Back to Journals » Drug Design, Development and Therapy » Volume 20
The Impact of Polymers in Amorphous Solid Dispersion on the Bioavailability of Sulfonylureas and Meglitinides
Authors Aulifa DL ![]() , Wibisono TTDW, Ningsih JD, Christy GDAB, Pertiwi DS, Praja HM
, Wibisono TTDW, Ningsih JD, Christy GDAB, Pertiwi DS, Praja HM ![]() , Gazzali AM, Amaliah S
, Gazzali AM, Amaliah S ![]() , Budiman A
, Budiman A
Received 19 March 2026
Accepted for publication 2 June 2026
Published 8 June 2026 Volume 2026:20 610513
DOI https://doi.org/10.2147/DDDT.S610513
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leonidas D. Panos
Diah Lia Aulifa,1 Tribuana Tungga Dewi Wansha Wibisono,1 Jahraema Deagustia Ningsih,1 Gabriel Daendya Amalia Bintang Christy,1 Dwi Sekar Pertiwi,2 Hilman Maulana Praja,2 Amirah Mohd Gazzali,3 Salma Amaliah,2 Arif Budiman2
1Department of Pharmaceutical Analysis and Medicinal Chemistry, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, 45363, Indonesia; 2Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, 45363, Indonesia; 3Discipline of Pharmaceutical Technology, School of Pharmaceutical Sciences, Universiti Sains Malaysia, Penang, Malaysia
Correspondence: Arif Budiman, Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Universitas Padjadjaran, Sumedang, 45363, Indonesia, Email [email protected]
Abstract: Diabetes mellitus (DM) represents a major and growing global health challenge. Among antidiabetic medications, sulfonylureas and meglitinides remain clinically relevant insulin secretagogues for the management of type 2 DM (T2DM). However, many drugs in these classes belong to Biopharmaceutics Classification System (BCS) Class II, which are characterized by low aqueous solubility and high membrane permeability. These physicochemical constraints result in slow dissolution, variable oral absorption, suboptimal bioavailability and inadequate glycemic control. Converting drugs into their amorphous form can enhance solubility and dissolution; however, amorphous drugs are inherently unstable and prone to recrystallization, limiting their practical use. Amorphous solid dispersion (ASD), which incorporates poorly soluble antidiabetic drugs into a polymeric matrix, offers a promising strategy to overcome these limitations. The polymeric carrier stabilizes the drug in its high-energy amorphous state, improving solubility, dissolution, and ultimately, bioavailability. In ASD systems, drug molecules are dispersed within the polymer matrix at the molecular level, forming extremely fine dispersion domains that may be even smaller than conventional nanoparticle systems, thereby enhancing apparent solubility and dissolution. This review provides a comprehensive overview of ASD applications in antidiabetic therapy, discussing the principles of ASD, commonly used polymeric carriers, fabrication methods, and recent in vitro and in vivo findings. Studies consistently show that ASD formulations of antidiabetic drugs such as glimepiride, repaglinide, gliclazide, gliquidone, and glyburide enhance insulin secretion and contribute to more effective glycemic control, showing the potential of ASD in improving the therapeutic performance of poorly soluble antidiabetic agents. By enhancing solubility, stability, and bioavailability, ASD technology holds significant promise for developing more potent and effective antidiabetic therapies, ultimately supporting better patient care and addressing the global burden of diabetes mellitus. ASD enhances sulfonylureas/meglitinides dissolution, bioavailability, insulin release and glucose control.The diagram illustrates the process involving amorphous solid dispersion (ASD) of sulfonylureas/meglitinides. It begins with ASD, leading to an improved dissolution profile shown in a graph with dissolution percentage on the y-axis and time on the x-axis. The graph compares pure drug and ASD, indicating higher dissolution for ASD. Next, improved bioavailability is depicted with a graph showing Cmax and AUC (Area Under Curve) for pure drug and ASD, with ASD having higher values. This process results in increased insulin secretion and decreased blood glucose levels, as indicated by the final icons.
Keywords: diabetes mellitus, polymer, poorly water-soluble drugs, amorphous solid dispersion, bioavailability, insulin secretion
Introduction
Diabetes presents a rapidly escalating global health challenge, imposing a growing burden on individuals, families, and healthcare systems worldwide. Globally, 11.1% of the adult population—equating to 1 in 9 adults aged 20–79—is currently living with Type 2 Diabetes Mellitus (T2DM), in which 4 in 10 of these individuals are undiagnosed and are unaware of their condition.1 Indonesia Also contributes substantially to the global burden of T2DM.2 A wide range of anti-diabetic drugs with diverse mechanisms are available for the treatment of T2DM and this includes sulfonylureas, meglitinides, thiazolidinediones, biguanides, and α-glucosidase inhibitors. More recently, incretin-based therapies like dipeptidyl peptidase-4 (DPP-4) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists have been developed.3,4 Despite this extensive selection of medications, sulfonylureas such as glibenclamide, glimepiride, gliclazide, and gliquidone continue to be the most commonly used treatment for T2DM due to their proven efficacy in stimulating insulin secretion.5 However, the therapeutic effectiveness of drugs from this group is often hampered by their intrinsic biopharmaceutical properties, in which many of these drugs are classified Biopharmaceutical Classification System (BCS) Class II, which is characterized by low water solubility despite having high intestinal absorption.6,7
The main limitation arising from this low solubility is the slow dissolution rate in the gastrointestinal tract. This results in erratic and often incomplete bioavailability of the drug, which can reduce therapeutic effectiveness and require a higher daily dose.8,9 In addition to the biopharmaceutical challenges, its clinical use also faces the risk of significant side effects especially hypoglycaemia, which is often reported to be due to the fluctuations in drug absorption.10 To address these issues, various formulation approaches have been developed, including lipid-based systems, self-emulsifying formulations, particle size reduction, cyclodextrin complexation, and amorphous solid dispersion (ASD) technology. Lipid-based systems, such as lipid nanoemulsions, can improve oral absorption by solubilizing poorly water-soluble drugs within lipid phases and facilitating their dispersion in gastrointestinal fluids. However, ASD provides a distinct solid-state strategy that directly modifies the physical state of the drug by converting the crystalline form into a high-energy amorphous form and stabilizing it within a polymeric matrix. A recent comparative study using a poorly water-soluble non-antidiabetic model drug showed that ASD and lipid nanoemulsion could both improve oral bioavailability, supporting ASD as a competitive formulation approach among solubility-enhancement technologies.11 In the context of sulfonylureas and meglitinides, ASD is particularly relevant because it targets the main biopharmaceutical limitation of these BCS Class II drugs, namely poor dissolution, through amorphization, molecular-level dispersion, polymer-mediated stabilization, and supersaturation maintenance.
