")
Back to Journals » Vascular Health and Risk Management » Volume 18
The Klippel-Trénaunay Syndrome in 2022: Unravelling Its Genetic and Molecular Profile and Its Link to the Limb Overgrowth Syndromes
Authors Harnarayan P, Harnanan D
Received 27 January 2022
Accepted for publication 24 March 2022
Published 2 April 2022 Volume 2022:18 Pages 201—209
DOI https://doi.org/10.2147/VHRM.S358849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Pietro Scicchitano
Patrick Harnarayan, Dave Harnanan
Department of Clinical Surgical Sciences, University of The West Indies, St. Augustine, Trinidad & Tobago, West Indies
Correspondence: Patrick Harnarayan, Department of Clinical Surgical Sciences, University of The West Indies, St. Augustine, Trinidad & Tobago, West Indies, Email [email protected]
Abstract: The Klippel-Trénaunay syndrome is an unusual syndrome of vascular and dermatologic manifestation in which patients demonstrate hemihypertrophy of the soft tissue and bones of one limb, cutaneous haemangiomas and varicosities in anatomically abnormal positions. Described in 1900 by two French physicians, the etiology remained unclear until recently, when evidence emerged that there was a genetic basis for this sporadic disorder. Genes that encoded pathological angiogenic factors and caused vascular dysmorphogenesis, explaining the molecular bases of this syndrome, were identified. Several angiogenic genes were identified but one gene, the AGGF1 (formerly VG5Q) gene, was seen in mutations involving patients diagnosed with Klippel-Trénaunay syndrome. Furthermore, this syndrome was also noted to have overlapping clinical features linked with the “overgrowth syndromes,” in which genetic mutations along somatic lines were identified. These involved The PI3K enzyme which forms part of the phosphoinositide 3–kinase pathway which is encoded by the PIK3CA-gene. This enzyme mediates embryonic cellular growth in-utero and diseases involved in this pathway are classified as members of the PIK3CA-related overgrowth syndrome. This paper reviews the status of what is now known about the molecular genetics of this unusual, but clinically challenging disorder and its differentiation from similar diseases, linked with the PIK3CA-gene and the related overgrowth syndromes.
Keywords: Klippel-Trénaunay, mosaic transmission, PIK3CA-gene, limb-overgrowth spectrum
Introduction
The Klippel-Trénaunay syndrome (KTS) was described by Maurice Klippel, a French neurologist and Paul Trénaunay, a Parisian physician in 1900. It is a congenital, sporadic disease with a triad of cutaneous haemangiomas (capillary malformations), varicosities in anatomically abnormal positions and limb hypertrophy including soft tissues and bones.1 Two of these three cardinal features are needed to make a phenotypical diagnosis of KTS.2 The vascular abnormalities described are the slow-flow type, devoid of significant arterio-venous fistulas in comparison to the Parkes-Weber Syndrome, which is a high-flow disorder.3 We herein look at the molecular and genetic basis of this disease regarding its clinical presentation in terms of angiogenesis, cutaneous involvement and limb tissue overgrowth. Other diseases which may be confused with KTS and are part of the limb overgrowth syndromes are also discussed.
Distinguishing Clinical Features
KTS is a complex, combined disorder made up of capillary, lymphatic and venous malformations with overgrowth of the affected limb.4 These capillary malformations also called “port-wine stains” and are regarded as the most common vascular cutaneous malformation in KTS, seen in 98% of cases2 [Figure 1]. These are abnormal ectactic capillaries in the papillary dermis, with the capillary walls being very thin.4 Varicose veins occur in 72% of patients with KTS,2,4 the prominent feature being the persistent (embryonic) lateral vein present in 56% of patients,2 which can be considered a pathognomonic feature [Figures 2 and 3]. There are significantly large valveless truncal veins presenting as enormous varicosities, but many other anomalies may exist, such as compressive fibrous bands, aneurysmal dilatation, duplication, hypoplasia, atresia and aplasia.5–7 Due to venous stasis in these large valveless veins, deep vein thrombosis (DVT) and associated pulmonary embolism are well known complications of KTS.
