Back to Journals » International Journal of General Medicine » Volume 15
The Key Genes Underlying Pathophysiology Correlation Between the Acute Myocardial Infarction and COVID-19
Authors You H ![]() , Zhao Q, Dong M
, Zhao Q, Dong M ![]()
Received 23 December 2021
Accepted for publication 23 February 2022
Published 4 March 2022 Volume 2022:15 Pages 2479—2490
DOI https://doi.org/10.2147/IJGM.S354885
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Scott Fraser
Hongjun You,1 Qianqian Zhao,2 Mengya Dong1
1Department of Cardiovascular Medicine, Shaanxi Provincial People’s Hospital, Xi’an, 710068, Shaanxi, People’s Republic of China; 2Department of Clinical Immunology, The First Affiliated Hospital, Air Force Military Medical University, Xi’an, 710032, Shaanxi, People’s Republic of China
Correspondence: Mengya Dong, Department of Cardiovascular Medicine, Shaanxi Provincial People’s Hospital, 256 West Youyi Road, Xi’an, Shaanxi, 710068, People’s Republic of China, Tel +86– 15802943974, Email [email protected]
Introduction: Accumulating evidences disclose that COVID-19, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has a marked effect on acute myocardial infarction (AMI). Nevertheless, the underlying pathophysiology correlation between the AMI and COVID-19 remains vague.
Materials and Methods: Bioinformatics analyses of the altered transcriptional profiling of peripheral blood mononuclear cells (PBMCs) in patients with AMI and COVID-19 were implemented, including identification of differentially expressed genes and common genes between AMI and COVID-19, protein–protein interactions, Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses, TF-genes and miRNA coregulatory networks, to explore their biological functions and potential roles in the pathogenesis of COVID-19-related AMI.
Conclusion: Our bioinformatic analyses of gene expression profiling of PBMCs in patients with AMI and COVID-19 provide us with a unique view regarding underlying pathophysiology correlation between the two vital diseases.
Keywords: AMI, SARS-CoV-2, differentially expressed genes, DEGs, bioinformatics, pathophysiology
Introduction
So far, acute myocardial infarction (AMI) still has a soaring morbidity and mortality globally, whose survival rate is approaching malignant tumors, a severe menace to public health. The major latent pathological alteration acknowledged are coronary inflammation, atherosclerotic plaques’ tendency to instability and rupture, endothelial dysfunction, platelet activation, coagulation disorder and acute coronary thrombosis event. With the propagation of COVID-19 attributed to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), accumulating evidences disclose that the lethal virus can also invade the cardiovascular system, debilitating endothelial barrier, causing coronary thromboembolic events and acute myocardial injury.1 The endotheliocytes and myocardial cells highly express angiotensin converting enzyme 2 (ACE2) receptors, via which SARS-CoV-2 could smite into cardiovascular system leading to endothelial malfunction, vessel or cardiomyocytes inflammation, and subsequent plaque rupture and extemporary heart attacks.2,3 It has been reported by Huang et al4 that approximate 12% of sufferers of COVID-19 were diagnosed acute myocardial injury, characterized by elevated levels of high-sensitive troponin I. Once attacked by SARS-CoV-2, systemic inflammation storm will perturb body’s immune system balance, prone to coronary plaque rupture, thrombogenesis and even acute stent thrombosis in cardiovascular diseases (CVD) sufferers.5 What’s more, vaccination against SARS-CoV-2, which mimics a mild viral infection and is crucial to eradicate the pandemic, was reported to be implicated into acute myocardial infarction.6 Whether and how COVID-19 serves as a trigger for acute myocardial infarction and the detailed pathogenic molecular mechanism remains vague. Hence, it’s of tremendous clinical significance to investigate and decipher the underlying pathophysiology relevancy between the acute myocardial infarction and COVID-19.
Both acute myocardial infarction and COVID-19 could provoke a transformation of original gene expression profiling of hematological cells. This prerequisite taken into account, we comprehensively analyze the altered gene expression profiles of the two vital diseases via bioinformatic methods, to disclose their possible relevance. In our current study, we integrate altered gene expression status of peripheral blood mononuclear cells (PBMCs) in patients with acute myocardial infarction and novel coronavirus infection, identify their common differentially expressed genes (DEGs) and hub genes, probe their function and involved molecular mechanisms, to probe whether COVID-19 plays a causative role in the onset of AMI. The workflow for the current research is shown in Figure 1.
 |
Figure 1 Workflow for the current study. The DEGs for GSE62646 dataset of AMI and GSE164805 dataset of COVID-19 were identified using the GEO2R. Common DEGs of the two diseases was obtained via the “VennDiagram”. GO and KEGG analyses were conducted for all common DEGs by the R programming language. From all common DEGs, a PPI network was created, and hub genes identification and enrichment analysis were performed. TF-miRNA coregulatory network for hub genes were conducted by NetworkAnalyst. |
Methods
Dataset’s Collection
Dataset (GSE62646) expounds the transcriptional profiling of PBMCs in patients with AMI and dataset (GSE164805) elucidates the specifical transcriptional signatures of PBMCs in COVID-19 patients. Both datasets were extracted from GEO database of the National Center for Biotechnology Information platform (www.ncbi.nlm.nih.gov/geo).7–9 GPL6244 (Affymetrix Human Gene 1.0 ST Array) platform and GPL26963 (Agilent-085982 Arraystar human lncRNA V5 microarray) platform are for the GSE62646 dataset and GSE164805 dataset, respectively. GSE62646 dataset was contributed by Kiliszek et al,10 in which 14 patients with stable coronary artery disease, without history of myocardial infarction and 28 patients suffering acute ST-segment elevation myocardial infarction (STEMI) are included. Drugs prescription’s perturbation taken into consideration, bloods samples collected on admission were applied in the current study, while blood sampling on discharge and 6 months after MI were excluded. Zhang Q et al furnished the dataset GSE164805, containing 5 healthy subjects and 10 patients suffering from COVID-19.11 The study complied with the Declaration of Helsinki and was approved by the Ethics Committee of Shaanxi Provincial People’s Hospital, Xi’an, Shaanxi, China.