ASD’s relevance as a solution lies in its ability to change the physical state of a drug from crystalline (which is poorly soluble) to amorphous (which is more soluble) by dispersing it within a polymer matrix.12,13 Within ASD systems, drug molecules are distributed throughout the polymer matrix at a molecular scale, resulting in extremely fine dispersion domains that significantly increase the effective surface area exposed to the dissolution medium. In some cases, this dispersion scale may even approach or surpass the level of particle size reduction achieved in conventional nanoparticle systems. This transformation significantly increases the free energy of the drug molecules, resulting in a drastic increase in solubility and dissolution rate.14
The performance of ASD systems is largely governed by the interaction between the drug and the polymeric carrier.15 Polymers function as stabilizing matrices that maintain the drug in an amorphous or molecularly dispersed state.16 Through intermolecular interactions such as hydrogen bonding, dipole–dipole interactions, and hydrophobic interactions, polymers can reduce drug molecular mobility, increase the glass transition temperature of the system, and inhibit nucleation and crystal growth during storage and dissolution.17,18 These properties are essential because the amorphous form has higher free energy than the crystalline form, making it thermodynamically unstable and more susceptible to recrystallization.15 In addition, appropriate polymers can maintain drug supersaturation in gastrointestinal fluids and delay precipitation, thereby increasing the concentration gradient available for absorption and improving oral bioavailability.19,20
The primary advantage of ASD systems lies in their ability to create and stabilize a state of drug supersaturation in the gastrointestinal tract.21 This promotes faster and more consistent drug absorption, ultimately improving overall bioavailability.22 This increased bioavailability potentially allows for lower drug doses, which can reduce the risk of dose-related adverse events such as hypoglycemia.23 Therefore, the purpose of this review article is to comprehensively examine the application of ASD technology for antidiabetic drugs in the sulfonylurea and meglitinide classes, which act by increasing insulin secretion.24 This review will focus on improving biopharmaceutical properties, specifically analyzing how ASD formulations can improve the solubility profile, dissolution rate, and ultimately, oral bioavailability of these drugs, as an effort to optimize therapeutic outcomes in patients with T2DM.25,26
Diabetes Mellitus
Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycaemia due to impaired insulin secretion or action, or both.27 Symptoms include polyuria, polydipsia, weight loss, and loss of consciousness.28 Individuals without diabetes typically have an HbA1c level between 4.0% and 5.6%. Levels between 5.7% and 6.4% indicate prediabetes, and a reading of 6.5% or higher indicate that the patients may suffer from DM. Due to the numerous health complications associated with the disease, it is recommended that people with diabetes adopt a healthy diet and exercise regimen to maintain their HbA1c below 7.0%.29 This goal could be achieved by ensuring proper insulin function, a peptide hormone produced by pancreatic β-cells, that is responsible for regulating blood glucose levels.30
Insulin itself is a peptide hormone produced by pancreatic β-cells, which plays an important role in regulating energy storage and release during feeding and fasting conditions, thereby controlling blood glucose levels.31 This hormone functions to increase glucose uptake by muscle cells and adipose tissue through the Glucose Transporter Type 4 (GLUT4) mechanism. This glucose transport takes place following a concentration gradient through facilitated diffusion without the need for ATP.32 Insulin receptors recognize the insulin hormone and facilitate the movement of glucose across the cell membrane.
Diabetes has several subclassifications, with the two main types being the Type 1 Diabetes Mellitus (T1DM) and T2DM. T1DM is a chronic autoimmune disorder characterized by insufficient insulin production leading to hyperglycemia.33 T1DM results from autoimmune damage to pancreatic beta cells, leading to complete insulin deficiency.34 The main therapy for T1DM is insulin therapy, in which synthetic insulin is administered into the body, although this method is often unable to achieve optimal blood glucose control in many individuals.35
T2DM on the other hand develops when pancreatic β-cells fail to compensate for insulin resistance in peripheral tissues, particularly in skeletal muscle and liver.36 Current therapeutic approaches emphasize the importance of achieving early glycaemic control to preserve β-cell function and improve long-term clinical outcomes in the affected patients.37 T2DM is associated with various risk factors such as high blood glucose levels, obesity, hypertriglyceridemia, poor diet, lack of physical activity, advanced age, family history, stress, anxiety, and depression.38 Insulin therapy, combined with oral glucose-lowering agents, is essential in the treatment and management of T2DM.39
However, T2DM pathogenesis extends far beyond β-cell dysfunction. The “egregious eleven” include α-cell dysregulation (causing hyperglucagonemia), insulin resistance in adipose tissue, muscle, and liver, CNS-mediated appetite dysregulation, gut microbiota alterations, incretin hormone deficiency, and heightened renal glucose reabsorption.40 To address these multiple pathological mechanisms, current management strategies employs multiple drugs to achieve targeted therapeutic outcomes. Medications from sulfonylureas, meglitinides, and GLP-1 agonist groups act by enhancing the β-cell function; GLP-1 receptor agonists, DPP-4 inhibitors, and amylin suppress glucagon secretion; and thiazolidinediones mitigate insulin resistance in adipose and muscle tissues.41,42 In addition, metformin specifically acts by suppressing gluconeogenesis in the liver and enhancing insulin sensitivity in muscle tissues.43 Additional pharmacological agents, including bromocriptine address insulin resistance within the central nervous system, whereas acarbose functions by inhibiting the enzyme α-glucosidase in the gastrointestinal tract. SGLT-2 inhibitors on the other hand decrease glucose reabsorption from the renal system, whilst probiotics and prebiotics enhance microbiota composition and managing systemic inflammation, which is integral to the long-term therapeutic strategy.40 Among these strategies, insulin secretagogues—particularly sulfonylureas remain clinically vital for addressing the pancreatic β-cell dysfunction (Figure 1).44–46
