Back to Journals » Journal of Pain Research » Volume 14
The Increased Channel Activity of N-Methyl-D-Aspartate Receptors at Extrasynaptic Sites in the Anterior Cingulate Cortex of Neonatal Rats Following Prolonged Ketamine Exposure
Received 18 May 2021
Accepted for publication 15 July 2021
Published 8 August 2021 Volume 2021:14 Pages 2381—2389
DOI https://doi.org/10.2147/JPR.S320674
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Jianhui Jin,1,2 Ruirui Wang,2 Qing Lin2
1Department of pain Management, Beijing Tiantan Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Department of Psychology, The University of Texas at Arlington, Arlington, TX, USA
Correspondence: Qing Lin
Department of Psychology, The University of Texas at Arlington, Arlington, TX, 76019, USA
Tel +1 817-272-0154
Email [email protected]
Background: Ketamine is a dissociative anesthetic, commonly used for analgesia and anesthesia in a variety of pediatric procedures. It acts as a non-competitive antagonist to block ion channels of the N-methyl-D-aspartate receptors (NMDARs). Our previous study showed that repeated ketamine exposure developed a compensatory increase in NMDAR-mediated currents in neurons of the anterior cingulate cortex (ACC) of neonatal rats, and this increase was largely mediated by the GluN2B subunit-containing receptors, a predominant type of NMDARs during embryonic and early development of the brain. These data provide the molecular evidence to support that immature neurons are highly vulnerable to the development of apoptotic cell death after prolonged ketamine exposure.
Methods: Using whole-cell patch-clamp electrophysiology in an in vitro preparation of rat forebrain slices containing the ACC, the present study aimed at further determining whether GluN2B-containing NMDARs at extrasynaptic sites of immature neurons were the major target of ketamine for developing a compensatory increase in NMDAR-mediated synaptic transmission.
Results: Our major findings were that GluN2B subunits played a significant role in mediating ketamine-induced blockade of NMDAR-mediated currents in neonatal neurons and GluN2B-containing NMDARs expressed at extrasynaptic sites in neonatal neurons were the major player in compensatory enhancement of NMDAR-mediated currents after repeated ketamine exposure.
Conclusion: These results provide new evidence to strongly indicate that GluN2B-containing NMDARs at extrasynaptic sites are the key molecule contributing to the high vulnerability of the neonatal brain to ketamine-induced neurotoxic effects.
Keywords: immature neurons, ketamine, extrasynaptic NMDA receptors, GluN2B-containing NMDA receptors
Introduction
Ketamine is well-known as an antagonist of the N-methyl-D-aspartate glutamate receptors (NMDARs) with a myriad of clinical uses.1,2 Particularly, it is commonly used in pediatric patients to induce analgesia and general anesthesia.3–6 However, over the last 20 years, experimental studies by our and other groups have shown that prolonged or repeatedly systemic (in vivo) administration of ketamine triggered neuronal apoptotic death in the developing brain.7–10 Major areas affected are the frontal cortex (including the anterior cingulate cortex, ACC), parietal cortex, hippocampus and amygdala.7,11 This neurodegenerative change is significantly linked to the subsequent deficits of learning and memory later in adulthood.10,12,13 An in vitro electrophysiological study by our group suggests that the impairment of long-term potentiation of the synaptic transmission in the ACC contributes to the cognitive deficits.14 However, the mechanisms still remain unclear underlying the selective vulnerability of the neonatal brain to ketamine-induced neuroapoptosis.
Structurally, NMDARs are ligand-gated ion channels that exist as heterotetramers, which consist of two obligatory GluN1 subunits and two regulatory subunits of either GluN2 (GluN2A-2D) or GluN3 (GluN3A-B) subtype to comprise diheteromeric and/or triheteromeric receptors.15,16 Accumulating evidence showed that the regulatory GluN2 composition, mainly GluN2A/GluN2B ratio, changes across the brain development. During embryonic and early development, the central nervous system (CNS) in particular the forebrain is predominated by GluN2B-containing NMDARs.17–19 Our previous study reported a higher sensitivity of NMDARs to ketamine block in immature neurons than in mature neurons.20 We then further demonstrated that after repeated exposure to ketamine, the synaptic transmission mediated by GluN2B-containing NMDARs was compensatorily increased in neonatal neurons of the ACC.21 NMDARs are located not only at synaptic but also at extrasynaptic sites. In neonatal cortical neurons, extrasynaptic NMDARs make up over three-fourth of all NMDARs.22 At extrasynaptic sites, GluN1/GluN2B are the major subunits comprising NMDARs that are expressed significantly higher in neonatal pups.23–25 It has been proposed that active extrasynaptic NMDARs promote excitotoxicity, increasing neuronal apoptosis.26,27 We have proposed that such a compensatory increase in synaptic transmission mediated by GluN2B-containing NMDARs has high potential to cause ketamine-induced apoptosis in immature neurons.28
The present study is the logical next step to build on our preceding studies.20,21 We aim to further examine if sensitivity of NMDARs to ketamine is predominantly regulated by GluN2B subunits in immature neurons of the ACC, and then to evaluate the effects of repeated ketamine exposure on the NMDAR channel activity at extrasynaptic sites in these neurons. The results obtained will provide further evidence that GluN2B-containing NMDARs are the key molecule contributing to the high vulnerability of the developing brain to ketamine-induced neurotoxic effects.
