")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
The limits of oral therapy in pulmonary arterial hypertension management
Received 22 August 2014
Accepted for publication 16 September 2015
Published 23 November 2015 Volume 2015:11 Pages 1731—1741
DOI https://doi.org/10.2147/TCRM.S49026
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Garry Walsh
Qian-Qian Liu,1,2 Zhi-Cheng Jing1,3
1Department of Cardio-Pulmonary Circulation, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China; 2Department of Echocardiography, Fu Wai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China; 3State Key Laboratory of Cardiovascular Disease, Fu Wai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China
Abstract: Pulmonary arterial hypertension (PAH) is a devastating disease in which remodeling of the small pulmonary arteries leads to a progressive increase in pulmonary vascular resistance and right-sided heart failure. Over the past decade, new treatments for PAH, such as the use of ERAs, PDE-5 inhibitors and prostacyclin analogs, have brought about dramatic improvements in clinical outcomes. Epoprostenol infusion therapy has been shown to improve hemodynamics, functional status, and survival, and it remains the gold standard for treatment of patients with severe PAH. Many agents, approved for PAH are always delivered in pill form. Although oral therapy occupies an important position, it has some drawbacks and limitations in PAH management. For patients in World Health Organization functional class IV and with severe right heart failure, there are few data on the long-term survival of patients treated with oral medications. Further research, exploration, and clinical experience with oral therapy in severe PAH and combination therapy will redefine its position in PAH management.
Keywords: pulmonary arterial hypertension, right heart failure, oral therapy, survival
Introduction
Pulmonary arterial hypertension (PAH) is a series of diseases characterized by elevated pulmonary vascular resistance (PVR) and may lead to right heart failure and death, especially in patients with World Health Organization functional class (WHO-FC) III or IV.1–4 Progress in medical treatment over the past decade has considerably improved the overall quality of life and outcome of PAH patients.5–8 The clinical use of epoprostenol improves clinical condition and survival in PAH patients, opening the modern management era.9–11 Oral therapy for PAH patients, such as bosentan, sildenafil, tadalafil, et al, because of its stability and convenient administration route, occupies an important position in the PAH management. However, when comparing with other routes of administration, oral therapy shows a number of drawbacks and limitations in the PAH management, especially in patients with PAH crisis and severe right heart failure. This review will focus on the advantages and disadvantages of orally administered PAH-specific drugs versus intravenous, subcutaneous, and inhaled drugs. Furthermore, we will discuss the combination therapy strategy and treatment choices.
PAH-specific drugs and administration routes
Therapy with PAH-specific drugs needs to be initiated in PAH patients who are not long-term vasoreactive or are vasoreactive but not responding appropriately to calcium channel blockers. A brief description of PAH-approved drugs, according to the related pharmacological pathway is provided subsequently. Classical compounds include ERAs, PDE-5 inhibitors, sGC stimulators, and prostacyclin analogs. There are new agents targeting established vasodilatory pathways. Compounds for this new approach include TKIs and selective prostacyclin receptor (IP receptor) agonists. The treatment algorithm includes four different routes of administration (oral, inhaled, subcutaneous, and intravenous).12 In addition to intravenous, inhaled and subcutaneous prostacyclin analogs and intravenous sildenafil, other compounds which have been approved for PAH therapy are always delivered in pill form.
In the modern treatment era, PAH-specific drugs have brought about more available choices.
The orally administered dual ETA and ETB receptors antagonist bosentan improved exercise capacity and cardiopulmonary hemodynamics in patients with PAH.13 The selective ETA receptor antagonist ambrisentan, leading to release of vasodilators and antiproliferative substances,14 has been approved for the treatment of WHO-FC II and III patients, and has been demonstrated to be effective on symptoms, exercise capacity, hemodynamics, and time to clinical worsening (TTCW) of patients with idiopathic PAH and PAH associated with connective tissue disease and HIV infection.15,16 Macitentan is characterized by sustained receptor binding and enhanced tissue penetration.17,18 In the SERAPHIN study, macitentan significantly reduced the composite endpoint of morbidity and mortality among patients with PAH and also increased exercise capacity.19 Sitaxentan, a highly selective ETA receptor antagonist, was withdrawn from the market in 2010 because of an increasing number of deaths attributed to acute liver toxicity.20
The oral PDE-5 inhibitors approved for PAH include sildenafil and tadalafil.7 Vardenafil is a PDE-5 inhibitor that was approved for the treatment of erectile dysfunction in 2005 and was not approved in either the United States or Europe for PAH. Favorable effects with vardenafil treatment in patients with PAH have been reported.21–23 The three PDE-5 inhibitors inhibit PDE-5, an enzyme that metabolizes cGMP, thereby enhancing the cGMP-mediated relaxation and growth inhibition of vascular smooth-muscle cells, including those in the lung. Sildenafil was the first PDE-5 inhibitor approved for the treatment of PAH and its efficacy was demonstrated in the SUPER-1 randomized controlled trial (RCT) which showed that three doses (20 mg, 40 mg, and 80 mg) were associated with similar treatment effects in 6-minute walk distance (6MWD).24 Tadalafil, an orally administered, once-daily dosing, selective inhibitor of PDE-5, was assessed in the PHIRST-1 RCT for 16 weeks and tadalafil 40 mg was well tolerated and improved exercise capacity and quality of life measures and reduced clinical worsening.25
Riociguat stimulates sGC directly, independent of nitric oxide (NO) availability. In an RCT – PATENT-1, 443 PAH patients were treated with riociguat up to 2.5 mg three times daily and has shown favorable results on exercise capacity, hemodynamics, WHO-FC, and TTCW.26 Based on the benefits from clinical trials for both PAH and chronic thromboembolic pulmonary hypertension, riociguat was approved by Health Canada and the United States Food and Drug Administration as the first medical therapy for patients with chronic thromboembolic pulmonary hypertension who are deemed inoperable or have residual/recurrent pulmonary hypertension after pulmonary endarterectomy, and as a novel treatment option for patients with PAH.27,28
Epoprostenol (intravenous), iloprost (inhaled), and treprostinil (subcutaneous or intravenous) are approved prostacyclin analogs for PAH patients with WHO-FC II–IV. Epoprostenol, a synthetic prostacyclin analog administered as a continuous intravenous infusion, was the first targeted PAH therapy to be approved.10,29–31 Alternatives to epoprostenol include subcutaneously administered treprostinil, which is also available for intravenous and inhaled use in the USA, and inhaled iloprost.32 Oral treprostinil, a tricyclic benzidine analog of epoprostenol, has been evaluated in two RCTs in PAH patients on background therapy with bosentan and/or sildenafil (FREEDOM C1 and C2) and in both the primary endpoint 6MWD did not reach statistical significance.33,34 An additional RCT of oral treprostinil in PAH naïve patients showed improvement in 6MWD by 26 m at peak dose.35 However, all studies thus far with oral prostacyclin analogs, such as beraprost, have failed to show a persistent treatment effect as determined by the primary efficacy end-point.36,37 Beraprost, an orally active prostanoid, has not been approved in the United States or Europe for PAH due to lack of long-term efficacy but was approved in Japan and South Korea.