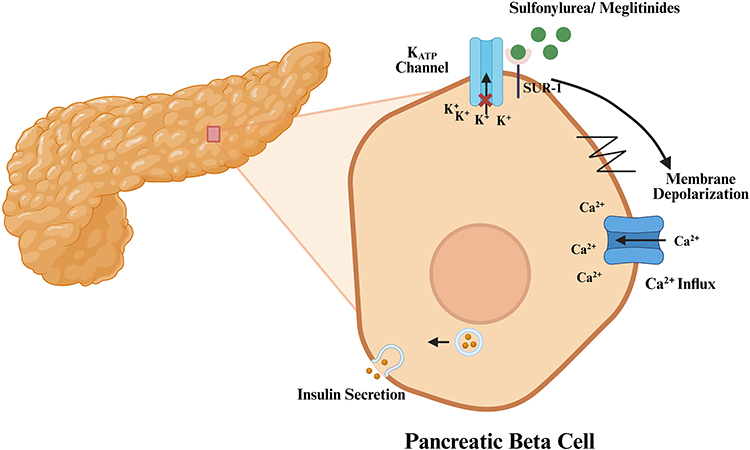 |
Figure 1 Mechanism of Sulfonylurea/Meglitinides. Sulfonylureas and meglitinides, represented by green circles, bind to sulfonylurea receptor-1 (SUR-1) on the ATP-sensitive potassium (KATP) channel. The red cross symbol indicates inhibition or closure of the KATP channel, thereby blocking K+ efflux from the beta cell. This inhibition leads to membrane depolarization, represented by the zig-zag membrane signal and the black directional arrow. Membrane depolarization subsequently opens voltage-dependent Ca2+ channels, shown as the blue channel on the right side of the cell, allowing Ca2+ influx into the cytoplasm. The increased intracellular Ca2+ concentration promotes the movement of insulin-containing secretory granules toward the plasma membrane and triggers insulin exocytosis, shown by the small vesicles, Orange dots, and directional arrows near the cell membrane. |
Sulfonylureas and meglitinides are known to demonstrate poor aqueous solubility (Table 1). These limitations have an impact on therapeutic variability, such as low bioavailability, high dose requirements, and the potential for increased side effects. This inherent physicochemical deficiency of the drug molecules themselves creates a compelling rationale for formulating them as ASDs. The high-dose requirements are a direct consequence of poor dissolution; a significant portion of each administered dose remains undissolved and is never absorbed, necessitating a higher initial dose to achieve a therapeutic effect. This, in turn, increases the drug load in the gastrointestinal tract, raising the risk of dose-dependent concentration-related side effects. Furthermore, the erratic and incomplete dissolution leads to high inter- and intra-patient variability in drug absorption. This unpredictable bioavailability makes it challenging to achieve consistent glycemic control, as the same dose can produce vastly different blood glucose-lowering effects from one administration to the next or between different patients. ASD technology directly targets this root cause by fundamentally enhancing the drug’s dissolution properties, thereby mitigating these critical limitations and enabling more reliable, effective, and potentially safer therapy.47
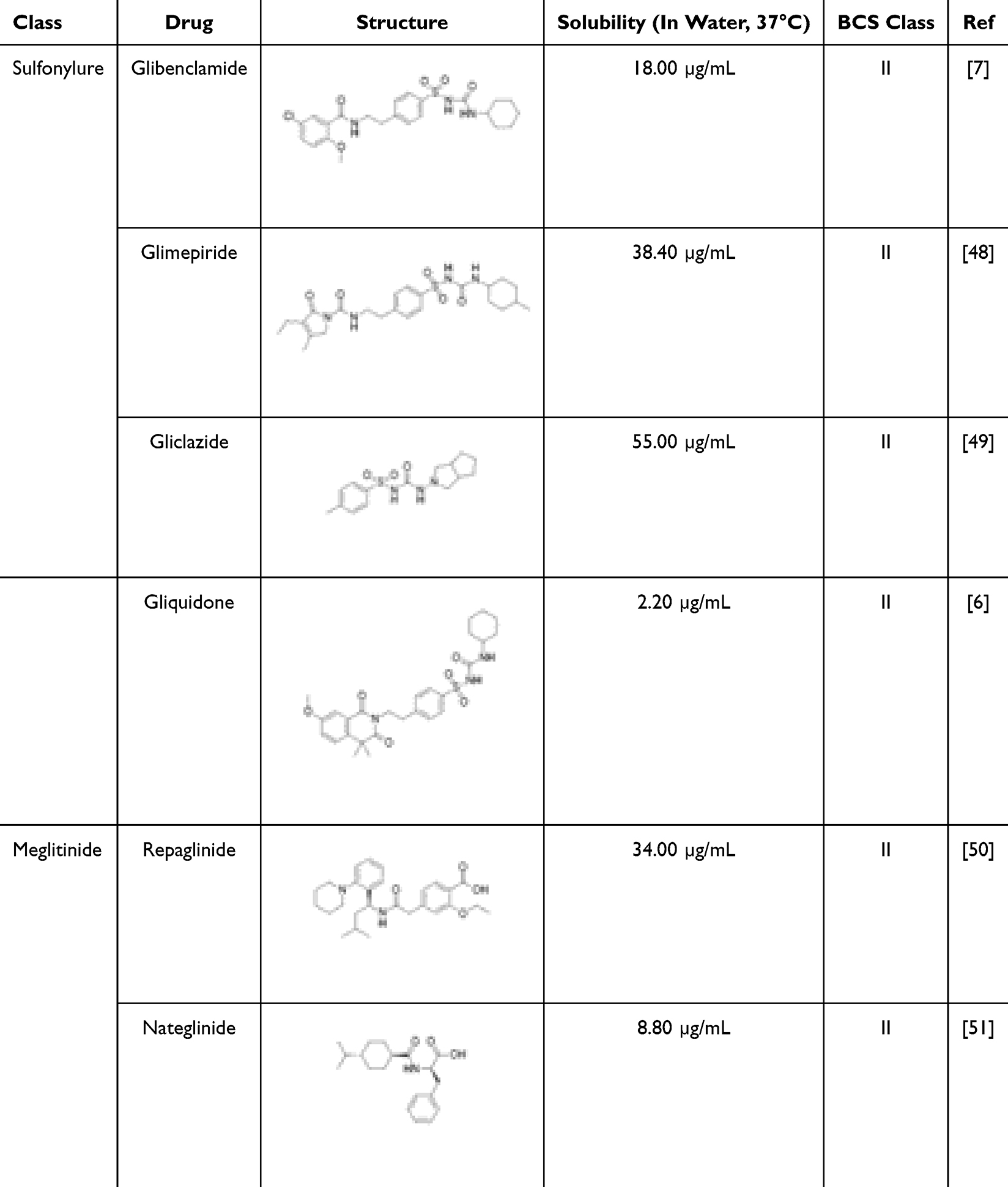 |
Table 1 Oral Hypoglycemic Agents from the Sulfonylurea and Meglitinide Classes |
Therefore, the transformation of these crystalline drugs into an amorphous state via ASD is an essential formulation strategy to overcome their intrinsic pharmacokinetic limitation, ensuring a sufficient fraction of the dose is dissolved and absorbed to achieve a predictable and potent therapeutic response, while minimizing the risks associated with high and variable dosing.
Amorphous Solid Dispersion
Definition
ASD is a molecular mixture of poorly water-soluble drugs and hydrophilic carriers. They modulate the drug release profile and are characterised by the reduction of drug particle size to a molecular level, facilitating the solubilisation or co-dissolution of the drug within the soluble carriers.52 In this system, the drug is dispersed homogeneously within the polymer matrix, forming extremely fine dispersion domains approaching the scale of individual molecules. Unlike nanoparticle-based delivery systems, where drug remains as discrete particles in the nanometer range, ASD systems distribute drug as molecules uniformly within the carrier matrix. As a result, the effective dispersion scale can be even smaller than conventional nanoparticles, which contributes to enhanced apparent solubility and dissolution behavior. Overall, enhanced wettability and dispersibility are achieved as the drug exists in a supersaturated state resulting from forced solubilisation in hydrophilic carriers.53–57 Solid dispersions are categorized into first, second, or third generation.58 First generation involves the creation of crystalline solid dispersions in which a molecule of a crystalline carrier substitutes for one drug molecule within its crystalline structure. The second generation involves the creation of ASD utilising polymeric carriers. The third generation consists of ASD formed by a combination of amorphous carriers and, ideally, surfactants, which demonstrate improved drug release, long-term stability, and increased bioavailability.58,59 ASD employs designated carriers to amorphize the drug substance and maintain its stability in the solid state.60–62
The Mechanism of Amorphous Solid Dispersions
ASD are often employed to formulate drugs with low water solubility and/or bioavailability, particularly those classified as Biopharmaceutics Classification System (BCS) Class II and IV compounds.63–65 As illustrated in Figure 2, ASD formation involves the conversion of a crystalline drug into an amorphous form and its incorporation into a hydrophilic polymer matrix.14,66 Within this system, the polymer serves as a carrier that disperses the drug at the molecular level.67 In contrast to nanosuspension or nanoparticle formulations, where the drug remains as discrete particles, ASDs maintain drug molecules in a molecularly dispersed state within the polymer matrix. This homogeneous dispersion eliminates particle boundaries and significantly increases the effective surface area available for dissolution, thereby promoting rapid drug release and supersaturation in aqueous media. Various types of polymers with diverse physicochemical and thermochemical properties can be used to formulate ASDs. The selection of an appropriate polymer is critical, as it influences the stability, solubility, dissolution rate, manufacturability, and ultimately the bioavailability of the solid dispersion.68,69
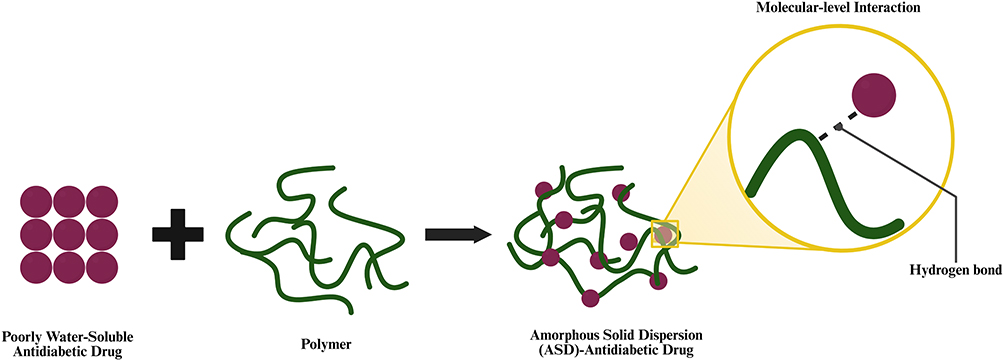 |
Figure 2 Schematic illustration of molecular drug dispersion within a polymeric matrix during ASD formation. The purple circles represent the poorly water-soluble antidiabetic drug, and the green irregular lines represent the polymer. The black arrow indicates the dispersion process. In the final structure, the dispersed purple circles within the green polymeric network represent drug molecules distributed in the amorphous solid dispersion systems. |
The Impact of Amorphous Solid Dispersions on Dissolution