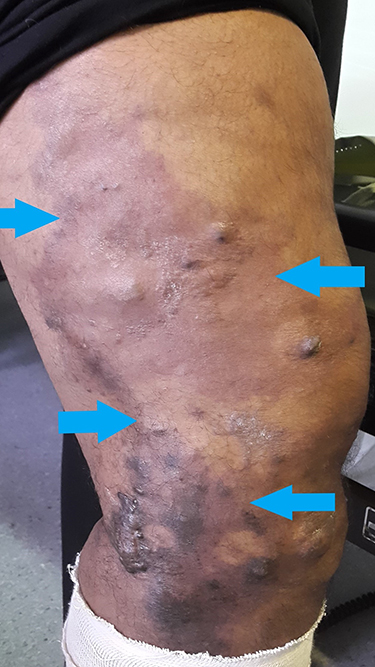 |
Figure 1 Cutaneous haemangioma seen on left thigh of a patient with Klippel-Trénaunay syndrome (limit of edges outlined by blue arrows). |
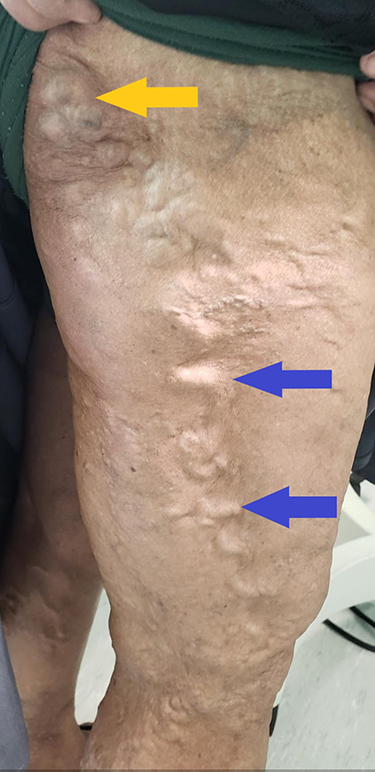 |
Figure 2 Lateral marginal vein (blue arrows) and gluteal vein (yellow arrow) seen in patient with Klippel-Trénaunay syndrome. The lateral marginal vein is considered pathognomonic of the disease. |
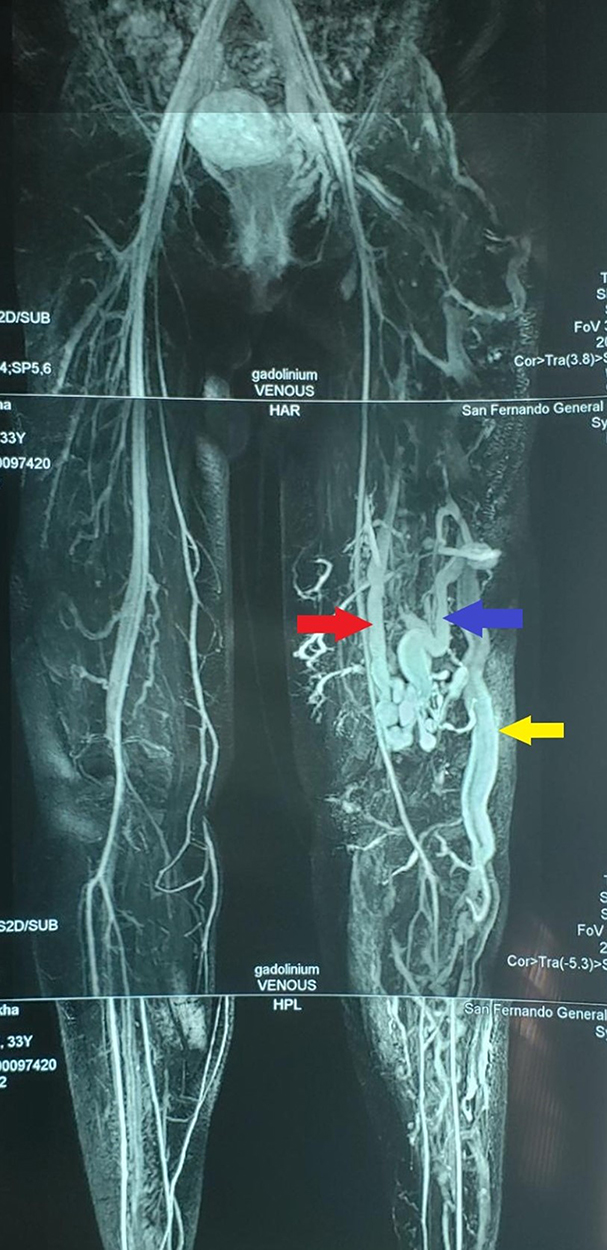 |
Figure 3 MRI showing lateral marginal vein (yellow arrow), perforator vein (blue arrow) and an isolated segment of a vestigial or hypoplastic femoral vein (red arrow). |
Lymphatic malformations, which are more common than expected, occur in 11% of KTS patients. They consist of dilated vessels filled with clear protinaceous fluid, but they do not connect to normal lymphatic vessels lying in cutaneous and subcutaneous tissue.8 The lymphatic system has a very close developmental, structural and functional relationship with the venous system,9–11 and plays an important role in the symptomatology and progress of these patients. It is best imaged by MRI- lymphangiography.9
Limb hypertrophy occurs in 67% of KTS patients, with 88% involving the lower limb2 and 71.5% involving a single limb6 [Figure 4]. Though bilateral and truncal involvement are rare, extensive venous networks extend to spread across viscera of the pelvis and spinal cord.12 Visceral vascular malformations can be seen in the liver, bladder, rectum, retropeitoneum and pericardium.4 Patients can therefore present with a variety of symptoms including internal haemorrhage and rectal bleeding.13
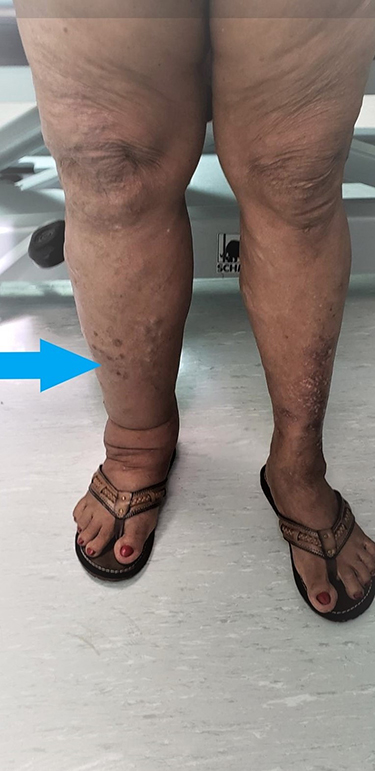 |
Figure 4 Hemi-hypertrophy of right lower limb (blue arrow) in patient with Klippel-Trénaunay syndrome. |
The Genetic Profile of KTS Patients-The Susceptibility Gene Theory
