Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
The interaction of sterically stabilized magnetic nanoparticles with fresh human red blood cells
Authors Pham B ![]() , Jain N, Kuchel P, Chapman B, Bickley S, Jones S, Hawkett B
, Jain N, Kuchel P, Chapman B, Bickley S, Jones S, Hawkett B
Received 28 July 2015
Accepted for publication 19 September 2015
Published 23 October 2015 Volume 2015:10(1) Pages 6645—6655
DOI https://doi.org/10.2147/IJN.S93225
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Binh TT Pham,1 Nirmesh Jain,1 Philip W Kuchel,2 Bogdan E Chapman,2,† Stephanie A Bickley,3 Stephen K Jones,3 Brian S Hawkett1
1School of Chemistry, University of Sydney, 2School of Molecular Bioscience, University of Sydney, 3Sirtex Medical Limited, Sydney, NSW, Australia
†Bogdan E Chapman passed away on August 16, 2014
Abstract: Sterically stabilized superparamagnetic iron oxide nanoparticles (SPIONs) were incubated with fresh human erythrocytes (red blood cells [RBCs]) to explore their potential application as magnetic resonance imaging contrast agents. The chemical shift and linewidth of 133Cs+ resonances from inside and outside the RBCs in 133Cs nuclear magnetic resonance spectra were monitored as a function of time. Thus, we investigated whether SPIONs of two different core sizes and with three different types of polymeric stabilizers entered metabolically active RBCs, consuming glucose at 37°C. The SPIONs broadened the extracellular 133Cs+ nuclear magnetic resonance, and brought about a small change in its chemical shift to a higher frequency; while the intracellular resonance remained unchanged in both amplitude and chemical shift. This situation pertained over incubation times of up to 90 minutes. If the SPIONs had entered the RBCs, the intracellular resonance would have become broader and possibly even shifted. Therefore, we concluded that our SPIONs did not enter the RBCs. In addition, the T2 relaxivity of the small and large particles was 368 and 953 mM-1 s-1, respectively (three and nine times that of the most effective commercially available samples). This suggests that these new SPIONs will provide a superior performance to any others reported thus far as magnetic resonance imaging contrast agents.
Keywords: 133Cs NMR spectroscopy, bulk magnetic susceptibility, fresh human erythrocyte, particle stability, paramagnetic shift, superparamagnetic iron oxide nanoparticles, SPIONs
Introduction
In the past decade, attention has been given to nanometer-sized colloidal particles (nanoparticles [NPs]) as tools for biomedical applications including diagnostic imaging and therapy.1,2 Among the various types of NPs used for these purposes, those with magnetic cores have been the most extensively studied.3–13 The range of applications of magnetic NPs includes magnetic drug targeting, induced hyperthermia, and as contrast agents (CAs) in magnetic resonance imaging (MRI).
It is well documented that iron oxide NPs enhance nuclear magnetic resonance (NMR) proton relaxation at their site of accumulation,14 making them a useful tool for contrast enhancement in MRI, particularly for non-invasive diagnosis of human soft tissue pathologies.15 In the past 2 decades, superparamagnetic iron oxide nanoparticles (SPIONs) and ultra-small SPIONs dispersed in an aqueous phase have been explored as contrast-enhancing probes in MRI.
Products such as Endorem™ (also called Feridex®; Guerbet, Villepinte, France), Feraheme® (ferumoxide-AMI-25) (AMAG Pharmaceuticals Inc., Cambridge, MA, USA), Primovist™ and Eovist™ (Bayer AG, Leverkusen, Germany), are currently used as CAs, while products such as Supravist™ (SHU555C) (Bayer AG) and VSOP-C184 (Ferropharm GmbH, Teltow, Germany), may enter the market in the future.16 Much effort has been expended on designing iron oxide NPs that are effective at lower concentrations, and that have an increased circulation time in the blood, thus enhancing the probability of selective transport (endocytosis) of the NPs across plasma membranes.17–19
Some publications20 have reported studies on the interaction of targeted tissues or cells with NPs. However, it is unclear whether these particles exhibit non-specific reactions with various cell types; eg, NPs in the blood stream may adhere to the surface of red blood cells (RBCs), thus preventing them from targeting other cells, and from entering the target cells. The uptake of NPs by RBCs has been reported by Geiser et al21 and Rothen-Rutishauser et al.22 In both cases, the NPs were thought to affect the physiological properties of the cells; eg, iron oxide NPs increased the membrane permeability of human microvascular endothelial cells.23
Surface modification of NPs imparts biocompatibility and specific functional groups for particular biological and medical applications. A common method for such modification is “steric stabilization” of magnetic NPs using biocompatible polymers.24–32 Surface coating of SPIONs with poly(ethylene glycol) (PEG) or dextran was proposed to prolong the circulation time of NPs by shielding the surface charge of the particles.24,33,34 A PEG coating also ensured a decreased uptake of the NPs by macrophages, leading to an increase in circulation time, and therefore an increase in the passive accumulation in tumors.35–40 Dextran coated NPs were extensively studied and have US Food and Drug Administration approval for use in patients.41 However, high molecular weight dextran contributes significantly to the overall hydrodynamic size of the particle, and consequently the average density and composition of the metal in the particle is lowered. This may be disadvantageous for MRI.
Reversible addition fragmentation chain transfer (RAFT) polymerization is a versatile tool for synthesizing polymers of a defined size and composition with a narrow molecular weight distribution.42 Short chain block copolymers have been designed and synthesized using RAFT-mediated polymerization. These polymers have previously been used as steric stabilizers to produce iron oxide NPs that are stable in water and physiological solutions, even at high salt concentrations.30
In the present work, we studied SPIONs with two different core sizes viz, 10–15 and 20–25 nm. The particles were sterically stabilized with short chain block copolymers prepared by RAFT polymerization. The block copolymers used in this study are methoxypolyethylene glycol-block-poly(monoacryloxyethyl phosphate)10 macro-RAFT agent (RAFT-MAEP10-MPEG, with 17 units of ethylene oxide in the MPEG block), poly(monoacryloxyethyl phosphate)10-block-poly(acrylamide)20 macro-RAFT agent (RAFT-MAEP10-AAm20), and poly(monoacryloxyethyl phosphate)10-block-poly(acrylamide)60 macro-RAFT agent (RAFT-MAEP10-AAm60). The aim was to investigate if these sterically stabilized SPIONs entered metabolically active, fresh human RBCs. To do this non-invasively, we exploited 133Cs NMR and the known feature that populations of intra- and extracellular 133Cs+ have different resonance frequencies.43
Materials and methods
Chemicals
Fe(II) chloride tetrahydrate (99%) and Fe(III) chloride hexahydrate (98%) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA) and used as supplied. Fe(III) nitrate nonahydrate (99%) was from EMD Millipore, Billerica, MA, USA and used as supplied. Ammonium hydroxide (28% NH3 in water, w/w) was purchased from Ajax Finechem (Thermo Fisher Scientific, Waltham, MA, USA). RAFT agents 2-{[(butylsulfanyl)carbonothioyl]sulfanyl}propanoic acid (RAFT-COOH) and methoxy polyethylene glycol modified 2-{[(butylsulfanyl)carbonothioyl]sulfanyl} propanoic acid (RAFT-MPEG) were provided by Dr Algi Serelis (DuluxGroup, Melbourne, Australia). Fluka – 1,4-dioxane was distilled under reduced pressure. MAEP (Sigma-Aldrich Co.) was passed through an inhibitor removal column (Sigma-Aldrich Co.). AAm (Sigma-Aldrich Co.), 4,4′-azobis(4-cyanovaleric acid) (V-501; Sigma-Aldrich Co.), and sodium hydroxide (NaOH; Sigma-Aldrich Co.) were used as received. Milli-Q (MQ) water was used throughout the work. 2H2O was from the Australian Nuclear Science and Technology Organisation (ANSTO; Lucas Heights, NSW, Australia).