Identification of DEGs and Common Genes Identification Between AMI and COVID-19
Datasets GSE62646 and GSE164805 were analyzed via GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) web tool which takes advantage of limma package of R programming language for identifying differentially expressed genes (DEGs). Benjamini-Hochberg was applied for both the datasets for controlling of false discovery rate (FDR).12 Cut-off criteria was set as adjusted P-value < 0.05 and log2-fold change (absolute) > 0.0 for dataset GSE62646, while adjusted P-value < 0.05 and log2-fold change (absolute) > 1.0 for dataset GSE164805. The DEGs were visualized in a volcano plot using the R package “ggplot2” (https://cran.r-project.org/web/packages/ggplot2/). The common genes identification between DEGs of GSE62646 and GSE164805 datasets was acquired by virtue of the R programming language and visualized through the “VennDiagram” (https://cran.r‑project.org/web/packages/VennDiagram/).
GO and KEGG Pathway Enrichment Analysis of DEGs
Gene Ontology (GO)13,14 and Kyoto Encyclopedia of Genes and Genomes (KEGG)15,16 pathway enrichment analysis of common DEGs or hub genes were performed by the R package “clusterProfiler” (http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html).17 The enrichment analysis results of GO categories including biological process (BP), cellular component (CC) and molecular function (MF) were obtained to elucidate the molecular activity, cellular role and the distribution in a cell where the genes exert their functionality.18 To illustrate and understand the metabolic or signaling pathways which the DEGs might convolved in, KEGG enrichment analysis was simultaneously implemented. The significance threshold was P-adjusted<0.05.
PPIs Network Construction and Hub Genes Identification
Protein–protein interactions (PPIs) network can furnish luxuriant information on the functional interactions between proteins in cellular biology study.19,20 The common DEGs were input into Search Tool for the Retrieval of Interacting Genes (STRING) (https://string-db.org/) online for coming into being a PPIs network.21 STRING transmits experimental and predictive interaction-founded information and the interaction generated via the online tool is defined with 3-dimensional structures, auxiliary biomedical information and confidence score.22 The confidence score in the current study was set as 0.900 considered to be a highest confidence score utilizing the STRING platform. Subsequently, the acquired PPIs network was reimported into Cytoscape for further analysis (https://cytoscape.org/), which is identified as the most potent instrument when it relates to integration with larger databases of genetic interactions, protein–protein and protein-DNA interactions.23,24 Cytoscape software plugin cytoHubba uses multiple scoring methods to dissect the PPIs network. Top 30 genes in the topological algorithm of Degree were identified as hub genes.
TF-miRNA Coregulatory Network
TFs and miRNAs reciprocity with the hub genes might participate a vital part in regulating the expression of hub genes. TF-miRNA coregulatory network for current hub genes was established by feat of NetworkAnalyst (https://www.networkanalyst.ca/), a powerful online-based platform for comprehensive analyses, systematic interpretation and deciphering of gene expression25,26 and RegNetwork repository, a synthetical database of transcriptional and post-transcriptional regulating networks.27
Results
Identification of DEGs and Common Genes Between AMI and COVID-19
In dataset GSE62646, 4035 DEGs were up-regulated, while 3366 DEGs were down-regulated. For dataset GSE164805, 6983 DEGs were up-regulated, while 6246 DEGs were down-regulated. The DEGs were visualized in a volcano plot, respectively (Figure 2A and B). As shown in Figure 2C, there were 2529 DEGs overlapped among the 7401 AMI DEGs and the 13228 COVID-19 DEGs.
 |
Figure 2 (Left) Volcano plot of the distributions of all DEGs for GSE62646 dataset of AMI, mapping 4035 upregulated genes (red dots) and 3366 downregulated genes (blue dots). |Log2FC|>0 and P-adjusted <0.05 were the cut-off criteria. (Middle) Volcano plot of the distributions of all DEGs for GSE164805 dataset of COVID-19, mapping 6983 upregulated genes (red dots) and 6246 downregulated genes (blue dots). |Log2FC|>1 and P-adjusted <0.05 were the cut-off criteria. (Right) Venn diagram. The two datasets for AMI and COVID-19 showed an overlap of 2529 common DEGs. |
GO and KEGG Pathway Enrichment Analysis of Common DEGs
GO and KEGG pathway enrichment analysis of 2529 common DEGs were performed. Top 10 terms of each category (BP, CC and MF) were visualized in bubble diagram (Figure 3A). KEGG analysis was showed in Figure 3B. GO and KEGG analyses information were presented in Supplementary Table 1 in detail. BP subset indicates common DEGs were highly enhanced in regulation of mRNA metabolic process, regulation of mRNA processing and proteasomal protein catabolic process; CC subsection mainly manifests spliceosomal complex, nuclear speck, chromosomal region and catalytic step 2 spliceosome; MF part expounds ribonucleoprotein complex binding, ubiquitin-protein transferase activity and ubiquitin protein ligase activity. KEGG pathway enrichment analyses clarify that common DEGs were significantly involved in spliceosome, yersinia infection, salmonella infection, platelet activation, Wnt signaling pathway, B cell receptor signaling pathway, T cell receptor signaling pathway, osteoclast differentiation, Fc gamma R-mediated phagocytosis, mTOR signaling pathway, insulin resistance and cellular senescence.