Imatinib is an antiproliferative agent developed to target the BCR-ABL tyrosine kinase in patients with chronic myeloid leukemia. The inhibitory effects of imatinib on PDGF receptors α and β and c-KIT make it efficacious in PAH.38–40 Imatinib improved pulmonary hemodynamics in a subgroup of patients with severe disease (defined as PVR >1,000 dyn·s·cm−5),41 the Phase III IMPRES RCT was subsequently conducted.42 Imatinib improved exercise capacity and hemodynamics in patients with advanced PAH, but importantly, treatment with imatinib was poorly tolerated and serious adverse events and study drug discontinuations were common, including the occurrence of subdural hematomas. The unfavorable risk-benefit ratio of imatinib has resulted in its cessation from further therapeutic development in PAH. Imatinib has been withdrawn due to unfavorable safety/efficacy profile for PAH.
Selexipag is a first-in-class orally available selective non-prostanoid IP receptor agonist. Selexipag is rapidly hydrolyzed to an active metabolite, ACT-333679. Both selexipag and its metabolite are highly selective for the IP receptor compared with other prostanoid receptors. This selectivity for the IP receptor offers the potential for improved tolerability with selexipag, as side effects (eg, nausea and vomiting) that might result from activation of the other prostanoid receptors may be minimized. Selexipag showed effects on pharmacodynamic endpoints obtained with right heart catheterization in a Phase II trial in patients with PAH,43 and is being evaluated in the ongoing Phase III trial (GRIPHON trial, Clinicaltrials.gov NCT01106014).
Advantages and disadvantages of orally administered drugs versus intravenous, subcutaneous, and inhaled drugs
The side effects and damages of major PAH-specific drugs
Prostacyclin analogs (intravenous, subcutaneous or inhaled) improved symptoms, exercise capacity, and clinical outcome in patients with PAH, but are limited by either parenteral or inhaled delivery systems, the need for laboratory monitoring, or frequent dosing schedules. Orally administered ERAs, PDE-5 inhibitors, and prostacyclin analogs have also demonstrated clinical and hemodynamic improvement.24,35,44,45 However, orally administered drugs, despite their chemical stability, steady-state plasma concentration, and convenient administration route, are inevitably associated with significant side effects. For PAH-specific oral administration, the most common adverse effects include headaches, flushing, liver enzyme abnormalities, dyspepsia, and diarrhea. The side effects and damages of major PAH-specific therapies were shown in Table 1.
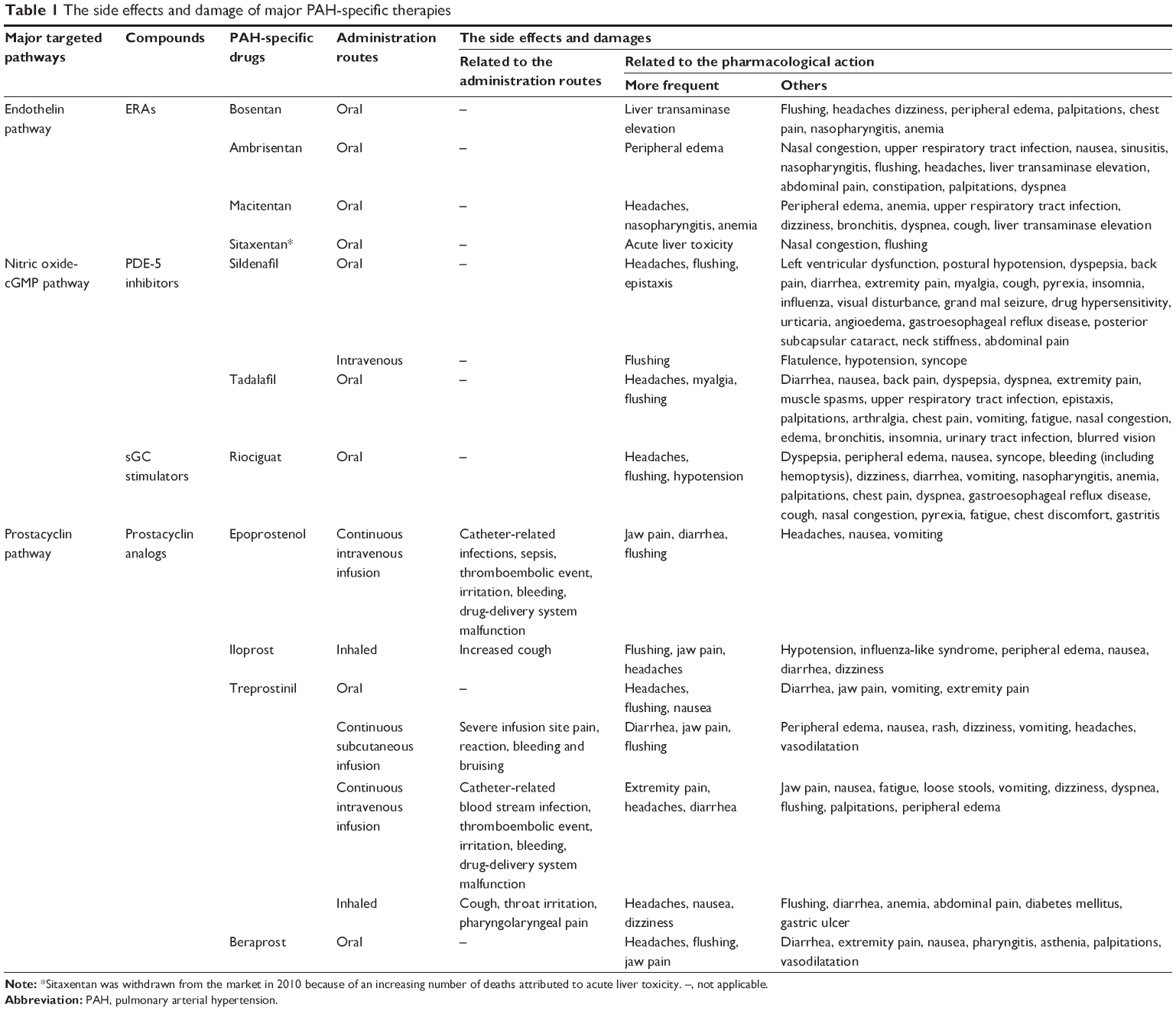 | Table 1 The side effects and damage of major PAH-specific therapies |
Liver enzyme abnormalities are the most outstanding side effects of oral ERA. Increases in hepatic aminotransferases occurred in approximately 10% of the subjects who take bosentan but were found to be dose-dependent and reversible after dose reduction or discontinuation.46,47 Sitaxentan was withdrawn from the market because of association with acute liver toxicity.20 The irreversible liver failure caused by sitaxentan and its withdrawal from global markets emphasized the importance of hepatic monitoring in patients treated with ERAs.48 In the event-driven macitentan SERAPHIN study,19 no liver toxicity was shown. Ambrisentan has been approved for the treatment of WHO-FC II and III patients and the incidence of abnormal liver function tests range from 0.8% to 3%. An increased incidence of peripheral edema has been reported with ambrisentan use.49
