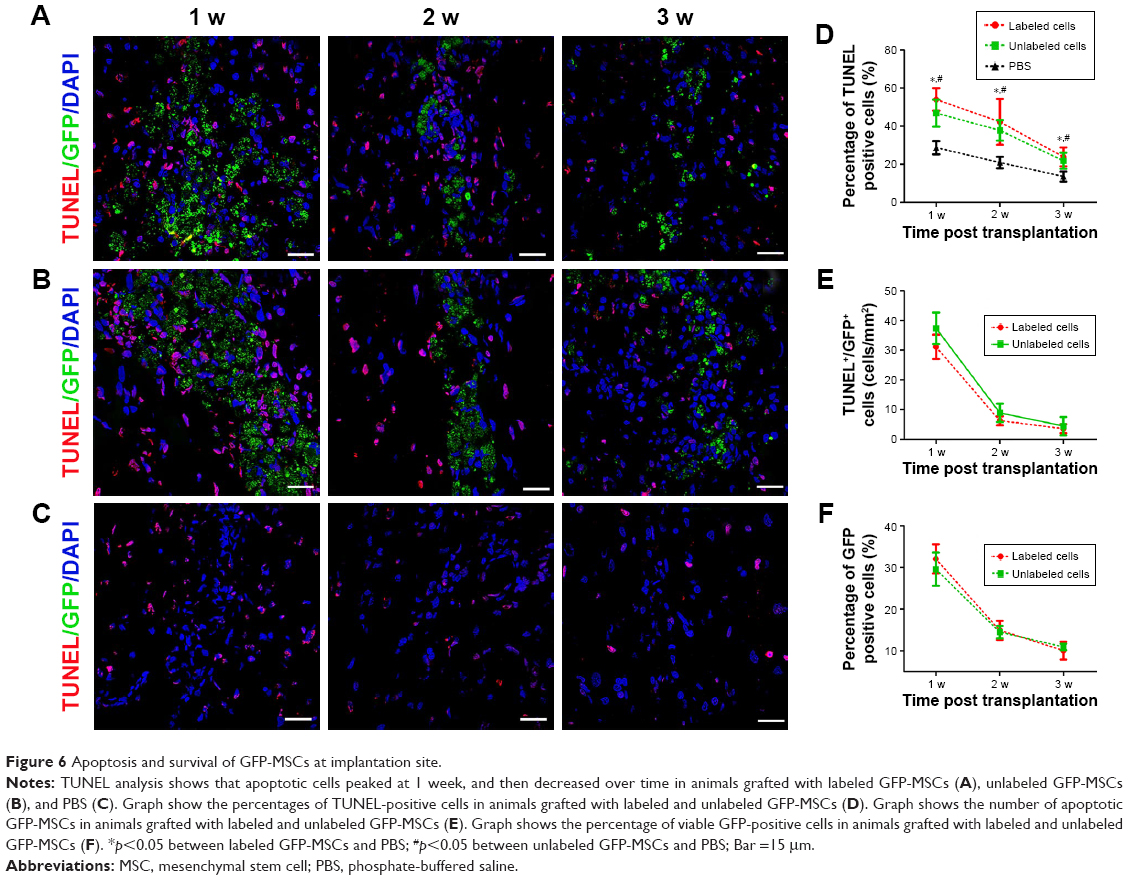 | Figure 6 Apoptosis and survival of GFP-MSCs at implantation site. |
Relationship between SPIONs and macrophages
Prussian blue staining showed that numerous positive cells were present in the injection site as were a small number of positive cells in the right corpus callosum in animals treated with labeled GFP-MSCs at 2 weeks after transplantation, which then gradually decreased over time (Figure 7). Iron and CD11b co-staining showed that a large fraction of iron-positive cells was also positive for CD11b both in the injection site and the corpus callosum, suggesting the uptake of SPION in reactive macrophages (Figure 7). The percentages of iron+/CD11b+ cells in the injection site and corpus callosum were 53.9%±5.4%, 48.1%±4.2%, and 42.0%±3.6%, and 19.2%±3.9%, 15.2%±3.2%, and 9.1%±2.9%, respectively, at 2, 4, and 8 weeks after transplantation. The percentages of iron+/CD11b+ cells in the injection site and corpus callosum was significantly reduced at 8 weeks than that at 2 weeks after transplantation (p<0.05). These findings suggest that most of the transplanted labeled MSCs did not survive, and their iron content was engulfed by resident macrophages. Intracerebral iron particles were cleared slowly with time.