The molecular genetic basis of KTS was first studied by searching for a susceptible gene. However, its sporadic occurrence and mosaic distribution14 could only be explained by a paradominant inheritance, or a model of autosomal dominant inheritance with incomplete penetrance.15 Though most cases were sporadic, symptoms of capillary malformations with varicose veins had occurred within family members of patients.3,4,16–18 This led to the quest for a germline input into its etiology, and three chromosomal abnormalities were subsequently observed in KTS patients. These were 2 chromosomal translocations and an extra-supernumerary ring chromosome, confirming that genetic factors contributed to the etiology of KTS.4,18–20 The presence of three separate chromosomal abnormalities also demonstrated that there was some degree of heterogeneity in its genetic constitution. Investigations showed that at the breakpoint of the chromosome (11p15.1), there was a promoter region of a novel Vascular Gene on 5q initially named VG5Q. This vascular gene was then formally named AGGF1 [Angiogenic Growth Factor 1], a potent angiogenesis factor lying in the domain of the vascular endothelial growth factors (VEGFs-A to F). VEGFA was already known to be an early embryonic marker for vasculature formation.15,21,22 Identification was therefore made of a susceptibility gene which, when mutated, caused a predilection to Klippel-Trenaunay Syndrome.15 This gene was identified in 5 of 130 patients with KTS (but not in 200 control patients). It was thought to be of great significance, since a genetic component was finally identified.15 This was important in the context of the many malformation syndromes that have a vascular component at the forefront of their clinical presentations, such as KTS, Parkes Weber Syndrome (PWS) and the Servelle Matorell Syndrome.
