Back to Journals » Patient Related Outcome Measures » Volume 9
The importance of patient-reported outcomes in clinical trials and strategies for future optimization
Authors Mercieca-Bebber R ![]() , King MT, Calvert MJ, Stockler MR
, King MT, Calvert MJ, Stockler MR ![]() , Friedlander M
, Friedlander M
Received 29 May 2018
Accepted for publication 11 July 2018
Published 1 November 2018 Volume 2018:9 Pages 353—367
DOI https://doi.org/10.2147/PROM.S156279
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lynne Nemeth
Rebecca Mercieca-Bebber,1 Madeleine T King,2,3 Melanie J Calvert,4,5 Martin R Stockler,1 Michael Friedlander1,6
1NHMRC Clinical Trials Centre, The University of Sydney, Sydney, NSW, Australia; 2Central Clinical School, Sydney Medical School, The University of Sydney, Sydney, NSW, Australia; 3School of Psychology, The University of Sydney, Sydney, NSW, Australia; 4Centre for Patient-Reported Outcomes Research and NIHR Birmingham Biomedical Research Centre, University of Birmingham, Edgbaston, Birmingham, UK; 5Institute of Applied Health Research, University of Birmingham, Edgbaston, Birmingham, UK; 6Prince of Wales Clinical School, University of New South Wales, Sydney, NSW, Australia
Abstract: Patient-reported outcomes (PROs) can be included in clinical trials as primary or secondary endpoints and are increasingly recognized by regulators, clinicians, and patients as valuable tools to collect patient-centered data. PROs provide unique information on the impact of a medical condition and its treatment from the patient’s perspective; therefore, PROs can be included in clinical trials to ensure the impact of a trial intervention is comprehensively assessed. This review first discusses examples of how PRO endpoints have added value to clinical trial interpretation. Second, it describes the problems with current practices in designing, implementing, and reporting PRO studies, and how these problems may be addressed by complying with guidance for protocol development, selecting appropriate PRO measures to match clinically motivated PRO hypotheses, minimizing the rates of avoidable missing PRO data, analyzing and interpreting PRO data, and transparently reporting PRO findings.
Keywords: patient-reported outcomes, quality of life, trial conduct, research practices, clinical trials as topic
Introduction
Patient-reported outcomes (PROs) are defined as “any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else” (Food and Drug Administration [FDA], p. 6).1 PRO is an umbrella term which may refer to patient-reported: 1) disease symptoms or treatment side effects, such as pain, fatigue, or anxiety; 2) functional outcomes such as physical, sexual, social, role, emotional, or cognitive functioning; or 3) multidimensional constructs such as health-related quality of life (HRQOL) or health utility. HRQOL is defined as “the subjective assessment of the impact of disease and treatment across the physical, psychological, social and somatic domains of functioning and well-being” (Revicki et al, p. 888).2 In the present review, we focus on PROs as clinical trial endpoints and differentiate PROs from other types of patient-reported data, such as patient-reported experiences or patient-reported behaviors, which may also be included as clinical trial endpoints.
PROs are assessed in trials using questionnaires, often referred to as “PRO measures.” Validated PRO measures are used in clinical trials, as opposed to asking participants open-ended questions about their outcomes, to ensure that the questions, response options, and the general approach to assessment are standardized for all participants. This enables the research team to attribute any differences between patient responses to real differences in perceptions of their outcomes, as opposed to methodological differences or biases. PRO measures are typically developed with input from clinicians, patients, and psychometric experts to ensure that the PRO measure assesses clinically relevant issues that are meaningful to patients in a robust manner.
PROs provide unique information on the impact of a medical condition and its treatment from the patients’ perspective;3,4 therefore, PROs can be included as trial endpoints to ensure that the impact of a trial intervention is comprehensively assessed. In palliative care or rehabilitation trials, PROs may be the primary endpoint of interest. In other trials, they may be included as secondary endpoints to support and help interpret the primary endpoint. In some contexts, PROs may be included as exploratory or tertiary endpoints, with the intention of generating hypotheses for testing in future studies. Including PROs in a clinical trial requires careful thought regarding the specific research questions to be addressed and the needs of all stakeholders, including patients, clinicians, trial sponsors, and regulatory authorities.
Importance of including PROs in clinical trials
Increased use and recognition of PROs
The use of PROs in clinical trials has increased over time. Consecutive reviews of ClinicalTrials.gov in 2004–20075 and 2007–20136 determined that use of PRO endpoints had increased from 14% to 27% of trials over that period. A more recent review of the Australian New Zealand Clinical Trials Registry determined that 45% of trials registered from 2005 to March 2017 included PROs and had a strong increase in the proportion of trials with PROs registered over time (r=0.74, P=0.009).7 Similarly, between 2000 and 2015, the Center for Devices and Radiological Health reported an increase of over 500% in pre-market submissions including PRO measures.8
Increases in the use of PROs in clinical trials may be attributable to top-down encouragement from professional societies and regulatory bodies.9 In oncology, for example, the American Society of Clinical Oncology (ASCO)10 and European Society for Medical Oncology (ESMO)11 have proposed standardized approaches to evaluate clinical trial results by using scores to evaluate the Magnitude of Clinical Benefit (ESMO MCB) or the Net Health Benefit (ASCO), which include survival endpoints in addition to toxicity and HRQOL.10,11 The ESMO MCB scores based on survival outcomes are upgraded if there is evidence to indicate improved, or delayed deterioration in, HRQOL using validated PRO measures or a substantial reduction in adverse events. The ESMO and ASCO recommendations are clearly important in evaluating new therapies and highlight the importance of including PRO endpoints in clinical trials.
Furthermore, the Australian government supports a Quality of Life Office to work with the 13 National Cancer Clinical Trials Groups to include PRO endpoints in investigator-initiated oncology trials.12 The European Medicines Agency released guidance in 2016 on the use of PROs in the evaluation of anti-cancer medicinal products.13 PROs have also been highlighted by professional oncology societies as important endpoints in specific oncology trial contexts, for example, in ovarian cancer clinical trials.14–17
Beyond oncology, the FDA-released guidance in 2009 on the evaluation of PRO measures used to support medical product labeling claims. This guidance aimed to improve the efficiency and transparency of discussions between sponsors and the FDA during the drug development process, to streamline the FDA’s review of PRO measures and associated clinical trial data, and to improve methods for considering patients’ perspectives in the medical product review process.1 The Organization for Economic Co-operation and Development recently highlighted the importance of collecting PROs to enable a more complete understanding of health system performance.4 In 2017, the Medicare Evidence Development and Coverage Advisory Committee, USA, determined that quality of life (QOL) measures were of particular interest and should be included as health outcomes in future heart failure studies.8,18
What value do PROs add to clinical trials?