The fundamental principle of ASD lies in dispersing the drug at a molecular level within a polymer matrix, thereby maintaining it in a high-energy, non-crystalline amorphous state.69,70 This molecular-level dispersion increases the apparent solubility and accelerates the dissolution rate.71 The choice of polymer is critical, as it prevents drug recrystallization - a common challenge in amorphous systems72–74,74 Consequently, ASD improve dissolution by enhancing drug wettability, reducing the effective particle size to the molecular level, and facilitating the formation of a supersaturated solution.75 The successful application of these principles has consistently demonstrated substantial improvements in the dissolution performance of a wide range of drugs. As illustrated in Figure 2, drug molecules are molecularly dispersed within the polymer matrix, resulting in enhanced dissolution behavior and supersaturation. This improved dissolution subsequently contributes to enhanced oral bioavailability. The dissolution-enhancing effect of ASD has been reported across several poorly water-soluble drugs outside the antidiabetic class, including pimobendan (PIMO), nintedanib (NIN), and erlotinib (ERL), which showed marked improvements in dissolution after ASD formulation.76,77 These examples provide broader mechanistic evidence that molecular dispersion, amorphization, and polymer-mediated stabilization can overcome dissolution-limited absorption. However, because the present review focuses on insulin secretagogues, these non-antidiabetic examples are used only to contextualize the general applicability of ASD technology. The following discussion therefore emphasizes ASD applications in sulfonylureas and meglitinides, where poor aqueous solubility similarly limits dissolution, oral bioavailability, and therapeutic consistency.77
Taken together, these findings present a compelling case for ASD as a robust and versatile platform for addressing the solubility limitations of crystalline drugs.78 The remarkable success of ASDs can be attributed to their foundational mechanisms: maintaining the drug in a high-energy amorphous state, inhibiting recrystallization through strong drug-polymer interactions, and sustaining supersaturation in the dissolution medium.79 Consequently, ASD technology has emerged as a cornerstone of modern pharmaceutical formulation science, enabling the effective delivery of promising yet poorly soluble therapeutic agents.78
The Impact of ASDs on Bioavailability and Pharmacological Activity
ASD are formulation systems that stabilize an active pharmaceutical ingredient (API) by incorporating it in the form of amorphous within a solid polymer matrix.80 This strategy is specifically designed to overcome the challenge of poor aqueous solubility, thereby enhancing the in vivo bioavailability of orally administered drugs. By maintaining the drug in a high-energy amorphous state - which has higher apparent solubility than its crystalline counterpart – an initial supersaturation can be achieved.81 The polymer component then plays a crucial role in stabilizing this supersaturated state, extending the duration that the molecule is available for absorption. This improvement in dissolution and absorption ultimately translates to increased bioavailability and, consequently, enhanced pharmacological activity.82,83 The bioavailability-enhancing effect of ASD has been reported in several poorly water-soluble drugs outside the antidiabetic class, including paliperidone, itraconazole, oxyberberine, and honokiol.81,84–86 These examples provide general proof-of-concept that amorphization, polymer-mediated stabilization, and supersaturation maintenance can improve systemic exposure and pharmacological performance.21,79 Furthermore, these examples support the broader applicability of ASD technology as a formulation strategy for poorly water-soluble drug and provide a mechanistic rationale for its application to sulfonylureas and meglitinides.87
ASDs of Antidiabetic Compound
According to the Biopharmaceutics Classification System (BCS), approximately 40% of marketed antidiabetic drugs suffer from low aqueous solubility, which limits their dissolution in gastrointestinal fluids and, consequently, their absorption.50 Class II drugs, such as glibenclamide, gliclazide, and repaglinide, are characterized by high membrane permeability but poor water solubility, resulting in reduced systemic availability and low bioavailability.(DEMIRTÜRK and ÖNER, 2004) For example, the solubility of glibenclamide in water is only 4 mg/L.88 Improving its solubility is therefore essential in enhancing drug absorption, and several formulation strategies such as complexation, salt formation, particle size reduction, cocrystallization, and solid dispersion have been developed to address this challenge. Among these, solid dispersion techniques have proven particularly effective in increasing the dissolution rate of antidiabetic drugs, as illustrated in Table 2 89
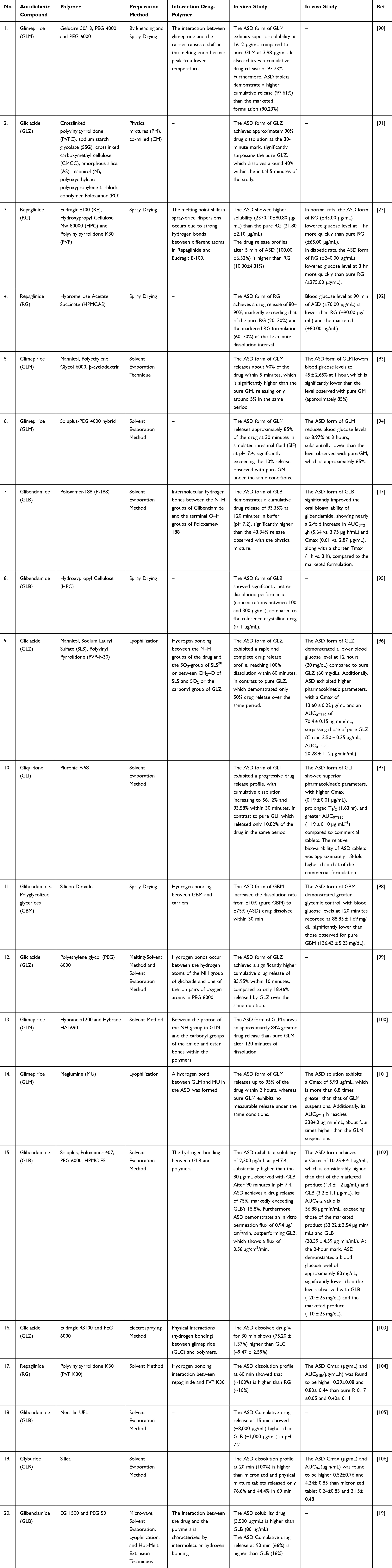 |
Table 2 The Impact of ASD System on in vitro and in vivo of Antidiabetic Compound |
The studies summarized in Table 2 indicate that ASD formulations generally improved the dissolution performance of sulfonylureas and meglitinides compared with their crystalline or pure drug forms. Several formulations achieved marked improvements, including substantially higher cumulative drug release, increased Cmax and AUC, and stronger glucose-lowering effects compared with pure drug or marketed formulations. The most frequently investigated compounds include glibenclamide/glyburide, glimepiride, gliclazide, repaglinide, and gliquidone, with polyvinylpyrrolidone (PVP), polyethylene glycol (PEG), Poloxamer, Soluplus, hydroxypropyl methylcellulose (HPMC) derivatives, Eudragit, Pluronic, and related polymeric carriers commonly used to enhance drug dispersion and stabilization. Across these studies, the improvement in dissolution was mainly associated with reduced crystallinity, enhanced wettability, molecular-level dispersion, supersaturation maintenance, and possible drug–polymer interactions, including hydrogen bonding. However, the extent of improvement varied depending on the drug, polymer type, drug–polymer ratio, preparation method, and dissolution conditions, indicating that ASD performance cannot be generalized across all insulin secretagogues without formulation-specific evaluation. More detailed case-by-case discussions of dissolution performance, oral bioavailability, and antidiabetic activity are provided in the following subsections.