Synthesis of the sterically stabilized magnetic NPs
Magnetic NPs of two different core diameters, 10–15 nm (small) and 20–25 nm (large), were used in this work. The small NPs (10–15 nm core diameter) were produced as previously described using the Massart method.30 The large NPs (20–25 nm core diameter) were obtained from Sirtex Medical Limited, Sydney, NSW, Australia. Sterically stabilized iron oxide NPs SPIONs were prepared by coating the iron oxide cores with the desired steric stabilizers.30,31 Figure 1A shows the structures of the polymeric stabilizers used in this work.
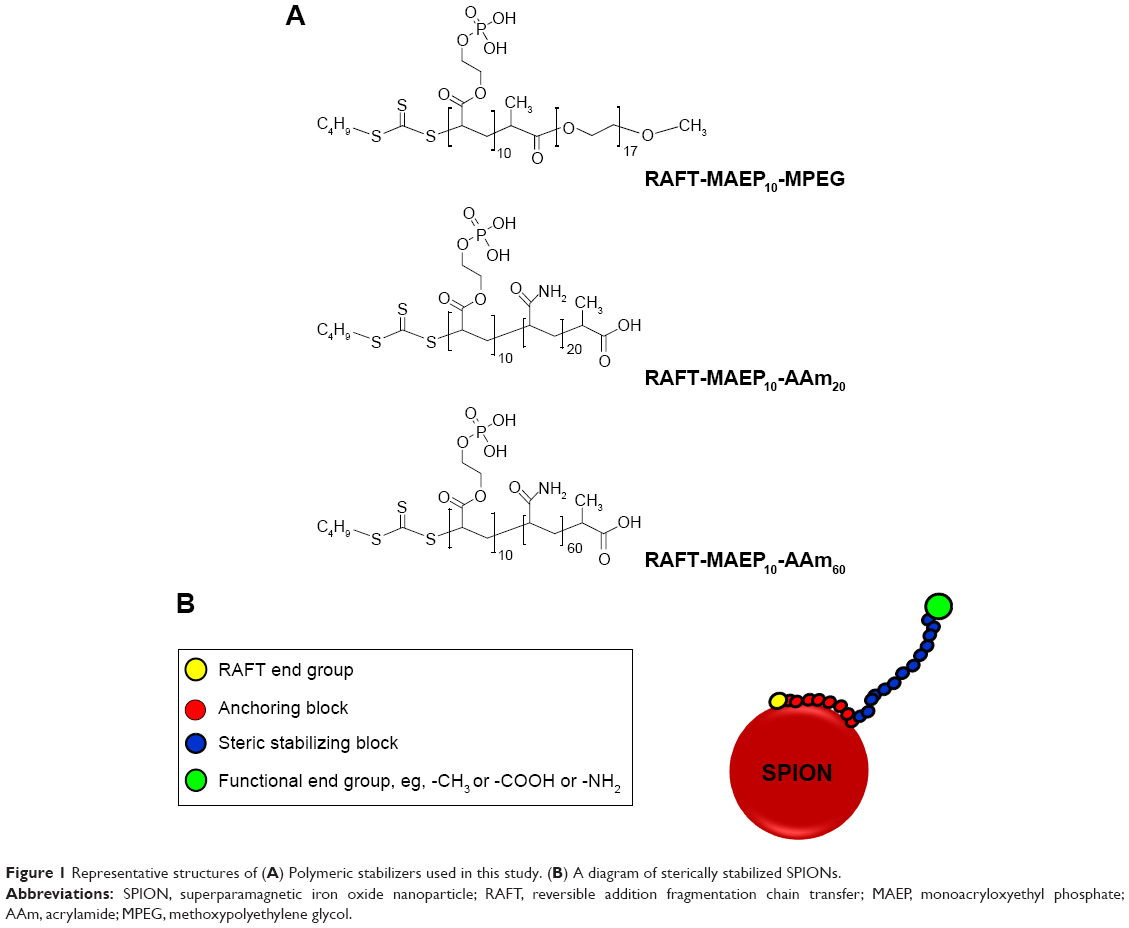 | Figure 1 Representative structures of (A) Polymeric stabilizers used in this study. (B) A diagram of sterically stabilized SPIONs. |
Preparation of RAFT-MAEP10-MPEG
A solution of RAFT-MPEG (3.60 g, 3.5 mmol), 4,4′-azobis(4-cyanovaleric acid) (0.20 g, 0.7 mmol), MAEP (6.89 g, 35 mmol) in dioxane (45 g) and water (22.5 g) was prepared in a 250 mL round bottom flask. This was stirred magnetically and purged with nitrogen for 15 minutes. The reaction was carried out at 70°C for 12 hours. The copolymer solution contained 15.1% solids. The copolymer solution was then diluted with MQ water to 0.7 wt%, and the pH adjusted to 5 using 0.1 M NaOH. See Figure 1A for the structure.
Preparation of RAFT-MAEP10-AAm20 using 2-{[(butylsulfanyl)-carbonothioyl]sulfanyl}propanoic acid
A solution of RAFT-COOH (3.2 g, 13.6 mmol), 4,4′-azobis(4-cyanovaleric acid) (0.19 g, 0.7 mmol), AAm (19.27 g, 271.1 mmol) in dioxane (45 g) and water (22.5 g) was prepared in a 250 mL round bottom flask. This was stirred magnetically, purged with nitrogen for 15 minutes, and the reaction carried out at 70°C for 4 hours. The homopolymer solution contained 32.0% solids. Fifteen grams of the obtained homopolymer solution, MAEP (4.50 g, 22.9 mmol), and 4,4′-azobis(4-cyanovaleric acid) (0.04 g, 0.2 mmol) were added to a 100 mL round bottom flask. The mixture was then deoxygenated by purging with nitrogen for 15 minutes and the flask stirred in a 70°C oil bath for 12 hours. The copolymer solution, which contained 40.4% solids, was then diluted with MQ water to 1.2 wt%. The pH of the diluted copolymer solution was adjusted to 5 with 0.1 M NaOH. See Figure 1A for the structure.
Preparation of RAFT-MAEP10-AAm60 using 2-{[(butylsulfanyl)-carbonothioyl]sulfanyl}propanoic acid
A solution of RAFT-COOH (0.36 g, 1.5 mmol), 4,4′-azobis(4-cyanovaleric acid) (0.08 g, 0.3 mmol), AAm (6.59 g, 92.8 mmol) in dioxane (20 g) and water (20 g) was prepared in a 100 mL round bottom flask. This mixture was then stirred magnetically, purged with nitrogen for 15 minutes, and the reaction carried out at 70°C for 4 hours. The obtained homopolymer solution, MAEP (3.03 g, 15.4 mmol), and 4,4′-azobis (4-cyanovaleric acid) (0.08 g, 0.3 mmol) were added to a 100 mL round bottom flask. The mixture was deoxygenated by purging with nitrogen for 15 minutes, and the flask stirred in a 70°C oil bath for 12 hours. The copolymer solution, which contained 26% solids, was then diluted with MQ water to 1.2 wt%. The pH of the diluted copolymer solution was adjusted to 5 with 0.1 M NaOH. See Figure 1A for the structure.
Preparation of RBCs from fresh human blood
Blood was obtained under Human Ethics Committee Clearance at the University of Sydney, from a healthy donor (PWK), anti-coagulated with heparin (~15 IU mL−1), and diluted 1:2 in a saline solution in which NaCl was largely substituted by CsCl, viz, 50 mM NaCl, 100 mM CsCl, 10 mM KCl, and 10 mM glucose. The diluted blood was centrifuged (10 minutes, 4°C, 3,000× g), after which the supernatant and “buffy coat” were aspirated. This procedure was repeated once more, followed by re-suspension of the RBC pellet, and bubbling it with CO for 5 minutes, to render the hemoglobin into a stable diamagnetic state that gives an optimal signal-to-noise ratio in the NMR spectra. The sample was centrifuged as above, and the pellet re-suspended in another saline solution using a 10% 2H2O/1H2O mixture. The tube containing the RBC suspension was placed on a sample rocker (~20 oscillations min−1) for ~16 hours. This was then centrifuged again as above to yield a pellet of RBCs. The hematocrit (Ht) of the pellet was measured with a capillary centrifuge (Clements, North Ryde, NSW, Australia), and the final value adjusted to 76%. Aliquots of the RBC suspension (3 mL each) were added to high-precision 10 mm glass NMR tubes (Wilmad-LabGlass, Vineland, NJ, USA).
Transmission electron microscopy (TEM)
The size and morphology of the NPs was investigated by TEM (Philips CM120 Biofilter, 120 kV; Philips, Amsterdam, the Netherlands). The ferrofluid dispersions were first diluted with deionized water to approximately 0.001 wt%, before being deposited on a carbon coated copper grid.
Dynamic light scattering
The size distribution of the NPs was measured by laser light scattering on a Zeta Sizer NanoS (Malvern Zeta, 173 scattering angle; Malvern Instruments, Malvern, UK) at 25°C. The ferrofluid dispersions were diluted to 0.1 wt%.
Thermal gravimetric analysis
The composition of the particles was determined by thermogravimetric analysis (TGA) using a TA Instruments TGA apparatus (Eugene, OR, USA) under an inert atmosphere, at a heating rate of 5°C/min from 100°C to 800°C.
Vibration sample magnetometry
The magnetic properties of the samples were measured in the solid state at room temperature using a vibrating sample magnetometer (Lake Shore 7300, with a 2 T electromagnet; Westerville, OH, USA). The magnetic moment of each dried sample was measured over a range of applied magnetic fields from −20 to +20 kOe, with a sensitivity of 0.1 emu. The saturation magnetization was obtained by fitting the experimental data to the Langevin equation.
133Cs NMR and RBCs
While cesium has 40 known isotopes, only 133Cs is stable. It is NMR receptive, with a spin-quantum number of 7/2.44 As an aside, this is an attribute that is well elicited in stretched gelatin gels.45 Although it is a quadrupolar nucleus, and may give broad NMR resonances as a result, this is not a major problem with modern high field NMR spectrometers. The fact that it is used as a potassium congener means that its concentration in cells is relatively high, second only to 23Na+, and as such, the NMR spectra are acquired rapidly, in a matter of seconds. As the 133Cs+ nucleus is orbited by 54 electrons that magnetically shield the nucleus, the nuclear magnetic environment (local magnetic induction) is very sensitive to the interactions of all these electrons with other ions, molecules in the solution (probing differences in the suspension medium and the cell’s cytoplasm), and with macromolecular and even macroscopic surfaces. In the case of RBCs, this leads to 133Cs+ having an intrinsic chemical shift difference between the inside and outside of the cells, which arises without the use of chemical shift reagents.43 It is because of its exquisite sensitivity to bulk magnetic susceptibility (BMS) variations,46,47 paramagnetic properties, and bulk-solution content, that 133Cs+ was chosen as a probe to investigate the possible entry of our SPIONs into RBCs. An amount of 7.2 μL of the SPION suspension (original concentration of 5 mg mL−1) was used to give a final concentration of 50 μg mL−1 in the extracellular space; temperature, 37°C. NMR spectra were acquired by averaging four transients, using a spectral width of 1,000 Hz, 2,048 time domain points, a 90° radiofrequency pulse width of 10 μs, and a relaxation delay between transients of 5 seconds.
NMR
NMR spectra were recorded at 400.13 MHz for 1H, and 52.5 MHz for 133Cs, on an Avance III spectrometer (Bruker Optik GmbH, Ettlingen, Germany) with a 9.4 T vertical wide-bore magnet (Oxford Instruments, Oxford, UK). A 10 mm broadband observe probe was used and the temperature was calibrated (Bruker script) using a sample of neat methanol, and set to 37°C for the time course experiments.
133Cs NMR data analysis
The NMR spectra were processed using TopSpin 3.0 (Bruker), and the spectra were extracted as text files that were read in a Mathematica program for subsequent analysis. All spectra were phase corrected and baseline adjusted prior to analysis. A line broadening factor of 3.0 Hz was used for all 133Cs NMR spectra.
Results and discussion
Characterization of the magnetic NPs
The small-core magnetic NPs (diameter 10–15 nm) were produced and characterized as reported in our previous work.30,48 These particles were coated with a diblock copolymer containing either 20 units of AAm or 17 units of ethylene oxide (EO) in the stabilizing block, and 10 units of MAEP in the anchoring block. The MAEP block anchors onto the NP surface via complexing with the surface hydroxyl groups; whereas, the water soluble AAm or EO blocks stretch out into the disperse phase preventing their agglomeration in the aqueous dispersion. The coated particles demonstrate excellent stability in salt solutions over a wide pH range.30
As the diameter of the superparamagnetic particles is increased beyond 10 nm,49 the generation of stable dispersions becomes problematic. The strong attractive magnetic interactions can overcome thermal energy fluctuations at room temperature. This is of concern in biomedical applications of large-core magnetic NPs. We overcame this problem by increasing the length of the stabilizing block polymer. Twenty AAm or 17 EO units in the stabilizing block were sufficient to impart good stability to the small-core magnetic NPs. However, the large cores (20–25 nm diameter) used in the present study required a minimum of 60 units of AAm in the stabilizing block (Figure 1A). Sixty units of AAm provided a thickness of approximately 14 nm to impart steric stabilization (Figure 2 and Table 1).
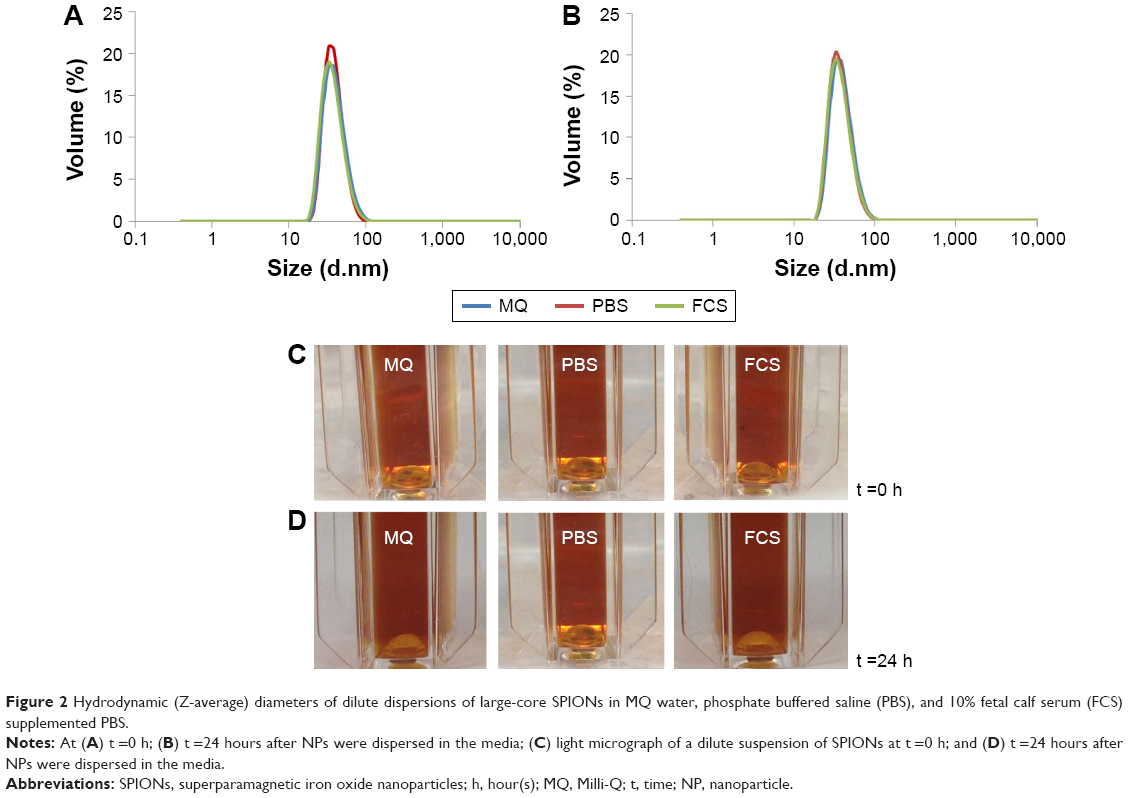 | Figure 2 Hydrodynamic (Z-average) diameters of dilute dispersions of large-core SPIONs in MQ water, phosphate buffered saline (PBS), and 10% fetal calf serum (FCS) supplemented PBS. |
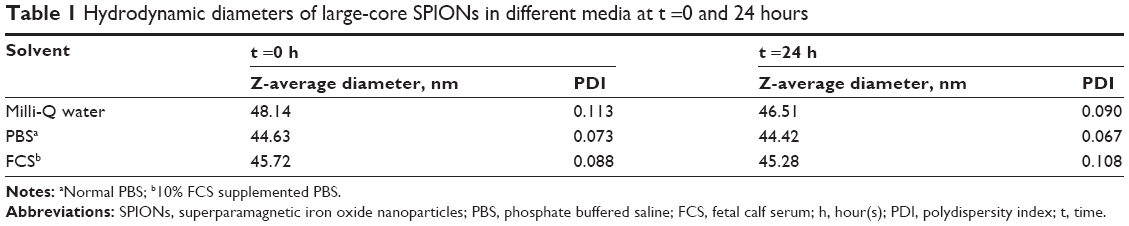 | Table 1 Hydrodynamic diameters of large-core SPIONs in different media at t =0 and 24 hours |
The coated NPs were characterized by TEM, dynamic light scattering, TGA, and magnetometry, as discussed in the Materials and methods section.
Figure 3A shows a typical TEM image of the large-core magnetic NPs, with a core diameter of 20–25 nm, and a very narrow size distribution. A small fraction of the magnetic NPs underwent aggregation during the stabilization process. This may have resulted from the attractive forces between the few large particles present in the initial dispersion of uncoated magnetic NPs. The suspension of coated magnetic NPs was centrifuged several times before use; this separated very small and large particles, producing the final suspension of SPIONs with a narrow size distribution, as shown in Figure 3. The TEM of small-core NPs (Figure 3B) is also included for comparison. These NPs are somewhat less monodisperse.
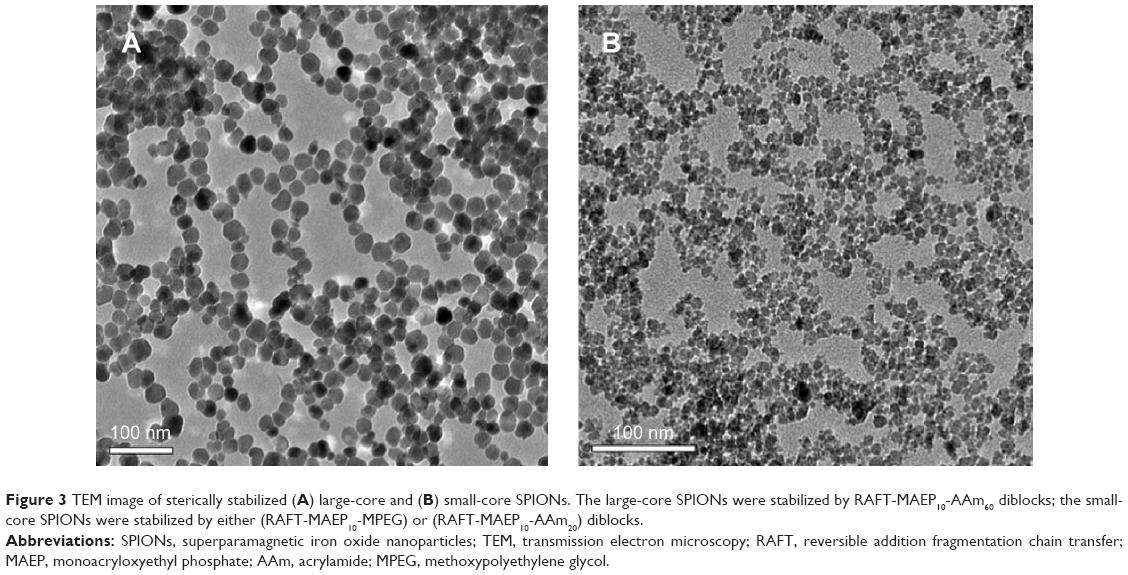 | Figure 3 TEM image of sterically stabilized (A) large-core and (B) small-core SPIONs. The large-core SPIONs were stabilized by RAFT-MAEP10-AAm60 diblocks; the small-core SPIONs were stabilized by either (RAFT-MAEP10-MPEG) or (RAFT-MAEP10-AAm20) diblocks. |
Figure 2A and B show the size distribution of the coated particles in various media measured by dynamic light scattering. The average hydrodynamic diameter of the stabilized large particles in water was 48 nm. That of the stabilized NPs was increased by ~20–25 nm relative to the core particles. This is in good agreement with the expected size of the steric stabilizing polymers. Sixty units of AAm provides a coating of ~10–12 nm for steric stabilization. This increase was due to solvation and extension of the poly(AAm) stabilizing block into the aqueous phase, which is approximately the same length as the fully extended poly(AAm) block. Similar results were obtained in our previous work on poly(acrylic acid) stabilized latex particles.30,31
The size distribution of the coated particles (Figure 2A) remained unchanged in various media including MQ water, phosphate buffered saline (PBS), and fetal calf serum (10%) supplemented PBS at time t =0 hour. This highlights their high stability in different biological media. Figure 2B shows their size distribution after 24 hours. The measured hydrodynamic diameters are shown in Table 1. The size distribution and average hydrodynamic diameter remained unchanged after 24 hours in all media (Table 1). Figure 2C and D are light micrographs of the NP dispersions in MQ water, PBS, and fetal calf serum supplemented PBS media taken at t =0 and 24 hours. We concluded from these size distribution measurements that the coated particles are very stable in all media tested.
The percentage composition of polymer and iron metal in the final large-core NP was determined from thermogravimetric analysis (TGA). The TGA trace (Figure 4) shows a gradual and almost single-step weight loss profile. The weight loss that resulted from the polymer degradation was ~14%. This indicated that there was ~86 wt% of iron oxide in the polymer coated NPs, which exceeded the highest value of ~76% previously achieved by our group,30 and the ~70 wt% reported by Lee et al.50
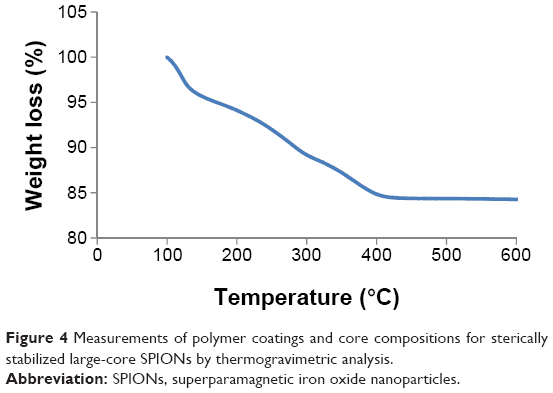 | Figure 4 Measurements of polymer coatings and core compositions for sterically stabilized large-core SPIONs by thermogravimetric analysis. |
Figure 5 shows the magnetization curves of aqueous dispersions of both the electrostatically and sterically stabilized large-core NPs determined by vibrating sample magnetometry at room temperature. The variation in magnetization, M, with applied field, H, showed no hysteresis in each case. Thus, both the remanence and coercivity were zero. The absence of hysteresis was confirmed by measurements of the magnetization of both samples at relatively weaker magnetic field strengths. The saturation magnetization of both the electrostatically stabilized NPs and polymer coated SPIONs were almost identical (Ms =66 emu g−1). However, the saturation magnetization was lower than that of pure maghemite (Ms =72 emu g−1)51 in both cases. This has been attributed elsewhere to the presence of nonmagnetic or “dead” surface layers resulting from a chemical reaction between the stabilizing surfactant and the ferrite particles.52
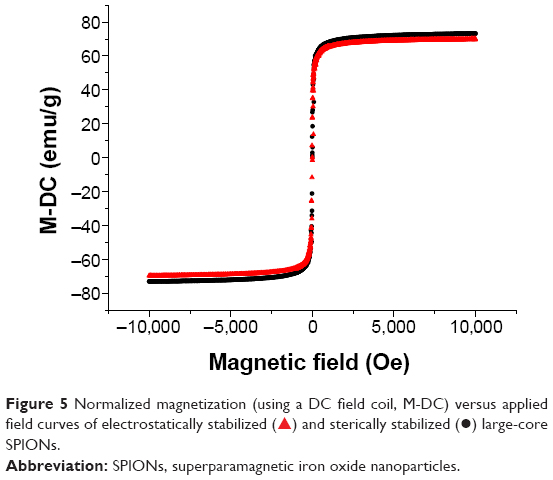 | Figure 5 Normalized magnetization (using a DC field coil, M-DC) versus applied field curves of electrostatically stabilized ( |
 ) and sterically stabilized (•) large-core SPIONs.
) and sterically stabilized (•) large-core SPIONs.The toxicity of both small-31 and large-core SPIONS have been tested in vitro in a number of cell lines using the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. The cell viability was not significantly affected in the presence of SPIONs at concentrations up to 500 mg/L (data not shown).
Relaxometric properties
To examine the MRI potential of the large SPIONS, 1H NMR relaxometry measurements were carried out at room temperature by measuring the transverse relaxation time, T2, at 400 MHz. This corresponded to an external magnetic field B0=11.7 Tesla (T). Figure 6 shows R2, and the transverse relaxivity (1/relaxation time constant) of dilute dispersions of large-core SPIONs within the concentration range 0–0.5 mM of Fe. In comparison to the R2 transverse relaxivity value in water, the negative contrast was observed to increase with increasing iron concentration. It can be seen from the data in Figure 6 that the large-core SPIONs showed a transverse R2 relaxivity of 953 mM−1-s−1, which is nine times higher than the commercial sample from Endorem™. Also, large-core SPIONs rendered a significantly higher relaxivity in comparison to the small particles (R2 relaxivity of 368 mM−1-s−1) reported by us in the past.48 An increase in R2 with an increase in the NP size has also been seen by Lartigue et al.53 This was attributed to a higher saturation magnetization of large-core SPIONs than that of small-core SPIONs. A lower magnetic field of 4.7 T (Figure 6) was also used to investigate the effect of the external magnetic field on the measured relaxivity. The values were almost identical. Similar apparent field-strength independence has also been reported by Lartigue et al.53 The findings in this study demonstrate that our SPIONS are promising for use as MRI CAs (see further comments in the Discussion).
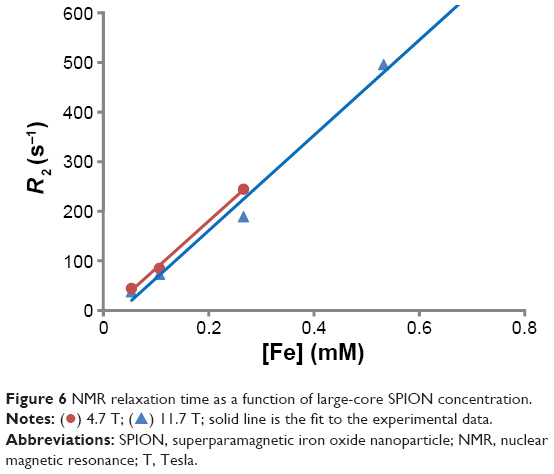 | Figure 6 NMR relaxation time as a function of large-core SPION concentration. |
 ) 4.7 T; (
) 4.7 T; ( ) 11.7 T; solid line is the fit to the experimental data.
) 11.7 T; solid line is the fit to the experimental data.Interaction of magnetic NPs with RBCs
133Cs+ in RBCs
The black spectra obtained for the three series shown in Figure 7 indicate that the RBCs were successfully loaded with Cs+ over the previous 16 hours. The spectra show a marked separation of 63 Hz between the resonances from the populations of the cation inside and outside the RBCs. Sample dilution decreased the volume of RBCs in the sample, and further reduced the area of the smaller resonance; the smaller resonance was therefore assigned to the intracellular 133Cs+. All three spectra were essentially identical, demonstrating the high level of reproducibility in the sample preparation that is achievable with human RBCs and CsCl.
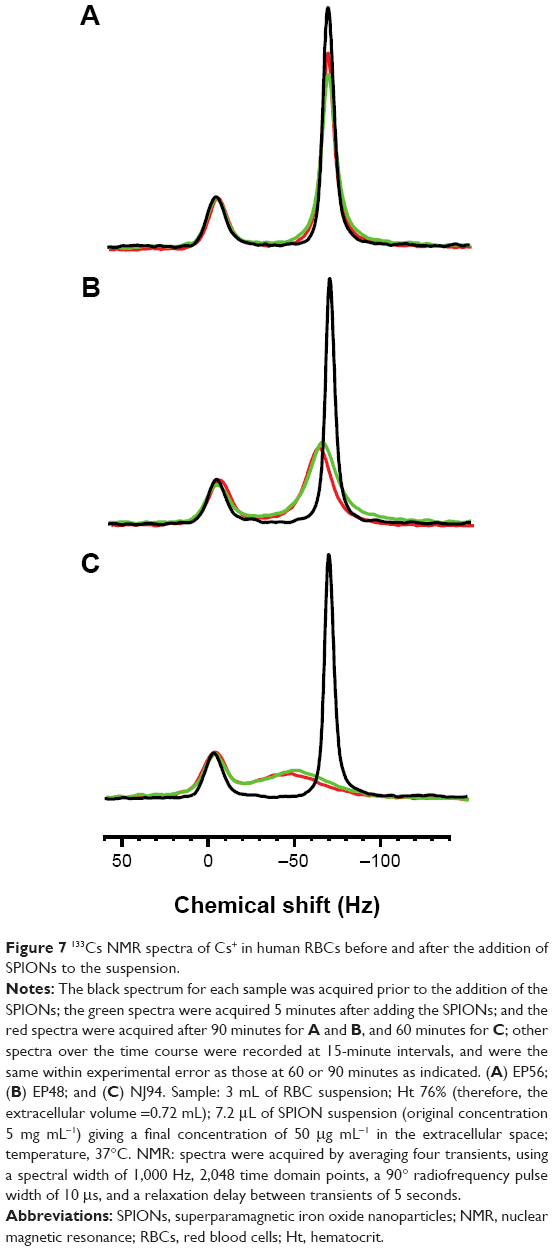 | Figure 7 133Cs NMR spectra of Cs+ in human RBCs before and after the addition of SPIONs to the suspension. |
SPIONs addition
The green and red spectra in Figure 7A were acquired at 5 and 90 minutes after the addition of SPIONs EP56 (NPs coated with RAFT-MAEP10-MPEG) to the RBC suspension, respectively. The immediate effect observed after the addition (green spectrum) was a decrease in the amplitude of the resonance from the extracellular 133Cs+, accompanied by an increase in its width. On the other hand, line broadening was not evident with the intracellular 133Cs+. There was a small amount of sharpening in the extracellular resonance over the subsequent 85 minutes, but no equivalent change for the intracellular resonance.
In comparison to the effect of EP56 (NPs coated with RAFT-MAEP10-MPEG) shown in Figure 8A, Figure 8B shows that the effect of the SPIONs EP48 (NPs coated with RAFT-MAEP10-AAm20) on the extracellular resonance was more dramatic. Not only did the resonance become significantly broadened, accompanied by a ~threefold decrease in the peak amplitude, but also the signal was shifted to a higher frequency by ~5 Hz. The broadening increased slightly (~1 Hz) in contrast with Figure 7A over the time course investigated, up to 90 minutes. However, only a slight broadening was observed for the intracellular resonance within this time.
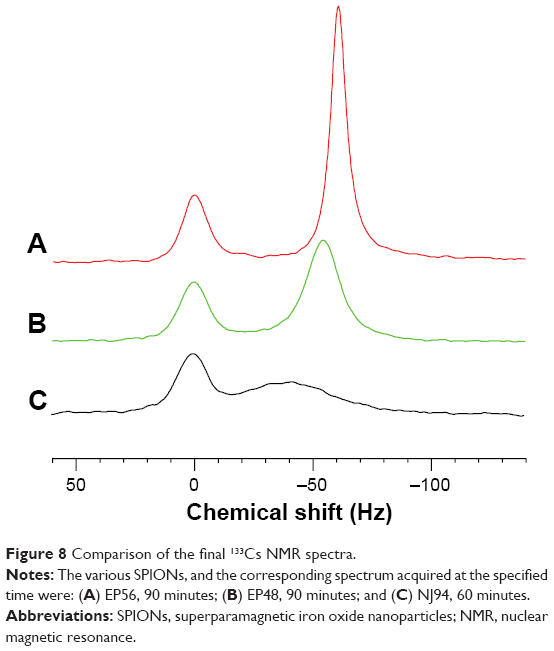 | Figure 8 Comparison of the final 133Cs NMR spectra. |
As seen in Figure 8C, the effect of the addition of SPIONs NJ94 (NPs coated with RAFT-MAEP10-AAm60) on the extracellular resonance was even more dramatic than for the other two SPIONs. The observed resonance broadening was significant, and this was also accompanied by a chemical shift of ~16 Hz. The broadened extracellular resonance encroached on the intracellular resonance, making it difficult to resolve the two peaks. However, the amplitude of the intracellular resonance did not change significantly over the 60-minute time course.
133Cs loading
The RBCs were successfully loaded with 133Cs+ under the conditions we employed, and the cells were kept in a metabolically active state by the ample supply of glucose throughout the entire Cs-loading procedure, as well as during the subsequent NMR experiments. The chemical shift of 133Cs+ is very sensitive to its local ionic environment because it is a large multi-electron ion whose nucleus therefore “senses” any changes in the local (molecular-scale) inhomogeneous electric field. Therefore, its chemical shift responds sensitively to changes in the electronic surroundings; consequently, a large transmembrane chemical shift difference of 63 Hz was observed in our experiments. Such a chemical shift difference has been seen in vivo.43,54–56 Therefore, the technical developments made in the present work are likely to be extendable to in vivo animal studies.
SPION effects
SPIONs, being Fe(III) complexes that are superparamagnetic, perturb the local magnetic field that arises from the difference in the BMS in various regions of an RBC suspension. As the SPIONs reside in the space between the RBCs, and perhaps on their surface, the BMS effect broadens the 133Cs+ resonance. On the other hand, the magnetic field perturbation does not extend greatly into the cytoplasm (the effect dies off as 1/r3 from a magnetic dipole, where r is distance from the center of the dipole), and therefore the resonance of intracellular 133Cs+ was not significantly affected. 133Cs+ that is in the same compartment as the SPIONs encounters BMS-induced magnetic field inhomogeneity that causes spectral line broadening. 133Cs+ can also interact with the surface polymer of the SPIONs particle. The rapid relaxation of the electrons in the Fe(III) brings about rapid relaxation of the high energy nuclear-magnetic state of the 133Cs+. This hyperfine interaction also contributes to broadening of the 133Cs+ line.
The first broadening mechanism produced by the BMS effect gives “inhomogeneous broadening”. This refers to the inhomogeneity of the magnetic field. The width-at-half-height, Δν1/2, of the 133Cs+ resonance (or any NMR peak for that matter) is equal to 1/(πT2*), where T2 is the transverse relaxation time, and the * denotes that this is an apparent rather than an intrinsic value. The smaller value of T2* relative to the intrinsic spin-spin relaxation time constant is the result of inhomogeneous line broadening due to the paramagnetic effect. The envelope of the free induction decay (FID) acquired in the NMR experiment is characterized by the apparent relaxation time constant 1/T2*; hence, the intrinsic T2 is generally measured by using the Hahn- or Carr-Purcell spin-echo pulse sequence. Thus, a comparison of the T2* and T2 values is used to estimate whether the line broadening is primarily due to a BMS effect, or if it arises from a direct ion-paramagnetic-interaction effect.
Another effect associated with the SPIONs is to change the average magnetic field of all the 133Cs+ ions uniformly within the medium of a shared compartment. The ions will be in a rapid exchange between the bulk medium and being transiently bound to the surface of the SPIONs. This overall average change in the magnetic field will bring about a change in the NMR resonance frequency. This effect was evident with the EP8 and NJ94 SPIONs (Figure 7B and C, respectively), but not for EP56 SPIONs.
Uptake of SPIONs into RBCs?
The motivation behind the experiments reported here was to determine whether SPIONs would penetrate the RBC plasma membrane, and if they may differentially broaden the NMR resonances (spectral lines) from a cation. The separation of the 133Cs+ NMR signals from inside and outside the cells allowed the distribution of the SPIONs in the sample to be recorded. If SPIONs entered the cells, then the previously narrow peak would become broader and perhaps even undergo a chemical shift. This was not observed over the 60- to 90-minute periods used. The system response, including the shifting and broadening of the extracellular resonance, eventually stabilized after the addition of the SPIONs, while there were no significant changes observed in the resonance associated with intracellular 133Cs+. The constant extracellular signals of 133Cs+ over the time course investigated strongly indicates that the SPIONs are highly stable in the biological environment.
Conclusion
The attractive magnetic interactions between the big-core (20–25 nm diameter) SPIONs were overcome by using larger block lengths of steric stabilizers to prevent aggregation. Sterically stabilized SPIONs showed excellent stability in all media. We also observed that the large-core SPIONs were efficient negative CAs, having a transverse relaxivity value of 958 mM−1s−1; this is nine times higher than any of the “best” commercially available reagents.
The three different samples of SPIONs (EP56, EP48, and NJ94) invoked a graded response from the extracellular 133Cs+ resonance (EP56<EP48<NJ94), but had no effect on the intracellular 133Cs+ resonance over the 60- to 90-minute time courses investigated. This indicated that under our incubation conditions, the metabolically active RBCs were not penetrated by the studied SPIONs.
Because of their different NMR spectral effects, the three types of SPIONs could be used either experimentally or in in vivo studies to investigate cation transport, particularly for 133Cs+, 23Na+, and other cations whose intra- and extracellular resonances are separated by a shift reagent, or to enhance MRI images based on their BMS and hence their enhancement of intrinsic transverse relaxation. If a suitable 133Cs model can be established for in vivo studies, with the stability of the SPIONs in the presence of RBCs as shown in this work, any changes in the observed intracellular signal would reflect changes in the ion concentration and hence the diffusion and/or transportation of ions or small molecules across the RBC membrane. Furthermore, the SPIONs were shown to reduce the extracellular signal, which would enhance the signal-to-noise ratio in imaging. This suggests that these SPIONs will be potentially valuable CAs for MRI.
Acknowledgments
The 133Cs NMR work was funded by an Australian Research Council Discovery Project Grant to PWK. Authors acknowledge financial support from Sirtex Medical Ltd. It is with considerable sadness that we record the death of Dr Bogdan Edward Chapman (1948–2014) on August 16, 2014. He made major contributions to the project, the results of which are reported here.
Disclosure
The authors report no conflicts of interest in this work.
References
Thassu D, Deleers M, Pathak Y, editors. Nanoparticulate Drug Delivery Systems. New York: Informa Healthcare USA, Inc; 2007. | ||
Li M, Al-Jamal KT, Kostarelos K, Reineke J. Physiologically based pharmacokinetic modeling of nanoparticles. ACS Nano. 2010;4(11):6303–6317. | ||
Laurent S, Forge D, Port M, et al. Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem Rev. 2008;108(6):2064–2110. | ||
Hao R, Xing R, Xu Z, Hou Y, Gao S, Sun S. Synthesis, functionalization, and biomedical applications of multifunctional magnetic nanoparticles. Adv Mater. 2010;22(25):2729–2742. | ||
Ho D, Sun X, Sun S. Monodisperse Magnetic Nanoparticles for Theranostic Applications. Acc Chem Res. 2011;44(10):875–882. | ||
Laurent S, Dutz S, Hafeli UO, Mahmoudi M. Magnetic fluid hyperthermia: focus on superparamagnetic iron oxide nanoparticles. Adv Colloid Interface Sci. 2011;166(1–2):8–23. | ||
Amstad E, Textor M, Reimhult E. Stabilization and functionalization of iron oxide nanoparticles for biomedical applications. Nanoscale. 2011;3(7):2819–2843. | ||
Yoo D, Lee JH, Shin TH, Cheon J. Theranostic magnetic nanoparticles. Acc Chem Res. 2011;44(10):863–874. | ||
Mahmoudi M, Hosseinkhani H, Hosseinkhani M, et al. Magnetic resonance imaging tracking of stem cells in vivo using iron oxide nanoparticles as a tool for the advancement of clinical regenerative medicine. Chem Rev. 2011;111(2):253–280. | ||
Yigit MV, Moore A, Medarova Z. Magnetic nanoparticles for cancer diagnosis and therapy. Pharm Res. 2012;29(5):1180–1188. | ||
Wahajuddin, Arora S. Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers. Int J of Nanomedicine. 2012;7:3445–3471. | ||
Hilger I, Kaiser WA. Iron oxide-based nanostructures for MRI and magnetic hyperthermia. Nanomedicine (Lond). 2012;7(9):1443–1459. | ||
Reddy LH, Arias JL, Nicolas J, Couvreur P. Magnetic nanoparticles: design and characterization, toxicity and biocompatibility, pharmaceutical and biomedical applications. Chem Rev. 2012;112(11):5818–5878. | ||
Bulte JW, Kraitchman DL. Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed. 2004;17(7):484–499. | ||
Rinck PA, editor. Magnetic Resonance in Medicine. 3rd ed. Oxford, UK: Blackwell; 1993. | ||
Wang YX. Superparamagnetic iron oxide based MRI contrast agents: Current status of clinical application. Quant Imaging Med Surg. 2011;1(1):35–40. | ||
Na HB, Song IC, Hyeon T. Inorganic Nanoparticles for MRI Contrast Agents. Adv Mater. 2009;21(21):2133–2148. | ||
Jun YW, Seo JW, Cheon J. Nanoscaling laws of magnetic nanoparticles and their applicabilities in biomedical sciences. Acc Chem Res. 2008;41(2):179–189. | ||
Peng E, Wang F, Xue JM. Nanostructured magnetic nanocomposites as MRI contrast agents. J Mater Chem B. 2015;3(11):2241–2276. | ||
Hillyer JF, Albrecht RM. Gastrointestinal persorption and tissue distribution of differently sized colloidal gold nanoparticles. J Pharm Sci. 2001;90(12):1927–1936. | ||
Geiser M, Rothen-Rutishauser B, Kapp N, et al. Ultrafine particles cross cellular membranes by nonphagocytic mechanisms in lungs and in cultured cells. Environ Health Perspect. 2005;113(11):1555–1560. | ||
Rothen-Rutishauser BM, Schürch S, Haenni B, Kapp N, Gehr P. Interaction of fine particles and nanoparticles with red blood cells visualized with advanced microscopic techniques. Environ Sci Technol. 2006;40(14):4353–4359. | ||
Apopa P, Qian Y, Shao R, et al. Iron oxide nanoparticles induce human microvascular endothelial cell permeability through reactive oxygen species production and microtubule remodeling. Part Fibre Toxicol. 2009;6(1):1. | ||
Karakoti AS, Das S, Thevuthasan S, Seal S. PEGylated Inorganic Nanoparticles. Angew Chem Int Ed Engl. 2011;50(9):1980–1994. | ||
Lee JH, Lee HB, Andrade JD. Blood compatibility of polyethylene oxide surfaces. Prog Polym Sci. 1995;20(6):1043–1079. | ||
Storm G, Belliot SO, Daemen T, Lasic DD. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Adv Drug Deliv Rev. 1995;17(1):31–48. | ||
Suh J, Choy KL, Lai SK, et al. PEGylation of nanoparticles improves their cytoplasmic transport. Int J Nanomedicine. 2007;2(4):735–741. | ||
Allen C, Dos Santos N, Gallagher R, et al. Controlling the physical behavior and biological performance of liposome formulations through use of surface grafted poly(ethylene glycol). Biosci Rep. 2002;22(2):225–250. | ||
Maruyama K. PEG-immunoliposome. Biosci Rep. 2002;22(2):251–266. | ||
Jain N, Wang Y, Jones SK, Hawkett BS, Warr GG. Optimized steric stabilization of aqueous ferrofluids and magnetic nanoparticles. Langmuir. 2010;26(6):4465–4472. | ||
Bryce NS, Pham BTT, Fong NW, et al. The composition and end-group functionality of sterically stabilized nanoparticles enhances the effectiveness of co-administered cytotoxins. Biomater Sci. 2013;1(12):1260–1272. | ||
Jain N, Zhang X, Hawkett BS, Warr GG. Stable and water-tolerant ionic liquid ferrofluids. ACS Appl Mater Interfaces. 2011;3(3):662–667. | ||
Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond). 2011;6(4):715–728. | ||
Allard-Vannier E, Cohen-Jonathan S, Gautier J, et al. Pegylated magnetic nanocarriers for doxorubicin delivery: A quantitative determination of stealthiness in vitro and in vivo. Eur J Pharm Biopharm. 2012;81(3):498–505. | ||
Huynh NT, Roger E, Lautram N, Benoit JP, Passirani C. The rise and rise of stealth nanocarriers for cancer therapy: passive versus active targeting. Nanomedicine (Lond). 2010;5(9):1415–1433. | ||
Larsen EK, Nielsen T, Wittenborn T, et al. Accumulation of magnetic iron oxide nanoparticles coated with variably sized polyethylene glycol in murine tumors. Nanoscale. 2012;4(7):2352–2361. | ||
Wang Z, Chui WK, Ho PC. Nanoparticulate delivery system targeted to tumor neovasculature for combined anticancer and antiangiogenesis therapy. Pharm Res. 2011;28(3):585–596. | ||
Kaminskas LM, McLeod VM, Kelly BD, et al. Doxorubicin-conjugated pegylated dendrimers show similar tumoricidal activity but lower systemic toxicity when compared to pegylated liposome and solution formulations in mouse and rat tumor models. Mol Pharm. 2012;9(3):422–432. | ||
Yoo HS, Park TG. Folate-receptor-targeted delivery of doxorubicin nano-aggregates stabilized by doxorubicin–PEG–folate conjugate. J Control Release. 2004;100(2):247–256. | ||
Harrington KJ, Lewanski C, Northcote AD, et al. Phase II study of pegylated liposomal doxorubicin (Caelyx™) as induction chemotherapy for patients with squamous cell cancer of the head and neck. Eur J Cancer. 2001;37(16):2015–2022. | ||
Tassa C, Shaw SY, Weissleder R. Dextran-coated iron oxide nanoparticles: a versatile platform for targeted molecular imaging, molecular diagnostics, and therapy. Acc Chem Res. 2011;44(10):842–852. | ||
Moad G, Rizzardo E, Thang SH. RAFT polymerization and some of its applications. Chem Asian J. 2013;8(8):1634–1644. | ||
Wellard RM, Shehan BP, Craik DJ, Adam WR. Factors affecting 133Cs chemical shifts in erythrocytes from cesium-fed rats. J Magn Reson B.1994;104(3):276–279. | ||
Power WP, Mooibroek S, Wasylishen RE, Cameron TS. Cesium-133 single-crystal NMR study of cesium chromate. J Phys Chem. 1994;98(6):1552–1560. | ||
Philp DJ, Naumann C, Kuchel PW. Relative intensities of components of quadrupolar-split multiplets in NMR spectra: Rationale for a simple rule. Concepts Magn Reson Part A. 2012;40A(2):90–99. | ||
Kuchel PW, Chapman BE, Bubb WA, Hansen PE, Durrant CJ, Hertzberg MP. Magnetic susceptibility: Solutions, emulsions, and cells. Concepts Magn Reson Part A. 2003;18A(1):56–71. | ||
Durrant CJ, Hertzberg MP, Kuchel PW. Magnetic susceptibility: Further insights into macroscopic and microscopic fields and the sphere of Lorentz. Concepts Magn Reson Part A. 2003;18A(1):72–95. | ||
Eamegdool SS, Weible MW 2nd, Pham BT, Hawkett BS, Grieve SM, Chan-ling T. Ultrasmall superparamagnetic iron oxide nanoparticle prelabelling of human neural precursor cells. Biomaterials. 2014;35(21):5549–5564. | ||
Philipse AP, van Bruggen MP, Pathmamanoharan C. Magnetic silica dispersions: preparation and stability of surface-modified silica particles with a magnetic core. Langmuir. 1994;10(1):92–99. | ||
Lee H, Lee E, Kim DK, Jang NK, Jeong YY, Jon S. Antibiofouling polymer-coated superparamagnetic iron oxide nanoparticles as potential magnetic resonance contrast agents for in vivo cancer imaging. J Am Chem Soc. 2006;128(22):7383–7389. | ||
Saravanan P, Alam S, Mathur GN. Comparative study on the synthesis of γ-Fe2O3 and Fe3O4 nanocrystals using high-temperature solution-phase technique. J Mater Sci Lett. 2003;22(18):1283–1285. | ||
Chantrell RW, Popplewell J, Charles S. Measurements of particle size distribution parameters in ferrofluids. Magnetics, IEEE Transactions. 1978;14(5):975–977. | ||
Lartigue L, Innocenti C, Kalaivani T, et al. Water-dispersible sugar-coated iron oxide nanoparticles. An evaluation of their relaxometric and magnetic hyperthermia properties. J Am Chem Soc. 2011;133(27):10459–10472. | ||
Shehan BP, Wellard RM, Adam WR, Craik DJ. The use of dietary loading of 133Cs as a potassium substitute in NMR studies of tissues. Magn Reson Med. 1993;30(5):573–582. | ||
Shehan BP, Wellard RM, Craik DJ, Adam WR. 133Cs relaxation times in rat tissues. J Magn Reson B. 1995;107(2):179–185. | ||
Wellard RM, Adam WR. Functional hepatocyte cation compartmentation demonstrated with 133Cs NMR. Magn Reson Med. 2002;48(5):810–818. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.