Materials and Methods
Animals
Sprague-Dawley rats (male and female) in age groups of postnatal day 7 (PND 7) and 4–5 week-old (adolescents) were used in this study. Rats were housed under a 12–12 h constant light/dark cycle in a temperature (22–25°C) and humidity (55–60%) controlled environment. Access to food and water was provided ad libitum. Experimental procedures were approved by the Institutional Animal Care and Use Committee of University of Texas at Arlington (#A13.008). All experiments were performed according to the National Institutes of health (NIH) guidelines for the care and use of laboratory animals.
Brain Slice Preparation
After decapitation under deep anesthesia with sodium pentobarbital (50 mg/kg, i.p.), the whole brain of a rat was quickly transferred to an ice-cold (0–4°C) bath of artificial cerebrospinal fluid (ACSF; composition in mM: NaCl 124, KCl 3.3, KH2PO4 1.2, CaCl2 2H2O 2.5, MgSO4 2.4, NaHCO3 26, and glucose 10, pH 7.3–7.4). The bath was continuously bubbled with 95% O2/5% CO2 gas mixture. After cooling for about 2 min, the anterior half of the forebrain (including the ACC) was isolated and glued onto the stage of a vibratome (DTK-1000, Dosaka EM. Co., Ltd., Japan). 350–400 μm of coronal slices were sectioned and immediately transferred to a chamber with oxygenated ACSF (95% O2/5% CO2) for recovery at room temperature (23–25°C) for 1.5–2 h. All electrophysiological patch-clamp recordings were done at room temperature.
In vitro Whole-Cell Patch-Clamp Electrophysiology
In vitro whole-cell patch-clamp recordings, data acquisition and equipment are similar with those in our previously published work14,20,21,29 and were briefly described as below. Recording electrodes tip openings of 1–2 um (2–4 MΩ) were prepared from borosilicate glass (1.2 mm outside diameter, 0.69 mm inside) using a horizontal electrode puller (P-87, Sutter, USA). Electrodes were filled with internal solution (composition in mM: Cs2SO4, 110; CaCl2, 0.5; MgCl2, 2; EGTA, 5; HEPES, 5; tetraethylammonium-Cl, 5; with pH adjusted to 7.2–7.4 by CsOH, and had an osmolarity of 290–320 mOsm). A brain slice was held down in the recording chamber with an anchor and immersed in oxygenated ACSF. The pyramidal neurons of the ACC were patched under voltage-clamp mode at a holding membrane potential of −70 mV to maintain physiological conditions. Neurons from layers II/III of the ACC were recognized and identified by their morphology using a 40×water-immersion lens. Previously described recording procedures were used to record evoked NMDAR-mediated currents.20,21 Briefly, the NMDAR-mediated currents were pharmacologically isolated by adding 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, a non-NMDAR antagonist, 10 μM; Sigma/Aldrich) and bicuculline (a GABAA receptor antagonist, 10 μM; Sigma/Aldrich) to the circulating ACSF, and recorded under voltage clamp mode at a holding voltage of +40 mV. NMDAR-mediated currents were evoked electrically by delivering a single electrical pulse (0.3 ms, 0.25–0.5 mA) to the layer V of the frontal cortex using a bipolar tungsten stimulating electrode20 and recorded with an Axon 200B amplifier (Molecular Devices, USA) connected to a Digidata interface (Digidata 1440A, Molecular Devices, USA).
Experimental Protocol
Recordings of NMDAR-Mediated Currents to Examine the Role of GluN2B Subunits in the Regulation of Sensitivity of Immature Neurons to Ketamine
The functional properties of NMDARs are regulated by GluN2 subunits that experience a developmental switch in alterations of the composition of these subunits (mainly GluN2A/GluN2B ratio) across developmental stages of neurons. In the first part of the study, NMDAR-mediated currents were recorded from brain slices of both PND 7 and adolescent rats. Using pharmacological manipulations, the currents regulated by GluN2A were isolated by bath application of a GluN2B antagonist, ifenprodil (IFEN, 3 μM; Sigma/Aldrich) and the currents regulated by GluN2B were isolated by bath application of a GluN2A antagonist, PEAQX (0.4 μM; Sigma/Aldrich), respectively.30,31 The drug was bath applied to the circulating ACSF for 5–10 min after a neuron was patched. Control NMDAR-mediated currents (without GluN2A and GluN2B antagonist treatments) were set as 100%. The contributions of GluN2A and GluN2B to NMDAR-mediated currents were then determined, respectively. Thereafter, the role of GluN2B in ketamine-induced channel blockade was examined by bath application of ketamine (from Medvet Inc.) at the concentration of 10 μM for 5 min20 to brain slices that had been pre-treated with IFEN. The current levels when slices were pre-treated with IFEN were set as 100%.
Recordings of Extrasynaptic NMDAR-Mediated Currents to Examine the Effects of Repeated Ketamine Exposure
Repeated administrations of ketamine in vivo at a dose of 20 mg/kg have been established and validated to induce significant neurotoxic injury to neurons as evidenced by the apoptotic neuronal death in neonatal, but not adolescent, rats.8 Our previous study has reported that repeated ketamine exposure enhanced the synaptic transmission mediated by GluN2B-containing NMDARs in immature, but not mature, neurons.21 Therefore, we wanted to use this neonatal rat model to further evaluate the contribution of extrasynaptic NMDARs to the enhanced channel activity after repeated ketamine exposure. Rat pups at the age of PND 7, obtained randomly from different litters, were assigned to ketamine and saline (control) treatment groups randomly. Briefly, ketamine hydrochloride solution was injected subcutaneously at a dose of 20 mg/kg for 6 times at 2 h intervals. Adolescent rats used to demonstrate age-dependent effects of repeated ketamine exposure on the NMDAR channel activity were also given the same treatment. An equal volume of 0.9% saline was given in vivo for control purposes. Animals were maintained at a light-anesthetized level during ketamine treatment as evidenced by lack of voluntary movement and minimal reaction to the physical stimulation with body temperature being maintained at 37°C using a thermostatical heating blanket. Between injections and also during drug washout time, the neonatal rats were returned back to their dams to provide warmth and reduce potential stress.
The extrasynaptic NMDAR-mediated current activity was pharmacologically isolated by selectively blocking the active synaptic NMDARs. Using the procedures established and validated by other groups,32,33 MK-801 (from Sigma-Aldrich) was added to the circulating ACSF at the concentration of 20 µM for 5 min and repeated electrical stimulation (0.125 Hz) was delivered to layer V of the cortex in the presence of MK-801. After washout of MK-801 for 10 min, the remaining currents were considered as extrasynaptic NMDAR-mediated currents. The proportion of extrasynaptic NMDAR-mediated currents in both neonatal and adolescent brain slices was estimated as percentage of current levels before MK-801 pre-treatment (set as 100%). The effects of prolonged ketamine exposure on currents mediated by extrasynaptic NMDARs were examined in the neonatal brain slices that had been repeatedly exposed to ketamine in vivo and pre-treated with MK-801 in vitro. Extrasynaptic NMDAR-mediated currents were recorded at 3, 8 and 12 h after the last in vivo ketamine dose. The evoked channel activity recorded 12 h after saline treatment in vivo served as a control.
Data Analysis and Statistics
Data were digitized using pCLAMP 10.3 (Molecular Devices, USA). Statistical significance before and after drug application was analyzed using paired t tests. A grouped t test was used to compare the differences in responses between groups with different ages, different treatments or different time points. Data are reported as mean±SEM with significance set at P<0.05.
Results
The Blocking Effect of Ketamine on Current Activity Mediated by GluN2B-Containing NMDARs in Immature Neurons
Because the ratio of GluN2A- and GluN2B-containing NMDARs changes cross the developmental stages of the neurons, we would like to evaluate the different contributions of GluN2A and GluN2B to the regulation of NMDAR channel activity of immature and mature neurons in the slices of the forebrain from both neonatal and adolescent rats. To this end, NMDAR-mediated currents were recorded from brain slices that were pre-treated with IFEN (a GluN2B antagonist) and PEAQX (a GluN2A antagonist), respectively. Figure 1A are grouped data of the amplitude of currents recorded from IFEN- and PEAQX-pretreated brain slices of neonatal and adolescent rats. The mean amplitudes of currents were 58.7±6.2% (n=9 cells/7 rats) and 45.9±5.6% (n=9 cells/8 rats) of baselines when bath application of IFEN and PEAQX, respectively, in neonatal brain slices. There was no statistical difference in the amplitudes between the IFEN- and PEAQX-treated groups in neonatal rats (P > 0.05). In contrast, the mean amplitudes of currents were 85.4±4.7% (n=8 cells/6 rats) and 17.3±3.7% (n=8 cells/5 rats) of baselines when bath application of IFEN and PEAQX, respectively, in adolescent brain slices. There was a significant statistical difference in the amplitude between the IFEN- and PEAQX-treated groups in adolescent rats. These data showed that NMDAR-mediated channel activity is regulated fairly equally by both GluN2A and GluN2B subunits in immature neurons of neonatal rats. In contrast, the channel activity is regulated much more predominantly by GluN2A subunits in mature neurons of adolescent rats. Thus, these results support the views that 1) GluN2B-containing NMDARs are abundantly expressed in immature neurons and the GluN2B subunits play a substantial role in channel regulation during the early postnatal period;17–19 and 2) GluN2A/GluN2B ratio increases as brain development progresses to maturity.34,35
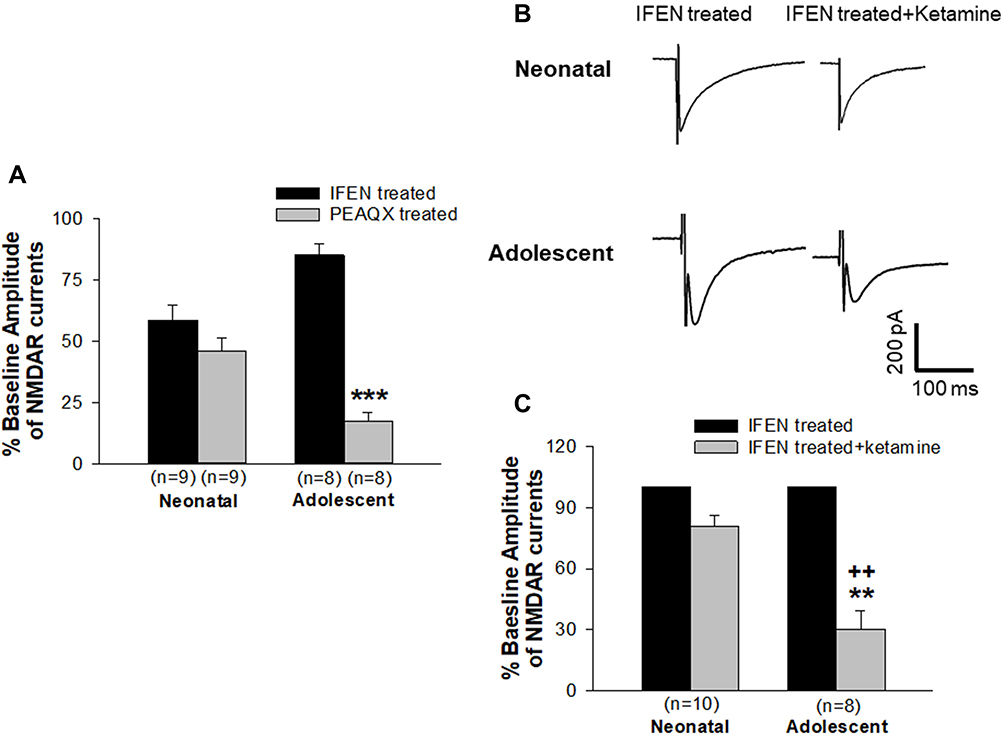 |
Figure 1 Contributions of GluN2A and GluN2B to the regulation of NMDAR-mediated currents and the role of GluN2B subunits in ketamine-induced blockade of currents in immature and mature neurons. (A) Proportion of NMDAR currents regulated by GluN2A and GluN2B were pharmacologically isolated by bath application of IFEN and PEAQX, respectively, and was presented as percentage of baseline currents (without antagonist treatment, set as 100%). ***P<0.001 compared with current levels treated with IFEN. (B and C) Ketamine-induced NMDAR channel blockade was tested by bath application of ketamine to brain slices that had been pre-treated with IFEN. Representative traces are shown in (B). The levels of currents when slices were pre-treated with IFEN were set as baseline controls (100%). The changes in currents following ketamine application were then compared with baseline controls (**P<0.01). A significant difference of the ketamine-induced reduction in channel currents between groups of neonatal and adolescent neurons was determined and indicated as ++P<0.01. |
Next, we examined whether the sensitivity of NMDARs to ketamine’s blocking effect is regulated by GluN2B subunits since our previous experiments have revealed that ketamine produced a greater and longer blocking effect on NMDAR channels in immature neurons than in mature neurons.20 The experiments were performed in the brain slices that had been pretreated with IFEN to test the effect of GluN2B inhibition on the blockade of channel activity produced by ketamine. The levels of NMDAR-mediated currents when slices were pre-treated with IFEN were set as the baseline (100%). Figure 1B shows representative traces of currents and their ketamine-induced changes. In the presence of ketamine, the mean amplitude of currents was 80.6±5.5% of baseline in neonatal brain slices (n=10 cells/9 rats) and 30.5±8.8% of baseline in adolescent brain slices (n=8 cells/6 rats), respectively, as seen in Figure 1C. Thus, inhibiting GluN2Bs significantly reduced the blocking effect of ketamine on channel current activity in immature neurons, as there was no statistically significant difference between groups of baseline control and ketamine treatment. In contrast, inhibition of GluN2Bs did not affect the ketamine-induced blockade of channel current activity in mature neurons, as there was a significant reduction in channel current activity compared with baseline control. This was also evidenced by the fact that the mean percent inhibition of current amplitude produced by ketamine was significantly lowered in mature neurons than in immature neurons (++, P<0.01). Thus, these results strongly suggest that the GluN2B subunit plays a much bigger role in the sensitivity of NMDARs to ketamine’s blocking effect in immature neurons compared to mature neurons.