Dissolution Study
Several studies have demonstrated that the incorporation of polymers in ASDs can significantly enhance drug dissolution. Varshosaz et al (2017) reported the formulation of repaglinide (RG) ASDs using Eudragit E100, hydroxypropyl cellulose (HPC), and polyvinylpyrrolidone K30 (PVP K30), observing a remarkable improvement in solubility. The Eudragit E100-based formulation achieved complete (100%) drug release within 5 minutes, compared with only 10.3% for the pure drug.23 Solubility studies confirmed that while physical mixtures provided slight improvements through limited polymer interactions, the enhancement was substantially greater in ASDs due to molecular-level drug dispersion, which effectively prevented recrystallization. Moreover, dissolution profiling showed that pure RG required approximately 20 minutes for complete release, whereas ASDs – particularly those containing Eudragit E100, achieved full release within 5 minutes. This improvement was particularly evident in spray-dried dispersions at a drug-to-polymer ratio of 1:3, resulting in up to a 100-fold increase in solubility.
Sakure et al (2020) formulated glimepiride (GLM) in ASDs using excipients such as Gelucire 50/13, PEG 4000, and PEG 6000. The resulting formulations showed a significant increase in drug release, reaching 93.73% compared to only 3.98% for the pure drug.90 This improvement was attributed to the ASD’s ability to increase surface area and apparent solubility, thereby accelerating drug release in the aqueous environment of the gastrointestinal tract. Additionally, excipients such as Gelucire and PEGs play a role in enhancing drug solubility by reducing crystallization tendency and promoting molecular dispersion. This significant enhancement in dissolution performance has the potential to improve bioavailability and glycemic control in diabetic patients, underscoring the effectiveness of ASD technology in optimizing drug delivery and therapeutic efficacy.
Bioavailability Study
Improved dissolution enhances therapeutic effectiveness by accelerating the onset of pharmacological action. Due to the fact that only the dissolved fraction of a drug can be absorbed, increased dissolution results in a higher amount of drug available for absorption, thereby improving systemic bioavailability. Mir et al (2024) reported that the AUC of glibenclamide (GLB) formulated with Poloxamer 188 increased by 50% compared to the pure drug, indicating a significant enhancement in bioavailability.47 This improvements was attributed to Poloxamer 188’s ability to enhance the wettability and solubilization of the lipophilic GLB, as well as its stabilizing effect on the amorphous form of the drug, preventing recrystallization during dissolution. The transformation of GLB from a crystalline to an amorphous state within the ASD contributed to higher apparent solubility and sustained supersaturation, resulting in improved absorption and a higher AUC.
Similarly, Mansour and Aly (2015) demonstrated that gliclazide (GLZ) formulated as an ASD with mannitol, sodium lauryl sulfate (SLS), and polyvinylpyrrolidone K-30 (PVP-K30) achieved an AUC more than 3-fold higher than of its pure crystalline form, reflecting a significant improvement in oral bioavailability.96 The observed increases in both Cmax and AUC0–12 indicate that the ASD formulation enhanced absorption efficiency without altering the rate of absorption onset. This improvement can be attributed to the conversion of GLZ into its amorphous state and the presence of hydrophilic excipients such as SLS and PVP K30, which improved drug wettability, solubilization, and maintenance of supersaturation during dissolution. Collectively, these findings highlight the potential of ASD technology to markedly enhance systemic exposure and therapeutic efficacy of poorly soluble antidiabetic drugs.
Diabetic Treatment
By improving solubility and dissolution rate, ASD formulations increase drug absorption, as reflected in higher AUC and Cmax values. This increased systemic exposure enables drugs to reach therapeutic plasma concentrations more effectively, thereby improving their hypoglycemic effect and achieving better blood glucose control. Among the various strategies available to overcome the poor bioavailability of diabetes drugs, ASD has proven particularly effective. Solid dispersion systems of repaglinide have demonstrated significant improvements in antidiabetic activity through enhanced solubility and bioavailability, as evidenced by several in vivo studies. In diabetic animal models, repaglinide formulated via spray drying or solvent evaporation methods using hydrophilic polymers such as Eudragit E100, hydroxypropyl cellulose (HPC, MW 80,000), and PVP K30 exhibited superior glucose-lowering effects compared to the pure drug and commercial formulations. These ASDs not only improved the dissolution rate but also produced markedly greater reductions in blood glucose levels.
Varshosaz et al (2017) reported that in vivo studies on male albino Wistar rats shows significant reduction of the blood glucose levels in normal rats in the repaglinide (RG) ASD group within 8 hours of sampling compared to the control group, with notable reductions observed at 30 and 60 minutes. The most pronounced decrease occurred after 2 hours. In both normal and diabetic rats, the ASD formulation reduced blood glucose more rapidly than the pure drug, with a faster effect at one hour in normal rats – approximately 89.3% greater than the pure drug – and at three hours in diabetic rats, showing an 85.7% greater reduction compared to RG alone.23
Reginald (2015) also compared a glimepiride (GLM) ASD formulation with the pure drug and a commercially available product, observing a more pronounced and sustained reduction in glucose levels in rats treated with the ASD. Effective blood glucose reduction began within one hour after oral administration, reaching a maximum Tmax at three hours. Blood glucose levels decreased from 100% to 9.81% and 8.97%, respectively. However, blood glucose levels did not return to the baseline levels after 24 hours of the study. In comparison with pure and commercial formulations, the GLM ASD formulation maintained normoglycemic levels for 9–12 hours, demonstrating that ASD technology can effectively and efficiently enhance the pharmacological performance of antidiabetic drugs.94
Similarly for another sulfonylurea compound, glyburide, Guan et al (2014) evaluated the oral exposure of ASD-formulated glyburide in dogs. The findings revealed significant differences in plasma concentrations and absorption rates between the test tablets (solid dispersion tablets) and reference commercial tablets. The ASD tablets produced markedly higher plasma levels after oral administration, with the AUC increased by approximate 97.7% compared to the reference product, indicating nearly double the oral bioavailability. These results confirm that enhancing solubility and dissolution through ASD – particularly when prepared by techniques such as the supercritical fluid (SCF) process – can substantially improve the oral bioavailability and therapeutic efficacy of glyburide.106
Discussion
The primary challenge in developing effective oral antidiabetic drugs lies in overcoming the pharmacokinetic limitations associated with their physicochemical properties. Most oral antidiabetic agents belong to the BCS Class II compounds, characterized by low solubility and high permeability. This inherent low solubility limits their dissolution in gastrointestinal fluids, thereby restricting absorption and reducing overall bioavailability and therapeutic efficacy. To address this fundamental challenge, ASD technology has emerged as a pivotal formulation strategy. The core principle of ASD is to transform the crystalline drug into a high-energy, metastable amorphous form within a polymer matrix. This transformation disrupts the rigid crystal lattice, drastically lowering the energy barrier for dissolution and thereby significantly enhancing the drug’s apparent solubility and dissolution rate, which are the key determinants of absorption for BCS Class II drugs.
The translation of these in vitro advantages into in vivo outcomes is evident in pharmacokinetic studies. Enhanced dissolution achieved through ASDs facilitates greater and more consistent absorption in the gastrointestinal tract, often resulting in a 2-fold or greater increase in the AUC compared to the pure drug or conventional formulations. Improved bioavailability represents the crucial link between formulation design and clinical response, producing a faster onset of action and a more pronounced reduction in blood glucose levels, with pharmacokinetic parameters such as increased Cmax and AUC confirm the enhanced absorption and systemic exposure achieved through ASD technology. Collectively, in vivo evidence consistently demonstrates that ASD formulations of antidiabetic drugs produce superior glucose-lowering effect, reflecting their ability to optimize pharmacokinetic profiles for both rapid and prolonged therapeutic action.
Pure crystalline drugs generally exhibit low solubility, which leads to slower dissolution, reduced absorption, lower oral bioavailability. This limitation may reduce systemic drug exposure and consequently lower plasma insulin concentrations. This in turn limit their therapeutic potential. By converting the active pharmaceutical compound from its crystalline to amorphous form, ASD technology significantly enhances solubility, dissolution rate, and bioavailability, thereby facilitating more efficient absorption. As illustrated in Figure 3, ASD formulations demonstrate higher apparent solubility, allowing the drug to dissolve faster and penetrate the apical membrane into systemic circulations. This improved absorption may result in greater systemic exposure of the active compound and, in the context of insulin secretagogues, may contribute to enhanced insulin secretion and more effective glycemic control. These effects are consistent with the in vivo findings summarized in Table 2. Thus, Figure 3 provides an integrated schematic link between molecular-level ASD performance and its expected pharmacokinetic and pharmacodynamic consequences in antidiabetic therapy.