Au et al3 reviewed Phase III oncology cancer trials led by the National Cancer Institute of Canada Clinical Trials Group and classified the benefits of including a PRO as a trial endpoint into three categories. First, PROs may assist clinicians and future patients to select the best treatment by providing a clearer picture of the costs and benefits of treatment.3 Second, PRO data can enrich our understanding of the patients’ experience with unique information that could not be gained from biomedical outcomes alone, as certain domains are difficult to observe (eg, pain and fatigue) and outcomes such as the degree of symptom bother are subjective and best collected through patient report.3 Third, PROs can help to improve future clinical trial methods.3 In contexts where certain PROs have confirmed prognostic significance, PROs can be used to stratify participants. Furthermore, careful examination of PRO assessment compliance can illuminate areas where methodological improvement is needed.3
Other applications of PRO data include informing regulatory decisions, cost-effectiveness analyses, and informing clinical guidelines and health policy. Patient advocacy groups promote the use of PROs as they enable patients to communicate their experience, assess whether their experience aligns with their expectations of treatment, and highlight any unmet needs or care areas that are in need of improvement.19 For these reasons, patient advocates are vocal about the importance of including PROs.19,20 In an era where consumer input into clinical trial protocols is highly valued and encouraged, advocacy for PRO endpoints may result in increased implementation of PRO endpoints in clinical trials.
Examples of how PROs have contributed to the interpretation of clinical trial results
Investigators should determine primary and secondary trial endpoints a priori according to what is appropriate for the interpretation of the individual clinical trial. As a secondary endpoint, PRO data add information to assist the interpretation of primary trial endpoints. In other clinical contexts, it may be appropriate for the PRO to be the primary trial endpoint. There is no “one-size” approach to interpret PRO data, and all clinical trial data including PROs should be interpreted in context. There are numerous examples where PRO data, interpreted in conjunction with other trial outcomes, have been fundamental in understanding patients’ perceptions of the risks and benefits of treatment and have impacted the interpretation of the trial results.
A clinical trial comparing prednisone with or without mitoxantrone in men with metastatic prostate cancer found that although there was no difference in overall survival (P=0.27), or serum prostate-specific antigen levels (P=0.11) between the two arms,21 the patients receiving mitoxantrone experienced significant improvement in PROs. The primary outcome was palliation of pain (response defined as a 2-point reduction in the 6-point pain intensity scale of the McGill-Melzack Pain Questionnaire, or complete loss of pain if the patient initially scored >1, maintained on two consecutive, three-weekly evaluations, without an increase in analgesic score.) Response rates were 29% (95% CI: 19%, 40%) in the mitoxantrone + prednisone group and 12% (95% CI: 6%, 22%) in the prednisone alone group (P=0.01). Furthermore, pain (severity, impact, and relief), fatigue, mood, aspects of functioning, global QOL, and other PROs improved with mitoxantrone + prednisone from the baseline (P<0.01).21,22 The PRO endpoints led to regulatory approval of mitoxantrone for this indication and wide implementation in the clinic.
In a trial of ruxolitinib compared to placebo for the treatment of intermediate- or high-risk myelofibrosis, ruxolitinib was found to improve fatigue, as assessed by the Patient Reported Outcomes Measurement Information System (PROMIS) Fatigue 7-item short form.23 PROMIS Fatigue is included on the ruxolitinib label accordingly, along with PRO data from previous studies which demonstrated that ruxolitinib was associated with a greater proportion of patients experiencing a reduction of ≥50% in patient-reported myelofibrosis symptoms (abdominal discomfort, pain under left ribs, early satiety, night sweats, itching, bone/muscle pain, and inactivity), as well as significant improvement (P<0.001) in these symptoms until 20 weeks, and until 24 weeks for abdominal discomfort, early satiety, and itching. Conversely, the placebo arm experienced a steady decline across all myelofibrosis symptoms over the 24 weeks.23,24
Gnanasakthy et al have published a series of papers detailing the FDA labels that include PRO data that support regulatory approvals.25–27 Between 2006 and 2010, 28 of 116 (24%) products approved by the FDA were granted PRO claims. Of these products, 71% included a PRO as the primary trial endpoint, and pain-related endpoints were most common (38%).27 Between 2011 and 2015, 30 of the 182 (17%) new drugs approved by the FDA received PRO labeling. Again, many of these trials had primary PRO endpoints (77%).26 Between 2010 and 2014, 40 drugs were approved by the FDA Office of Hematology and Oncology products, of which three (8%) were granted PRO labeling.25 A further 13 oncology trials included PROs on the Drug Approval Package (“a summary of clinical study reports and related documents written by the FDA staff after data from pivotal studies submitted by the study sponsors has been reviewed”) (Gnanasakthy et al,25 p. 2), but did not receive PRO labeling due to various reasons, including inappropriate choice of PRO measure, high rates of missing PRO data, and poor reporting of PROs.25
Strategies for future optimization
In this section, we summarize the current challenges with PRO use in trials and present solutions and guidance to address these issues.
Protocol development
Issues with current practice
There is good evidence that trial protocols are often incomplete with regard to PRO endpoints. A review of 75 trial protocols (all clinical areas) with PRO endpoints submitted to the United Kingdom (UK) National Institute for Health Research (NIHR) Health Technology Assessment program in 2012–2013 determined that only 8% of the 75 trials studied described a rationale for PRO assessment, 60% addressed PRO-specific quality assurance issues, and only 8% offered PRO data collection instructions.28 Overall, the protocols addressed a mean of 33% of recommended PRO items.28
A second review examined the PRO content of 26 protocols of Phase III international, ovarian cancer trials published between 2000 and 2016 against a list of PRO-specific items that would eventually inform the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT)-PRO Extension.29,30 Of the 26 trials, one had a co-primary PRO endpoint, and 25 had secondary PRO endpoints. This review demonstrated that overall coverage of recommended PRO items ranged from 8% to 66% (mean coverage 28%). More than half of the recommended and applicable items were addressed by only two protocols. Basic aspects of PRO research design, including the primary/secondary status of the PRO endpoints and assessment schedules, were addressed in the majority of ovarian cancer trial protocols: as 92% and 96%, respectively. However, key guidance about PRO administration and quality assurance was often incomplete or omitted. For example, only 23% of these trial protocols specified time windows for each scheduled PRO assessment.30 Assessment windows are particularly important in trials of chemotherapy because of its fluctuating and transient side effects. If the PRO assessment seeks to capture the acute impact of chemotherapy on HRQOL, the time windows specified in the protocol should capture the days where the effects of chemotherapy toxicity would be felt. Without time windows, trial staff may not realize the time-sensitive nature of the scheduled assessment, which may lead to uninformative PRO data or biased interpretation if the time window is missed. Further trial staff who do not administer the PRO measure on the targeted date may not realize that they could validly continue to pursue that PRO assessment for a few more days, leading to that PRO assessment being missed.
In addition, only 31% of protocols specified a PRO-specific objective and only 19% included a PRO hypothesis.30 This is poor practice and makes it difficult to assess whether the PRO study was designed around a clinically motivated and relevant research question, and whether the chosen PRO measures and assessment time points were appropriate and would provide meaningful data for subsequent interpretation of the trial findings alongside other clinical endpoints.
These examples illustrate the importance of providing clear and comprehensive PRO study content in trial protocols, as this ensures that high-quality data are collected using consistent data collection methods to inform any clinical recommendations resulting from the trial.
A third study is in progress which will review the PRO content of cancer trials (all cancers) submitted to the UK NIHR Portfolio;31 however, at the time of this review, the results were yet to be published.
PRO-specific protocol guidance
The SPIRIT statement was published in 201332,33 and provided an evidence-based list of items recommended for inclusion in trial protocols. Until recently, no such consensus-based guidance for PROs existed. In 2018 the SPIRIT-PRO guidance was released.29 SPIRIT-PRO is the product of years of methodological research. It was developed based on an international, expert consensus process that aimed to identify the essential items to include in PRO sections of clinical trial protocols. It should be used in conjunction with the SPIRIT 2013 statement32,33 when developing a research protocol for a trial with a primary or important secondary PRO endpoint. SPIRIT-PRO contains 16 items, 11 of which are new, PRO-specific “extensions” to the core SPIRIT 2013 checklist, and five PRO-specific “elaborations” on existing SPIRIT 2013 items. The SPIRIT-PRO Extension items address the PRO study rationale, objectives, eligibility criteria, PRO domains used to evaluate the intervention, assessment time points, selection of PRO measures, measurement properties of these PRO measures, the PRO data collection plan, translation of PRO measures to other languages, proxy completion, strategies to minimize missing PRO data, and whether PRO data will be monitored during the active trial phase to inform the clinical care of participants.29 The full checklist is available in the SPIRIT-PRO publication29 and on the EQUATOR (Enhancing the QUAlity and Transparency Of health Research) Network website.34
Appropriate choice of PROs and PRO measures
Issues with current practice
The suitability of PRO measures to address pre-specified hypotheses was evaluated in 66 head and neck cancer and thyroid (HNT) cancer clinical trials published between 2004 and 2015 (22 with primary PRO endpoints, 44 with secondary PRO endpoints).35 This study found that only eight of the 66 studies (12%) reported a PRO hypothesis, and eight of these chose an appropriate PRO measure, which assessed and produced scores for the constructs specified in their hypotheses. While the sample size was small (n=8), the result suggested that explicitly specifying a PRO hypothesis a priori engaged investigators to carefully consider the constructs of particular relevance to their clinical population and to select appropriate PRO measures. PRO measures that are not suited to the specific research question cannot yield the required information, resulting in a waste of resources and time in collecting PRO data. It is concerning that 58 (88%) of the HNT randomized controlled trials (RCTs) failed to report any PRO hypotheses, precluding assessment of the suitability of their chosen PRO measures.35
Furthermore, five (8%) of the 66 HNT RCT publications did not name the PRO measure that had been used. Many more RCTs modified validated PRO measures without specifying the nature of the modifications.35 Modifications may have involved rewording, removing or adding items, or altering scoring procedures and may have compromised the psychometric properties of the PRO measures. None of these RCTs assessed the impact of these modifications on the psychometric integrity and performance of the measure. A follow-on issue is that the publication of RCTs using modified questionnaires may set a precedent for future RCTs to use the modified version rather than the validated version, thereby potentially perpetuating the problem of use of poor quality PRO measures.
As noted earlier, the FDA has declined to provide PRO labeling on the basis that clinical trials have not selected an appropriate PRO measure or that the measure had not been appropriately developed and validated.25 This suggests that the problem occurs even among well-funded and well-resourced trials presented to the FDA.
Guidance available for the selection of PRO measures
Clinicians, patients, researchers, and other relevant stakeholders should be involved in discussions about important, clinically relevant PROs and, corresponding appropriate and valid PRO measures. Kluetz et al state that “The goal [of PRO measure selection] should be to achieve a comprehensive evaluation of the patient experience most affected by the therapy, while maximizing the relevance of individual questions and minimizing overall burden and duplication” (p. 1557).36
Snyder et al describe how to develop a measurement strategy for prospective labeling claims.37 This involves identifying relevant PRO domains, developing a conceptual framework around these domains, identifying approaches to measure these domains, and designing a measurement strategy based on this information. Snyder acknowledges that existing scales may not fit the purpose of some studies and advocates that modifications to PRO measures should be subject to reliability and validity tests prior to implementation.37
Luckett and King describe six guiding principles toward selecting a PRO measure in cancer clinical research; however, the principles apply more broadly. These principles state that researchers should
- consider PRO measures early in the study design,
- select a primary PRO that is proximal to the disease or treatment (ie, symptoms or direct treatment side effects as opposed to down-stream impact on HRQOL),
- ensure that the PRO items (individual questions) are appropriate to the study and consider how the items combine into summary scales,
- appraise evidence regarding the reliability and validity of the PRO measure,
- consider practicalities such as respondent burden, mode of administration, and the need for validated language translations, and
- take a minimalist approach to ad hoc items, that is, avoid adding to or modifying existing PRO measures.38
As these guidelines demonstrate, trial investigators are increasingly being encouraged to measure proximal PROs, such as symptoms, in preference to measuring more distal or multidimensional constructs such as HRQOL, as primary PRO endpoints. This is because more distal domains are more likely to be influenced by factors beyond the trial interventions, such as social context and other life events. The FDA advises against HRQOL alone as an outcome in the era of novel therapies and recommends a focused and flexible approach to PRO measure selection. The PROMIS suite and PRO-CTCAE (Patient Reported Outcome Common Terminology Criteria for Adverse Events) have been identified as being well-suited to this measurement approach.36
PRO administration
Trial staff experiences
A UK study about trial staff members’ experiences with PRO studies, across a range of health conditions, found that the trial team members felt they lacked guidance and training for PRO studies. A problematic issue relating to concerning PRO data, for example very high anxiety scores or participants’ unsolicited comments about their PRO scores on questionnaire sheets, was highlighted. Trial staff noted that trial protocols and training rarely addressed how to respond in these situations.39
An Australian study of 20 cancer clinical trial coordinators revealed that PRO administration procedures were often unclear regarding participants who were unable to complete questionnaires in English, handling participants’ family members who attempted to complete PRO measures, whether to approach participants who appeared unwell or distressed, how to handle concerning PRO responses, and being flexible to participants’ needs while also adhering to protocol procedures.40 Coordinators were uncertain how to respond to these challenges, particularly if they perceived a discord between their duty of care and their trial coordinator role, or if they had received conflicting instructions previously. For example, whether to act on a concerning PRO score if instructed not to check completed questionnaires.40
Poor PRO administration practices were common. Trial participants were often not fully informed about the nature of their research participation, for example, if PROs were not addressed at the trial consent stage, or if the purpose of PRO assessments was not discussed at all. The timing and mode of PRO administration often departed from the protocol. Some sites failed to train back-up personnel for when primary trial coordinators were absent from work, leading to missing PRO data during the primary coordinator’s absence.40
Furthermore, PRO training was a key concern. Only two of the 20 coordinators interviewed had received PRO-specific training, and two others received no PRO training whatsoever.40 PRO training was more frequently provided by colleagues or briefly addressed at trial start-up meetings. As a result, trial coordinators often received inconsistent guidance regarding PRO administration, leading to confusion and use of inconsistent methods. Despite this, 55% of trial coordinators felt they did not need further PRO administration training40 in contrast to the UK study,39 which may present a barrier to their future attendance of PRO training courses. The general lack of PRO training reported by many of the coordinators interviewed raises concerns about the extent to which trial sponsors are complying with the good clinical practice guideline that trial staff must be adequately trained.41
Modes of administration
There is an extensive literature regarding different modes of questionnaire administration, comparing both the methods (such as paper-based, electronic [computer, smartphone, or tablet], telephone interview, etc.) and setting (home and clinic) of administration. In our experience, paper-based and electronic modes of administration are most commonly used methods. Pros and cons of each method are presented in Table 1.
 | Table 1 Pros and cons of paper and electronic modes of administration Abbreviation: PRO, patient-reported outcomes. |
A recent meta-analysis found no difference in patient responses (bias) between electronic and paper-based methods when the patient self-completed questionnaires. The authors concluded that self-completed questionnaires originally developed for paper-based administration could safely be administered by electronic modes, or both modes could be used in any one study. However, when self-completed questionnaire data (paper or electronic administration) was compared to administered modes (ie, questionnaire administered by a researcher over the phone or face-to-face), there was a slight difference in responses according to the administration setting (at home vs in clinic). Therefore, the authors recommended that if using self-complete and administered modes, the setting of administration should be kept consistent.42 Future meta-analyses are needed to determine whether mode of administration impacts questionnaire return rate or participant retention and whether the questionnaire return method (ie, returning to a coordinating center rather than to the recruitment site) impacts responses or return rate.
Burden associated with PRO assessment
It is important that investigators consider the extent to which PRO assessment may become burdensome to participants, trial staff, and statisticians. Unnecessary burden must be minimized. PRO assessment may become burdensome in a number of ways, as detailed in Table 2.
 | Table 2 How PRO assessment can become unnecessarily burdensome for trial participants and staff Abbreviation: PRO, patient-reported outcomes. |
PRO administration guidance
Trial staff should receive clear PRO administration instructions in order for PROs to be administered consistently. Impressing the critical importance of PROs to the interpretation of trial results to trial coordinators and clinicians is required before the trial begins. Consensus guidelines for PRO administration are lacking, although many good examples of guidance developed by individual groups exist. For example, the European Organization for the Research and Treatment of Cancer (EORTC) guidelines,43 the South West Oncology Group (SWOG) Training Module, presented by Dr Lisa Hansen (available for SWOG trials), and the QOL Office Checklist of instructions for the administration of PRO measures.44 Future research should develop consensus-based PRO administration guidance and test the efficacy of accompanying training interventions.
Experienced clinical trial groups such as SWOG45 and the Epidemiology of Diabetes Interventions and Complications group46 have seen benefits including increased PRO compliance, improved work satisfaction, and participant retention, as a result of involving trial coordinators or researcher nurses more actively in the research process and investing more resources in their professional development. These groups also engage experienced coordinators in training new coordinators. A more formal approach to peer-training may be a feasible and acceptable approach to future trial coordinator training efforts for PRO administration.
Missing PRO data
Issues with current practice
It is important to minimize the rates of missing data in clinical trials. High rates of missing PRO data can reduce study power,47 inflate risk of type 2 error,48 bias interpretation,47 and potentially undermine randomization.49 Fielding et al determined that 18% of RCTs published in the four highest-impact medical journals between 2005 and 2006 had missing PRO data rates exceeding 20%, and a further 20% of trials failed to clearly report rates of missing PRO data.50 In many cases, the methods used to address missing PRO data in these trials were unclear or unsatisfactory.50 In an update of this review (2013–2014), ~8% of RCTs published in high-impact journals used complete case analysis for PROs51 despite the inappropriateness of this approach in many contexts.52,53 Furthermore, the rate of missing PRO data was unclear in 35% of studies, 10% of trials had >20% missing PRO data, and only 13% indicated the missing data mechanism51 (system for classifying missing data according to their cause).48,54,55 It is important to deduce and report the missing data mechanism because this allows researchers to handle missing data appropriately, to select appropriate analysis methods, and to interpret results with caution. Often the missing data mechanism has a stronger impact on the results than does the proportion of missing data.52,56 For example, if analysis methods assume that missing data are not related to the patients’ illness (eg, as with “complete case” analysis methods), when in fact the illness may have led to the data not being available, the results may falsely indicate that the outcome is favourable (resulting in bias). It is impossible to determine the true missing data mechanism, but information about the clinical status of the patient or the reasons for missing data, particularly whether these reasons are related to the patient’s worsening illness or not, can assist researchers in deducing the underlying mechanism.52
A review of the rates of missing PRO data in 36 ovarian cancer trials found similar problems to those noted above.57 Many of these trials included patients with advanced/recurrent ovarian cancer, which may be associated with early progression and high mortality. Hence, a degree of missing data or trial drop out is expected to be related to worsening health status of the trial participants. It is concerning that the reasons for missing PRO data were indiscernible for 72% of the publications studied. In the 10 RCTs (28%) that transparently reported PRO compliance rates (ie, those that differentiated between avoidable and informative sources of missing data), the percentages of avoidable missing PRO data were at times very high. The worst compliance rate was 59% (ie, 41% of trial participants had avoidable missing data (not illness-related). This suggested that trials did not sufficiently implement strategies to minimize avoidable missing PRO data. Analysis of the corresponding trial protocols for 26 of these 36 RCTs (72%) determined that trials that had less complete PRO protocol sections (ie, addressed the lowest number of recommended PRO protocol checklist items) had the highest rates of avoidable missing PRO data. This suggests a plausible link between the quality of PRO content of the protocol and the quality of PRO data collected, highlighting the importance of comprehensively addressing PROs in the trial protocol.57
Palmer et al developed a classification framework for factors associated with missing PRO data. This framework includes five categories: instrument (questionnaire content, length, language, etc.), participant (disease stage, physical impairment, refusal, etc.), center (infrastructure, location, etc.), staff (interpersonal skills, knowledge, motivation, etc.), and study-related factors (frequency and timing of assessment, mode of administration, type of treatment, etc.).58 Due to a majority of studies failing to report the reasons for missing PRO data, it is impossible to reliably estimate the rates of missing data attributed to these different categories. Research conducted in the late 1990s suggested that logistic and administrative errors caused a larger problem with respect to missing PRO data than patient-related and design factors.59
Guidance available on missing PRO data
Although methods exist to address problems with missing data statistically,60,61 it is better practice to prevent avoidable types of missing PRO data, such as staff failing to provide the questionnaires to patients, whenever possible, and to also clearly report the rates, reasons, and handling of missing PRO data in publications.55
A large systematic review of strategies for minimizing the problem of missing PRO data revealed how the whole trial team can actively reduce the problems of missing PRO data across all research stages.55 The strategies during study design include: specifying a clinically informative and feasible PRO assessment schedule with defined acceptable time windows and stopping rules, collecting auxiliary data (clinically relevant variables that are likely to be correlated to PRO data and recorded at the same time points) to facilitate unbiased interpretation of PRO data in the presence of missing data and inform statistical imputation of missing PRO data, specifying clear eligibility criteria for the PRO study including literacy and language requirements and the need for a valid baseline PRO assessment, ensuring that the PRO study is feasible and adequately resourced, ensuring the mode of questionnaire administration is feasible and acceptable, minimizing participant burden, selecting a clinically relevant and validated PRO measure, incorporating PROs into all relevant trial documents, involving patient partners and trial staff in the design of PRO studies, ensuring that the trial team is committed to the PRO study, developing quality assurance procedures, ensuring that the PRO sample size is representative and sufficient for planned analyses, and involving a multidisciplinary team into PRO study design.55
Implementation strategies include: developing standardized PRO administration procedures, educating and engaging participants, maintaining patient records, appointing a site coordinator responsible for PRO assessment at each recruiting site, ensuring that the trial team remains engaged and committed to the PRO study, and training trial staff.55
Reporting strategies for addressing the problem of missing PRO data include: clearly reporting study and PRO analysis methods, describing the sample -including baseline PRO scores, defining and providing PRO compliance rates, comparing participants with and without missing PRO data, providing reasons for missing data, and discussing the impact of missing data on generalizability.55
Analysis
Issues with analysis of PRO studies
A major challenge for the analysis of PRO data is that many different analytic approaches are available, which has caused confusion for researchers, statisticians, clinicians, and others about the most appropriate methods and how results should be interpreted. The methodology to be used should be determined a priori and included in the protocol with more detailed elaboration of analysis methods provided in the statistical analysis plan.
PRO analysis guidance
The ongoing “Setting International Standards in Analyzing Patient-Reported Outcomes and Quality of Life Endpoints Data” (SISAQOL)62 initiative is developing recommendations for standardized analysis and interpretation of PROs in cancer clinical trials, based on international and multidisciplinary consensus, which should become available within the next few years. This initiative acknowledges the challenges in using analysis methods that are suited to the PRO research question, statistically correct, handle missing data appropriately, transparent to clinicians, and comparable with results of other trials in systematic reviews.62
Reporting
Issues with reporting of PRO studies
Readers must be able to appraise the design, analysis, and interpretation of a PRO study from its publication; therefore, transparent reporting is essential. In recent years, numerous studies have scrutinized the reporting of PROs in clinical trials, using various checklists of recommended reporting items,35,57,63–69 as summarized in Table 3. Table 3 shows variation across various RCT cohorts studied as to how comprehensively studies were reported, (overall and at the item level). Items that were repeatedly poorly reported included: PRO hypotheses with PRO domains, approaches to handling missing PRO data, and discussions of limitations of the PRO study. In contrast, the reviews found that evidence of the validity of the PRO measures was typically provided.
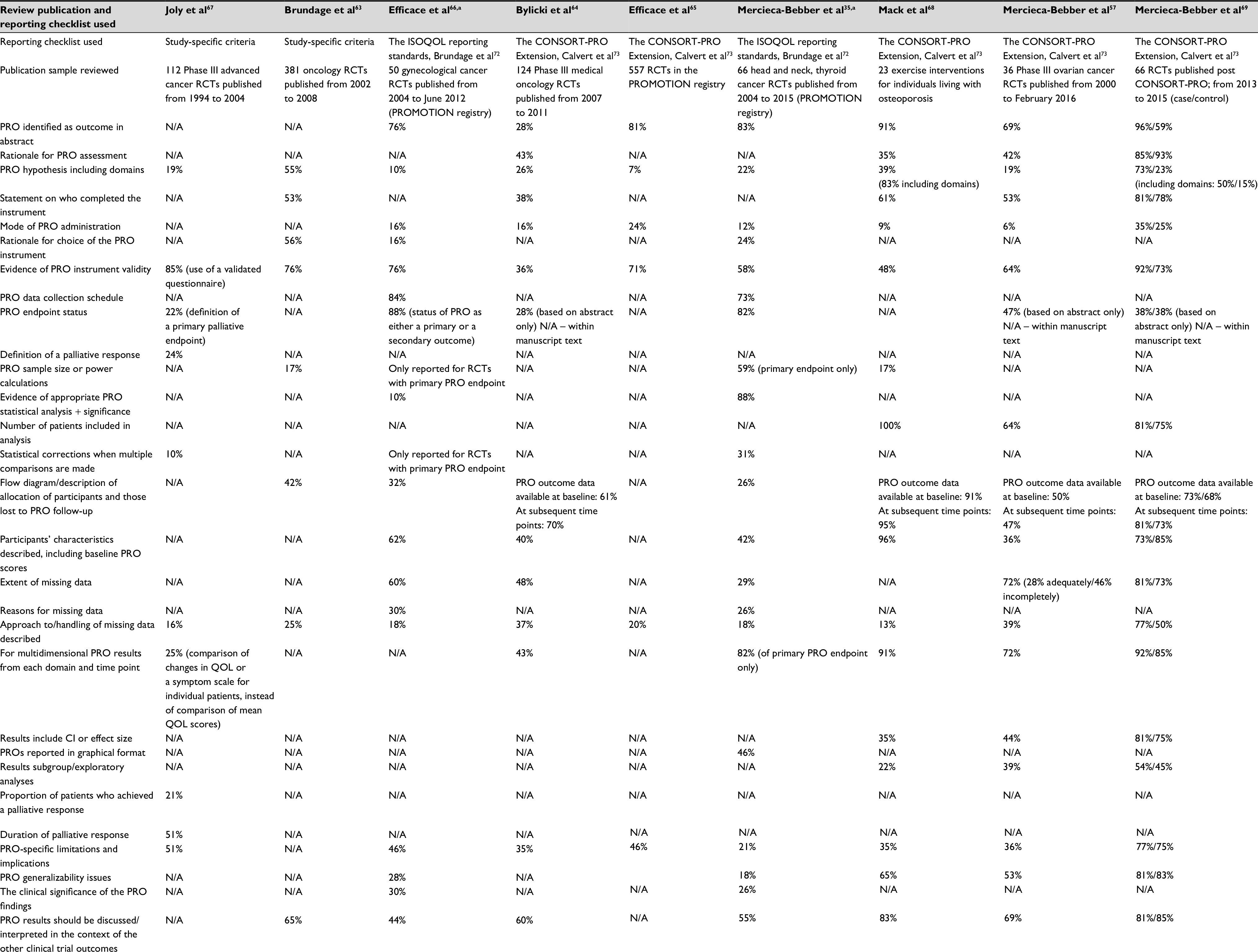 | Table 3 Completeness of PRO reporting in RCTs – results from nine evaluations Notes: Percentages reported indicate the percentage of trials from each review that addressed that criterion; N/A, not assessed; aReporting criteria for trials with a primary PRO endpoint only are not reported here. Abbreviations: PRO, patient-reported outcome; QOL, quality of life; RCT, randomized controlled trial; ISOQOL, International Society for Quality of Life Research; CONSORT-PRO Extension, Consolidated Standards of Reporting Trials – Patient-reported outcome Extension; PROMOTION, patient reported outcomes over time in oncology. |
Failure to report PRO endpoints is also an area of concern. Schandelmaier et al found that only 20% of 173 oncology RCTs with a PRO endpoint listed in the protocol subsequently reported PRO findings.70 The authors suggested that trial discontinuation was significantly associated with both failure to report any PRO data and failure to report PROs as specified in the protocol.70 Schandelmaier et al’s study was based on RCTs approved by six ethics committees from three countries, and results may therefore not represent the rate of PRO reporting internationally.70 However, the issue of non-reporting of PROs is emerging and requires attention, as it is unethical to waste the funding, resources, and patients’ efforts expended in PRO data collection.71
PRO reporting guidance
The International Society for Quality of Life Research (ISOQOL) published comprehensive reporting standards in 2013,72 consisting of 29 items, 11 of which are only relevant to RCTs with primary PRO endpoints and 18 are relevant to trials with either primary or secondary PRO endpoints. This work informed the development of a CONSORT-PRO Extension (Consolidated Standards of Reporting Trials), which can be considered the minimum reporting items for PRO endpoints (primary or key secondary) in RCTs.73 CONSORT-PRO consists of 14 items, five of which were newly developed PRO-specific reporting recommendations (eg, the PRO should be identified in the abstract as primary or secondary outcome, provide evidence of validity of PRO measures, and report statistical approaches for dealing with missing PRO data). The remaining nine CONSORT-PRO items are elaborated from the CONSORT-201074 statement to specifically address PROs.
Table 3 shows that RCTs published after the release of CONSORT-PRO73 generally had higher rates of reporting on most criteria, particularly those RCTs which cited CONSORT-PRO.69 This suggests a preliminary benefit of CONSORT-PRO in improving the standard of reporting; however, given that some RCTs that used CONSORT-PRO had incomplete reporting, additional knowledge transfer efforts are needed.69 It is encouraging to note that a number of high-ranking journals now request evidence of compliance with the CONSORT-PRO checklist for publications reporting PRO endpoints.
Guidance across PRO research stages
We encourage readers to access these key resources, which provide excellent guidance across research stages:
- Fairclough DL. Design and Analysis of Quality of Life Studies in Clinical Trials. 2nd ed. boca raton, FL: Taylor and Francis Group; 2010.
- Fayers P, Machin D. Quality of Life: The Assessment, Analysis and Interpretation of Patient-Reported Outcomes. 2nd ed. Chichester, UK: John Wiley & Sons; 2007.
- The Center for Patient Reported Outcomes Research (CPROR) PROlearn resources: Available from: www.birmingham.ac.uk/prolearn.
Conclusion
Ongoing methodological research is important for determining what constitutes best practice in PRO research and adhering to those standards. High-quality clinical trials inform clinical practice and policy. Therefore, researchers and clinical trial investigators must implement evidence-based strategies to promote high-quality PRO data collection, analysis, and reporting of PRO evidence. This process must begin with improved, PRO-specific education for trial team members about the core principles and methods for PRO research, with particular focus on strategies to minimize the frequency and effects of missing data. PRO aspects of trial protocols should be developed in accordance with the SPIRIT-PRO guidance.29 Multidisciplinary teams, including patient partners, must be involved in the design of PRO aspects of clinical trials to ensure that scientific, logistic, and resource considerations are addressed with high-quality, complete data collection in mind. Clinically informative, valid, and reliable PRO measures and time points for assessment should be carefully chosen to address the PRO hypotheses. A plan to facilitate handling of unavoidable and/or informative missing data should be employed, particularly in trials involving participants with advanced disease who are unlikely to complete all scheduled follow-up assessments. Trials should incorporate ongoing quality assurance measures. PRO findings should be published according to CONSORT-PRO and ISOQOL PRO reporting guidelines,72,75 in a timely manner. If the PRO results are to be published separately, the primary trial publication should reference, or at least acknowledge the forthcoming PRO publication. Each trial publication should be clearly labeled with the trial name/number for ease of identification and to facilitate joint interpretation of PRO and other trial outcomes. Funders, ethics committees, journals, and trial sponsors should proactively ensure that PRO studies are designed, conducted, analyzed, and reported to these standards; this can only be achieved with their active engagement in such efforts. If all these stakeholders play their role, the result will be high-quality PRO evidence, integrated with other trial outcomes, to better inform clinical decision-making and health care policy.
Acknowledgments
RMB is funded by a NHMRC Early Career Fellowship. MK is funded by the Australian Government courtesy of Cancer Australia. MC is partly funded by the NIHR Birmingham Biomedical Research Centre and the NIHR Surgical Reconstruction and Microbiology Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham. The views expressed are those of the author and not necessarily those of the NHMRC, Cancer Australia, NHS, the NIHR or the Department of Health.
Disclosure
Unrelated to the current work:
- Professor Calvert has received personal fees from Astellas Pharmaand Ferring and chairs the ISOQOL Best Practice for PROs in Trials Taskforce and Standards and Best Practice Committee.
- Professor Stockler’s institution has received research funding from Astellas Pharma, Celgene, Bayer, Bionomics, Medivation, Sanofi,Pfizer, AstraZeneca, Bristol-Myers Squibb, Roche, Amgen, Merck Sharp& Dohme (MSD), and Tilray and has received travel support from Medivation/Pfizer.
- Professor Friedlander has received honoraria for advisory boards for Astra Zeneca and MSD.
The authors report no other conflicts of interest in this work.
References
Guidance for Industry: patient-reported outcome measures: use in medical product development to support labelling claims. Food and Drug Administration; 2009. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf. Accessed January 10, 2015. | ||
Revicki DA, Osoba D, Fairclough D, et al. Recommendations on health-related quality of life research to support labeling and promotional claims in the United States. Qual Life Res. 2000;9(8):887–900. | ||
Au HJ, Ringash J, Brundage M, et al. Added value of health-related quality of life measurement in cancer clinical trials: the experience of the NCIC CTG. Expert Rev Pharmacoecon Outcomes Res. 2010;10(2):119–128. | ||
Organisation for Economic Co-operation and Development (OECD). Strengthening the international comparison of health system performance through patient-reported indicators. Recommendations to OECD Ministers of Health from the High Level Reflection Group on the Future of Health Statistics; 2017. Available from: https://www.oecd.org/els/health-systems/Recommendations-from-high-level-reflection-group-on-the-future-of-health-statistics.pdf. Accessed May 07, 2017. | ||
Scoggins JF, Patrick DL. The use of patient-reported outcomes instruments in registered clinical trials: evidence from ClinicalTrials.gov. Contemp Clin Trials. 2009;30(4):289–292. | ||
Vodicka E, Kim K, Devine EB, et al. Inclusion of patient-reported outcome measures in registered clinical trials: evidence from ClinicalTrials.gov (2007-2013). Contemp Clin Trials. 2015;43:1–9. | ||
Mercieca-Bebber R, Williams D, Tait MA, et al. Trials with patient-reported outcomes registered on the Australian New Zealand Clinical Trials Registry (ANZCTR). Qual Life Res. 2018:27(10);2581–2591. | ||
Food and Drug Administration. Value and Use of Patient-Reported Outcomes (PROs) in Assessing Effects of Medical Devices CDRH Strategic Priorities. CDRH Strategic Priorities 2016–2017; 2017. Available from: https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHVisionandMission/UCM588576.pdf. Accessed February 22, 2018. | ||
Kluetz PG, O’Connor DJ, Soltys K. Incorporating the patient experience into regulatory decision making in the USA, Europe, and Canada. Lancet Oncol. 2018;19(5):e267–e274. | ||
Schnipper LE, Davidson NE, Wollins DS, et al. Updating the American Society of Clinical Oncology Value Framework: revisions and reflections in response to comments received. J Clin Oncol. 2016;34(24):2925–2934. | ||
Cherny NI, Sullivan R, Dafni U, et al. A standardised, generic, validated approach to stratify the magnitude of clinical benefit that can be anticipated from anti-cancer therapies: the European Society for Medical Oncology Magnitude of Clinical Benefit Scale (ESMO-MCBS). Ann Oncol. 2015;26(8):1547–1573. | ||
Cancer Australia. Support for Cancer Clinical Trials; 2013. Available from: http://canceraustralia.gov.au/research-data/support-clinical-trials. Accessed November 22, 2017. | ||
European Medicines Agency. Appendix 2 to the guideline on the evaluation of anticancer medicinal products in man: the use of patient-reported outcome (PRO) measures in oncology studies. London, UK; 2016. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Other/2016/04/WC500205159.pdf. Accessed December 05, 2017. | ||
du Bois A, Quinn M, Thigpen T, et al. consensus statements on the management of ovarian cancer: final document of the 3rd International Gynecologic Cancer Intergroup Ovarian Cancer Consensus Conference (GCIG OCCC 2004). Ann Oncol. 2004;16:viii7–viii12. | ||
Food and Drug Administration. Ovarian Cancer Endpoints Workshop April 26, 2006, Meeting Summary; 2006. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/CancerDrugs/ucm094598.pdf accessed 09 August 2016 https://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ucm120657.pdf .Accessed 14 August 2017.v | ||
Friedlander M, Trimble E, Tinker A, et al. Clinical trials in recurrent ovarian cancer. Int J Gynecol Cancer. 2011;21(4):771–775. | ||
Stuart GC, Kitchener H, Bacon M, et al. 2010 Gynecologic Cancer InterGroup (GCIG) consensus statement on clinical trials in ovarian cancer: report from the Fourth Ovarian Cancer Consensus Conference. Int J Gynecol Cancer. 2011;21(4):750–755. | ||
Centres for Medicare and Medicaid Services. MEDCAC Meeting 3/22/2017 – Health Outcomes in Heart Failure Treatment Technology Studies; 2017. Available from: https://www.cms.gov/medicare-coverage-database/details/medcac-meeting-details.aspx?MEDCACId=73. Accessed October 10, 1017. | ||
Schorr A. Patient-reported outcomes and where patient advocacy groups can help. Change Together; 2017. Available from: https://www.changetogether.com/general/patient-reported-outcomes-patient-advocacy-groups-can-help. August 08, 2017. | ||
Wilson R. Patient led PROMs must take centre stage in cancer research. Res Involv Engagem. 2018;4:7. | ||
Tannock IF, Osoba D, Stockler MR, et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J Clin Oncol. 1996;14(6):1756–1764. | ||
Osoba D, Tannock IF, Ernst DS, Neville AJ. Health-related quality of life in men with metastatic prostate cancer treated with prednisone alone or mitoxantrone and prednisone. J Clin Oncol. 1999;17(6):1654–1654. | ||
Mesa RA, Gotlib J, Gupta V, et al. Effect of ruxolitinib therapy on myelofibrosis-related symptoms and other patient-reported outcomes in COMFORT-I: a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2013;31(10):1285–1292. | ||
Food and Drug Administration: JAKAFI® (ruxolitinib) tablets for oral use; 2017. | ||
Gnanasakthy A, Demuro C, Clark M, et al. Patient-reported outcomes labeling for products approved by the Office of Hematology and Oncology Products of the US Food and Drug Administration (2010-2014). J Clin Oncol. 2016;34(16):1928–1934. | ||
Gnanasakthy A, Mordin M, Evans E, Doward L, Demuro C. A review of patient-reported outcome labeling in the United States (2011-2015). Value Health. 2017;20(3):420–429. | ||
Gnanasakthy A, Mordin M, Clark M, et al. A review of patient-reported outcome labels in the United States: 2006 to 2010. Value Health. 2012;15(3):437–442. | ||
Kyte D, Duffy H, Fletcher B, et al. Systematic evaluation of the patient-reported outcome (PRO) content of clinical trial protocols. PLoS One. 2014;9(10):e110229. | ||
Calvert M, Kyte D, Mercieca-Bebber R, et al. Guidelines for inclusion of patient-reported outcomes in clinical trial protocols: The SPIRIT-PRO Extension. JAMA. 2018;319(5):483–494. | ||
Mercieca-Bebber R, Friedlander M, Kok PS, et al. The patient-reported outcome content of international ovarian cancer randomised controlled trial protocols. Qual Life Res. 2016;25(10):2457–2465. | ||
Ahmed K, Kyte D, Keeley T, et al. Systematic evaluation of patient-reported outcome (PRO) protocol content and reporting in UK cancer clinical trials: the EPiC study protocol. BMJ Open. 2016;6(9):e012863. | ||
Chan AW, Tetzlaff JM, Altman DG, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):2002013–2207. | ||
Chan AW, Tetzlaff JM, Gøtzsche PC, et al. SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. BMJ. 2013;346:2013. | ||
EQUATOR Network. Enhancing the QUAlity and Transparency Of health Research. Available from: https://www.equator-network.org. Accessed July 12, 2017. | ||
Mercieca-Bebber RL, Perreca A, King M, et al. Patient-reported outcomes in head and neck and thyroid cancer randomised controlled trials: a systematic review of completeness of reporting and impact on interpretation. Eur J Cancer. 2016;56:144–161. | ||
Kluetz PG, Slagle A, Papadopoulos EJ, et al. Focusing on core patient-reported outcomes in cancer clinical trials: symptomatic adverse events, physical function, and disease-related symptoms. Clin Cancer Res. 2016;22(7):1553–1558. | ||
Snyder CF, Watson ME, Jackson JD, et al. Patient-reported outcome instrument selection: designing a measurement strategy. Value Health. 2007;10(Suppl 2):S76–S85. | ||
Luckett T, King MT. Choosing patient-reported outcome measures for cancer clinical research – practical principles and an algorithm to assist non-specialist researchers. Eur J Cancer. 2010;46(18):3149–3157. | ||
Kyte D, Ives J, Draper H, Keeley T, Calvert M. Inconsistencies in quality of life data collection in clinical trials: a potential source of bias? Interviews with research nurses and trialists. PLoS One. 2013;8(10):e76625. | ||
Mercieca-Bebber R, Calvert M, Kyte D, Stockler M, King MT. The administration of patient-reported outcome questionnaires in cancer trials: Interviews with trial coordinators regarding their roles, experiences, challenges and training. Contemp Clin Trials Commun. 2018;9:23–32. | ||
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH): Integrated Addendum to ICH E6 (R1): Guideline for Good Clinical Practice E6 (R2), (ed 4); 2016. Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Step_4_2016_1109.pdf. Accessed February 02, 2017. | ||
Rutherford C, Costa D, Mercieca-Bebber R, et al. Mode of administration does not cause bias in patient-reported outcome results: a meta-analysis. Qual Life Res. 2016;25(3):559–574. | ||
Young T, de Haes D, Curran D, et al. Guidelines for assessing Quality of Life in EORTC clinical trials. Brussels: EORTC Quality of Life Group; 2002. | ||
QOL Office. Checklist of instructions for the administration of Patient Reported Outcome Measures. 2015. Available from: http://www.pocog.org.au/docview.aspx?id=355. Accessed February 02, 2017. | ||
Hansen LK, Moinpour CM, Ermete RB. Lessons learned: enhancing nurse contributions to cancer clinical trials in SWOG. Semin Oncol Nurs. 2014;30:26–31. | ||
Larkin ME, Lorenzi GM, Bayless M, et al. Evolution of the study coordinator role: the 28-year experience in Diabetes control and complications trial/epidemiology of Diabetes interventions and complications (DCCT/EDIC). Clin Trials. 2012;9(4):418–425. | ||
Fairclough DL, Peterson HF, Cella D, Bonomi P. Comparison of several model-based methods for analysing incomplete quality of life data in cancer clinical trials. Stat Med. 1998;17(5–7):781–796. | ||
Fairclough DL, Peterson HF, Chang V. Why are missing quality of life data a problem in clinical trials of cancer therapy? Stat Med. 1998;17(5–7):667–677. | ||
Bell ML, Fiero M, Horton NJ, Hsu CH. Handling missing data in RCTs; a review of the top medical journals. BMC Med Res Methodol. 2014;14:118. | ||
Fielding S, Maclennan G, Cook JA, Ramsay CR. A review of RCTs in four medical journals to assess the use of imputation to overcome missing data in quality of life outcomes. Trials. 2008;9:51. | ||
Fielding S, Ogbuagu A, Sivasubramaniam S, Maclennan G, Ramsay CR. Reporting and dealing with missing quality of life data in RCTs: has the picture changed in the last decade? Qual Life Res. 2016;25(12):2977–2983. | ||
Bell ML, Fairclough DL. Practical and statistical issues in missing data for longitudinal patient-reported outcomes. Stat Methods Med Res. 2014;23(5):440–459. | ||
Bell ML, Olivier J, King MT. Scientific rigour in psycho-oncology trials: why and how to avoid common statistical errors. Psychooncology. 2013;22(3):499–505. | ||
Curran D, Bacchi M, Schmitz SF, Molenberghs G, Sylvester RJ. Identifying the types of missingness in quality of life data from clinical trials. Stat Med. 1998;17(5–7):739–756. | ||
Mercieca-Bebber R, Palmer MJ, Brundage M, et al. Design, implementation and reporting strategies to reduce the instance and impact of missing patient-reported outcome (PRO) data: a systematic review. BMJ Open. 2016;6(6):e010938. | ||
Mercieca-Bebber R, Palmer MJ, Brundage M, et al. Design, implementation and reporting strategies to reduce the instance and impact of missing patient-reported outcome (PRO) data: a systematic review. BMJ Open. 2016;6(6):e010938. | ||
Mercieca-Bebber R, Friedlander M, Calvert M, et al. A systematic evaluation of compliance and reporting of patient-reported outcome endpoints in ovarian cancer randomised controlled trials: implications for generalisability and clinical practice. J Patient Rep Outcomes. 2017;1:5. | ||
Palmer MJ, Mercieca-Bebber R, King M, et al. A systematic review and development of a classification framework for factors associated with missing patient-reported outcome data. Clin Trials. 2018;15(1):95–106. | ||
Bernhard J, Cella DF, Coates AS, et al. Missing quality of life data in cancer clinical trials: serious problems and challenges. Stat Med. 1998;17(5-7):517–532. | ||
Fielding S, Fayers P, Ramsay CR. Analysing randomised controlled trials with missing data: choice of approach affects conclusions. Contemp Clin Trials. 2012;33(3):461–469. | ||
Post WJ, Buijs C, Stolk RP, de Vries EG, Le Cessie S. The analysis of longitudinal quality of life measures with informative drop-out: a pattern mixture approach. Qual Life Res. 2010;19(1):137–148. | ||
Bottomley A, Pe M, Sloan J, et al. Analysing data from patient-reported outcome and quality of life endpoints for cancer clinical trials: a start in setting international standards. Lancet Oncol. 2016;17(11):e510–e514. | ||
Brundage M, Bass B, Davidson J, et al. Patterns of reporting health-related quality of life outcomes in randomized clinical trials: implications for clinicians and quality of life researchers. Qual Life Res. 2011;20(5):653–664. | ||
Bylicki O, Gan HK, Joly F, et al. Poor patient-reported outcomes reporting according to CONSORT guidelines in randomized clinical trials evaluating systemic cancer therapy. Ann Oncol. 2015;26(1):231–237. | ||
Efficace F, Fayers P, Pusic A, et al. Quality of patient-reported outcome reporting across cancer randomized controlled trials according to the CONSORT patient-reported outcome extension: a pooled analysis of 557 trials. Cancer. 2015;121(18):3335–3342. | ||
Efficace F, Jacobs M, Pusic A, et al. Patient-reported outcomes in randomised controlled trials of gynaecological cancers: investigating methodological quality and impact on clinical decision-making. Eur J Cancer. 2014;50(11):1925–1941. | ||
Joly F, Vardy J, Pintilie M, Tannock IF. Quality of life and/or symptom control in randomized clinical trials for patients with advanced cancer. Ann Oncol. 2007;18(12):1935–1942. | ||
Mack DE, Wilson PM, Santos E, Brooks K. Standards of reporting: the use of CONSORT PRO and CERT in individuals living with osteoporosis. Osteoporos Int. 2018;29(2):305–313. | ||
Mercieca-Bebber R, Rouette J, Calvert M, et al. Preliminary evidence on the uptake, use and benefits of the CONSORT-PRO extension. Qual Life Res. 2017;26(6):1427–1437. | ||
Schandelmaier S, Conen K, von Elm E, et al. Planning and reporting of quality-of-life outcomes in cancer trials. Ann Oncol. 2015;26(9):1966–1973. | ||
Glasziou P, Altman DG, Bossuyt P, et al. Reducing waste from incomplete or unusable reports of biomedical research. Lancet. 2014;383(9913):267–276. | ||
Brundage M, Blazeby J, Revicki D, et al. Patient-reported outcomes in randomized clinical trials: development of ISOQOL reporting standards. Qual Life Res. 2013;22(6):1161–1175. | ||
Calvert M, Blazeby J, Altman DG, et al. Reporting of patient-reported outcomes in randomized trials: the CONSORT PRO extension. JAMA. 2013;309(8):814–822. | ||
Schulz KF, Altman DG, Moher D; CONSORT Group. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340:c332. | ||
Calvert M, Brundage M, Jacobsen PB, Schünemann HJ, Efficace F. The CONSORT Patient-Reported Outcome (PRO) extension: implications for clinical trials and practice. Health Qual Life Outcomes. 2013; 11:184. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.