Most side effects of PDE-5 inhibitor (sildenafil, tadalafil, and vardenafil) are mild to moderate and mainly related to vasodilation (headaches, flushing or epistaxis).21,24,25,35,50 Considering the potential hypotension, the concurrent use of PDE-5 inhibitors with riociguat is not recommended.
Sildenafil is not used in the United States for pediatric PAH, but approved in the European Union. The double-blind, placebo-controlled STARTS-1 study assessed sildenafil in pediatric patients with PAH; improved hemodynamics and exercise capacity occurred in medium- and high-dose groups.51 STARTS-2 was the extension study.52 In STARTS-2, sildenafil-treated patients continued STARTS-1 dosing; placebo-treated patients were randomized to one of the three sildenafil dose groups. As of August 2011, all children received ≥3 years of treatment (unless discontinued) from STARTS-1 baseline. Although children randomized to higher comparing with lower sildenafil doses had an unexplained increased mortality, all sildenafil dose groups displayed favorable survival for children with PAH. Survival in all dose groups was favorable compared with other reports in children using PAH-specific drugs that were only approved for adult PAH.53–59 Most patients reported adverse events during STARTS-1 and -2. The most frequently reported adverse events were upper respiratory tract infection, headaches, and vomiting. The majority of adverse events were of mild or moderate intensity. The most common treatment-related adverse events were headaches (15%) and vomiting (6%). Five patients had serious adverse events that were assessed as treatment related (low-dose group: enterocolitis, n=1; medium-dose group: convulsion, n=1; high-dose group: hypersensitivity and stridor, n=1; hypoxia, n=1; ventricular arrhythmia, n=1).
Prostacyclin analogs also have drawbacks with respect to frequent dosing (iloprost), injection site pain (subcutaneous treprostinil), recurrent infections of the intravenous catheter (intravenous epoprostenol), and typical prostanoid-associated side-effects such as headaches, flushing, diarrhea, and jaw pain.32,60
Pharmacokinetic characteristics
Compared with subcutaneous, intravenous, and inhaled prostacyclin analogs, orally administered compounds have a much longer plasma half-life, Table 2. Weakness associated with the longer plasma half-life is that, when adverse reactions occur, such as hypotension and pulmonary edema, it is difficult to reverse in a short time.
 | Table 2 Half-life of PAH-specific drugs |
Absorption refers to the transport of orally administered drugs across membranes of the epithelial cells in the gastrointestinal tract. In severe right heart failure patients, gastrointestinal congestion may affect the drug absorption.