Non-Inherited Gene Mutations
Vascular morphogenesis is vital for normal embryonic development,5 so the finding of genes to explain the vascular anomalies was critical. The presence of the limb overgrowth with the vascular abnormalities in KTS was compared with other congenital leg-length discrepancy syndromes, which had co-existing vascular morphogenic anomalies.12 This was necessary since the KTS mutations appeared to enhance increased angiogenesis.
Although KTS was considered to be a distinct entity,23 there were very similar syndromes in which patients had hemi-megalencephaly, seizures, developmental delay as well as syndactyly and macrodactyly. This suggested a multisystem disease existed, which could be part of a spectrum of disorders.23 However, KTS is a sporadic disease almost all of the time with no distinct family history, and it arises from non-inherited gene mutations, which are not germline, but somatic mutations.14,20,24,25 They are known to arise in cells in the embryonic stage prior to birth, and as development continues, cell proliferation occurs. Those cells arising from that original mutated cell, will carry the disease (the mutation), but the other cells do not carry this abnormality. This is referred to as mosaicism, where there is a mingling of cells, with and without mutation.26
The PIK3 CA-Induced Limb Overgrowth Syndromes
Many syndromes were also noted to have overlapping features with KTS.27–29 Vascular anomaly syndromes were recognised, especially in children where they sometimes coexisted with leg-length discrepancies, referred to as “limb overgrowth syndromes.”12 These syndromes not only have very unusual genetic and phenotypical features but have a somatic mosaicism that is sporadic.14 This coexistence led the finding of a gene mutation occurring along somatic lines, involving the phosphoInositide 3–kinase pathway (PI3K or PIK3).30 The PI3K enzyme is responsive to the body’s growth factors, such as the vascular endothelial growth factor, the epidermal growth factor receptor, and insulin. The Catalytic Alpha sub-unit of the PI3K enzyme is encoded by the gene called the PIK3 CA gene and it is the PIK3 CA-pathway that mediates initial cellular growth in the embryo.31 Signals from this PIK3CA pathway coordinate cell growth by regulating ribosomal protein synthesis.12
This PIK3CA cell-signalling pathway mediates cell growth as well as angiogenesis, so that mutations at certain points in this pathway lead to the leg-length discrepancy, or overgrowth. Vascular anomalies which characterize these syndromes have significant phenotypic overlap.
The PIK3CA Gene and the-Overgrowth Syndromes [MCAP, FAH and CLOVES]
The first sporadic growth disorder with cerebral, epidermal and musculoskeletal abnormalities was discovered in 1997.32 Researchers described the megalencephaly capillary malformation (MCAP) and the megalencephaly-polymicrogyria-polydectyly-hyrocepahlus (MPPH) syndromes. Interest centered on the fact that there was a somatic mutation in the PIK3CA gene which was noted in all twenty cases. In 2007, the congenital lipomatous overgrowth, vascular anomalies, epidermal naevi and scoliosis/spinal deformities (CLOVES) syndrome was discovered in seven patients, followed by the Fibroadipose hyperplasia syndrome (FAH) in 2012.34,35 Some of these patients were found to share overlapping features with KTS.33 For example, CLOVES is often misdiagnosed as KTS, Proteus and PWS.15
It is this description that led to the term PIK3CA-related overgrowth spectrum (PROS), disorders all involving somatic mutations in PIK3CA gene.30 Abnormal stimulation of the signalling pathway results in both tissue overgrowth and vascular abnormalities.12 PROS acts as a general term to group all limb overgrowth syndromes that have a somatic mutation in PIK3CA, and are typically characterized by vascular anomalies and limb hypertrophy.