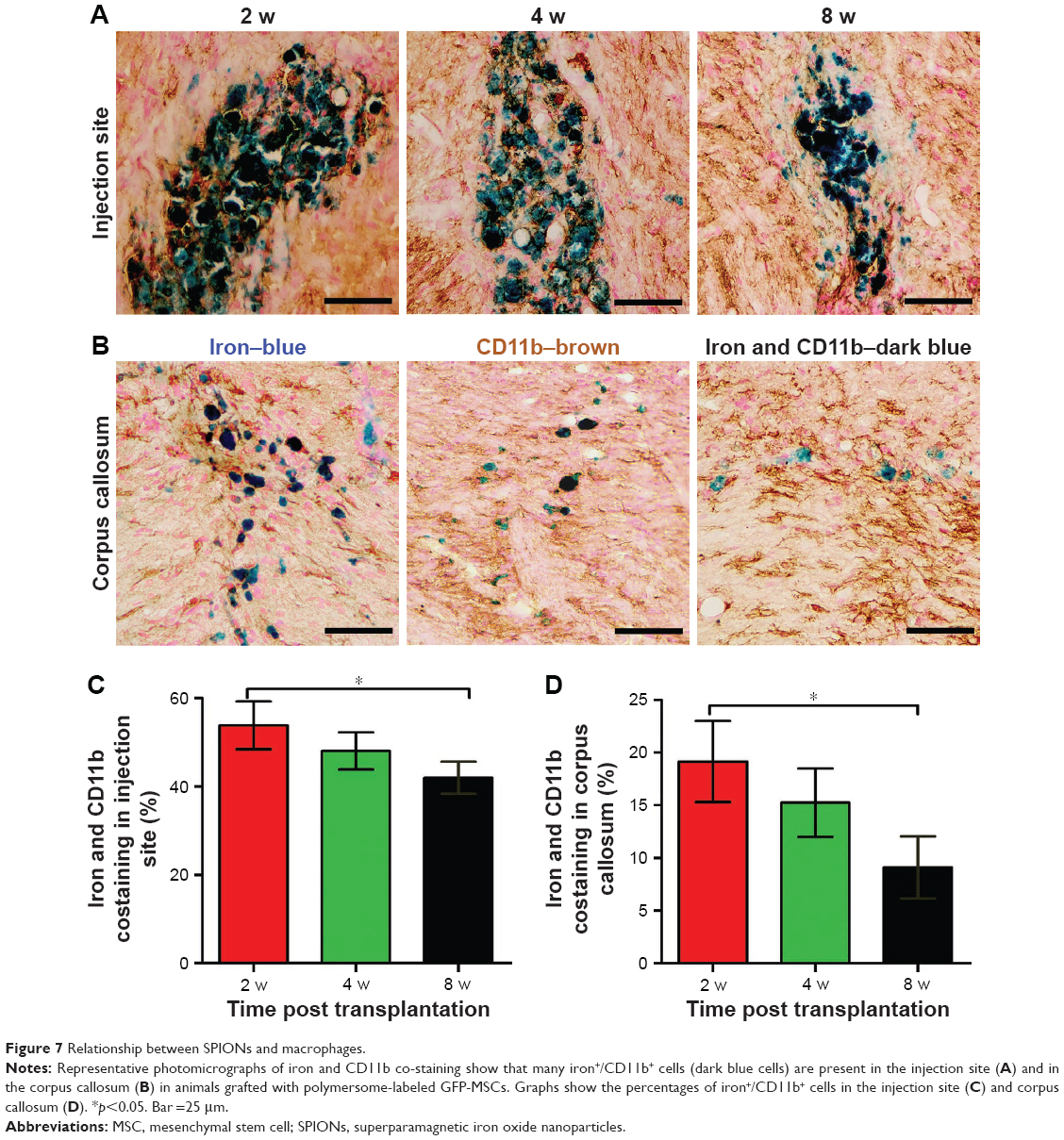 | Figure 7 Relationship between SPIONs and macrophages. |
Survival, differentiation, and phagocytosis of grafted GFP-MSCs
Immunofluorescence staining showed that GFP-MSCs survived in the implantation site (Figure 8). The percentages of surviving GFP-MSCs were 13.67%±2.10%, 5.77%±1.61%, and 2.17%±0.51% in animals grafted with labeled GFP-MSCs and 14.73%±2.32%, 6.56%±1.62%, and 2.30%±0.59% in animals grafted with unlabeled GFP-MSCs, respectively, at 2, 4, and 8 weeks after transplantation. The percentage of surviving GFP-MSCs that were positive for NeuN, GFAP, and CD11b in the injection site were 0%, 4.9%, and 50% at 2 weeks; 0%, 3.2%, and 45% at 4 weeks; and 0%, 1.5%, and 35% at 8 weeks after transplantation, respectively. There was a gradual decline in the number of surviving GFP-MSCs, as well as in the differentiation and phagocytosis of GFP-MSCs over time. No significant difference was found in survival, differentiation, and phagocytosis between the labeled and unlabeled GFP-MSCs groups (p<0.05). These results indicated that most of the grafted GFP-MSCs were dead and phagocytosed by macrophages, only a few viable cells were differentiated into GFAP-positive astrocytes, while no viable GFP-MSCs were differentiated into NeuN-positive neurons (Figure 8). A small portion of GFP-MSCs migrated into the right corpus callosum. Only a few GFP-positive cells were positive for GFAP and CD11b in the right corpus callosum, indicating that migrating cells were not differentiated into neurons, and were phagocytosed by macrophages (Figure 9).