The lack of pulmonary selectivity results in systemic side effects, and drug tolerance leads to progressive increases in dose. Systemic side effects and drug tolerance are the most common problems of oral therapy. Inhaled therapy for pulmonary hypertension may provide selectivity of the hemodynamic effects to the lung vasculature, thus decreasing systemic side effects. As an alternative, inhaled nitric oxide possesses pulmonary selectivity, but it is less potent than prostacyclin in the pulmonary vasculature.69,70
Iloprost is a stable analog of prostacyclin with a longer duration of vasodilatation. When administered during a short aerosolization maneuver to patients with pulmonary hypertension, its pulmonary vasodilatory potency was similar to that of prostacyclin, but its effects lasted for 30 to 90 minutes, as compared with 15 minutes.71–73 Inhaled iloprost therapy has a proven clinical efficacy in PAH, and this therapy necessitates six to nine inhalation sessions per day.74 In contrast to epoprostenol, treprostinil is chemically stable at room temperature and neutral pH and has a longer half-life (about 4 hours) permitting continuous subcutaneous infusion rather than continuous intravenous infusion, avoiding the risks of severe infection and thrombosis.75 Inhaled treprostinil exerts sustained pulmonary vasodilation with excellent tolerability at relatively low doses and may be inhaled in a few breaths.32,74,76
Voswinckel et al conducted randomized controlled pilot studies comparing the hemodynamic and systemic side effects of inhaled treprostinil with inhaled iloprost.74 Inhaled treprostinil had maximal effects occurring later than iloprost (18±2 minutes versus 8±1 minutes, P<0.001), and the effects lasted longer (P<0.0001): while the hemodynamic effects of iloprost had vanished within 1 hour, they persisted for >2 hours after treprostinil inhalation. A transient drop in systemic arterial pressure occurred after iloprost inhalation but not with treprostinil.64,74
Inhaled iloprost and treprostinil, combining the beneficial effects of prostacyclin with those of an inhalational application, is associated with a longer duration of vasodilatation than intravenous epoprostenol and higher pulmonary vascular selectivity. The advantages of intermittent inhaled therapy over intravenous and oral therapy, coupled with the improvement in a number of clinically meaningful variables, suggest that inhaled therapy may be a suitable alternative to continuous intravenous prostacyclin, especially in patients who do not derive a clear survival benefit with oral therapy.77,78
The relevant studies with PAH-specific drugs regarding disease severity and treatment choices
According to the guideline (National Institute for Health and Care Excellence, 2013), WHO-FC, as assessed through the symptoms, may be considered as the most important treatment indicator.79 Similarly, physical exercise capability, biochemical markers, and other hemodynamic parameters are also important indicators.
For patients with WHO-FC II, ambrisentan, bosentan, macitentan, riociguat, sildenafil, and tadalafil was recommended at Grade I and level of Evidence of A or B. For patients with WHO-FC III, intravenous epoprostenol, inhaled iloprost, subcutaneous and inhaled treprostinil were added. For patients with WHO-FC IV, intravenous epoprostenol was the only specific drug which was recommended at Grade I and level of Evidence of A or B.7
WHO-FC status of patients and primary endpoints in studies with PAH-specific drugs were shown in Table 3. In studies on macitentan and epoprostenol, morbidity and mortality are the primary endpoints.19,31 The traditional primary endpoint of the studies about oral therapy performed in PAH has always been the 6MWD to assess the exercise capacity, but unable to assess the long-term survival. The effect on survival of most of the oral PAH-specific compounds has not been appropriately assessed in individual trials because of small sample size and short duration.80 Oral therapy has almost been shown to improve exercise capacity, hemodynamics, and quality of life in most patients with severe PAH. There are few data on the long-term survival of patients with PAH treated with oral medicines, especially in WHO-FC IV.
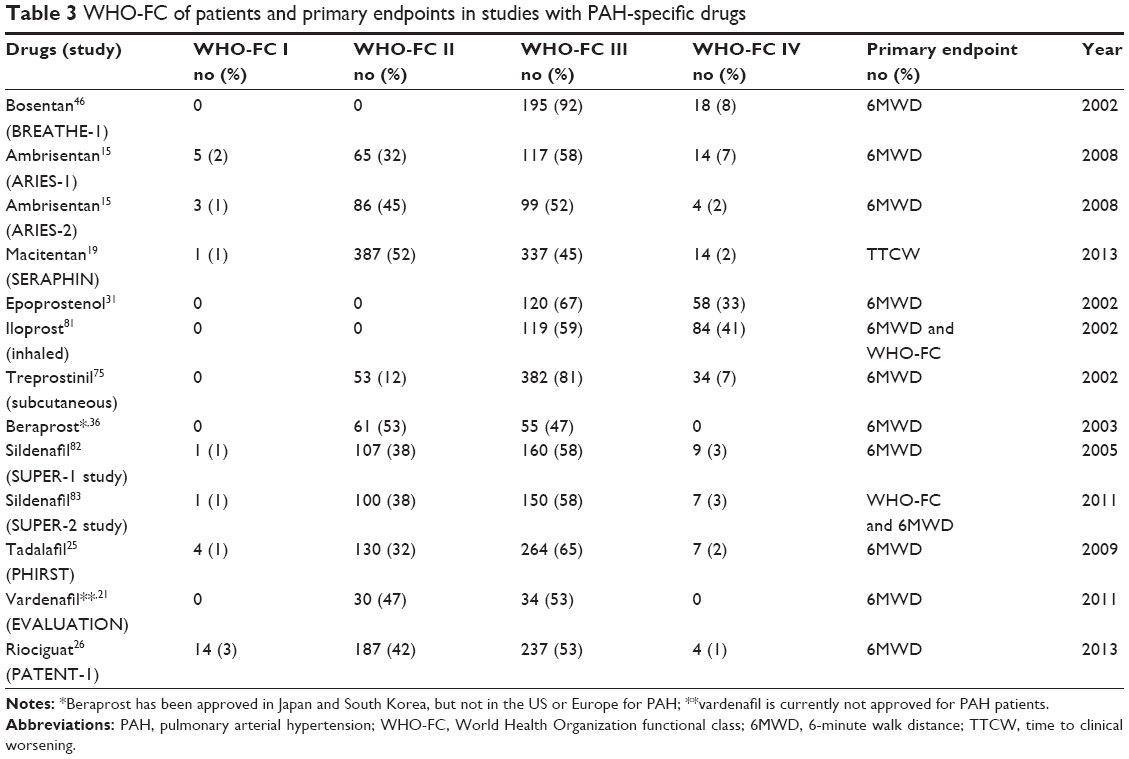 | Table 3 WHO-FC of patients and primary endpoints in studies with PAH-specific drugs |
However, Sitbon et al described significantly improved survival in an analysis of 178 patients with PAH in WHO-FC III or IV treated with intravenous epoprostenol infusion over a 10-year period.31 For comparison, survival data are also shown for a historical control group of 135 patients with PAH matched for WHO-FC and who never received intravenous epoprostenol therapy. In the group of patients treated with epoprostenol, the overall survival rates at 1, 2, 3, and 5 years were 85%, 70%, 63%, and 55%, respectively, as compared with 58%, 43%, 33%, and 28% in the historical control group (P<0.0001 by the Cox–Mantel log-rank test). The efficacy of continuous intravenous administration of epoprostenol on survival has been proven in another two studies in patients with idiopathic PAH.9,10
Two retrospective, multicenter, uncontrolled trials have investigated the long-term outcomes of treprostinil. Overall, both trials demonstrated long-term benefits of subcutaneous treprostinil on exercise capacity, functional class, and survival.83,84 Both trials enrolled subjects with WHO-FC II-IV, with the majority being class III, and allowed open-label treprostinil management titrated at the discretion of the physician based on symptoms.