These syndromes with overlapping features include vascular abnormalities with soft tissue, cutaneous and bone abnormalities, some being tissue-specific, others multi-organ systems.36 Many have been found to have heterozygous somatic mutations in the PIK3CA gene in a mosaic pattern,37 which means that all the cells do not have, nor demonstrate, the effect of the genetic mutation.23,33,34
Similar Syndromes: The Parkes-Weber Syndrome
There are also some syndromes which are very similar to KTS with vascular and growth abnormalities, but are caused by mutation of different genes. A distinction between them can be made when patients possess lower limb hypertrophy, ipsilateral cutaneous haemangioma and varicosities with arterio-venous fistulas. This represents a different disorder and Lindenauer suggested calling it the Parkes Weber Syndrome, because Weber had (in 1907) described cases with arteriovenous fistulas, as well as those similar to what Klippel and Trénaunay had originally described.38 The Parkes Weber Syndrome (PWS) is characterized by high-flow vascular malformations,39 including significant arterio-venous shunts, which are absent in KTS,3 but lymphatic malformations are rare.4 The disease is now known to be caused by mutations of another gene, the RASA-1 gene.40
Genetic Mutations in Other Members of Overgrowth Syndrome (PROS)
Another KTS-like disorder is the Proteus syndrome, which was originally thought to be a hamatomatous disorder.41 Clinical findings include epidermal naevi, asymmetrical limb growth and adipose tissue abnormalities. Imaging features are those of asymmetrical somatic overgrowth with capillary, venous and possible lymphatic malformations. This is a progressive disease with the phenotypic appearance worsening over time, so it was named after the Greek deity, Proteus, who was shape-shifting in disposition.42 It is associated with mutation of the AKT1 gene.36,37
Targeted Genetic Treatment Options-The MTOR Gene and Rapamycin
The catalytic subunit of the PIK3 enzyme, PIK3CA, is known to initiate cellular growth in the embryo, mediating proliferation and angiogenesis. It activates the AKT pathway which then activates mTOR, the mechanistic target of rapamycin (a macrolide used to treat graft rejection).43 The PIK3CA-AKT-mTOR axis lies in sequence so that rapamycin, which inhibits mTOR, has an upstream effect on PIK3CA.23 Rapamycin adheres to a binding protein and this gain-of-function complex acts as a specific inhibitor of mTOR. When it inactivates the mTOR-complex, it inhibits cellular proliferation with the cell cycle being immobilized in the G1 phase, mid to late cycle.23 Since this action affects PIK3CA, the mutation found in KTS, it may be useful in the early stages of the disease, inhibiting unwanted cell proliferation and tissue overgrowth in patients with KTS.44 In addition, members of the PROS, especially CLOVES, MCAP and FAH have heterozygous gain-of-function mutations in the PIK3CA gene in a mosaic pattern,34,35 and may also be the beneficiaries of targeted treatment.
Conclusion
The Klippel-Trenaunay Syndrome (KTS) can be diagnosed clinically by the experienced physician since it is a disease of recognition, based on distinct clinical findings and specific features on multimodal imaging. It belongs to a spectrum of limb-overgrowth syndromes (PROS), with whose members it shares many similarities. Genetic research has confirmed that a mutation in the PIK3CA gene has been implicated in KTS, and members of the related limb overgrowth spectrum (PROS). The existence of other genes such as the RASA-1 gene in Parkes Weber Syndrome and AKT1 gene in Proteus Syndrome has been noted, allowing researchers a better and more detailed understanding of role of molecular genetics in KTS and the PROS.
Genetic testing can now be extrapolated to patients and their families, and the identification of angiogenesis factors and vascular abnormalities may allow for therapeutic intervention with genetically directed options being available. The possibility of treatment at an early stage of the disease, and prevention of transmission to successive generations is currently at the forefront of genetic research in this field.
Consent & Ethical Approval
Informed consent was obtained from all the patients whose photographs and MRI scans are displayed. Institutional Review was sought from the Hospital/Health Authority’s Bioethics Committee but is not required for this review article. This study was conducted in accordance with The Declaration of Helsinki.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
The authors received no funding for this article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Klippel M, Trénaunay P. Du naevus variquex osteohypertrophique. Arch Genet Med Paris. 1900;3:611–672.
2. Jacob AG, Driscoll DJ, Shaughnessy WJ, Stanson AW, Clay RP, Gloviczki P. Klippel-Trénaunay syndrome: spectrum and management. Mayo Clin Proc. 1998;73(1):28–36. doi:10.1016/S0025-6196(11)63615-X
3. Lindenauer SM. The Klippel-Trénaunay syndrome: varicosity, hypertrophy and hemangioma with no arteriovenous fistula. Ann Surg. 1965;162(2):303–314. doi:10.1097/00000658-196508000-00023
4. Timur AA, Driscoll DJ, Wang Q. Biomedicine and diseases: the Klippel-Trenaunay syndrome, vascular anomalies and vascular morphogenesis. Cell Mol Life Sci. 2005;62(13):1434–1447. doi:10.1007/s00018-005-4523-7
5. Paes EH, Vollmar JF. Aneurysmal transformation in congenital venous angiodysplasias in lower extremities. Int Angiol. 1990;9:90–96.