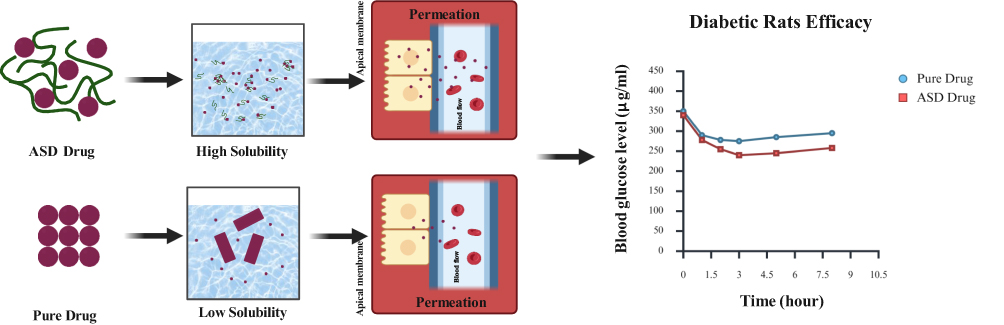 |
Figure 3 Amorphous solid dispersions ASD mechanism in body. The ASD drug is represented by purple drug particles dispersed within green polymeric chains, whereas the pure drug is represented by aggregated purple particles. The black arrows indicate the sequence from formulation to solubility, intestinal permeation, and antidiabetic efficacy. The high-solubility panel shows dispersed ASD drug particles in the dissolution medium, while the low-solubility panel shows poorly dispersed pure drug particles. The apical membrane represents the intestinal absorption barrier, and the arrows toward the blood flow compartment indicate drug permeation. In the efficacy graph, blue circles represent the pure drug and red squares represent the ASD drug. The lower blood glucose levels observed in the ASD drug group indicate a greater glucose-lowering effect over time compared with the pure drug in diabetic rats. |
The principle mechanism responsible for the improved bioavailability in ASD systems is the generation of a supersaturated state, which enables higher apparent solubility and faster passive diffusion of the drug into the bloodstream. In addition, the molecular dispersion of drug molecules within the polymer matrix produces extremely fine dispersion domains approaching the molecular scale, effectively increasing the surface area exposed to the dissolution medium and facilitating rapid drug release. This rapid drug dissolution promotes the formation of a supersaturated solution in gastrointestinal fluids, which increases the thermodynamic driving force for membrane permeation. Additionally, molecular interactions between the drug and the polymer matrix – such as via hydrogen bonding – stabilize the supersaturated state, prevent recrystallization, and maintain the amorphous form during dissolution. The elevated thermodynamic activity of the amorphous drug further enhances membrane permeation, collectively resulting in improved in vitro and in vivo performance compared with the crystalline form. These mechanisms collectively enhance both the physicochemical stability and pharmacological efficacy of the compound.
Although ASD technology offers clear advantages in improving the dissolution and oral bioavailability of poorly water-soluble sulfonylureas and meglitinides, several limitations and risks should be acknowledged. The amorphous form is thermodynamically unstable and may recrystallize during storage or upon exposure to gastrointestinal fluids, thereby reducing the dissolution and solubility advantages of the formulation. The stabilizing effect of polymers is highly drug-specific; therefore, a polymer that effectively maintains supersaturation for one compound may not provide the same benefit for another insulin secretagogue. High polymer content, hygroscopicity, moisture sensitivity, phase separation, and limited drug loading may also affect dosage form performance, manufacturability, and long-term physical stability. These issues indicate that ASD development requires careful evaluation of drug–polymer miscibility, glass transition behavior, storage stability, and precipitation inhibition.
The translation of ASD performance from in vitro dissolution studies to in vivo and clinical outcomes also remains challenging. Many studies on sulfonylureas and meglitinides are still limited to laboratory-scale formulations, animal models, or short-term pharmacokinetic evaluations. Therefore, improved dissolution, Cmax, or AUC should be interpreted as biopharmaceutical advantages rather than direct evidence of sustained glycemic control, lower hypoglycemia risk, or superior patient outcomes in clinical settings. Large-scale manufacturing processes, particularly spray drying and hot-melt extrusion, require strict control of processing parameters to ensure reproducibility and may introduce additional challenges, including thermal stress, residual solvent control, batch-to-batch variability, and higher production costs compared with conventional dosage forms. Continued optimization of polymer selection, stability testing, scale-up processes, and clinical validation is therefore essential before ASD systems can be considered clinically reliable for oral antidiabetic therapy.
Expert Opinion and Future Perspectives
ASD technology represents a promising formulation strategy for improving the biopharmaceutical performance of poorly water-soluble insulin secretagogues, particularly sulfonylureas and meglitinides. These drugs remain clinically relevant in the management of type 2 diabetes because of their ability to stimulate insulin secretion; however, their oral performance is frequently limited by poor aqueous solubility, slow dissolution, and variable absorption. By converting the crystalline drug into an amorphous form and stabilizing it within a polymeric matrix, ASD systems may enhance apparent solubility, dissolution rate, supersaturation, and systemic exposure. Nevertheless, these benefits should be interpreted with caution, as improved dissolution or bioavailability does not automatically ensure sustained glycemic control, reduced hypoglycemia risk, or superior clinical outcomes without further pharmacokinetic, pharmacodynamic, and clinical validation.
Future development of ASD-based sulfonylurea and meglitinide formulations should prioritize rational polymer selection, long-term physical stability, and reliable maintenance of supersaturation under gastrointestinal conditions. Because the amorphous form is thermodynamically unstable, recrystallization during storage or dissolution remains one of the major barriers to successful ASD translation. Therefore, future studies should place greater emphasis on drug–polymer miscibility, intermolecular interactions, glass transition behavior, moisture sensitivity, phase separation, and precipitation inhibition. The development of ternary ASD systems containing polymers, surfactants, or precipitation inhibitors may provide additional advantages for maintaining supersaturation and improving dissolution performance. However, these systems should be evaluated systematically, as improved in vitro dissolution alone is insufficient to confirm superior in vivo performance.
From a translational perspective, ASD technology may serve as a bridge between formulation science and clinical application, but stronger evidence is still required. Many available studies on sulfonylureas and meglitinides remain limited to laboratory-scale formulations, animal models, or short-term pharmacokinetic evaluations. Although several ASD-based drug products have reached clinical use in other therapeutic areas, the clinical translation of ASD formulations for sulfonylureas and meglitinides remains limited. At present, no ASD-based formulation of sulfonylureas or meglitinides has been clearly established for routine clinical use based on the studies reviewed. Therefore, improved dissolution, Cmax, or AUC should be interpreted as biopharmaceutical advantages rather than direct evidence of sustained glycemic control, reduced hypoglycemia risk, or superior long-term clinical outcomes. Future studies should also adopt more standardized reporting of formulation composition, drug–polymer ratio, dissolution conditions, stability data, and pharmacokinetic endpoints to allow more reliable comparison across sulfonylurea and meglitinide ASD formulations. Future research should therefore include stability-indicating studies under relevant storage conditions, scalable manufacturing assessment using spray drying or hot-melt extrusion, and comparative pharmacokinetic/pharmacodynamic studies that link formulation performance with glycemic outcomes. Clinical validation is particularly important because sulfonylureas and meglitinides require a careful balance between glucose-lowering efficacy and hypoglycemia risk. Given the increasing burden of type 2 diabetes, including in countries such as Indonesia, improving existing oral antidiabetic formulations through ASD technology may offer practical value, provided that stability, manufacturability, cost-effectiveness, regulatory acceptance, and patient-centered clinical outcomes are adequately addressed. Ultimately, ASD should be regarded not merely as a solubility-enhancement technique, but as a formulation platform whose clinical relevance depends on rigorous optimization and translational validation.