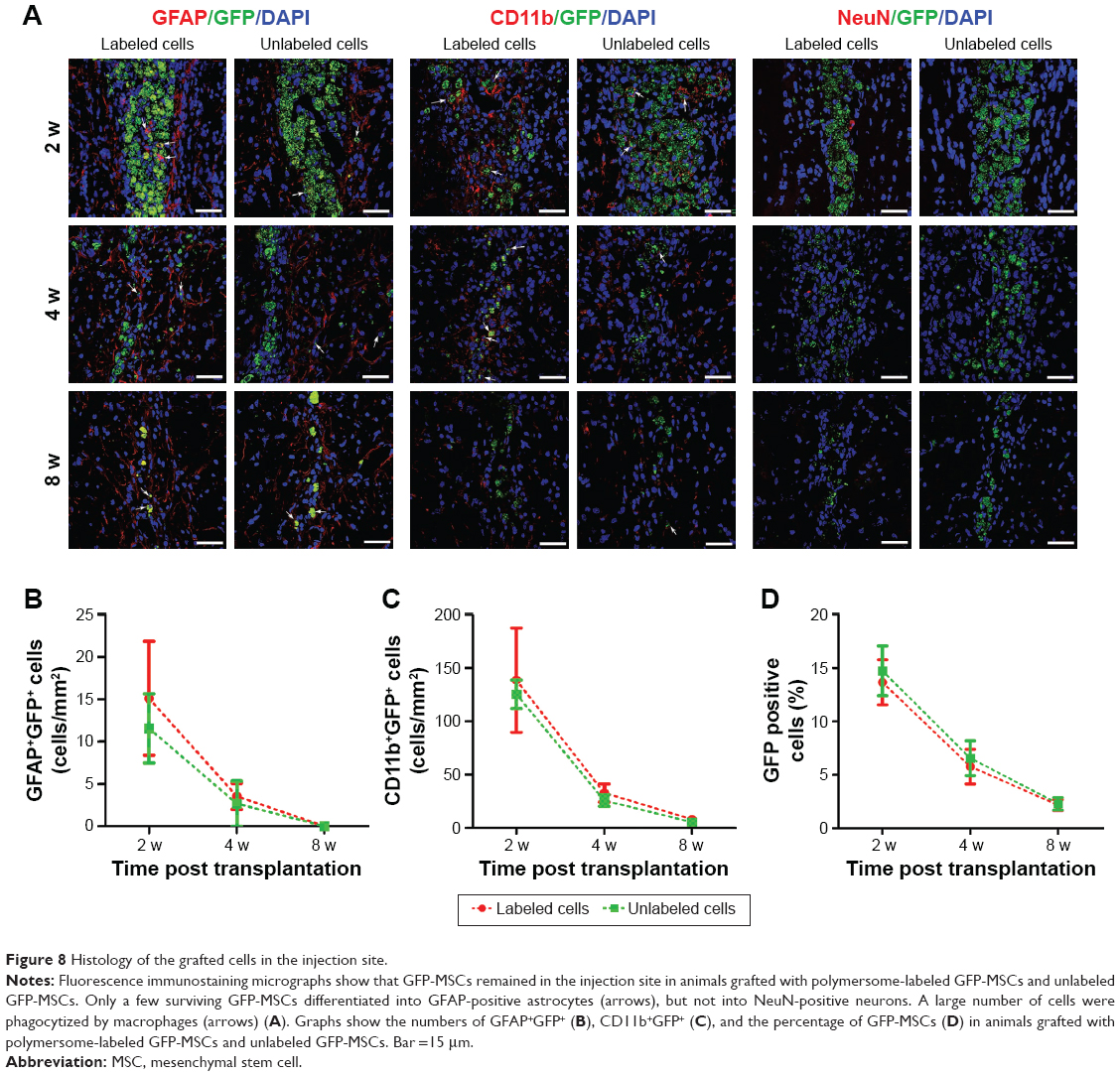 | Figure 8 Histology of the grafted cells in the injection site. |
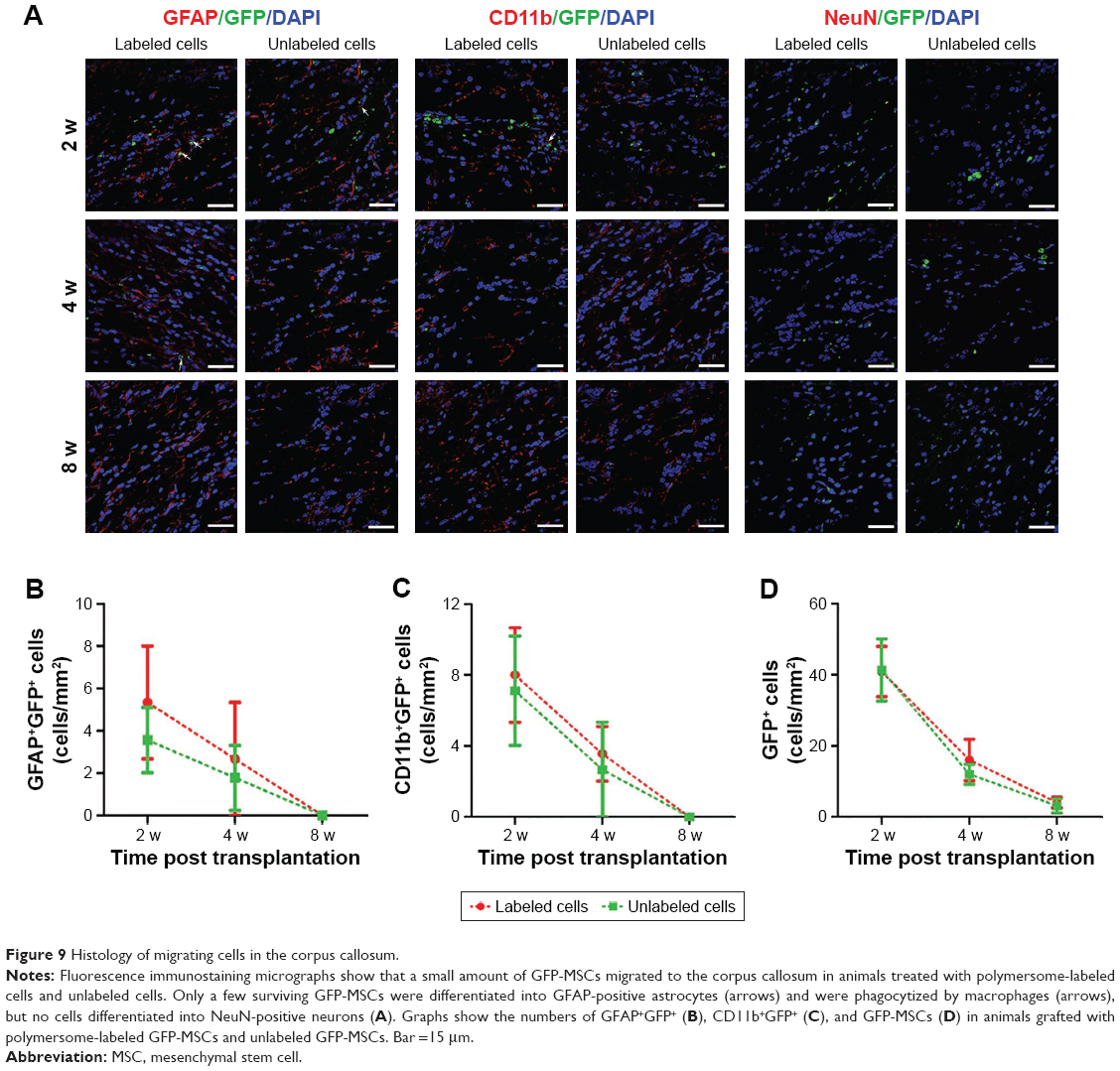 | Figure 9 Histology of migrating cells in the corpus callosum. |
ELISA
The secretion levels of growth factors (BDNF and GDNF), angiogenic factors (VEGF), and cytokines (IL-6) in three groups are shown in Figure 10. The secretion levels of BDNF, GDNF, VEGF, and IL-6 in labeled and unlabeled GFP-MSCs groups were significantly higher than that in the PBS group at each time point (p<0.05), but no significant difference was found between the labeled and unlabeled GFP-MSCs groups (p>0.05). In addition, all of these secretions peaked at 2 weeks, but decreased at 4 and 8 weeks after transplantation in labeled and unlabeled GFP-MSCs groups.
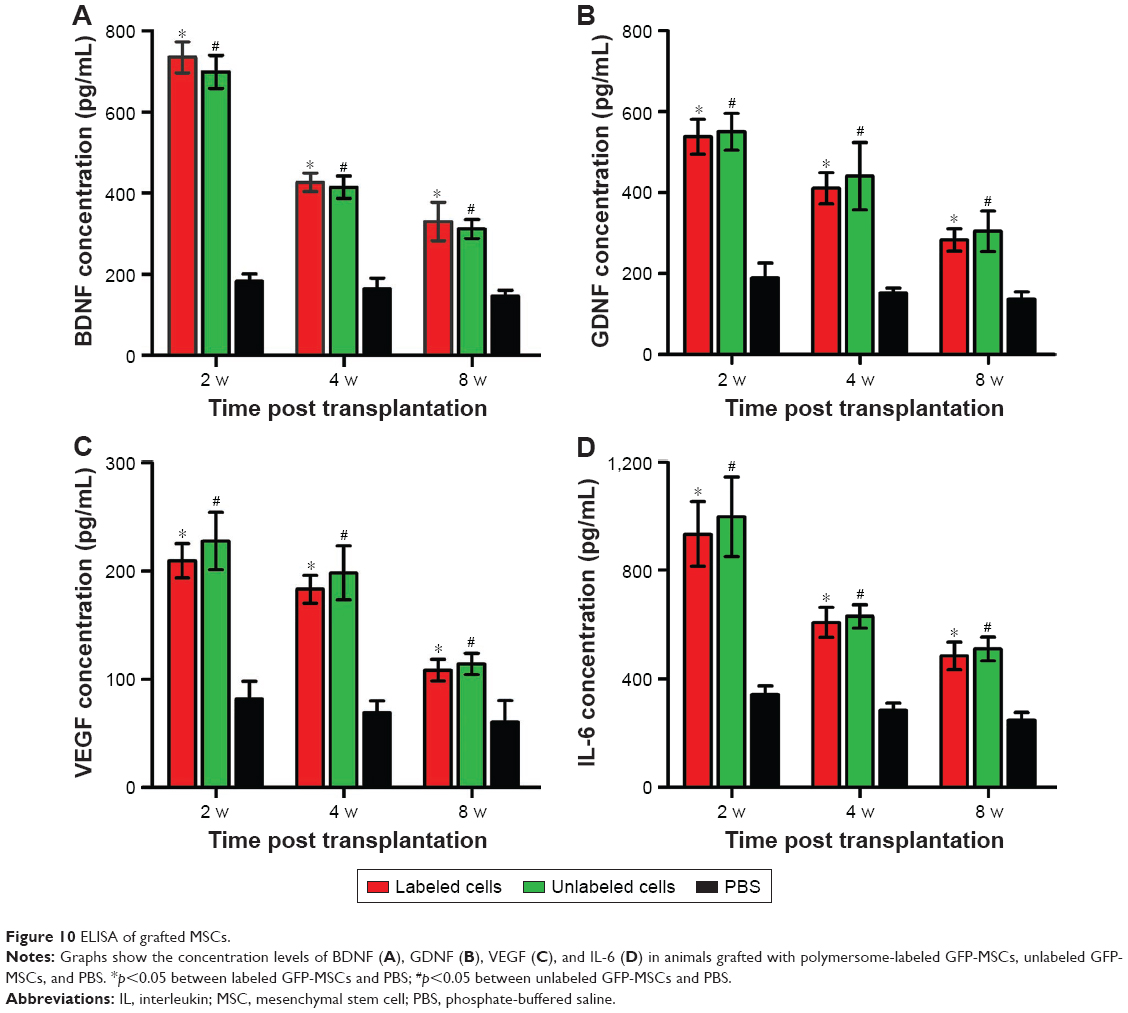 | Figure 10 ELISA of grafted MSCs. |
Discussion
Our results showed that transplantation of MSCs could improve the functional outcome and reduce the infarct volume of stroke brain. In vivo MRI can verify the biodistribution and migration of grafted cells when pre-labeled with SPION-loaded polymersomes. The dynamic change of low signal volume on MRI can reflect the tendency of cell survival and apoptosis, but may overestimate long-term graft survival owing to the presence of iron-laden macrophages around cell grafts. Most of the grafted MSCs died early; only a small fraction of grafted cells survived up to 8 weeks after transplantation. A minority of surviving cells differentiated into astrocytes, but not into neurons. MSCs might exert their therapeutic effect via paracrine factors rather than in vivo transdifferentiation into neuronal and/or glial phenotypes.18,19
Maintaining long-term survival, accurate homing, and migration of stem cells must first be accomplished before clinical applications in humans.27 MRI has been the most widely investigated imaging modality for in vivo tracking of the biodistribution and migration of stem cells in a stroke.9–11,23 In our study, MSCs were labeled by using SPION-loaded cationic polymersomes with a relatively low SPION concentration and an obviously short labeling period. This labeling had no adverse effects on cell viability, apoptosis and oxidative stress. The grafted, labeled MSCs was detected as a persistent hypointense signal in the injection site up to 8 weeks after transplantation. Histology showed that a small portion of grafted cells migrated into the corpus callosum. These cells appeared as a slight hypointense signal on MRI as well. A previous study has revealed that a hypointense signal outside the primary implantation site indicated the migration of grafted stem cells.11 The migrational behavior of biologically active cells can be accurately tracked by MRI, based on the emergence of such a developing signal. Taken together, MRI is well-suited for detection and verification of the cell distribution and migration with high image resolution.
However, whether MRI tracking of stem cells can reliably evaluate long-term cell engraftment and cell survival remains controversial.9,13,16,17 It has been reported that SPION-based MRI cannot provide reliable information on long-term viability and might overestimate the survival of SPION-labeled stem cells in myocardial infarction.13,14 In our study, the dynamic change of low signal volume on MRI was consistent with the change pattern of the number of viable cells. Notably, a remarkable drop in low signal volume occurred at 2 weeks after transplantation. This finding has not been reported in previous studies on myocardial infarction.13,14,16 As histology revealed a remarkable cell apoptosis at 1 week after transplantation, the apoptosis of grafted cells might mainly contribute to the drop of such low signal volume. Our results also showed that a small fraction of MSCs can survive for a long period of 8 weeks after transplantation. Along with the surviving cells in the implantation site, there were reactive macrophages that phagocytosed dead, grafted cells. This results in overestimation of the true size of the viable stem cell mass with MRI, which is consistent with the findings observed in acute myocardial infarction.13,14,16 Taken together, our results indicate that MRI of SPION-labeled stem cells can be used to verify the successful implantation of stem cells, visualize the biodistribution, and alert substantial loss of viable stem cell mass.
At present, the cause of the persistent hypointensity on MRI, especially after much of the grafted labeled cells died, remains controversial. Hypointensities detected on MRI refer only to iron oxide particles themselves, but cannot give any information on the type or viability of labeled cells.15 Besides iron particles retained in labeled cells, the persistence of hypointensities may be affected by the imbalance between the massive iron oxide particles released from the dead labeled stem cells and the relatively restricted iron oxide clearance capacity of the brain.17 It is well known that the iron oxide clearance was mainly through endogenous macrophages/microglia engulfment, which are then taken away by cerebral blood flow.16,17 In contrast, persistent hypo-signal intensities on MRI after stem cell transplantation were found to mainly arise from hematogenous macrophages that engulfed SPION in myocardial infarction,13,14 or extracellular iron particles entrapped in the myocardial interstitial space.17 The lack of monocyte-macrophage-like cells and blood flow in the ischemic or necrotic myocardium caused slow clearance of iron oxide that is released from dead SPION-labeled stem cells in the ischemic or necrotic region. The residual SPIONs in the myocardial interstitial space, thus, mainly resulted in the hypo-signal intensity on MRI.17 In our study, histology revealed that persistent hypointensities mainly resulted from SPION-engulfed macrophages, rather than the extracellular free iron oxide in the interstitial space.
MSCs have shown great promise in regenerating the injured brain and restoring its function in preclinical studies.2 However, several clinical trials have shown that the therapeutic effect was transient and marginal.6,28 Low cell survival and poor engraftment at the site of injury are thought to be major limiting factors mediating unsatisfactory outcomes in MSC-based therapeutics. A survival rate of 1%–32% of implanted cells in myocardial infarction25 and less than 3% engraftment rate in heart and liver injury models have been reported.7,29 In our study, only 13.67%–14.73% of grafted MSCs survived in the injection site, and the number of viable MSCs reduced gradually over time. Most of grafted MSCs died, initially, because of apoptosis and, subsequently, because of phagocytosis by macrophages. A small portion of grafted MSCs survived even at 8 weeks after transplantation. The survival rate of grafted MSCs in our study was slightly higher than in previous studies on the normal brain, stroke, and myocardial infarction.7,29,30 This may be attributed to the nature of the ischemic brain and the different experiment models. The intact adult brain has a limited capacity to direct the differentiation of grafted stem cells,30 whereas the injured brain expresses morphogenic cues that appear to foster donor plasticity.31
The exact causes of low cell survival and poor engraftment remains unclear. Possible reasons might include hypoxia, serum deprivation-induced apoptosis, necrosis, mechanical damage, immunological rejection, and infiltration of inflammatory microglia.7,8,32 Among them, hypoxia- and/or serum deprivation-induced apoptosis and immunological rejection are the major factors.30,32 In our study, the total number of apoptotic GFP-MSCs peaked at 1 week, and then gradually decreased over time. It indicates that protection of grafted stem cells against apoptosis in the acute phase is critical for successful cellular therapy. In addition, immunological rejection is another major obstacle for the survival and stable integration of stem cells grafted to the adult brain.30 In our study, reactive microglia were present after cell transplantation. Consistent with reports in previous studies,30,33 most of these microglia engulfed iron particles from dead MSCs, which peaked at 2 weeks and persisted at least 8 weeks after transplantation, Traditionally, MSCs have been considered to have multiple immunosuppressive properties, which would not stimulate alloreactivity, but could exert suppressive effects on T-cell proliferation, stimulation, and mixed lymphocyte reactions in host tissues.34 However, such an immune-privileged status of MSCs was challenged in in vivo applications.30 A recent study showed that autologous and allogeneic MSCs did not activate microglia themselves in vitro, but highly activated microglia after implantation in vivo.33 The reason might be that the dynamic interaction between immune cells and the injured neural parenchyma associated with surgical cell transplantation enables the brain to vigorously respond to the grafted stem cells, resulting in poor survival of the majority of transplanted MSCs.33
The precise mechanisms underlying MSC-mediated functional recovery in ischemia stroke are still unclear. A number of divergent mechanisms have been proposed, such as differentiation into cell types relevant to repair and cell replacement, modulation of the immune system, secretion of neuroprotective and neurotrophic factors, and promotion of angiogenesis and neurogenesis.18,19 In our study, only a few viable MSCs were differentiated into astrocytes, but not into neurons. Both labeled and unlabeled GFP-MSCs offered a therapeutic benefit on stroke. This indicates that cell replacement might not mainly be responsible for the beneficial effect of stem cells. MSCs can secrete a vast array of cytokines, chemokines, angiogenic factors, and growth factors in the host, which might play an essential role in brain regeneration in the treatment of stroke.19 In our study, expression levels of BDNF, GDNF, VEGF, and IL-6 in animals grafted with labeled and unlabeled MSCs increased markedly and showed a peak at 2 weeks, followed by a gradual decrease at 4 and 8 weeks after transplantation. This dynamic secretion change was well matched with the gradual decrease in the number of surviving stem cells. BDNF and GDNF are essential neurotrophic factors that can mediate neuroprotective effects on ischemic brain injury.35 VEGF expression promotes angiogenesis in the ischemic brain.36 IL-6 plays a vital role in immune regulation mediated by MSCs in the treatment of ischemic stroke.37,38 Therefore, the main mechanisms of MSCs in stroke therapy are probably associated with a paracrine effect in neuroprotective, angiogenic, and immunoregulatory ways via BDNF, GDNF, VEGF, and IL-6. However, further in-depth investigations are needed to elucidate the exact mechanisms underlying MSC-mediated tissue regeneration and functional recovery in ischemia stroke.
In conclusion, our results suggest that MRI of stem cells labeled with SPION-loaded polymersomes can be used to verify the biodistribution and migration of grafted stem cells and to imply the massive viable cell loss. However, this technique may also overestimate the survival of labeled stem cells because of the coexistence of immunoreaction. MSCs might exert their therapeutic effects via paracrine action, rather than direct cell replacement through differentiation into neuronal and/or glial phenotypes.
Acknowledgments
This work is supported by the Project Supported by Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2017), National Natural Science Foundation of China (Grant Nos: 81071028, 81371607, 81571739), National Basic Research Program of China (Grant No: 2015CB755500), Natural Science Foundation of Guangdong Province of China (Grant No: 2014A030312018), and Elite Young Scholars Program of Sun Yat-sen Memorial Hospital (J201403).
Disclosure
The authors report no conflicts of interest in this work.
References
Feigin VL, Forouzanfar MH, Krishnamurthi R, et al; Global Burden of Diseases, Injuries, and Risk Factors Study 2010 (GBD 2010) and the GBD Stroke Experts Group. Global and regional burden of stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet. 2014;383(9913):245–254. | ||
Steinberg GK, Kondziolka D, Wechsler LR, et al. Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: a phase 1/2a study. Stroke. 2016;47(7):1817–1824. | ||
Vu Q, Xie K, Eckert M, Zhao W, Cramer SC. Meta-analysis of preclinical studies of mesenchymal stromal cells for ischemic stroke. Neurology. 2014;82(14):1277–1286. | ||
Prasad K, Sharma A, Garg A, et al; InveST Study Group. Intravenous autologous bone marrow mononuclear stem cell therapy for ischemic stroke: a multicentric, randomized trial. Stroke. 2014;45(12):3618–3624. | ||
Bang OY, Lee JS, Lee PH, Lee G. Autologous mesenchymal stem cell transplantation in stroke patients. Ann Neurol. 2005;57(6):874–882. | ||
Ikegame Y, Yamashita K, Nakashima S, et al. Fate of graft cells: what should be clarified for development of mesenchymal stem cell therapy for ischemic stroke? Front Cell Neurosci. 2014;8:322. | ||
Hyun JS, Tran MC, Wong VW, et al. Enhancing stem cell survival in vivo for tissue repair. Biotechnol Adv. 2013;31(5):736–743. | ||
Shafiq M, Jung Y, Kim SH. Insight on stem cell preconditioning and instructive biomaterials to enhance cell adhesion, retention, and engraftment for tissue repair. Biomaterials. 2016;90:85–115. | ||
Kircher MF, Gambhir SS, Grimm J. Noninvasive cell-tracking methods. Nat Rev Clin Oncol. 2011;8(11):677–688. | ||
Li L, Jiang W, Luo K, et al. Superparamagnetic iron oxide nanoparticles as MRI contrast agents for non-invasive stem cell labeling and tracking. Theranostics. 2013;3(8):595–615. | ||
Hoehn M, Küstermann E, Blunk J, et al. Monitoring of implanted stem cell migration in vivo: a highly resolved in vivo magnetic resonance imaging investigation of experimental stroke in rat. Proc Natl Acad Sci U S A. 2002;99(25):16267–16272. | ||
Janowski M, Walczak P, Kropiwnicki T, et al. Long-term MRI cell tracking after intraventricular delivery in a patient with global cerebral ischemia and prospects for magnetic navigation of stem cells within the CSF. PLoS One. 2014;9(2):e97631. | ||
Terrovitis J, Stuber M, Youssef A, et al. Magnetic resonance imaging overestimates ferumoxide-labeled stem cell survival after transplantation in the heart. Circulation. 2008;117(12):1555–1562. | ||
Amsalem Y, Mardor Y, Feinberg MS, et al. Iron-oxide labeling and outcome of transplanted mesenchymal stem cells in the infarcted myocardium. Circulation. 2007;116(Suppl 11):I38–I45. | ||
Winter EM, Hogers B, van der Graaf LM, Gittenberger-de Groot AC, Poelmann RE, van der Weerd L. Cell tracking using iron oxide fails to distinguish dead from living transplanted cells in the infarcted heart. Magn Reson Med. 2010;63(3):817–821. | ||