6. Glovinski P, Stanson AW, Stickler GB, et al. Klippel-Trénaunay syndrome: the risks and benefits of vascular interventions. Surgery. 1991;110:469–479.
7. Gorenstein A, Shifrin E, Gordon RL, Katz S, Schiller M. Congenital aplasia of the deep veins of the lower extremities of children: the role of ascending functional phlebography. Surgery. 1986;99:414–420.
8. Vikkula M, Boon LM, Mulliken JB, Olsen BR. Molecular basis of vascular anomalies. Trends Cardiovasc Med. 1998;8:281–292. doi:10.1016/S1050-1738(98)00024-3
9. Liu NF, Lu Q, Yan ZX. Lymphatic malformation is a common component of Klippel-Trenaunay syndrome. J Vasc Surg. 2010;52(6):1557–1563. doi:10.1016/j.jvs.2010.06.166
10. Sabin FR. On the origin of the lymphatic system from the veins, and the development of the lymph hearts and thoracic duct in the pig. Am J Anat. 1902;1:367–389. doi:10.1002/aja.1000010310
11. Sabin FR. On the development of the superficial lymphatics in the skin of the pig. Am J Anat. 1904;3:183–195. doi:10.1002/aja.1000030205
12. Bertino F, Braithwaite KA, Hawkins CM, et al. Congenital limb overgrowth syndromes associated with vascular anomalies. Radiographics. 2019;39(2):491–515. doi:10.1148/rg.2019180136
13. Cha SH, Romeo MA, Neutze JA. Visceral manifestations of Klippel-Trénaunay syndrome. Radiographics. 2005;25(6):1694–1697. doi:10.1148/rg.256055042
14. Happel R. Klippel-Trénaunay syndrome: is it a paradominant trait? Br J Dermatol. 1993;1228:465–466.
15. Tian X-L, Kadaba R, You SA, et al. Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trénaunay syndrome. Nature. 2004;427(6975):640–645. doi:10.1038/nature02320
16. Aelvoet GE, Jorens PG, Roelen LM. Genetic aspects of the Klippel-Trenaunay syndrome. Br J Dermatol. 1992;126:603–607. doi:10.1111/j.1365-2133.1992.tb00107.x
17. Ceballos-Quintal JM, Pinto-Escalante D, Castillo-Zapata I. A new case of Klippel-Trenaunay-Weber (KTW) syndrome: evidence of autosomal dominant inheritance. Am J Med Genet. 1996;63:426–427. doi:10.1002/(SICI)1096-8628(19960614)63:3<426::AID-AJMG2>3.0.CO;2-P
18. Lorda-Sanchez I, Prieto L, Rodriguez-Pinilla E, Martinez-Frias ML. Increased parental age and number of pregnancies in Klippel-Trenaunay-Weber syndrome. Ann Hum Genet. 1998;62:235–239. doi:10.1046/j.1469-1809.1998.6230235.x
19. Whelan AJ, Watson MS, Porter FD, Steiner RD, Steiner RD. Klippel-Trenaunay-Weber syndrome associated with a 5:11 balanced translocation. Am J Med Genet. 1995;59:492–494. doi:10.1002/ajmg.1320590416
20. Wang Q, Timur AA, Szafranski P, et al. Identification and molecular characterization of de novo translocation t (8;14) (q22.3;q13) associated with a vascular and tissue overgrowth syndrome. Cytogenet Cell Genet. 2001;95:183–188. doi:10.1159/000059343
21. Ferrara N, Carver-Moore K, Chen H, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi:10.1038/380439a0
22. Shalaby F, Rossant J, Yamaguchi TP, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi:10.1038/376062a0
23. Vahidnezhad H, Youssefian L, Uitto J. Klippel-Trenaunay syndrome belongs to the PIK3CA-related overgrowth spectrum (PROS). Exp Dermatol. 2016;25(1):17–19. doi:10.1111/exd.12826
24. Happle R. Cutaneous manifestation of lethal genes. Hum Genet. 1986;72:820. doi:10.1007/BF00291899
25. Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899–906. doi:10.1016/S0190-9622(87)80249-9
26. Hu Y, Li L, Seidelmann SB, et al. Identification of association of common AGGF1 variants with susceptibility for Klippel-Trenaunay syndrome using the structure association program. Ann Hum Genet. 2008;72(Pt 5):636–643. doi:10.1111/j.1469-1809.2008.00458.x