 |
Figure 3 (Left) Partial visualization of GO enrichment analysis for all common DEGs. (Right) Partial visualization of KEGG pathway enrichment analysis for all common DEGs. |
PPIs Network and Hub Genes Identification
The PPIs network contains 364 nodes and 4216 edges, and top 30 hub genes were identified based on Degree topological algorithm, picturized and highlighted in Figure 4. Top 30 identified hub genes are RAC1, CUL1, BTRC, NHP2L1, MAGOH, ANAPC10, PLK1, SRSF1, ALYREF, SNRPE, RHOA, FBXW5, SRSF7, SNRPF, SF3B1, FBXL3, CPSF2, SNRPD3, SNRPD1, SNW1, PIK3R1, VAMP8, KBTBD7, HIST1H2BL, KLHL20, FBXO2, KBTBD13, FBXL13, ASB18 and FBXO11. Topological result exploration for hub genes identification was presented in Supplementary Table 2.
 |
Figure 4 Identification of hub genes from the PPIs network of all common DEGs. The 30 highlighted hub genes based on the degree topological algorithm are RAC1, CUL1, BTRC, NHP2L1, MAGOH, ANAPC10, PLK1, SRSF1, ALYREF, SNRPE, RHOA, FBXW5, SRSF7, SNRPF, SF3B1, FBXL3, CPSF2, SNRPD3, SNRPD1, SNW1, PIK3R1, VAMP8, KBTBD7, HIST1H2BL, KLHL20, FBXO2, KBTBD13, FBXL13, ASB18 and FBXO11. The PPI network contains 442 nodes and 4951 edges. |
GO and KEGG Enrichment Analysis of Hub Genes
To further probe the role of hub genes of common genes between AMI and COVID-19, functional enrichment analysis of hub genes was implemented. The top 10 terms of BP, CC and MF were visualized in a bubble diagram (Figure 5A). Figure 5B shows us the KEGG analysis outcome. And the concrete information was displayed in Supplementary Table 3. GO enrichment analyses illustrate that hub genes were significantly involved in RNA splicing, post-translational protein modification, catalytic step 2 spliceosome, cullin-RING ubiquitin ligase complex, spliceosomal complex, ubiquitin-protein transferase activity, protein phosphorylated amino acid binding and ubiquitin-like protein transferase activity, etc. Interrelations between GO terms and hub genes were partially displayed in Figure 6A–C. With regard to KEGG pathway enrichment analyses, hub genes were significantly involved in spliceosome, circadian rhythm, oocyte meiosis, shigellosis, ubiquitin mediated proteolysis, Wnt signaling pathway, bacterial invasion of epithelial cells, colorectal cancer, viral carcinogenesis, mRNA surveillance pathway, human immunodeficiency virus 1 infection, progesterone-mediated oocyte maturation, leukocyte transendothelial migration, sphingolipid signaling pathway, neurotrophin signaling pathway, cell cycle, platelet activation, yersinia infection, fluid shear stress and atherosclerosis and hedgehog signaling pathway.
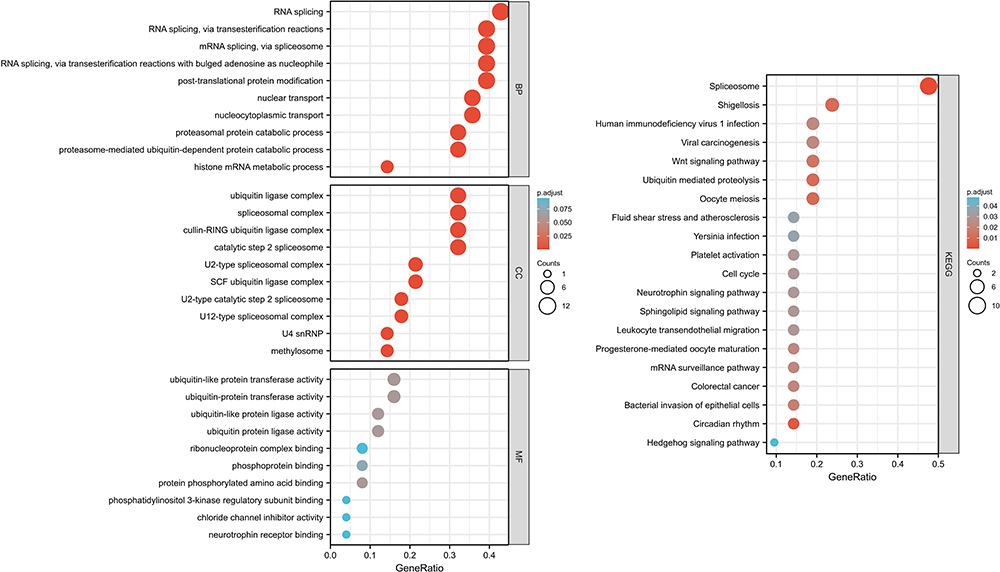 |
Figure 5 (Left) Partial visualization of GO enrichment analysis for hub genes. (Right) Partial visualization of KEGG pathway enrichment analysis for hub genes. |
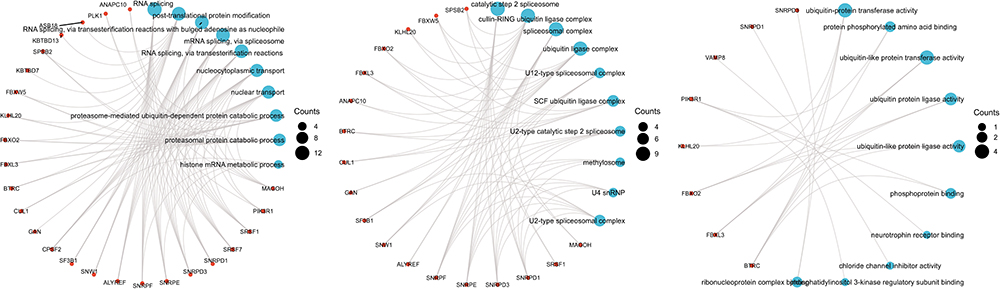 |
Figure 6 (Left) Partial visualization of interrelations between BP terms and hub genes. (Middle) Partial visualization of interrelations between CC terms and hub genes. (Right) Partial visualization of interrelations between MF terms and hub genes. |
TF-miRNA Coregulatory Network
The TF-miRNA coregulatory network was shown in Figure 7, constituting 473 nodes, 766 edges and 28 hub genes. The complicated coregulatory network includes abundant interactions between TFs and miRNAs and hub genes. TFs, namely, MYC, E2F1, EGR1, YY1, SNRPF, MAX, MEF2A, NFYA5, CTCF5 and TFAP2C interact with 9, 8, 7, 7, 6, 6, 6, 5, 5 and 5 hub genes, respectively. Hsa-miR-495, hsa-miR-16, hsa-miR-429, hsa-miR-200c and hsa-miR-300 interacts with 8, 5, 5, 5 and 5 hub genes simultaneously.
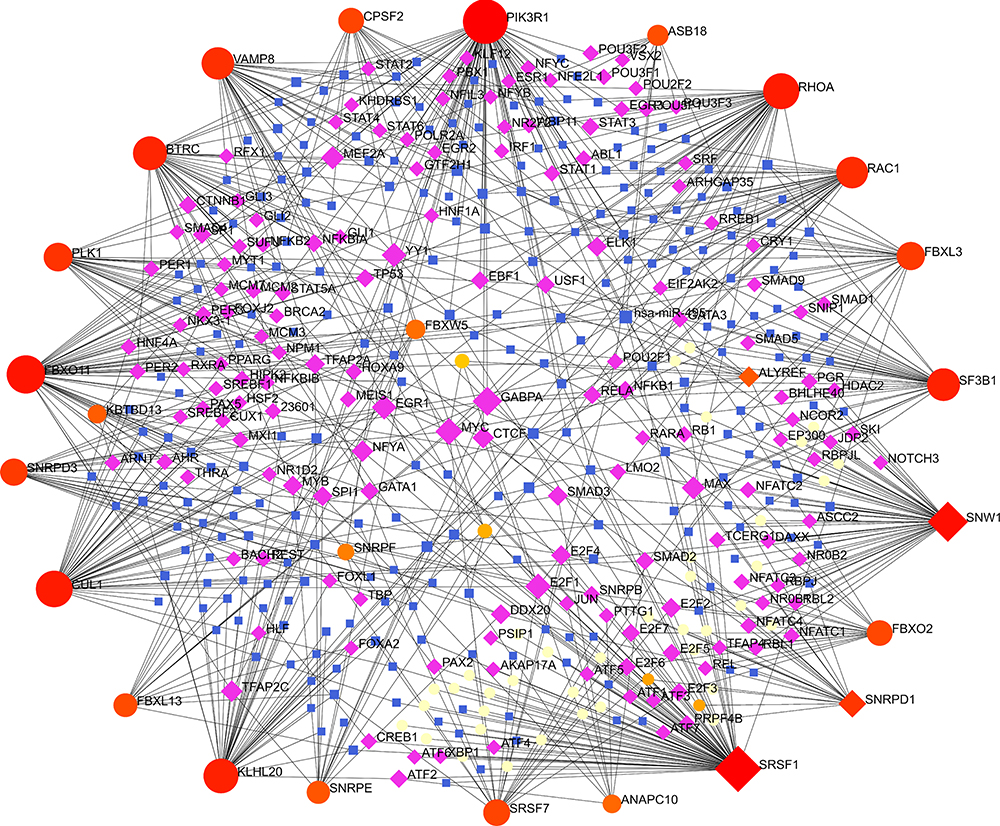 |
Figure 7 The TF-miRNA coregulatory network. The network constitutes of 473 nodes and 766 edges, including 28 hub genes. The nodes in red or orange are the hub genes, a blue node represents miRNA and purple nodes indicate TF genes. |
Discussion
With the accumulation of clinical data, cardiovascular complications induced by COVID-19 have attracted more and more attention. AMI, as an acute manifestation of cardiovascular diseases, is not only a common complication of SARS-CoV-2 viral infection, but also an important cause of poor prognosis in patients with COVID-19.1,4 However, the underlying mechanisms of how this viral infection leading to AMI are still blurry.
In the present study, we used two datasets, which expounded the transcriptional profiling of PBMCs in patients with AMI and COVID-19 respectively and performed a series of bioinformatics analysis trying to illustrate the pathogenesis of AMI in patients with SARS-CoV-2 infection. First, we identified common DEGs between these two diseases and explored functional annotations of these DEGs by means of GO and KEGG pathway enrichment analysis. Second, we constructed PPIs network and picked hub genes out among the whole common DEGs and carried on the functional analysis of hub genes similarly. Third, we established TF-miRNA coregulatory network in order to find out the targets of prevention and therapy of AMI in patients with COVID-19.
First of all, GO and KEGG analysis were conducted to preliminarily analyze the functional annotation and pathway enrichment of common DEGs. For GO, proteasomal protein catabolic process was listed at the top in the GO terms. The maintenance of cellular homeostasis depends on the proteostasis network to a great extent, which mainly includes autophagy and the ubiquitin-proteasome system. The network removes dysfunctional cellular components while maintains or replenishes normal and functional molecules via synthesizing. Therefore, the dysregulation of the proteostasis network results in several severe life-threatening diseases including COVID-19.28 Nevertheless, this kind of dysregulated metabolism might resulted in the activation of numerous pro-inflammatory cytokines and a hyper-inflammatory state, which has been a well-recognized mechanism of coronary artery disease.29,30 Zhang et al have reported that PA28γ, the 11S proteasomal activator, could decrease levels of the SARS-CoV-2 N protein nucleocapsid protein of SARS-CoV-2 through increasing 20S proteasome activity.31 Gassen et al also found that autophagy-inducing compounds such as spermidine, MK-2206 and niclosamide could inhibit SARS-CoV-2 propagation.32 These results indicated that the ubiquitin-proteasome system and autophagy induction to limit the infection and inflammatory response might be a therapeutic potential as an effective treatment for COVID-19 and subsequent cardiovascular injuries.
For KEGG, platelet activation was of great concern. Clinical data has indicated the relationship between COVID-19 and a significant risk of various thrombotic complications.33 SARS-CoV-2 infection increased platelet activity partially by means of MAPK pathway activation and thromboxane generation.34 In addition, SARS-CoV-2 could induce platelet activation via the bind of spike to ACE2 causing thrombosis.35 Besides platelet aggregation and thrombus formation, platelets were reported to modulate critical leukocyte responses such as monocyte expression of tissue factor, which was an essential trigger of coagulation and thrombosis.36,37 This effect has been proved in patients with severe COVID-19.37–39 Platelet activation and it-mediated release of tissue factor might play a critical role in the development of coagulopathy and thrombosis in patients with COVID-19, which shared a similar process with the fracture of the atherosclerotic plaque causing occlusion of the vessel and leading to an acute ischemic event.30 Our analysis revealed that common DEGs also enriched in Wnt signaling pathway. The Wnt/β-catenin/HIF-1α pathway has been confirmed to be activated in COVID-19 patients and caused pulmonary injury probably through inducing inflammation and a cytokine storm.30,40 Meanwhile, the persistent activation of Wnt was related to the pathological process of MI, including inflammatory response, myocardial apoptosis, granulation tissue formation and fibrosis.41,42 In addition, mTOR signaling pathway was also indicated in the pathogenesis of COVID-19 and AMI. The mTOR signaling pathway has been well-known in regulating apoptosis, cell survival and host transcription and translation and was activated in SARS-CoV-2 infection. Researchers has proved that MK-2206 could reduce SARS-CoV-2 production through inhibition of mTOR signaling pathway.43–45 The mTOR pathway also played a key role in cardiovascular physiology and pathology. Activation of mTOR increased myocardial damage, induced apoptosis, and cardiac rupture after AMI.46–49 These pathways mentioned above probably participated the occurrence and development of AMI after SARS-CoV-2 infection.
Secondly, we identified hub genes via the construction of the PPIs network. We retrieved all 30 hub genes in PubMed. Several genes have been researched in both COVID-19 and AMI. For example, reduced RAC1 activity was associated with acute lung injury in COVID-19 through endothelial barrier dysfunction.50 RAC1 has also been implicated in cardiovascular disorders including AMI. The adverse effect of the up-regulation of RAC1 on AMI was probably related with the increasing of ischemia/reperfusion injury, inflammation, proliferation and migration of endothelial cells and myocardial stunning.51–54 SRSF1, as an exonic splicing enhancer, played a significant role in genome stability. SRSF1 depletion led to infection spread to adjacent pneumocytes and other tissue and organs in SARS-CoV-2 infection.55 In AMI, SRSF1 took part in myocardial ischemia-reperfusion injury.56 Some genes were only studied in COVID-19. For instance, VAMP8, as an important component of the STX17-SNAP29-VAMP8 SNARE complex, mediated autophagosome/amphisome fusion with lysosomes. However, SARS-CoV-2 inhibited the assembly of the complex in order to escape from lysosomal destruction.57 Other genes were also indicated in COVID-19 through bioinformatics methods, such as PLK1, RHOA and PIK3R1.58–60 How the latter two kinds of hub genes triggered AMI in patients with COVID-19 needed further researches.
Then we carried on the functional analysis of hub genes and the results were widely consistent with mentioned above. Specially, hub genes were involved in the pathway of leukocyte transendothelial migration via KEGG enrichment analysis. A part of COVID-19 patients died from acute respiratory distress syndrome (ARDS), which was characterized by a massive migration of leukocytes to the lungs. Researchers has reported GT-73, a blocker of leukocyte transendothelial migration from blood to the peripheral tissues, might serve as a candidate drug for the treatment of ARDS in COVID-19 patients.61 It was well-known that leukocyte transendothelial migration was a crucial process in atherogenesis.30 This common pathogenesis probably accounted for AMI in COVID-19 patients.
Thirdly, we constructed the TF-miRNA coregulatory network in order to find the regulatory molecules of hub genes. Previous studies have proposed that MYC, a transcription factor, was one of key cellular factors to mediate and promote the glutamine metabolic reprogramming to meet the demand of energy and biosynthetic requirements during the infection cycle in SARS-CoV-2 infected cells.62 Others also suggested that MYC was a potential drug target for COVID-19, which were consistent with ours.63,64 Meanwhile, MYC also played a critical role in the control of the mitochondrial function in hypoxic ischemic injury and the protection of cardiomyocytes through enhancing the bone marrow mesenchymal stem cell viability, migration and anti-apoptosis effects in AMI.65,66 The dysfunction of MYC was probably a common pathway in the pathogenesis of COVID-19 and AMI. As for miRNA, Li et al has reported that hsa-miR-16 was the top one upregulated miRNA in the peripheral blood in patients with COVID-19 compared to that of the healthy controls.67 Furthermore, Wicik et al found that hsa-miR-16 might mediate the interactions between SARS-CoV-2 and angiotensin-converting enzyme 2 functional networks, which was an important regulator in AMI.68 TF-miRNA-hub genes were a complicated regulatory network. These TF and miRNAs might become therapeutic targets for patients with COVID-19 and high risk for AMI.
Lots of biomarkers have been reported in COVID-19 patients complicated with ischemic heart diseases. For example, a decrease in fibrinogen and an increase in D-dimer, which indicated systemic hypercoagulable state, might have predictive values in these patients. And anticoagulation therapy based on these two indicators might improve the prognosis of patients with COVID-19 and cardiovascular complications.69 Moreover, most researchers consider that there is a close association between this troponin increasing and the worse prognosis in COVID-19 patients no matter with ischemic heart injury or not.70,71 However, it is also worth noting that there is still no evidence of direct infection of SARS-CoV-2 in myocardium. Myocardial injury, defined by a troponin elevation in patients with COVID-19, is to a great extent related to severe hypoxia, sepsis, systemic inflammation, pulmonary thrombosis embolism, and also probably related to stress cardiomyopathy, myocarditis.72 Hence, the pathogenesis of AMI in COVID-19 still need to be addressed in further studies.
Conclusion
Our bioinformatic analyses of gene expression profiling of PBMCs in patients with AMI and COVID-19 provide us with a unique view regarding underlying pathophysiology correlation between the two vital diseases.
Abbreviations
ACE2, angiotensin converting enzyme 2; AMI, acute myocardial infarction; ARDS, acute respiratory distress syndrome; BP, biological process; CC, cellular component; CVD, cardiovascular diseases; DEGs, differentially expressed genes; FDR, false discovery rate; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MF, molecular function; PBMCs, peripheral blood mononuclear cells; PPIs, protein–protein interactions; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; STEMI, ST-segment elevation myocardial infarction.
Funding
There is no funding to report.
Disclosure
All authors report no conflicts of interest for this work.
References
1. Guo T, Fan Y, Chen M, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5(7):811–818. doi:10.1001/jamacardio.2020.1017
2. Froldi G, Dorigo P. Endothelial dysfunction in Coronavirus disease 2019 (COVID-19): gender and age influences. Med Hypotheses. 2020;144:110015. doi:10.1016/j.mehy.2020.110015
3. Yau JW, Teoh H, Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord. 2015;15:130. doi:10.1186/s12872-015-0124-z
4. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi:10.1016/S0140-6736(20)30183-5
5. Xiong TY, Redwood S, Prendergast B, Chen M. Coronaviruses and the cardiovascular system: acute and long-term implications. Eur Heart J. 2020;41(19):1798–1800. doi:10.1093/eurheartj/ehaa231
6. Özdemir İH, Özlek B, Özen MB, Gündüz R, Bayturan Ö. Type 1 Kounis syndrome induced by inactivated SARS-COV-2 vaccine. J Emerg Med. 2021;61(4):e71–e76. doi:10.1016/j.jemermed.2021.04.018
7. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi:10.1093/nar/30.1.207
8. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(Database issue):D991–995. doi:10.1093/nar/gks1193
9. Clough E, Barrett T. The gene expression omnibus database. Methods Mol Biol. 2016;1418:93–110.
10. Kiliszek M, Burzynska B, Michalak M, et al. Altered gene expression pattern in peripheral blood mononuclear cells in patients with acute myocardial infarction. PLoS One. 2012;7(11):e50054. doi:10.1371/journal.pone.0050054
11. Zhang Q, Meng Y, Wang K, et al. Inflammation and antiviral immune response associated with severe progression of COVID-19. Front Immunol. 2021;12:631226. doi:10.3389/fimmu.2021.631226
12. Korthauer K, Kimes PK, Duvallet C, et al. A practical guide to methods controlling false discoveries in computational biology. Genome Biol. 2019;20(1):118. doi:10.1186/s13059-019-1716-1
13. Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47(D1):D330–D338. doi:10.1093/nar/gky1055
14. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi:10.1038/75556
15. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–353D361. doi:10.1093/nar/gkw1092
16. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27
17. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
18. Doms A, Schroeder M. GoPubMed: exploring PubMed with the Gene Ontology. Nucleic Acids Res. 2005;33(Web Server issue):W783–786. doi:10.1093/nar/gki470
19. Ben-Hur A, Noble WS. Kernel methods for predicting protein-protein interactions. Bioinformatics. 2005;21(Suppl 1):i38–46. doi:10.1093/bioinformatics/bti1016
20. Ewing RM, Chu P, Elisma F, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi:10.1038/msb4100134
21. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–452. doi:10.1093/nar/gku1003
22. Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–568. doi:10.1093/nar/gkq973
23. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
24. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8 Suppl 4(Suppl4):S11. doi:10.1186/1752-0509-8-S4-S11
25. Xia J, Gill EE, Hancock RE. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc. 2015;10(6):823–844. doi:10.1038/nprot.2015.052
26. Zhou G, Soufan O, Ewald J, Hancock R, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47(W1):W234–234W241. doi:10.1093/nar/gkz240
27. Liu ZP, Wu C, Miao H, Wu H. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database (Oxford). 2015;2015:bav095. doi:10.1093/database/bav095
28. Pehote G, Vij N. Autophagy augmentation to alleviate immune response dysfunction, and resolve respiratory and COVID-19 exacerbations. Cells. 2020;9(9):1952. doi:10.3390/cells9091952
29. Jamwal S, Gautam A, Elsworth J, Kumar M, Chawla R, Kumar P. An updated insight into the molecular pathogenesis, secondary complications and potential therapeutics of COVID-19 pandemic. Life Sci. 2020;257:118105. doi:10.1016/j.lfs.2020.118105
30. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17(11):1410–1422. doi:10.1038/nm.2538
31. Zhang H, Tu J, Cao C, Yang T, Gao L. Proteasome activator PA28γ-dependent degradation of coronavirus disease (COVID-19) nucleocapsid protein. Biochem Biophys Res Commun. 2020;529(2):251–256. doi:10.1016/j.bbrc.2020.06.058
32. Gassen NC, Papies J, Bajaj T, et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat Commun. 2021;12(1):3818. doi:10.1038/s41467-021-24007-w
33. McFadyen JD, Stevens H, Peter K. The emerging threat of (Micro)Thrombosis in COVID-19 and its therapeutic implications. Circ Res. 2020;127(4):571–587. doi:10.1161/CIRCRESAHA.120.317447
34. Manne BK, Denorme F, Middleton EA, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136(11):1317–1329. doi:10.1182/blood.2020007214
35. Zhang S, Liu Y, Wang X, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020;13(1):120. doi:10.1186/s13045-020-00954-7
36. Stevens H, McFadyen JD. Platelets as central actors in thrombosis-reprising an old role and defining a new character. Semin Thromb Hemost. 2019;45(8):802–809. doi:10.1055/s-0039-1698829
37. Lindmark E, Tenno T, Siegbahn A. Role of platelet P-selectin and CD40 ligand in the induction of monocytic tissue factor expression. Arterioscler Thromb Vasc Biol. 2000;20(10):2322–2328. doi:10.1161/01.ATV.20.10.2322
38. Eslamifar Z, Behzadifard M, Soleimani M, Behzadifard S. Coagulation abnormalities in SARS-CoV-2 infection: overexpression tissue factor. Thromb J. 2020;18(1):38. doi:10.1186/s12959-020-00250-x
39. Mackman N, Grover SP, Antoniak S. Tissue factor expression, extracellular vesicles, and thrombosis after infection with the respiratory viruses influenza A virus and coronavirus. J Thromb Haemost. 2021;19(11):2652–2658. doi:10.1111/jth.15509
40. Vallée A, Lecarpentier Y, Vallée JN. Interplay of opposing effects of the WNT/β-catenin pathway and PPARγ and implications for SARS-CoV2 treatment. Front Immunol. 2021;12:666693. doi:10.3389/fimmu.2021.666693
41. Liu FJ. LncRNA-P21 suppresses apoptosis of myocardial cells in rats with acute myocardial infarction via regulating Wnt/β-ca tenin signaling pathway. Eur Rev Med Pharmacol Sci. 2020;24(19):10078–10085. doi:10.26355/eurrev_202010_23227
42. Sun HY, Wang XL, Ma LC, et al. Influence of MiR-154 on myocardial apoptosis in rats with acute myocardial infarction through Wnt/β-catenin signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23(2):818–825. doi:10.26355/eurrev_201901_16896
43. Appelberg S, Gupta S, Svensson Akusjärvi S, et al. Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg Microbes Infect. 2020;9(1):1748–1760. doi:10.1080/22221751.2020.1799723
44. Maiese K. The mechanistic target of rapamycin (mTOR): novel considerations as an antiviral treatment. Curr Neurovasc Res. 2020;17(3):332–337. doi:10.2174/1567202617666200425205122
45. Karam BS, Morris RS, Bramante CT, et al. mTOR inhibition in COVID-19: a commentary and review of efficacy in RNA viruses. J Med Virol. 2021;93(4):1843–1846. doi:10.1002/jmv.26728
46. Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114(3):549–564. doi:10.1161/CIRCRESAHA.114.302022
47. Yan L, Guo N, Cao Y, et al. miRNA‑145 inhibits myocardial infarction‑induced apoptosis through autophagy via Akt3/mTOR signaling pathway in vitro and in vivo. Int J Mol Med. 2018;42(3):1537–1547. doi:10.3892/ijmm.2018.3748
48. Ning S, Li Z, Ji Z, et al. MicroRNA‑494 suppresses hypoxia/reoxygenation‑induced cardiomyocyte apoptosis and autophagy via the PI3K/AKT/mTOR signaling pathway by targeting SIRT1. Mol Med Rep. 2020;22(6):5231–5242. doi:10.3892/mmr.2020.11636
49. Hua J, Liu Z, Liu Z, et al. Metformin increases cardiac rupture after myocardial infarction via the AMPK-MTOR/PGC-1α signaling pathway in rats with acute myocardial infarction. Med Sci Monit. 2018;24:6989–7000. doi:10.12659/MSM.910930
50. Wang T, Yegambaram M, Gross C, et al. RAC1 nitration at Y(32) IS involved in the endothelial barrier disruption associated with lipopolysaccharide-mediated acute lung injury. Redox Biol. 2021;38:101794. doi:10.1016/j.redox.2020.101794
51. Su Q, Liu Y, Lv XW, et al. Inhibition of lncRNA TUG1 upregulates miR-142-3p to ameliorate myocardial injury during ischemia and reperfusion via targeting HMGB1- and Rac1-induced autophagy. J Mol Cell Cardiol. 2019;133:12–25. doi:10.1016/j.yjmcc.2019.05.021
52. Su J, Gao C, Wang R, Xiao C, Yang M. Genes associated with inflammation and the cell cycle may serve as biomarkers for the diagnosis and prognosis of acute myocardial infarction in a Chinese population. Mol Med Rep. 2018;18(2):1311–1322. doi:10.3892/mmr.2018.9077
53. Gallo S, Sala V, Gatti S, Crepaldi T. Cellular and molecular mechanisms of HGF/Met in the cardiovascular system. Clin Sci (Lond). 2015;129(12):1173–1193. doi:10.1042/CS20150502
54. Talukder MA, Elnakish MT, Yang F, et al. Cardiomyocyte-specific overexpression of an active form of Rac predisposes the heart to increased myocardial stunning and ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2013;304(2):H294–302. doi:10.1152/ajpheart.00367.2012
55. Rogan PK, Mucaki EJ, Shirley BC. A proposed molecular mechanism for pathogenesis of severe RNA-viral pulmonary infections. F1000Res. 2020;9:943. doi:10.12688/f1000research.25390.1
56. Sun Y, Hu ZQ. LncRNA HOTAIR aggravates myocardial ischemia-reperfusion injury by sponging microRNA-126 to upregulate SRSF1. Eur Rev Med Pharmacol Sci. 2020;24(17):9046–9054. doi:10.26355/eurrev_202009_22850
57. Miao G, Zhao H, Li Y, et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell. 2021;56(4):427–442.e5. doi:10.1016/j.devcel.2020.12.010
58. Auwul MR, Rahman MR, Gov E, Shahjaman M, Moni MA. Bioinformatics and machine learning approach identifies potential drug targets and pathways in COVID-19. Brief Bioinform. 2021;22(5). doi:10.1093/bib/bbab120
59. He B, Garmire L. Prediction of repurposed drugs for treating lung injury in COVID-19. F1000Res. 2020;9:609. doi:10.12688/f1000research.23996.2
60. Bharne D. A protein interactions map of multiple organ systems associated with COVID-19 disease. Genomics Inform. 2021;19(2):e14. doi:10.5808/gi.20078
61. Blum E, Margalit R, Levy L, et al. A potent leukocyte transmigration blocker: GT-73 showed a protective effect against LPS-induced ARDS in mice. Molecules. 2021;26(15):4583. doi:10.3390/molecules26154583
62. Bharadwaj S, Singh M, Kirtipal N, Kang SG. SARS-CoV-2 and glutamine: SARS-CoV-2 triggered pathogenesis via metabolic reprogramming of glutamine in host cells. Front Mol Biosci. 2020;7:627842. doi:10.3389/fmolb.2020.627842
63. Selvaraj G, Kaliamurthi S, Peslherbe GH, Wei DQ. Identifying potential drug targets and candidate drugs for COVID-19: biological networks and structural modeling approaches. F1000Res. 2021;10:127. doi:10.12688/f1000research.50850.2
64. Banaganapalli B, Al-Rayes N, Awan ZA, et al. Multilevel systems biology analysis of lung transcriptomics data identifies key miRNAs and potential miRNA target genes for SARS-CoV-2 infection. Comput Biol Med. 2021;135:104570. doi:10.1016/j.compbiomed.2021.104570
65. Ham PB
66. Lin M, Liu X, Zheng H, et al. IGF-1 enhances BMSC viability, migration, and anti-apoptosis in myocardial infarction via secreted frizzled-related protein 2 pathway. Stem Cell Res Ther. 2020;11(1):22. doi:10.1186/s13287-019-1544-y
67. Li C, Hu X, Li L, Li JH. Differential microRNA expression in the peripheral blood from human patients with COVID-19. J Clin Lab Anal. 2020;34(10):e23590. doi:10.1002/jcla.23590
68. Wicik Z, Eyileten C, Jakubik D, et al. ACE2 interaction networks in COVID-19: a physiological framework for prediction of outcome in patients with cardiovascular risk factors. J Clin Med. 2020;9(11):3743. doi:10.3390/jcm9113743
69. Hayıroğlu Mİ, Çınar T, Tekkeşin Aİ. Fibrinogen and D-dimer variances and anticoagulation recommendations in Covid-19: current literature review. Rev Assoc Med Bras. 2020;66(6):842–848. doi:10.1590/1806-9282.66.6.842
70. Bivona G, Agnello L, Ciaccio M. Biomarkers for prognosis and treatment response in COVID-19 patients. Ann Lab Med. 2021;41(6):540–548. doi:10.3343/alm.2021.41.6.540
71. Çınar T, Hayıroğlu Mİ, Çiçek V, et al. Prognostic significance of cardiac troponin level in Covid-19 patients without known cardiovascular risk factors. Am J Emerg Med. 2021;45:595–597. doi:10.1016/j.ajem.2020.12.033
72. Imazio M, Klingel K, Kindermann I, et al. COVID-19 pandemic and troponin: indirect myocardial injury, myocardial inflammation or myocarditis? Heart. 2020;106(15):1127–1131. doi:10.1136/heartjnl-2020-317186
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.