The Changes in Channel Currents Mediated by Extrasynaptic NMDARs in Immature Neurons After Repeated Ketamine Exposure
In immature neurons, GluN2B-containing receptors are expressed significantly higher at extrasynaptic sites22–25 and GluN2B plays a more significant role than GluN2A in mediating the age-dependent compensatory upregulation of NMDAR channel activity induced by repeated ketamine exposure.21 Thus, we wanted to specifically examine the contribution of extrasynaptic NMDARs to such an age-dependent enhancement of channel activity due to repeated ketamine exposures. Extrasynaptic NMDAR-mediated currents were isolated by bath application of a use-dependent and irreversible NMDAR blocker, MK-801, to pharmacologically remove active synaptic NMDAR channels as described in Materials and Methods. Current levels before MK-801 pre-treatment were set as baseline (100%). After MK-801 treatment, the current amplitudes were 65.6±3.8% of baseline in neonatal slices (n=10 cells/8 rats) and 37.0±4.6% of baseline in adolescent ones (n=10 cells/6 rats), respectively. There was a significant statistical difference in the amplitudes between neonatal and adolescent group (++, P<0.01). These data showed that extrasynaptic NMDARs contributed over 65% to the mediation of current activity in neonatal neurons compared to about 37% contributing to current activity in adolescent neurons (Figure 2A). These are generally consistent with a previous report that extrasynaptic NMDARs represented up to three-quarters of all NMDARs in immature brain neurons.22 Together with the data from adolescent neurons (Figure 2A), the results demonstrated an age-dependent difference in the contribution of extrasynaptic NMDARs to channel current activity. Thus, the next study was conducted only on neonatal neurons that had been repeatedly exposed to ketamine in vivo and pretreated with MK-801 in vitro. As shown in Figure 2B, repeat exposures of neonatal rats to ketamine led to a significant time-dependent increase in the amplitude of the extrasynaptic NMDAR-mediated currents, measured at 3, 8 and 12 h after the last ketamine exposure. The mean amplitude of currents recorded at 12 h after saline injection in vivo was set as 100% (baseline, n=10 cells/7 rats). At 3 h after the last ketamine injection, the current amplitude was 105.1±8.8% (n=10 cells/7 rats). The amplitude increased to 148.0±5.3% (n=10 cells/10 rats) of baseline level at 8 h after the last ketamine injection and to 150.1±7.6% (n=9 cells/9 rats) of baseline at 12 h (Figure 2B). These results clearly revealed that extrasynaptic NMDARs predominantly expressed in immature neurons become active and are significantly involved in the compensatory enhancement of excitatory glutamate-mediated events following prolonged ketamine exposure.
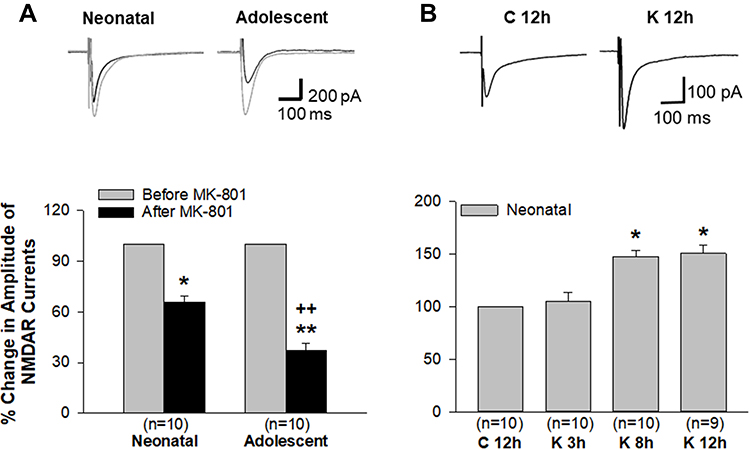 |
Figure 2 A time-dependent enhancement of extrasynaptic NMDAR-mediated channel current activity developed in neonatal brain neurons after ketamine was withdrawn from in vivo repeated administration. (A) In both neonatal and adolescent brain slices, extrasynaptic NMDAR-mediated currents were isolated from synaptic currents by MK-801 treatment followed by wash out. The contribution of extrasynaptic NMDARs to currents was presented as percentage of baseline current levels (100%, before MK-801 treatment). *P<0.05; **P<0.01, compared with baseline levels. A statistically significant difference of the extrasynaptic NMDAR’s contribution to currents between groups of neonatal and adolescent neurons was determined and indicated as ++P<0.01. (B) Extrasynaptic NMDAR-mediated currents were recorded only from neonatal brain slices at 3, 8 and 12 h after the last dose of ketamine administration. *, P<0.05, compared with the current level recorded from brain slices at 12 h after last saline injection. Representative traces of currents are shown in the upper panels of (A and B). |
Discussion
In the present study, we have provided electrophysiological evidence to indicate that the GluN2B subunits contribute substantially to the mediation of ketamine-induced blockade of NMDAR channel activity in the immature neurons of the developing brain, and that the compensatory enhancement of NMDAR current activity in immature neurons following repeated ketamine exposure is significantly regulated by NMDARs at extrasynaptic sites where GluN2B-containing receptors are predominantly expressed. These results together with our preceding published data strongly suggest a pathophysiological link to the higher vulnerability to the triggering of neuroapoptosis specifically seen in the developing brain.
NMDARs with different compositions of regulatory GluN2 subunits confer distinct electrophysiological and pharmacological properties on the receptors and couple them with different regulatory mechanisms.15,16 Of these, GluN2A- and GluN2B-containing receptor subtypes have distinct roles in influencing the direction of synaptic plasticity.36,37 The expression of GluN2A or GluN2B in NMDARs varies depending on the developmental stage of the neurons. During early development, the CNS is predominated by GluN2B-containing NMDARs and the expression of them peaks at PND 7 particularly in forebrain neurons.38,39 Using GluN2A and GluN2B antagonists in the present study, we measured the proportion of channel currents mediated by GluN2A-containing and GluN2B-containing NMDARs, respectively, in brain slices of neonatal and adolescent rats. Our results suggest that the GluN2B subunit is involved in the regulation of NMDAR channel activity in neonatal neurons, which is consistent with the view that the CNS is predominated by GluN2B-containing NMDARs that substantially regulate the channel activity during early brain development.38,39 These results prompted us to examine whether the effectiveness of ketamine-induced channel blockade is GluN2B-dependent in neonatal neurons. The results indicate that GluN2B-containing NMDARs are more sensitive than GluN2A-containing NMDARs to ketamine, evidenced by the facts that the blocking effect of ketamine on channel currents in immature neurons was dramatically reduced when GluN2Bs were inhibited by IFEN and inhibition of GluN2Bs did not affect the ketamine-induced channel blockade in mature neurons. Thus, a higher sensitivity of such a type of NMDARs to ketamine is likely to cause compensatory upregulation selectively in immature neurons after prolonged ketamine exposure.
Data obtained from neurochemical and molecular studies using animal models have shown that the expression of GluN1 subunits was increased in neonatal neurons following prolonged and/or repeated ketamine exposure in vivo.8,11,40,41 Such a compensatory upregulation of NMDARs corresponded in the time-course to the development of apoptotic neuronal death in the immature brain evident at 4–6 h after the repeated ketamine exposure was withdrawn.7,8 Further, a recent study by our group21 has shown that a) In neonatal neurons, repeated ketamine administration led to a time-dependent increase in NMDAR-mediated currents beginning at 2 h after the last ketamine dose and peaking at 6–8 h, consistent with the timeline of ketamine-induced neuroapoptosis; b) This increased NMDAR-mediated synaptic transmission was also consistent with a significantly higher expression of GluN2B proteins, as well as an increase in the GluN1 expression as shown in molecular studies by other groups;8,11,41 c). More importantly, the above increased changes were GluN2B-dependent and seen selectively in immature neurons. The slower decay kinetics of GluN2B-containing NMDARs confer higher capacity for transfer of Ca2+ into neurons.39 When the receptors develop compensatory up-regulation, a massive influx of Ca2+ would trigger the apoptosis selectively in immature neurons.42 These findings have demonstrated the age-dependent effects of ketamine and provided pathophysiological proof of ketamine’s ability to affect the cellular and molecular responses of immature neurons differently from their mature counterparts.
During the CNS development, extrasynaptic NMDARs outnumber synaptic NMDARs, comprising up to three-quarters of all NMDARs. GluN2B-containing NMDARs are expressed more abundantly than GluN2A-containing NMDARs at extrasynaptic sites.22–25 It has been documented that NMDAR activity has the potential to promote the process of survival or death in CNS neurons depending on whether synaptic or extrasynaptic NMDARs and GluN2A- or GluN2B-containing NMDARs are involved.26,27,43,44 Studies by our and other groups supported this view and further suggested that compensatory up-regulation of NMDARs induced by repeated or prolonged ketamine exposure is age-dependent, selectively seen in neonatal neurons.8,11,21,40,41 Using the neonatal rat model of repeated ketamine administration, we wanted to test whether the NMDARs at extrasynaptic sites contribute crucially to such a compensatory increase in synaptic transmission due to prolonged ketamine exposure. Data obtained from brain slices where extrasynaptic NMDARs were isolated by blocking active synaptic NMDARs with MK-801 showed that extrasynaptic NMDARs contributed over 65% to the mediation of current activity in immature neurons and over 37% to that in mature neurons, which generally confirmed the age-dependent difference in the contribution of extrasynaptic NMDARs to channel activity.22 We then further examined if prolonged ketamine exposure-induced enhancement of NMDAR current activity in immature neurons involved the contribution of extrasynaptic NMDARs. In brain slices of neonatal rats that had been repeatedly exposed to ketamine in vivo and pretreated with MK-801 in vitro that blocked active synaptic NMDARs, an enhancement of currents after withdrawal from ketamine exposure was still evident as a 48–50% increase, compared to the control group. It has also been proposed that NMDARs are mobile and can shuttle between the synaptic and extrasynaptic membrane. The major function of extrasynaptic NMDARs would be to form a reserve pool waiting to replace synaptic receptors.45 However, a debate still remains on whether such exchange is operated rapidly within minutes.46,47 A recent finding appears to be supportive of our current data showing that the exchange of receptors between synaptic and extrasynaptic sites is mediated primarily by GluN2B-containing NMDARs.47 Thus, it is strongly suggested that GluN2B-containing NMDARs abundantly expressed at extrasynaptic sites in immature neurons contribute as a major player to the compensatory enhancement of NMDAR current activity following prolonged ketamine exposure. Future work on this study will determine whether pro-death signaling is triggered by the upregulated extrasynaptic NMDARs of immature neurons contributing to such an age-dependent neurotoxic effect.
In summary, new information provided by the present study supports the view that GluN2B-containing NMDARs predominantly expressed in immature neurons are upregulated progressively during the course of ketamine withdrawal from its prolonged administration in neonatal rodents. Further, the evidence specifically indicates that NMDAR-mediated signaling at extrasynaptic sites becomes significantly enhanced due to prolonged ketamine exposure. Thus, these findings have advanced our understanding why the immature neurons are more vulnerable to ketamine’s neurotoxic effects. In addition, these experimental data could be the basis on which new therapeutic strategies aimed at avoiding or reducing ketamine-induced adverse effects can be developed.
Acknowledgments
This work was supported by a Startup Fund of the University of Texas at Arlington to Q.L. We would like to thank Capital Medical University of China for the collaboration by sending Dr. Jianhui Jin as a visiting scholar to complete this study. We would also like to thank Dr. Michael Quast, Ph.D. for his helpful editing of the English language while preparing the manuscript.
Author Contributions
All authors contributed to study design, data acquisition and analysis, and drafting the manuscript. All authors have agreed to be accountable for all aspects of the work, agreed on the journal to which the article will be submitted, and given final approval of the version to be published.
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Zanos P, Moaddel R, Morris PJ, et al. Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev. 2018;70:621–660. doi:10.1124/pr.117.015198
2. Yang Y, Maher DP, Cohen SP. Emerging concepts on the use of ketamine for chronic pain. Expert Rev Clin Pharmacol. 2020;13:135–146. doi:10.1080/17512433.2020.1717947
3. Roelofse JA. The evolution of ketamine applications in children. Pediatr Anesth. 2010;20:240–245. doi:10.1111/j.1460-9592.2009.03145.x
4. Gorlin AW, Rosenfeld DM, Ramakrishna H. Intravenous subanesthetic ketamine for perioperative analgesia. J Anaesthesiol Clin Pharmacol. 2016;32:160–167. doi:10.4103/0970-9185.182085
5. Vadivelu N, Schermer E, Kodumudi V, Belani K, Urman RD, Kaye AD. Role of ketamine for analgesia in adults and children. J Anaesthesiol Clin Pharmacol. 2016;32:298–306. doi:10.4103/0970-9185.168149
6. Abdel-Ghaffar HS, Abdel-Wahab AH, Roushdy MM, Osman AMM. Preemptive nebulized ketamine for pain control after tonsillectomy in children: randomized controlled trial. Rev Bras Anestesiol. 2019;69:350–357. doi:10.1016/j.bjan.2019.03.007
7. Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi:10.1126/science.283.5398.70
8. Zou X, Patterson TA, Sadovova N, et al. Potential neurotoxicity of ketamine in the developing rat brain. Toxicol Sci. 2009;108:49–158. doi:10.1093/toxsci/kfn270
9. Brambrink AM, Evers AS, Avidan MS, et al. Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology. 2012;116:372–384. doi:10.1097/ALN.0b013e318242b2cd
10. Lyu D, Tang N, Womack AW, He Y-J, Lin Q. Neonatal ketamine exposure-induced hippocampal neuroapoptosis in the developing brain impairs adult spatial learning ability. Neural Regen Res. 2020;15:880–886. doi:10.4103/1673-5374.268929
11. Liu F, Paule MG, Ali S, Wang C. Ketamine-induced neurotoxicity and changes in gene expression in the developing brain. Curr Neuropharmacol. 2011;9:256–261. doi:10.2174/157015911795017155
12. Huang L, Liu Y, Jin W, Ji X, Dong Z. Ketamine potentiates hippocampal neurodegeneration and persistent learning and memory impairment through the PKCγ-ERK signaling pathway in the developing brain. Brain Res. 2012;1476:164–171. doi:10.1016/j.brainres.2012.07.059
13. Stevens RA, Butler BD, Kokane S, Womack A, Lin Q. Neonatal inhibition of Na+-K+-2Cl− cotransporter prevents ketamine induced spatial learning and memory impairments. Neurotoxic Teratol. 2017;60:82–86. doi:10.1016/j.ntt.2016.11.001
14. Wang RR, Jin JH, Womack AW, et al. Neonatal ketamine exposure causes impairment of long-term synaptic plasticity in the anterior cingulate cortex of rats. Neuroscience. 2014;268:309–317. doi:10.1016/j.neuroscience.2014.03.029
15. Stroebel D, Casado M, Paoletti P. TriheteromericNMDA receptors: from structure to synaptic physiology. Curr Opin Physiol. 2018;2:1–12. doi:10.1016/j.cophys.2017.12.004
16. Sun Y, Xu Y, Cheng X, et al. The differences between GluN2A and GluN2B signaling in the brain. J Neurosci Res. 2018;96:1430–1443. doi:10.1002/jnr.24251
17. Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi:10.1038/368144a0
18. Bloodgood BL, Sabatini BL. NMDA receptor-mediated calcium transients in dendritic spines. In: Dongen V, editor. Biology of the NMDA Receptor, Chapter 9. NIH, CRC Press, NCBI Bookshelf; 2009:Bookshelf ID: NBK5276. doi:10.1201/9781420044157.ch9.
19. Mckay S, Ryan T, McQueen J, et al. The developmental shift of NMDA receptor composition proceeds independently of GluN2 subunit-specific GluN2 C-terminal sequences. Cell Rep. 2018;25:841–851,e4. doi:10.1016/j.celrep.2018.09.089
20. Jin J, Gong K, Zou X, Wang R, Lin Q, Chen J. The blockade of NMDA receptor ion channels by ketamine is enhanced in developing rat cortical neurons. Neurosci Lett. 2013;539:11–15. doi:10.1016/j.neulet.2013.01.034
21. Kokane SS, Gong K, Jin J, Lin Q. Prolonged ketamine exposure induces increased activity of the GluN2B-containing N-methyl-D-aspartate receptor in the anterior cingulate cortex of neonatal rats. Neurotoxicol Teratol. 2017;63:1–8. doi:10.1016/j.ntt.2017.07.005
22. Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi:10.1523/JNEUROSCI.19-10-04180.1999
23. Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507:13–24. doi:10.1111/j.1469-7793.1998.013bu.x
24. Petralia RS. Distribution of extrasynaptic NMDA receptors on neurons. Sci World J. 2012;2012:267120. doi:10.1100/2012/267120
25. Jiang X, Knox R, Pathipati P, Ferriero D. Developmental localization of NMDA receptors, Src and MAP kinases in mouse brain. Neurosci Lett. 2011;503:215–219. doi:10.1016/j.neulet.2011.08.039
26. Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi:10.1038/nn835
27. Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signaling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi:10.1038/nrn2911
28. Kokane SS, Lin Q. Impact of early life ketamine exposure on the developing brain and cognitive sequelae: a discussion of apoptotic neurodegeneration mechanisms. In: Preedy VR, editor. The Neuropathology of Drug Addictions and Substance Misuse (Chapter 54). London: Kings College; 2016:581–592. doi:10.1016/B978-0-12-800212-4.00054-6
29. Gong K, Cao F, He Y, et al. Enhanced excitatory and reduced inhibitory synaptic transmission contribute to persistent pain-induced neuronal hyper-responsiveness in anterior cingulated cortex. Neuroscience. 2010;171:1314–1325. doi:10.1016/j.neuroscience.2010.10.028
30. Schreiber JA, Schepmann D, Frehland B, et al. A common mechanism allows selective targeting of GluN2B subunit-containing N-methyl-D-aspartate receptors. Commun Biol. 2019;2:420. doi:10.1038/s42003-019-0645-6
31. McQuail JA, Beas BS, Kelly KB, et al. NR2A-containing NMDARs in the prefrontal cortex are required for working memory and associated with age-related cognitive decline. J Neurosci. 2016;36:12537–12548. doi:10.1523/JNEUROSCI.2332-16.2016
32. Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi:10.1523/JNEUROSCI.0203-11.2011
33. Yang Q, Zhu G, Liu D, et al. Extrasynaptic NMDA receptor dependent long-term potentiation of hippocampal CA1 pyramidal neurons. Sci Rep. 2017;7:3045. doi:10.1038/s41598-017-03287-7
34. Hoffmann H, Gremme T, Hatt H, Gottmann K. Synaptic activity-dependent developmental regulation of NMDA receptor subunit expression in cultured neocortical neurons. J Neurochem. 2000;75:1590–1599. doi:10.1046/j.1471-4159.2000.0751590.x
35. Sun Y, Cheng X, Zhang L, et al. The Functional and molecular properties, physiological functions, and pathophysiological roles of GluN2A in the central nervous system. Mol Neurobiol. 2017;54:1008–1021. doi:10.1007/s12035-016-9715-7
36. Liu L, Wong TP, Pozza MF, et al. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi:10.1126/science.1096615
37. Massey PV, Johnson BE, Moult PR, et al. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi:10.1523/JNEUROSCI.1697-04.2004
38. Paoletti P. Molecular basis of NMDA receptor functional diversity. Eur J Neurosci. 2011;33:1351–1365. doi:10.1111/j.1460-9568.2011.07628.x
39. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi:10.1038/nrn3504
40. Wang C, Sadovova N, Fu X, et al. The role of the N-methyl-D-aspartate receptor in ketamine-induced apoptosis in rat forebrain culture. Neuroscience. 2005;132:967–977. doi:10.1016/j.neuroscience.2005.01.053
41. Lecointre M, Vézier C, Bénard M, et al. Age-dependent alterations of the NMDA receptor developmental profile and adult behavior in postnatally ketamine-treated mice. Dev Neurobiol. 2015;75:315–333. doi:10.1002/dneu.22232
42. Wang C, Liu F, Patterson TA, Paule MG, Slikker W. Relationship between ketamine-induced developmental neurotoxicity and NMDA receptor-mediated calcium influx in neural stem cell derived neurons. Neurotoxicology. 2017;60:254–259. doi:10.1016/j.neuro.2016.04.015
43. Liu Y, Wong TP, Aarts M, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi:10.1523/JNEUROSCI.0116-07.2007
44. Wittmann M, Bengtson CP, Bading H. Extrasynaptic NMDA receptors: mediators of excitotoxic cell death. In: Krieglstein J, Klumpp S, editors. Pharmacology of Cerebral Ischemia. Stuttgart: Medpharm Scientific Publishers; 2004:253–266. doi:10.11588/heidok.00005390
45. Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi:10.1016/s0896-6273(02)00658-x
46. Harris AZ, Pettit DL. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol. 2007;584:509–519. doi:10.1113/jphysiol.2007.137679
47. McQuate A, Barria A. Rapid exchange of synaptic and extrasynaptic NMDA receptors in hippocampal CA1 neurons. J Neurophysiol. 2020;123:1004–1014. doi:10.1152/jn.00458.2019
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.