27. Toriello HV, Mulliken JB. Accurately renaming macrocephaly-cutis marmorata telangiectasia congenita (M-CMTC) as macrocephaly-capillary malformation (M-CM). Am J Med Genet A. 2007;143A(24):3009. doi:10.1002/ajmg.a.31971
28. Mirzaa GM, Conway RL, Gripp KW, et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158A(2):269–291. doi:10.1002/ajmg.a.34402
29. Mirzaa GM, Dobyns WB. The “megalencephaly-capillary malformation” (MCAP) syndrome: the nomenclature of a highly recognizable multiple congenital anomaly syndrome. Am J Med Genet A. 2013;161A(8):2115. doi:10.1002/ajmg.a.35940
30. Loconte DC, Grossi V, Bozzao C, et al. Molecular and functional characterization of three different postzygotic mutations in PIK3CA-related overgrowth spectrum (PROS) patients: effects on PI3K/AKT/mTOR signaling and sensitivity to PIK3 inhibitors. PLoS One. 2015;10(4):e0123092. doi:10.1371/journal.pone.0123092
31. Kang HC, Baek ST, Song S, Gleeson JG. Clinical and genetic aspects of the segmental overgrowth spectrum due to somatic mutations in PIK3CA. J Pediatr. 2015;167(5):957–962. doi:10.1016/j.jpeds.2015.07.049
32. Moore CA, Toriello HV, Abuelo DN, et al. Macrocephaly-cutis marmorata telangiectatica congenita: a distinct disorder with developmental delay and connective tissue abnormalities. Am J Med Genet. 1997;70(1):67–73. doi:10.1002/(SICI)1096-8628(19970502)70:1<67::AID-AJMG13>3.0.CO;2-V
33. Sapp JC, Turner JT, van de Kamp JM, van Dijk FS, Lowry RB, Biesecker LG. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. Am J Med Genet A. 2007;143A(24):2944–2958. doi:10.1002/ajmg.a.32023
34. Lindhurst MJ, Parker VE, Payne F, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44(8):928–933. doi:10.1038/ng.2332
35. Kurek KC, Luks VL, Ayturk UM, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90(6):1108–1115.d. doi:10.1016/j.ajhg.2012.05.006
36. Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, et al. Clinical delineation and natural history of the PIK3CA-related overgrowth spectrum. Am J Med Genet A. 2014;164A(7):1713–1733. doi:10.1002/ajmg.a.36552
37. Nathan N, Keppler-Noreuil KM, Biesecker LG, Moss J, Darling TN. Mosaic disorders of the PI3K/PTEN/AKT/TSC/mTORC1 signalling pathway. Dermatol Clin. 2017;35(1):51–60. doi:10.1016/j.det.2016.07.001
38. Weber FP. Angioma formation in connection with hypertrophy of limbs and hemihypertrophy. Br J Dermatol. 1907;19:231–235.
39. ISSVA Classification of Vascular Anomalies. International society for the study of vascular anomalies. Available from: www.issva.org/UserFiles/file/ISSVA-Classification-2018.pdf.
40. Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation arteriovenous malformation, a new clinical and genetic disorder caused by the RASAI mutations. Am J Hum Genet. 2003;73:1240–1249. doi:10.1086/379793
41. Cohen MM, Hayden PW. A newly recognized hamartomatous syndrome. Birth Defects Orig Artic Ser. 1979;15(5B):291–296.
42. Wiedemann HR, Burgio GR, Aldenhoff P, Kunze J, Kaufmann HJ, Schirg E. The proteus syndrome: partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, macrocephaly or other skull anomalies and possible accelerated growth and visceral affections. Eur J Pediatr. 1983;140(1):5–12. doi:10.1007/BF00661895
43. Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12(3–4):112–124. doi:10.1016/j.drudis.2006.12.008
44. Youssefian L, Vahidnezhad H, Baghdadi T, et al. Fibroadipose hyperplasia versus Proteus syndrome: segmental overgrowth with a mosaic mutation in the PIK3CA gene. J Invest Dermatol. 2015;135(5):1450–1453. doi:10.1038/jid.2015.15
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.