In randomized, double-blind, placebo-controlled trials for oral therapy in PAH, almost all enrolled patients were in WHO-FC II or III, and few trials included patients in WHO-FC IV. The traditional primary endpoint of the RCTs performed in PAH has been the 6MWD that assesses the exercise capacity, and secondary endpoints have included hemodynamics and TTCW, a composite endpoint including death, hospitalization, and disease progression.85 The effect on survival of these oral compounds has not been appropriately assessed in PAH individual trials.
The role of oral administration in PAH-specific combination therapy strategy
After initial therapy, the next steps are based on the clinical response.7 If the clinical response is considered not adequate, combination therapy is considered. Intravenous or subcutaneous therapies are more appropriate than oral therapies for advanced patients, but, in fact, most patients with severe PAH are usually not on monotherapy throughout. Combination therapy strategy that includes multiple classes of PAH-specific drugs is an attractive option for the management of end-stage PAH. The specific combination therapies include three separate signaling pathways involved in the disease: the endothelin pathway, the NO pathway, and the prostacyclin pathway. The ways of combination include sequential combination therapy and upfront combination therapy.86
The use of combination therapy to achieve predefined treatment goals has emerged as a major theme to improve long-term outcomes in PAH. Main aspects and characteristics of studies with patients exposed to combination therapy of approved PAH drugs were shown in Table 4.
 | Table 4 Characteristics of studies with patients exposed to combination therapy of approved PAH drugs |
Almost all combination therapies included oral administration drugs, and the primary endpoint was 6MWD. Additional oral therapy may reduce the need to up-titrate the intravenous and inhaled prostacyclin analogs or other oral drugs’ dose and, therefore, potentially decrease the dose-related side-effects. However, the primary endpoint of combination therapy studies has not consistently been met in the STEP,87 COMBI,88 FREEDOM-C,33 FREEDOM-C2,34 and BREATHE-289 studies.
In TRIUMPH-1 study,90 the efficacy and safety of inhaled treprostinil or placebo in PAH patients receiving therapy with either bosentan or sildenafil were assessed. The primary endpoint was peak 6MWD at 12 weeks and it has met the defined statistical significance.
The 16-week, double-blind, placebo-controlled, parallel group study, the addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial (PACES Study).91 Trial demonstrated that the addition of sildenafil to background intravenous epoprostenol therapy improved 6MWD and delayed TTCW, but this treatment sequence does not often correspond to clinical practice because oral therapy is usually initiated first.
The two completed RCTs of macitentan (SERAPHIN trial)19 and riociguat (PATENT-1 trial)26 have substantially contributed to the evidence base of combination therapy. About 63% of enrolled subjects in SERAPHIN trial and 50% in PATENT-1 trial, were on background therapy with predominantly oral PAH agents. In both of these RCTs, the primary endpoint of the studies has been met.
The experience of BREATHE-2 study89 with initial combination therapy failed to demonstrate any significant difference between patients treated initially with the combination epoprostenol and bosentan as compared with epoprostenol alone. In a recent study, 23 treatment naïve PAH patients were treated with the initial combination of epoprostenol and bosentan and compared with a matched historical control group treated with epoprostenol.92 The study showed a statistically significantly greater decrease in PVR in the initial combination therapy group but this hemodynamic benefit did not translate into a statistically significant difference in survival, or in transplant-free survival. In a pilot study, the efficacy and safety of a triple upfront combination therapy regimen with epoprostenol, bosentan, and sildenafil in patients with severe PAH who present in WHO-FC III/IV together with severe hemodynamic impairment was investigated.93 At a median follow-up of 39.2 months, 18 of 19 patients had sustained clinical improvement and remained in WHO-FC I/II. 6MWD increased dramatically from 227 to 514 m (P<0.01) and PVR declined from 1,718 to 492 dyn·s·cm−5 (P<0.01). This pilot study provides preliminary evidence of the long-term benefits of upfront triple combination therapy in patients with severe PAH.
From Table 4, we can see that the majority of patients in combination studies, except for the triple combination study, were WHO-FC III. For WHO-FC IV PAH patients in case of non-availability of intravenous prostanoids, initial combination therapy has been allocated a grade of recommendation IIb and level of Evidence C.7
Insufficient initial therapy and transposition to interventional or surgical procedures
In the modern treatment era, exercise capacity, hemodynamics, quality of life, and survival of patients with PAH have improved. However, some patients do not benefit from the medication treatment, especially oral therapy, and must be considered for balloon atrial septostomy or lung transplantation.94
Novel strategies, including atrial septostomy or the pumpless Novalung® lung assist device with conduits, from the pulmonary artery to the left atrium, can be considered as a bridge to transplant for patients with rapid clinical decline, despite maximal medical therapy.95
Lung transplantation should continue to be an important option for those who fail on medication treatment programs and remain in WHO-FC III or IV.30,31,96 It is reasonable to consider eligibility for lung transplantation after an inadequate clinical response to the initial monotherapy and to refer the patient soon after the inadequate clinical response is confirmed on maximal combination therapy.97 Most of the end-stage PAH patients, who underwent lung transplantation, had been ever treated with epoprostenol.30,31 In this regard, oral therapy is not as crucial as epoprostenol in the perioperative management of severe PAH and right heart failure before lung transplantation. In the pharmacological management of perioperative PAH patients, intravenous epoprostenol or inhaled iloprost play an important role in pulmonary vessel diastole.
Conclusion
Orally administered ERAs, PDE-5 inhibitors, sGC stimulator, and prostacyclin analogs have demonstrated clinical and hemodynamic improvement in patients with PAH. However, there are limits in the efficacy and safety profile of oral therapy. Especially for PAH patients with WHO-FC IV and with severe right heart failure, there are few data on the long-term survival of patients treated with oral medications. Intravenous epoprostenol is still considered the gold standard therapy in severe PAH and has reduced the need for lung transplantation in patients with advanced disease. Medicinal chemistry programs must be embarked upon aiming at the identification of novel compounds for PAH with high oral efficacy. Further research, exploration, and clinical experience with oral therapy in severe PAH and combination therapy will redefine its position in PAH management.
Disclosure
Z-C Jing serves as a consultant and scientific advisor to Actelion, Bayer Schering, AstraZeneca, Pfizer and United Therapeutics, in addition to being an investigator in trials sponsored by these companies. Q-Q Liu declares no conflict of interest.
References
Rubin LJ. Primary pulmonary hypertension[J]. N Engl J Med. 1997; 336(2):111–117. | ||
Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension[J]. J Am Coll Cardiol. 2013; 62(25 Suppl):D34–D41. | ||
Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study[J]. Ann Intern Med.1987;107(2): 216–223. | ||
D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry[J]. Ann Intern Med. 1991;115(5):343–349. | ||
Galie N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT)[J]. Eur Heart J. 2009;30(20):2493–2537. | ||
Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era[J]. Circulation. 2010; 122(2):156–163. | ||
Galie N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension[J]. J Am Coll Cardiol. 2013;62(25 Suppl):D60–D72. | ||
Galie N, Manes A, Negro L, et al. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension[J]. Eur Heart J. 2009;30(4):394–403. | ||
Rubin LJ, Mendoza J, Hood M, et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial[J]. Ann Intern Med. 1990; 112(7):485–491. | ||
Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension[J]. N Engl J Med. 1996;334(5): 296–301. | ||
Badesch DB, Tapson VF, Mcgoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial[J]. Ann Intern Med. 2000;132(6):425–434. | ||
Galie N, Seeger W, Naeije R, et al. Comparative analysis of clinical trials and evidence-based treatment algorithm in pulmonary arterial hypertension[J]. J Am Coll Cardiol. 2004;43(12 Suppl S):81S–88S. | ||
Channick RN, Simonneau G, Sitbon O, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study[J]. Lancet. 2001;358(9288):1119–1123. | ||
Galie N, Badesch D, Oudiz R, et al. Ambrisentan therapy for pulmonary arterial hypertension[J]. J Am Coll Cardiol. 2005;46(3):529–535. | ||
Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2[J]. Circulation. 2008;117(23):3010–3019. | ||
Mcgoon MD, Frost AE, Oudiz RJ, et al. Ambrisentan therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function test abnormalities[J]. Chest. 2009;135(1):122–129. | ||
Bolli MH, Boss C, Binkert C, et al. The discovery of N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-p ropylsulfamide (Macitentan), an orally active, potent dual endothelin receptor antagonist[J]. J Med Chem. 2012;55(17): 7849–7861. | ||
Iglarz M, Binkert C, Morrison K, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist[J]. J Pharmacol Exp Ther. 2008;327(3):736–745. | ||
Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension[J]. N Engl J Med. 2013;369(9):809–818. | ||
Lavelle A, Sugrue R, Lawler G, et al. Sitaxentan-induced hepatic failure in two patients with pulmonary arterial hypertension[J]. Eur Respir J. 2009;34(3):770–771. | ||
Jing ZC, Yu ZX, Shen JY, et al. Vardenafil in pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled study[J]. Am J Respir Crit Care Med. 2011;183(12):1723–1729. | ||
Jing ZC, Jiang X, Wu BX, et al. Vardenafil treatment for patients with pulmonary arterial hypertension: a multicentre, open-label study[J]. Heart. 2009;95(18):1531–1536. | ||
Ghofrani HA, Voswinckel R, Reichenberger F, et al. Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension: a randomized prospective study[J]. J Am Coll Cardiol. 2004;44(7): 1488–1496. | ||
Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension[J]. N Engl J Med. 2005;353(20): 2148–2157. | ||
Galie N, Brundage BH, Ghofrani HA, et al. Tadalafil therapy for pulmonary arterial hypertension[J]. Circulation. 2009;119(22):2894–2903. | ||
Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension[J]. N Engl J Med. 2013;369(4): 330–340. | ||
Mielniczuk LM, Swiston JR, Mehta S. Riociguat: a novel therapeutic option for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension[J]. Can J Cardiol. 2014;30(10):1233–1240. | ||
Bishop BM. Riociguat for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension[J]. Am J Health Syst Pharm. 2014;71(21):1839–1844. | ||
Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension[J]. N Engl J Med. 1996; 334(5):296–301. | ||
Mclaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy[J]. Circulation. 2002;106(12):1477–1482. | ||
Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival[J]. J Am Coll Cardiol. 2002;40(4):780–788. | ||
Badesch DB, Mclaughlin VV, Delcroix M, et al. Prostanoid therapy for pulmonary arterial hypertension[J]. J Am Coll Cardiol. 2004;43 (12 Suppl S):56S–61S. | ||
Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial[J]. Chest. 2012;142(6):1383–1390. | ||
Tapson VF, Jing ZC, Xu KF, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial[J]. Chest. 2013;144(3):952–958. | ||
Jing ZC, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial[J]. Circulation. 2013;127(5):624–633. | ||
Barst RJ, Mcgoon M, Mclaughlin V, et al. Beraprost therapy for pulmonary arterial hypertension[J]. J Am Coll Cardiol. 2003;41(12): 2119–2125. | ||
Galie N, Humbert M, Vachiery JL, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial[J]. J Am Coll Cardiol. 2002;39(9):1496–1502. | ||
Montani D, Perros F, Gambaryan N, et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension[J]. Am J Respir Crit Care Med. 2011;184(1):116–123. | ||
Perros F, Montani D, Dorfmuller P, et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension[J]. Am J Respir Crit Care Med. 2008;178(1):81–88. | ||
Schermuly RT, Dony E, Ghofrani HA, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition[J]. J Clin Invest. 2005;115(10):2811–2821. | ||
Ghofrani HA, Morrell NW, Hoeper MM, et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy[J]. Am J Respir Crit Care Med. 2010;182(9):1171–1177. | ||
Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study[J]. Circulation. 2013;127(10):1128–1138. | ||
Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension[J]. Eur Respir J. 2012;40(4):874–880. | ||
Liu C, Chen J. Endothelin receptor antagonists for pulmonary arterial hypertension[J]. Cochrane Database Syst Rev. 2006;(3):D4434. | ||
Mclaughlin VV, Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension[J]. Eur Respir J. 2005;25(2):244–249. | ||
Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension[J]. N Engl J Med. 2002;346(12):896–903. | ||
Galie N, Rubin L, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial[J]. Lancet. 2008;371(9630):2093–2100. | ||
Owen K, Cross DM, Derzi M, et al. An overview of the preclinical toxicity and potential carcinogenicity of sitaxentan (Thelin(R)), a potent endothelin receptor antagonist developed for pulmonary arterial hypertension[J]. Regul Toxicol Pharmacol. 2012;64(1):95–103. | ||
Mcgoon MD, Frost AE, Oudiz RJ, et al. Ambrisentan therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function test abnormalities[J]. Chest. 2009;135(1):122–129. | ||
Sastry BK, Narasimhan C, Reddy NK, et al. Clinical efficacy of sildenafil in primary pulmonary hypertension: a randomized, placebo-controlled, double-blind, crossover study[J]. J Am Coll Cardiol. 2004;43(7):1149–1153. | ||
Barst RJ, Ivy DD, Gaitan G, et al. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension[J]. Circulation. 2012;125(2):324–334. | ||
Barst RJ, Beghetti M, Pulido T, et al. STARTS-2: long-term survival with oral sildenafil monotherapy in treatment-naive pediatric pulmonary arterial hypertension[J]. Circulation. 2014;129(19):1914–1923. | ||
Yung D, Widlitz AC, Rosenzweig EB, et al. Outcomes in children with idiopathic pulmonary arterial hypertension[J]. Circulation. 2004;110(6):660–665. | ||
Lammers AE, Hislop AA, Flynn Y, et al. Epoprostenol treatment in children with severe pulmonary hypertension[J]. Heart. 2007; 93(6):739–743. | ||
Hislop AA, Moledina S, Foster H, et al. Long-term efficacy of bosentan in treatment of pulmonary arterial hypertension in children[J]. Eur Respir J. 2011;38(1):70–77. | ||
Ivy DD, Rosenzweig EB, Lemarie JC, et al. Long-term outcomes in children with pulmonary arterial hypertension treated with bosentan in real-world clinical settings[J]. Am J Cardiol. 2010;106(9):1332–1338. | ||
Haworth SG, Hislop AA. Treatment and survival in children with pulmonary arterial hypertension: the UK Pulmonary Hypertension Service for Children 2001–2006[J]. Heart. 2009;95(4):312–317. | ||
Barst RJ, Mcgoon MD, Elliott CG, et al. Survival in childhood pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management[J]. Circulation. 2012;125(1):113–122. | ||
van Loon RL, Roofthooft MT, Delhaas T, et al. Outcome of pediatric patients with pulmonary arterial hypertension in the era of new medical therapies[J]. Am J Cardiol. 2010;106(1):117–124. | ||
Barst RJ, Rubin LJ, Mcgoon MD, et al. Survival in primary pulmonary hypertension with long-term continuous intravenous prostacyclin[J]. Ann Intern Med. 1994;121(6):409–415. | ||
Prakash A, Perry CM. Bosentan[J]. Am J Cardiovasc Drugs. 2002;2(5): 335–343. | ||
Billman GE. Ambrisentan (Myogen)[J]. Curr Opin Investig Drugs. 2002;3(10):1483–1486. | ||
Shapiro SM, Oudiz RJ, Cao T, et al. Primary pulmonary hypertension: improved long-term effects and survival with continuous intravenous epoprostenol infusion[J]. J Am Coll Cardiol. 1997;30(2):343–349. | ||
Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension[J]. Eur Respir J. 2008;31(4):891–901. | ||
Laliberte K, Arneson C, Jeffs R, et al. Pharmacokinetics and steady-state bioequivalence of treprostinil sodium (Remodulin) administered by the intravenous and subcutaneous route to normal volunteers[J]. J Cardiovasc Pharmacol. 2004;44(2):209–214. | ||
Walker DK, Ackland MJ, James GC, et al. Pharmacokinetics and metabolism of sildenafil in mouse, rat, rabbit, dog and man[J]. Xenobiotica. 1999;29(3):297–310. | ||
Palmieri EA, Affuso F, Fazio S, et al. Tadalafil in primary pulmonary arterial hypertension[J]. Ann Intern Med. 2004;141(9):743–744. | ||
Frey R, Muck W, Unger S, et al. Single-dose pharmacokinetics, pharmacodynamics, tolerability, and safety of the soluble guanylate cyclase stimulator BAY 63-2521: an ascending-dose study in healthy male volunteers[J]. J Clin Pharmacol. 2008;48(8):926–934. | ||
Warren JB, Higenbottam T. Caution with use of inhaled nitric oxide[J]. Lancet. 1996;348(9028):629–630. | ||
Hoeper MM, Olschewski H, Ghofrani HA, et al. A comparison of the acute hemodynamic effects of inhaled nitric oxide and aerosolized iloprost in primary pulmonary hypertension. German PPH study group[J]. J Am Coll Cardiol. 2000;35(1):176–182. | ||
Olschewski H, Walmrath D, Schermuly R, et al. Aerosolized prostacyclin and iloprost in severe pulmonary hypertension[J]. Ann Intern Med. 1996;124(9):820–824. | ||
Olschewski H, Ghofrani HA, Walmrath D, et al. Inhaled prostacyclin and iloprost in severe pulmonary hypertension secondary to lung fibrosis[J]. Am J Respir Crit Care Med. 1999;160(2):600–607. | ||
Gessler T, Schmehl T, Hoeper MM, et al. Ultrasonic versus jet nebulization of iloprost in severe pulmonary hypertension[J]. Eur Respir J. 2001;17(1):14–19. | ||
Voswinckel R, Enke B, Reichenberger F, et al. Favorable effects of inhaled treprostinil in severe pulmonary hypertension: results from randomized controlled pilot studies[J]. J Am Coll Cardiol. 2006; 48(8):1672–1681. | ||
Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial[J]. Am J Respir Crit Care Med. 2002;165(6):800–804. | ||
Galie N, Manes A, Branzi A. Prostanoids for pulmonary arterial hypertension[J]. Am J Respir Med. 2003;2(2):123–137. | ||
Lopez-Meseguer M, Berastegui C, Monforte V, et al. Inhaled iloprost plus oral sildenafil in patients with severe pulmonary arterial hypertension delays the need for lung transplantation[J]. Transplant Proc. 2013; 45(6):2347–2350. | ||
Onen ZP, Sayin T, Akkoca YO, et al. [Efficacy of long-term oral monotherapy and additional effect of inhaled iloprost in patients with severe idiopathic pulmonary arterial hypertension][J]. Anadolu Kardiyol Derg. 2010;10(5):458–460. | ||
Baldi F, Fuso L, Arrighi E, et al. Optimal management of pulmonary arterial hypertension: prognostic indicators to determine treatment course[J]. Ther Clin Risk Manag. 2014;10:825–839. | ||
Macchia A, Marchioli R, Marfisi R, et al. A meta-analysis of trials of pulmonary hypertension: a clinical condition looking for drugs and research methodology[J]. Am Heart J. 2007;153(6):1037–1047. | ||
Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension[J]. N Engl J Med. 2002;347(5):322–329. | ||
Rubin LJ, Badesch DB, Fleming TR, et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: the SUPER-2 study[J]. Chest. 2011;140(5):1274–1283. | ||
Lang I, Gomez-Sanchez M, Kneussl M, et al. Efficacy of long-term subcutaneous treprostinil sodium therapy in pulmonary hypertension[J]. Chest. 2006;129(6):1636–1643. | ||
Barst RJ, Galie N, Naeije R, et al. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil[J]. Eur Respir J. 2006;28(6):1195–1203. | ||
Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension[J]. J Am Coll Cardiol. 2009;54(1 Suppl):S55–S66. | ||
Humbert M, Lau EM, Montani D, et al. Advances in therapeutic interventions for patients with pulmonary arterial hypertension[J]. Circulation. 2014;130(24):2189–2208. | ||
Mclaughlin VV, Oudiz RJ, Frost A, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension[J]. Am J Respir Crit Care Med. 2006;174(11): 1257–1263. | ||
Hoeper MM, Leuchte H, Halank M, et al. Combining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertension[J]. Eur Respir J. 2006;28(4):691–694. | ||
Humbert M, Barst RJ, Robbins IM, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2[J]. Eur Respir J. 2004;24(3):353–359. | ||
Mclaughlin VV, Benza RL, Rubin LJ, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial[J]. J Am Coll Cardiol. 2010;55(18): 1915–1922. | ||
Simonneau G, Rubin LJ, Galie N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial[J]. Ann Intern Med. 2008;149(8):521–530. | ||
Kemp K, Savale L, O’Callaghan DS, et al. Usefulness of first-line combination therapy with epoprostenol and bosentan in pulmonary arterial hypertension: an observational study[J]. J Heart Lung Transplant. 2012;31(2):150–158. | ||
Sitbon O, Jais X, Savale L, et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study[J]. Eur Respir J. 2014;43(6):1691–1697. | ||
Keogh AM, Mayer E, Benza RL, et al. Interventional and surgical modalities of treatment in pulmonary hypertension[J]. J Am Coll Cardiol. 2009;54(1 Suppl):S67–S77. | ||
Lordan JL, Corris PA. Pulmonary arterial hypertension and lung transplantation[J]. Expert Rev Respir Med. 2011;5(3):441–454. | ||
Granton J, Mercier O, De Perrot M. Management of severe pulmonary arterial hypertension[J]. Semin Respir Crit Care Med. 2013;34(5): 700–713. | ||
Hoeper MM, Granton J. Intensive care unit management of patients with severe pulmonary hypertension and right heart failure[J]. Am J Respir Crit Care Med. 2011;184(10):1114–1124. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.