Conclusion
Current evidence consistently demonstrates that polymer-based amorphous solid dispersion (ASD) systems represent a promising strategy to overcome the major biopharmaceutical limitations of poorly water-soluble insulin secretagogues, particularly sulfonylureas and meglitinides. Across the reviewed studies, ASD formulations showed improved dissolution performance, enhanced oral bioavailability, and more consistent therapeutic outcomes compared with conventional crystalline formulations. These findings highlight the critical role of polymer selection and supersaturation stabilization in determining ASD performance and long-term formulation success. Despite these promising advances, several challenges continue to limit the widespread clinical and industrial application of ASD systems, including physical instability, recrystallization risk, scale-up complexity, and limited long-term clinical validation. Future progress in ASD technology will depend on the development of more stable and scalable formulations, deeper understanding of drug–polymer interactions, and stronger translational studies bridging laboratory findings with clinical applications. Overall, ASD technology holds strong potential to support the next generation of oral antidiabetic formulations with improved therapeutic efficiency and patient outcomes.
Data Sharing Statement
All data generated during this study are included in the manuscript.
Acknowledgments
This publication charge is funded by Universitas Padjadjaran through the Indonesian Endowment Fund for Education (LPDP) on behalf of the Indonesian Ministry of Higher Education, Science and Technology and managed under the EQUITY Program (Contract No. 4303/ B3/DT.03.08/2025 and 3927/UN6. RKT/HK.07.00/2025).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study is funded by Universitas Padjadjaran through the Indonesian Endowment Fund for Education (LPDP) on behalf of the Indonesian Ministry of Higher Education, Science and Technology and managed under the EQUITY Program (Contract No. 4303/ 83/ DT.03.08/ 2025 and 3927/ UN6.RKT/HK.07.00/2025) through scheme Hibah Article Review Equity awarded to Arif Budiman (No. 3899/UN6.3.1/PT.00/2025).
Disclosure
The authors report no conflicts of interest in this work.
References
1. IDF. 11th Edition | 2025 Diabetes Atlas. Magliano DJ, Boyko EJ, Genitsaridi I, Piemonte L, Riley P, Salpea P, eds.;2025.
2. Nuridzin DZ, Aghram N, Mawarni A, Retnowati R. Determinants of diabetes mellitus prevalence in Indonesia: a multiple linear regression model in an ecological analysis of adults aged 15 years and oldeR. Jurnal Manajemen Kesehatan Indonesia. 2024;12(3):327–19. doi:10.14710/jmki.12.3.2024.327-338
3. Desse TA, Namara K, Manias E. Patient-Perceived challenges to type 2 diabetes self-management in Sub-Saharan Africa: a Qualitative Exploratory Study. Sci Diabet Self-Management Care. 2024;50(6):456–468. doi:10.1177/26350106241279809
4. Gieroba B, Kryska A, Sroka-Bartnicka A. Type 2 diabetes mellitus – conventional therapies and future perspectives in innovative treatment. Biochem Biophys Rep. 2025;42:102037. doi:10.1016/j.bbrep.2025.102037
5. Allyhiani M, Kurdi A, Abdulaziz A, et al. Prescribing patterns of antidiabetics in type 2 diabetes and factors affecting them. Saudi Pharm J. 2022;30(2):112–119. doi:10.1016/j.jsps.2021.12.019
6. El-Araby M, El-Gizawy SA, Ashmawy SM, El Maghraby GM. Cinnamon oil-based self-emulsifying system for augmented dissolution and hypoglycemic efficacy of gliquidone. J Drug Deliv Sci Technol. 2024;97:105821. doi:10.1016/j.jddst.2024.105821
7. Maharjan R, Jeong J, Bhujel R, et al. Correlation of solubility thermodynamics of glibenclamide with recrystallization and in vitro release profile. Molecules. 2022;27(4):1392. doi:10.3390/molecules27041392
8. Budiman AR, Husni PA, Shafira AT. The development of glibenclamide-saccharin cocrystal tablet formulations to increase the dissolution rate of the drug. Int J Appl Pharm. 2019;11(4):359–364.
9. Budiman A, Aulifa DL. Characterization of drugs with good glass formers in loaded-mesoporous silica and its theoretical value relevance with mesopores surface and pore-filling capacity. Pharmaceuticals. 2022;15(1):93.
10. Ahmed TA, Alotaibi HA, Alharbi WS, Safo MK, El-Say KM. Development of 3D-Printed, liquisolid and directly compressed glimepiride tablets, loaded with black seed oil self-nanoemulsifying drug delivery system: in vitro and in vivo characterization. Pharmaceuticals. 2022;15(1):68. doi:10.3390/ph15010068
11. Meola TR, Kamath S, Elz AS, Prestidge CA, Wignall A, Joyce P. Contrasting the pharmacokinetic performance and gut microbiota effects of an amorphous solid dispersion and lipid nanoemulsion for a poorly water-soluble anti-psychotic. Eur J Pharm Biopharm. 2024;203:114453. doi:10.1016/j.ejpb.2024.114453
12. Pugliese A, Tobyn M, Hawarden LE, Abraham A, Blanc F. New development in understanding drug–polymer interactions in pharmaceutical amorphous solid dispersions from solid-state nuclear magnetic resonance. Mol Pharm. 2022;19(11):3685–3699. doi:10.1021/acs.molpharmaceut.2c00479
13. Iyer R, Petrovska Jovanovska V, Berginc K, et al. Amorphous Solid Dispersions (ASDs): the influence of material properties, manufacturing processes and analytical technologies in drug product development. Pharmaceutics. 2021;13(10):1682. doi:10.3390/pharmaceutics13101682
14. den Mooter G V. The use of amorphous solid dispersions: a formulation strategy to overcome poor solubility and dissolution rate. Drug Discov Today Technol. 2012;9(2):e79–e85. doi:10.1016/j.ddtec.2011.10.002
15. Meng F, Paul SK, Borde S, Chauhan H. Investigating crystallization tendency, miscibility, and molecular interactions of drug–polymer systems for the development of amorphous solid dispersions. Drug Dev Ind Pharm. 2021;47(4):579–608. doi:10.1080/03639045.2021.1892747
16. Que C, Deac A, Zemlyanov DY, et al. Impact of Drug–Polymer intermolecular interactions on dissolution performance of copovidone-based amorphous solid dispersions. Mol Pharm. 2021;18(9):3496–3508. doi:10.1021/acs.molpharmaceut.1c00419
17. Deac A, Qi Q, Indulkar AS, et al. Dissolution mechanisms of amorphous solid dispersions: role of drug load and molecular interactions. Mol Pharm. 2023;20(1):722–737. doi:10.1021/acs.molpharmaceut.2c00892
18. Bookwala M, Buckner IS, Wildfong PLD. Implications of coexistent halogen and hydrogen bonds in amorphous solid dispersions on drug solubility, miscibility, and mobility. Mol Pharm. 2022;19(11):3959–3972. doi:10.1021/acs.molpharmaceut.2c00434
19. Pisay M, Bhaskar KV, Mehta CH, Nayak UY, Koteshwara KB, Mutalik S. Drug-Carrier miscibility in solid dispersions of glibenclamide and a novel approach to enhance its solubility using an effervescent agent. AAPS Pharm Sci Tech. 2022;23(8):284. doi:10.1208/s12249-022-02437-z
20. Budiman A, Citraloka ZG, Muchtaridi M, Sriwidodo S, Aulifa DL, Rusdin A. Inhibition of crystal nucleation and growth in aqueous drug solutions: impact of different polymers on the supersaturation profiles of amorphous drugs—the case of alpha-mangostin. Pharmaceutics. 2022;14(11):2386. doi:10.3390/pharmaceutics14112386
21. Schittny A, Huwyler J, Puchkov M. Mechanisms of increased bioavailability through amorphous solid dispersions: a review. Drug Deliv. 2020;27(1):110–127. doi:10.1080/10717544.2019.1704940
22. Gupta S, Kesarla R, Omri A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharm. 2013;2013:1–16. doi:10.1155/2013/848043
23. Varshosaz J, Minayian M, Ahmadi M, Ghassami E. Enhancement of solubility and antidiabetic effects of Repaglinide using spray drying technique in STZ-induced diabetic rats. Pharm Dev Technol. 2017;22(6):754–763. doi:10.3109/10837450.2016.1143001
24. Chaudhury A, Duvoor C, Reddy Dendi VS, et al. Clinical review of antidiabetic drugs: implications for type 2 diabetes mellitus management. Front Endocrinol. 2017:8. doi:10.3389/fendo.2017.00006
25. Tambe S, Jain D, Meruva SK, et al. Recent advances in amorphous solid dispersions: preformulation, formulation strategies, technological advancements and characterization. Pharmaceutics. 2022;14(10):2203. doi:10.3390/pharmaceutics14102203
26. Tripathi D, Mp BH, Sahoo J, Kumari J. Navigating the solution to drug formulation problems at research and development stages by amorphous solid dispersion technology. Recent Adv Drug Deliv Formul. 2024;18(2):79–99. doi:10.2174/0126673878271641231201065151
27. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2013;36(Supplement_1):S67–S74. doi:10.2337/dc13-S067
28. AL-Ishaq RK, Abotaleb M, Kubatka P, Kajo K, Büsselberg D. Flavonoids and their anti-diabetic effects: cellular mechanisms and effects to improve blood sugar levels. Biomolecules. 2019;9(9):430. doi:10.3390/biom9090430
29. Delahanty LM, Halford BN. The role of diet behaviors in achieving improved glycemic control in intensively treated patients in the diabetes control and complications trial. Diabetes Care. 1993;16(11):1453–1458. doi:10.2337/diacare.16.11.1453
30. American Diabetes Association. Standards of medical care in diabetes—2014. Diabetes Care. 2014;37(Supplement_1):S14–S80. doi:10.2337/dc14-S014
31. Rahman MS, Hossain KS, Das S, et al. Role of insulin in health and disease: an update. Int J Mol Sci. 2021;22(12):6403. doi:10.2337/dc14-S014
32. Dimitriadis G, Mitrou P, Lambadiari V, Maratou E, Raptis SA. Insulin effects in muscle and adipose tissue. Diabet Res Clin Pract. 2011;93:S52–S59. doi:10.1016/S0168-8227(11)70014-6
33. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet. 2018;391(10138):2449–2462. doi:10.1016/S0140-6736(18)31320-5
34. American Diabetes Association. Standards of medical care in diabetes—2022 abridged for primary care providers. Clin Diabetes. 2022;40(1):10–38. doi:10.2337/cd22-as01
35. Akil AAS, Yassin E, Al-Maraghi A, Aliyev E, Al-Malki K, Fakhro KA. Diagnosis and treatment of type 1 diabetes at the Dawn of the personalized medicine era. J Transl Med. 2021;19(1):137. doi:10.1186/s12967-021-02778-6
36. Cui D, Feng X, Lei S, et al. Pancreatic β-cell failure, clinical implications, and therapeutic strategies in type 2 diabetes. Chin Med J. 2024;137(7):791–805. doi:10.1097/CM9.0000000000003034
37. Galicia-Garcia U, Benito-Vicente A, Jebari S, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci. 2020;21(17):6275. doi:10.3390/ijms21176275
38. Kyrou I, Tsigos C, Mavrogianni C, et al. Sociodemographic and lifestyle-related risk factors for identifying vulnerable groups for type 2 diabetes: a narrative review with emphasis on data from Europe. BMC Endocr Disord. 2020;20(S1):134. doi:10.1186/s12902-019-0463-3
39. Bailey CJ, Day C. Treatment of type 2 diabetes: future approaches. Br Med Bull. 2018;126(1):123–137. doi:10.1093/brimed/ldy013
40. Schwartz SS, Epstein S, Corkey BE, Grant SFA, Gavin JR, Aguilar RB. The time is right for a new classification system for diabetes: rationale and implications of the β-cell–centric classification schema. Diabetes Care. 2016;39(2):179–186. doi:10.2337/dc15-1585
41. Alfaris N, Waldrop S, Johnson V, Boaventura B, Kendrick K, Stanford FC. GLP-1 single, dual, and triple receptor agonists for treating type 2 diabetes and obesity: a narrative review. EClinicalMedicine. 2024;75:102782. doi:10.1016/j.eclinm.2024.102782
42. Schroeder EB. Management of Type 2 Diabetes: Selecting Amongst Available Pharmacological Agents. South Dartmouth (MA): MDText.com, Inc.; 2022.
43. Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–546. doi:10.1038/nature13270
44. Saisho Y. β-cell dysfunction: its critical role in prevention and management of type 2 diabetes. World J Diabetes. 2015;6(1):109. doi:10.4239/wjd.v6.i1.109
45. Lebovitz HE. Oral therapies for diabetic hyperglycemia. Endocrinol Metab Clin North Am. 2001;30(4):909–933. doi:10.1016/S0889-8529(05)70221-8
46. Mahgoub MO, Ali II, Adeghate JO, Tekes K, Kalász H, Adeghate EA. An update on the molecular and cellular basis of pharmacotherapy in type 2 diabetes mellitus. Int J Mol Sci. 2023;24(11):9328. doi:10.3390/ijms24119328
47. Mir KB, Abrol V, Singh N, et al. Spectroscopic characterization and pharmacokinetic evaluation of amorphous solid dispersions of glibenclamide for bioavailability enhancement in Wistar rats. Biomed Chromatogr. 2024;38(8). doi:10.1002/bmc.5901
48. Ahmed TA, Alotaibi HA, Almehmady AM, Safo MK, El-Say KM. Influences of glimepiride self-nanoemulsifying drug delivery system loaded liquisolid tablets on the hypoglycemic activity and pancreatic histopathological changes in streptozotocin-induced hyperglycemic rats. Nanomaterials. 2022;12(22):3966. doi:10.3390/nano12223966
49. Talari R, Varshosaz J, Mostafavi SA, Nokhodchi A. Gliclazide microcrystals prepared by two methods of in situ micronization: pharmacokinetic studies in diabetic and normal rats. AAPS Pharm Sci Tech. 2010;11(2):786–792. doi:10.1208/s12249-010-9441-9
50. Albetawi S, Abdalhafez A, Abu-Zaid A. A review on recent controlled release strategies for oral drug delivery of repaglinide (a BCS class II drug). Pharm Nanotechnol. 2021;9(5):326–338. doi:10.2174/2211738510666211221165318
51. Swain RP, Subudhi BB. Solid dispersion of nateglinide in polyoxy ethylene-polyoxy propylene block copolymer: in vitro and in vivo evaluation. Indian J Pharm Educ Res. 2017;51(4):562–570. doi:10.5530/ijper.51.4.85
52. Budiman A, Ivana H, Huang KA, et al. Biocompatible natural polymer-based amorphous solid dispersion system improving drug physicochemical properties, stability, and efficacy. Polymers. 2025;17(15):2059. doi:10.3390/polym17152059
53. Damian F, Blaton N, Naesens L, et al. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur J Pharm Sci. 2000;10(4):311–322. doi:10.1016/S0928-0987(00)00084-1
54. Karataş A, Yüksel N, Baykara T. Improved solubility and dissolution rate of piroxicam using gelucire 44/14 and labrasol. Il Farmaco. 2005;60(9):777–782. doi:10.1016/j.farmac.2005.04.014
55. