")
Back to Journals » International Medical Case Reports Journal » Volume 12
The improvement of pulmonary artery pressure after bosentan therapy in patients with β-thalassemia and Doppler-defined pulmonary arterial hypertension
Authors Karami H, Darvishi-Khezri H, Kosaryan M, Akbarzadeh R, Dabirian M
Received 18 July 2018
Accepted for publication 24 November 2018
Published 17 December 2018 Volume 2019:12 Pages 1—7
DOI https://doi.org/10.2147/IMCRJ.S180602
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Hossein Karami,1 Hadi Darvishi-Khezri,2 Mehrnoush Kosaryan,1 Rosetta Akbarzadeh,2 Mojdeh Dabirian3
1Department of Pediatrics, Thalassemia Research Center (TRC), Hemoglobinopathy Institute, Mazandaran University of Medical Sciences, Sari, Iran; 2Student Research Committee, Thalassemia Research Center (TRC), Hemoglobinopathy Institute, Mazandaran University of Medical Sciences, Sari, Iran; 3Department of Cardiology, Cardiovascular Research Center, Mazandaran University of Medical Sciences, Sari, Iran
Introduction: Pulmonary arterial hypertension (PAH) is relatively prevalent in patients with thalassemia. PAH treatment is necessary as the prevalence of Doppler-estimated PAH and the resultant mortality is high in such patients.
Materials and methods: This study aimed at evaluating the effect of bosentan therapy on patients with thalassemia suspected of PAH. Based on pulsed Doppler echocardiography, all the cases were suspected of severe PAH. Consequently, bosentan was initiated at a dose of 62.5 mg twice a day for 4 weeks, which was increased to 62.5–125 mg twice a day, if no adverse side effects were observed.
Results: The results of this study showed that pulmonary artery pressure (PAP) decreased after the administration of bosentan in three cases, from 160 to 120, 110 to 65, and 60 to 25 mmHg; in other words, the PAP reduced in the mentioned cases by 25%, 36.4%, and 58.4%, respectively.
Conclusion: In this study, PAP improved after bosentan therapy in patients with β-thalassemia suspected of PAH; however, further studies are required to confirm the findings.
Keywords: β-thalassemia major, pulmonary arterial hypertension, pulsed Doppler echocardiography, bosentan, tricuspid regurgitation jet velocity, right ventricular pressure
Introduction
Patients with β-thalassemia and pulmonary arterial hypertension (PAH) commonly have high morbidity and mortality due to cardiac and pulmonary deterioration.1 PAH is defined as a mean pulmonary artery pressure (PAP) of over 25 mmHg based on the results of right heart catheterization performed at rest.2 Although right heart catheterization is the gold standard to diagnose PAH, its application is limited due to the invasive nature of this method to study PAH.3
Doppler echocardiography (ECHO) is frequently used to detect PAH as a noninvasive technique.3 The presence of PAH can also be determined by Doppler ECHO.2 Doppler-defined PAH is reported as prevalent as 2%–79% in cases with thalassemia (for thalassemia intermedia, the range is 40%–50%),4,5 although its accurate prevalence is not known.6 Currently, the prevalence of PAH diagnosis by right heart catheterization is reported ~2.1%.7
Although PAH has an unknown etiology, it is assumed that hemolysis, hypoxia, hypercoagulability, and high cardiac output can contribute to this condition in patients with thalassemia.8,9 Furthermore, concentration of neurohormone endothelin-1 (ET-1) is reported to be correlated with PAH severity and diagnosis,10 resulting in vasoconstriction and promotion of fibrosis, cell proliferation, remodeling, and inflammation.10 Patients with hemoglobinopathies also develop elevated plasma levels of ET-1.11,12 Bosentan, a dual endothelin receptor (ETR), acts through antagonistic effects on ETR-A and ETR-B.13,14 A few clinical trials using phosphodiesterase-5 inhibitors and endothelin antagonists are published, but nonrandomized, controlled trials reported that all currently available PAH-specific medications are advantageous in patients with hemoglobinopathies. Some PAH treatments recommended for hemoglobinopathies include anticoagulation,15 phosphodiesterase-5 inhibitors (such as sildenafil),16 ETR antagonists (such as bosentan),17 prostacyclin analogs,18 and hydroxyurea.18 A small amount of data are available on the management strategies for patients with chronic hemolytic anemia and PAH.
This study reported the cases of three patients with thalassemia suspected of PAH defined by ECHO successfully treated with the dual ET-1 receptor antagonist bosentan. To the best of authors’ knowledge, there are few published studies evaluating the effects of PAH-specific therapy in such patients.
Materials and methods
The current clinical study was conducted in the Thalassemia Research Center of Bu-Ali Sina Hospital affiliated to Mazandaran University of Medical Sciences in Sari, Iran, in 2017. Written informed consent was obtained from the patients to use their clinical data.
In this study, the patients were placed on chronic transfusion therapy (15 mL/kg of leukocyte-filtered packed cells) every 2–4 weeks, targeted to keep a hemoglobin level of >10 g/dL (Table 1). Hydroxyurea was generally started at about 5–10 mg/kg/day and slowly escalated as tolerated to 20 mg/kg/day (to a maximum of two capsules per day, each capsule containing 500 mg of hydroxyurea). One patient did not receive hydroxyurea before starting bosentan (patient 1).
 | Table 1 Demographic and basic data of β-thalassemia patients under treatment with bosentan Note: aNumber of received blood units and blood transfusion interval before bosentan were calculated at 6 months before bosentan administration. Abbreviations: Hb, hemoglobin; NTDT, nontransfusion-dependent thalassemia; T2*, T2-weighted magnetic resonance imaging; TDT, transfusion-dependent thalassemia. |
The dose and duration of the latest iron chelator before bosentan were also recorded. The mean levels of hemoglobin and ferritin were calculated for 6 months before bosentan administration. The patients were asked about possible clinical side effects of bosentan via monthly visits. The background and clinical data for each patient, including blood transfusion history, iron-chelating medications, T2-weighted magnetic resonance imaging (MRI T2*) of the heart, and concurrent and historical drugs related to the treatment of suspected PAH were also recorded. The cardiovascular magnetic resonance tools of Brompton were utilized to compute T2* MRI19 (Table S1).
PAP was estimated by Doppler ECHO using the modified Bernoulli equation, which calculates the pressure difference between the two chambers of the heart using jet velocity between them.20 PAP can be assessed relatively accurately using the Bernoulli equation to estimate the pressure gradient between the right ventricle and the right atrium.21 By this method, right ventricular (RV) pressure can be estimated by tricuspid regurgitation jet velocity that is equal to PAP if there is no stenosis at RV outflow.
Patients’ transfusion therapy was commenced at least 3 months prior to starting bosentan and was not modified during bosentan therapy. One patient was already on sildenafil as a primary therapy for suspected PAH, with bosentan added for PAH worsening after a period of 12 months. This patient continued on a reduced stable dose of sildenafil for the time period of the current report.
Results
Three patients (one male and two females) with a mean age of 41.6±5.8 years (ranging from 35 to 46 years) received bosentan. All the patients had been treated with deferiprone, deferasirox, and desferrioxamine in the past for various lengths of time. Cases 2 and 3 had no previous history of blood transfusion and were treated with regular transfusions after diagnosis of suspected PAH. The demographics, therapeutic state, cardiac MRI T2* status, and hemoglobin and ferritin levels of the patients are shown in Tables 1 and 2.
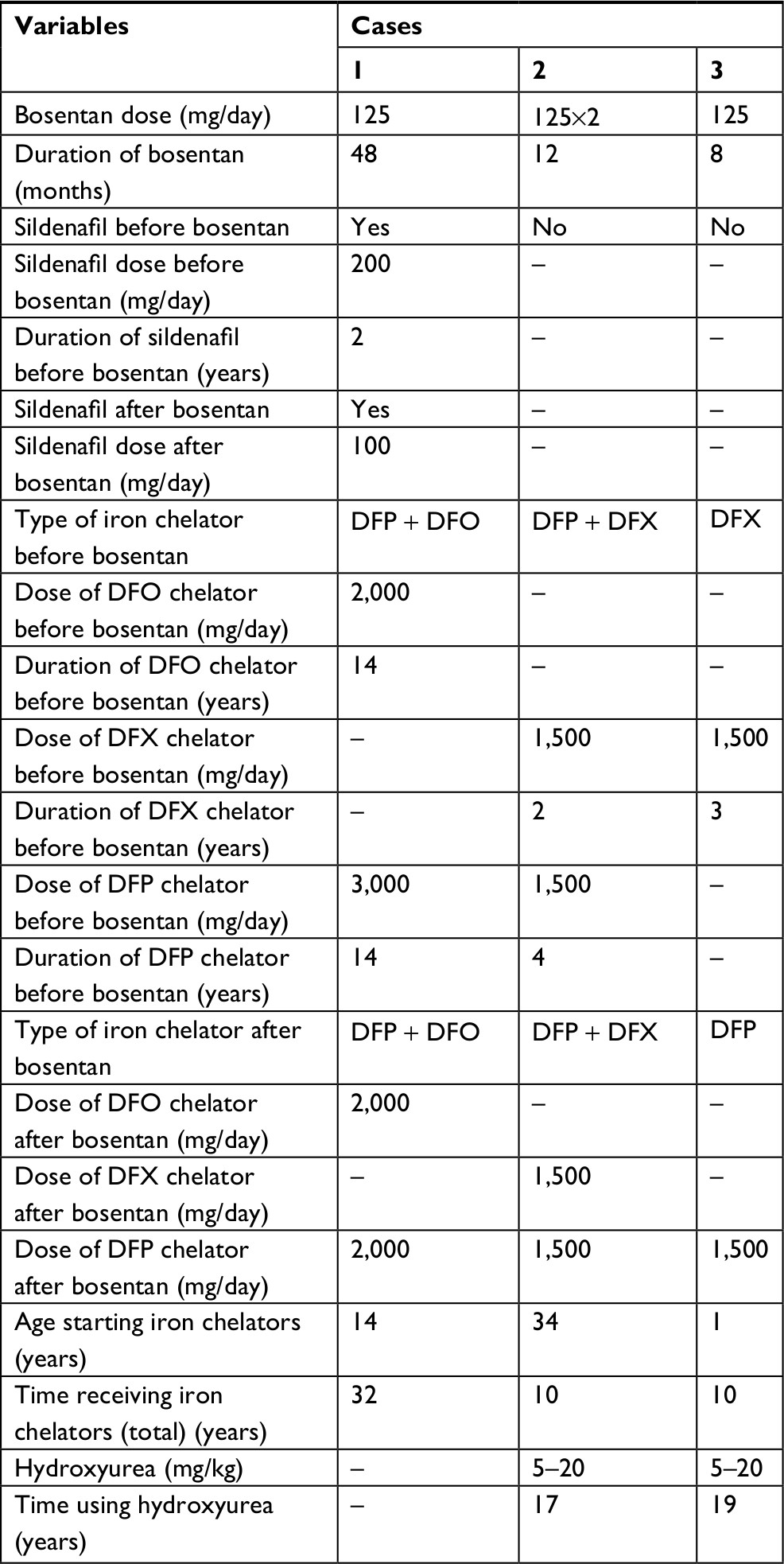 | Table 2 Therapeutic status in β-thalassemia patients under treatment with bosentan Abbreviations: DFO, desferrioxamine; DFP, deferiprone; DFX, deferasirox. |
Based on pulsed Doppler ECHO, severe PAH was suspected in all the cases (>60 mmHg). Consequently, bosentan was initiated at a dose of 62.5 mg twice a day for 4 weeks, which was increased to 62.5–125 mg twice a day if no side effects were noted (Table 2).
Cases 2 and 3 had no history of receiving blood transfusion before bosentan administration. Only one case received a combination therapy of bosentan with sildenafil; however, this case used sildenafil 2 years before starting bosentan. Deferiprone was a common medication in the new iron-chelating regimen in all cases suspected of severe PAH receiving bosentan. Cardiac MRI T2* status, including cardiac iron siderosis, before and after treatment with bosentan, was normal in all the patients with β-thalassemia.
PAP decreased after the administration of bosentan in all the three cases (case 1: from 160 to 120 mmHg, case 2: from 110 to 65 mmHg, and case 3: from 60 to 25 mmHg; Table 3). The percentages of PAP reduction in cases 1–3 were 25%, 36.4%, and 58.4%, respectively. No significant adverse effects were observed in the treatment of such patients suspected of PAH with bosentan.
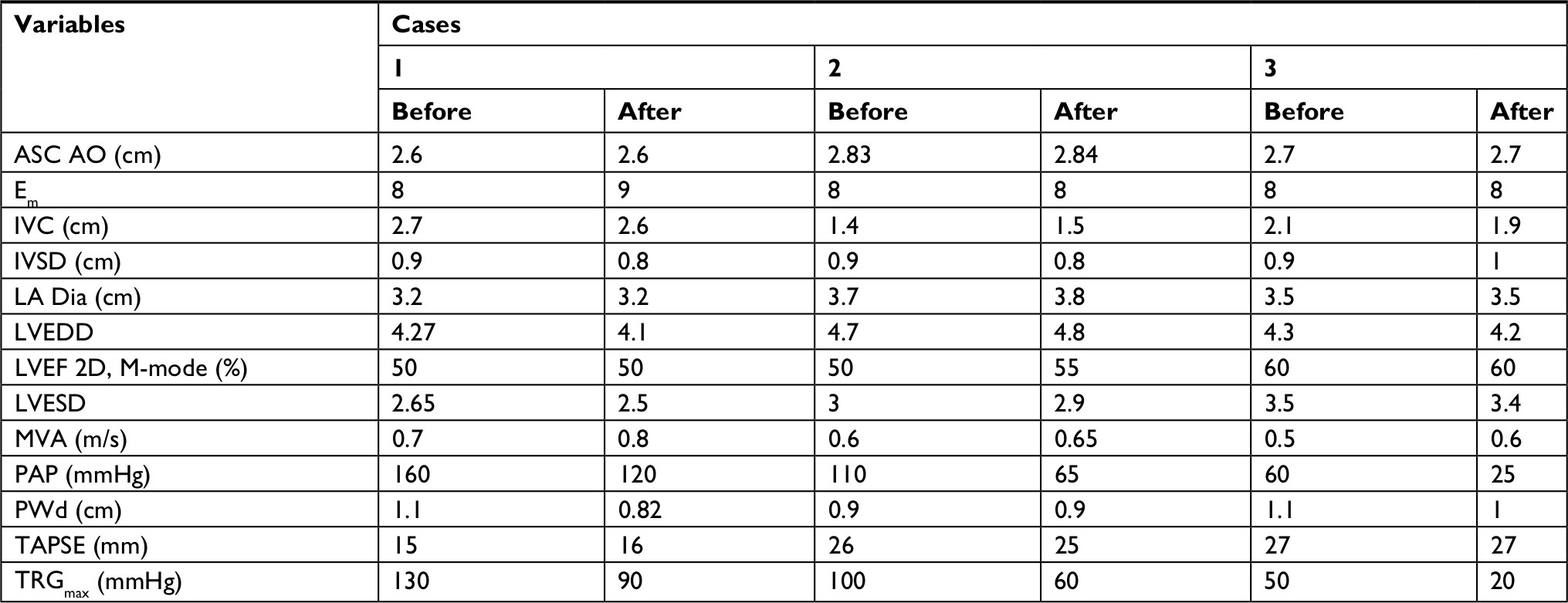 | Table 3 Echocardiography data in β-thalassemia patients under treatment with bosentan Abbreviations: ASC AO, ascending aorta; Em, peak early diastolic velocity at lateral mitral valve annulus; IVC, inferior vena cava diameter; IVSD, interventricular septum thickness at end-diastole; LA Dia, left atrium diameter; LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; LVESD, left ventricular end-systolic diameter; MVA, mitral valve area; PAP, pulmonary artery pressure; PWd, posterior wall dimension; TAPSE, tricuspid annular plane systolic excursion; TRGmax, tricuspid regurgitation gradient. |
Discussion
In this study, PAP improved after bosentan therapy in patients with β-thalassemia and Doppler-defined PAH. However, some echocardiographic and laboratory indices, such as plasma concentrations of ET-1, were not addressed in this study. Long-term observational studies with first-line bosentan are suggested to enhance survival in PAH22; however, no data based on controlled clinical trials are available in this regard.
Anthi et al initially reported that bosentan had a remedial impact on PAH in patients with thalassemia.17 According to them, patients’ positive responses to the treatment were somewhat caused by the clear hemodynamic identification prior to targeting PAH by some particular curative approaches.23
To the best of authors’ knowledge, no clinical trials are performed on patients with thalassemia to evaluate the effect of bosentan therapy as an efficient and safe treatment for PAH. However, two earlier studies were conducted on the treatment of patients with sickle cell disease (SCD).10,13 The impact of bosentan was appraised by Barst et al in PAH-associated SCD.10 The researchers observed no significant reductions in 6MWD, mean PAP, mean right arterial pressure, and pulmonary vascular resistance with bosentan in patients with situation-1 and situation-2 after 16 weeks (two simultaneous trials were designed in patients with SCD and precapillary pulmonary hypertension [situation-1] or pulmonary hypertension as mixed vasculopathy [situation-2]).10 In addition, bosentan led to a greater decrease in hemoglobin than placebo and no patient was withdrawn from the trial owing to deterioration of anemia.10 In the case report by Anthi et al,23 after 1-year follow-up, mean hemoglobin level dropped to 0.2 g/dL in patients with thalassemia and intermedia PAH treated with bosentan. This study results revealed no significant hemoglobin reductions following bosentan therapy.
Associations of bosentan therapy with PAH improvement, RV function, and remodeling in nonthalassemic chronic thromboembolic pulmonary hypertension were shown by Surie et al.15 In their pilot study (n=15), the change in mean right arterial pressure was not significantly different between the bosentan group and the controls (–8; –12–3 vs 0; –8–6, P=0.09).
Several risk factors are mentioned to develop PAH in patients with thalassemia such as chronic hypoxia, respiratory infections, and iron siderosis in tissue.24–26 Hypercoagulable state can be considered as one of the most important factors for thalassemia-related PAH incidence, which is abundantly observed in cases with splenectomized and intermedia thalassemia.12,27,28 Some alternations in red blood cells (RBCs) in patients with intermedia and splenectomized thalassemia increase adherence of RBCs to endothelial cells and thromboembolism events in multiple organs such as the lungs and pulmonary circulation and, in turn, promote the likelihood of occurrence of thalassemia-related PAH. These changes include RBCs’ membrane damage following the change of phospholipid molecules’ flip-flop in the cell membrane and damage to integral and cytoskeletal proteins of RBCs owing to the surplus of free β-globin chains as a result of reduced synthesis of β-globin.24,29 Decreased levels of protein C, protein S, and antithrombin-III, elevated platelet activator markers such as P-selectin, and high plasma level of thrombin–antithrombin complex are other potential causes that may contribute to a hypercoagulability in cases with thalassemia; however, its association with PAH incidence is not clear.25,27,30,31 Treatment with bosentan could improve thalassemia-related PAH via modification of the hypercoagulation state in such patients; more studies in this field are warranted. In addition, the results of studies on other conditions showed that bosentan improved arterial oxygenation in acute hypoxia and resulted in improved physical performance at altitude in a controlled laboratory setting.32
As the results showed, the patients had an increase in their blood transfusion rate after bosentan therapy; it should be noted that the raising transfusion rate was performed by a hematologist as a therapeutic approach to improve oxygenation and relieve workload of the lungs.33 Moreover, the results showed that the highest improvement was observed in the patient with lower time passed since splenectomy. Coagulation dysfunctions are among the known complications after splenectomy.34 Some studies indicated a possible relationship between hypercoagulable states and PAH in patients with thalassemia9–11; in this study, it was speculated that patients with higher time passed since splenectomy are more susceptible to PAH and may show poorer response to bosentan treatment.
Altogether, few studies with small sample sizes are performed on bosentan treatment in patients with β-thalassemia and PAH. Results of this study showed the therapeutic effects and safety of bosentan to relieve PAP in patients with β-thalassemia suspected with PAH; however, the lack of extensive hemodynamic data and the small number of patients were the limitations of this study. PAH diagnosis was made by ECHO alone (no right heart catheterization), and follow-up was also noninvasive. Furthermore, hemodynamic effects of bosentan at baseline and follow-up were not assessed in this study. It is suggested that clinical studies that aim to use bosentan in the treatment of PAH should measure other echo-derived parameters of RV dimensions and function and specific biomarkers that are dependent on treatment in such patients.
Conclusion
In this study, PAP improved after bosentan therapy in patients with β-thalassemia and Doppler-defined PAH. PAH associated with β-thalassemia is a prevalent clinical problem with significant morbidity, important prognostic implications, and a complex pathophysiology that necessitates particular management with clear guidelines and protocols. Further case–control studies with larger well-defined samples are recommended to evaluate the clinical efficacy, safety, and appropriate treatment doses and duration of bosentan therapy for severe PAP management.
Acknowledgments
Authors wish to thank Mazandaran University of Medical Sciences for providing all the necessary facilities. The authors also would like to thank the staff and patients at Bu-Ali Sina Hospital, Department of Thalassemia, Sari, Iran.
Disclosure
All authors declared no conflicts of interest in this work and that there was no source of extra institutional commercial funding, and the entire study was performed without external funding.
References
Aessopos A, Farmakis D, Karagiorga M, et al. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood. 2001;97(11):3411–3416. | ||
Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S55–S66. | ||
Chueamuangphan N, Wongtheptien W, Nawarawong W, Sukornthasarn A, Chuncharunee S, Tawichasri C, Patumanond J. Clinical indicators for pulmonary arterial hypertension in thalassemia. J Med Assoc Thai. 2012;95(1):16. | ||
Ussavarungsi K, Burger CD. Pulmonary arterial hypertension in a patient with β-thalassemia intermedia and reversal with infusion epoprostenol then transition to oral calcium channel blocker therapy: review of literature. Pulm Circ. 2014;4(3):520–526. | ||
Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–1892. | ||
Kalantari S, Gomberg-Maitland M. Group 5 pulmonary hypertension: the orphan’s orphan disease. Cardiol Clin. 2016;34(3):443–449. | ||
Derchi G, Galanello R, Bina P, et al; Webthal Pulmonary Arterial Hypertension Group*. Prevalence and risk factors for pulmonary arterial hypertension in a large group of β-thalassemia patients using right heart catheterization: a Webthal study. Circulation. 2014;129(3):338–345. | ||
Aessopos A, Farmakis D. Pulmonary hypertension in beta-thalassemia. Ann N Y Acad Sci. 2005;1054(1):342–349. | ||
Atichartakarn V, Chuncharunee S, Chandanamattha P, Likittanasombat K, Aryurachai K. Correction of hypercoagulability and amelioration of pulmonary arterial hypertension by chronic blood transfusion in an asplenic hemoglobin E/beta-thalassemia patient. Blood. 2004;103(7):2844–2846. | ||
Barst RJ, Mubarak KK, Machado RF, et al; ASSET study group*. Exercise capacity and haemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: results of the ASSET studies. Br J Haematol. 2010;149(3):426–435. | ||
Rybicki AC, Benjamin LJ. Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood. 1998;92(7):2594–2596. | ||
Singer ST, Kuypers F, Fineman J, et al. Elevated tricuspid regurgitant jet velocity in subgroups of thalassemia patients: insight into pathophysiology and the effect of splenectomy. Ann Hematol. 2014;93(7):1139–1148. | ||
Minniti CP, Machado RF, Coles WA, Sachdev V, Gladwin MT, Kato GJ. Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. Br J Haematol. 2009;147(5):737–743. | ||
Provencher S, Granton JT. Current treatment approaches to pulmonary arterial hypertension. Can J Cardiol. 2015;31(4):460–477. | ||
Surie S, Reesink HJ, Marcus JT, et al. Bosentan treatment is associated with improvement of right ventricular function and remodeling in chronic thromboembolic pulmonary hypertension. Clin Cardiol. 2013;36(11):698–703. | ||
Derchi G, Forni GL, Formisano F, et al. Efficacy and safety of sildenafil in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematologica. 2005;90(4):452–458. | ||
Anthi A, Tsangaris I, Hamodraka ES, Lekakis J, Armaganidis A, Orfanos SE. Treatment with bosentan in a patient with thalassemia intermedia and pulmonary arterial hypertension. Blood. 2012;120(7):1531–1532. | ||
Farmakis D, Aessopos A. Pulmonary hypertension associated with hemoglobinopathies: prevalent but overlooked. Circulation. 2011;123(11):1227–1232. | ||
Kosaryan M, Rahimi M, Darvishi-Khezri H, Gholizadeh N, Akbarzadeh R, Aliasgharian A. Correlation of pancreatic iron overload measured by T2*-weighted magnetic resonance imaging in diabetic patients with β-thalassemia major. Hemoglobin. 2017;41(3):151–156. | ||
Godara H. The Washington Manual of Medical Therapeutics. Philadelphia (PA): Lippincott Williams & Wilkins; 2013:62–63. | ||
Zipes DP, Libby P, Bonow RO, Mann DL, Tomaselli GF. Braunwald’s Heart Disease E-Book: A Textbook of Cardiovascular Medicine. Philadelphia: Elsevier Health Sciences; 2018:152. | ||
McLaughlin VV, Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J. 2005;25(2):244–249. | ||
Anthi A, Machado RF, Jison ML, et al. Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Respir Crit Care Med. 2007;175(12):1272–1279. | ||
Cappellini MD, Robbiolo L, Bottasso BM, Coppola R, Fiorelli G, Mannucci AP. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol. 2000;111(2):467–473. | ||
Singer ST, Kuypers FA, Styles L, Vichinsky EP, Foote D, Rosenfeld H. Pulmonary hypertension in thalassemia: association with platelet activation and hypercoagulable state. Am J Hematol. 2006;81(9):670–675. | ||
Factor JM, Pottipati SR, Rappoport I, Rosner IK, Lesser ML, Giardina PJ. Pulmonary function abnormalities in thalassemia major and the role of iron overload. Am J Respir Crit Care Med. 1994;149(6):1570–1574. | ||
Fayed MA, Abdel-Hady HE, Hafez MM, Salama OS, Al-Tonbary YA. Study of platelet activation, hypercoagulable state, and the association with pulmonary hypertension in children with β-thalassemia. Hematol Oncol Stem Cell Ther. 2018;11(2):65–74. | ||
Karimi M, Musallam KM, Cappellini MD, et al. Risk factors for pulmonary hypertension in patients with β thalassemia intermedia. Eur J Intern Med. 2011;22(6):607–610. | ||
Camaschella C, Cappellini MD. Thalassemia intermedia. Haematologica. 1995;80(1):58–68. | ||
Musumeci S, Leonardi S, Di Dio R, Fischer A, Di Costa G. Protein C and antithrombin III in polytransfused thalassemic patients. Acta Haematol. 1987;77(1):30–33. | ||
Eldor A, Durst R, Hy-Am E, et al. A chronic hypercoagulable state in patients with beta-thalassaemia major is already present in childhood. Br J Haematol. 1999;107(4):739–746. | ||
Olfert IM, Loeckinger A, Treml B, et al. Sildenafil and bosentan improve arterial oxygenation during acute hypoxic exercise: a controlled laboratory trial. Wilderness Environ Med. 2011;22(3):211–221. | ||
Hayes MM, Vedamurthy A, George G, et al; American Thoracic Society Implementation Task Force. Pulmonary hypertension in sickle cell disease. Ann Am Thorac Soc. 2014;11(9):1488–1489. | ||
Stewart GW, Amess JA, Eber SW, Kingswood C, Lane PA, Smith BD, Mentzer WC. Thrombo-embolic disease after splenectomy for hereditary stomatocytosis. Br J Haematol. 1996;93(2):303–310. |
Supplementary material
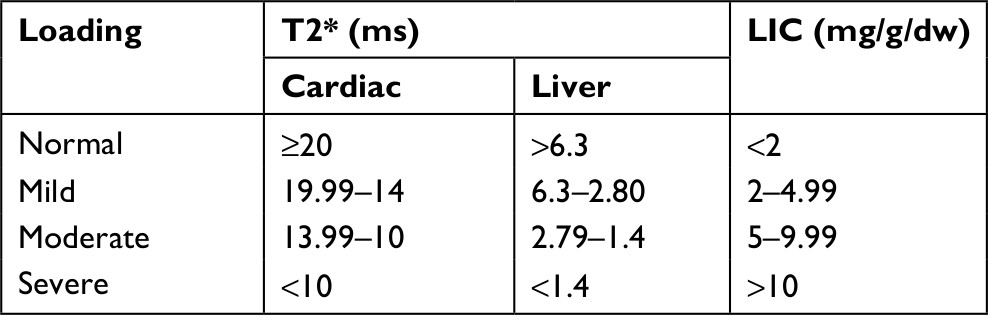 | Table S1 Interpretation and categorization of cardiac and liver MRI Abbreviations: LIC, liver iron concentration; T2*, T2-weighted magnetic resonance imaging. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.