Ma N, Cheng H, Lu M, et al. Magnetic resonance imaging with superparamagnetic iron oxide fails to track the long-term fate of mesenchymal stem cells transplanted into heart. Sci Rep. 2015;5:9058. | ||
Huang Z, Li C, Yang S, et al. Magnetic resonance hypointensive signal primarily originates from extracellular iron particles in the long-term tracking of mesenchymal stem cells transplanted in the infarcted myocardium. Int J Nanomedicine. 2015;10:1679–1690. | ||
Eckert MA, Vu Q, Xie K, et al. Evidence for high translational potential of mesenchymal stromal cell therapy to improve recovery from ischemic stroke. J Cereb Blood Flow Metab. 2013;33(9):1322–1334. | ||
Drago D, Cossetti C, Iraci N, et al. The stem cell secretome and its role in brain repair. Biochimie. 2013;95(12):2271–2285. | ||
Salgado AJ, Sousa JC, Costa BM, et al. Mesenchymal stem cells secretome as a modulator of the neurogenic niche: basic insights and therapeutic opportunities. Front Cell Neurosci. 2015;9:249. | ||
Duan X, Wang Y, Zhang F, et al. Superparamagnetic iron oxide-loaded cationic polymersomes for cellular MR imaging of therapeutic stem cells in stroke. J Biomed Nanotechnol. 2016;12(12):2112–2124. | ||
Li K, Qin J, Wang X, et al. Magnetic resonance imaging monitoring dual-labeled stem cells for treatment of mouse nerve injury. Cytotherapy. 2013;15(10):1275–1285. | ||
Wen X, Wang Y, Zhang F, et al. In vivo monitoring of neural stem cells after transplantation in acute cerebral infarction with dual-modal MR imaging and optical imaging. Biomaterials. 2014;35(16):4627–4635. | ||
Li Y, Chen J, Chen XG, et al. Human marrow stromal cell therapy for stroke in rat: neurotrophins and functional recovery. Neurology. 2002;59(4):514–523. | ||
Zhang M, Methot D, Poppa V, Fujio Y, Walsh K, Murry CE. Cardiomyocyte grafting for cardiac repair: graft cell death and anti-death strategies. J Mol Cell Cardiol. 2001;33(5):907–921. | ||
Modo M, Stroemer RP, Tang E, Patel S, Hodges H. Effects of implantation site of stem cell grafts on behavioral recovery from stroke damage. Stroke. 2002;33(9):2270–2278. | ||
Gu E, Chen WY, Gu J, Burridge P, Wu JC. Molecular imaging of stem cells: tracking survival, biodistribution, tumorigenicity, and immunogenicity. Theranostics. 2012;2(4):335–345. | ||
Moniche F, Gonzalez A, Gonzalez-Marcos JR, et al. Intra-arterial bone marrow mononuclear cells in ischemic stroke: a pilot clinical trial. Stroke. 2012;43(8):2242–2244. | ||
Rustad KC, Wong VW, Sorkin M, et al. Enhancement of mesenchymal stem cell angiogenic capacity and stemness by a biomimetic hydrogel scaffold. Biomaterials. 2012;33(1):80–90. | ||
Coyne TM, Marcus AJ, Woodbury D, Black IB. Marrow stromal cells transplanted to the adult brain are rejected by an inflammatory response and transfer donor labels to host neurons and glia. Stem Cells. 2006;24(11):2483–2492. | ||
Björklund A, Lindvall O. Cell replacement therapies for central nervous system disorders. Nat Neurosci. 2000;3(6):537–544. | ||
Zhu W, Chen J, Cong X, Hu S, Chen X. Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem cells. Stem Cells. 2006;24(2):416–425. | ||
Tambuyzer BR, Bergwerf I, De Vocht N, et al. Allogeneic stromal cell implantation in brain tissue leads to robust microglial activation. Immunol Cell Biol. 2009;87(4):267–273. | ||
Le Blanc K. Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy. 2003;5(6):485–489. | ||
Hao L, Zou Z, Tian H, Zhang Y, Zhou H, Liu L. Stem cell-based therapies for ischemic stroke. BioMed Res Int. 2014;2014:468748. | ||
Zacharek A, Chen J, Cui X, et al. Angiopoietin1/Tie2 and VEGF/Flk1 induced by MSC treatment amplifies angiogenesis and vascular stabilization after stroke. J Cereb Blood Flow Metab. 2007;27(10):1684–1691. | ||
Ma S, Xie N, Li W, Yuan B, Shi Y, Wang Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014;21(2):216–225. | ||
Djouad F, Charbonnier LM, Bouffi C, et al. Mesenchymal stem cells inhibit the differentiation of dendritic cells through an interleukin-6-dependent mechanism. Stem Cells. 2007;25(8):2025–2032. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.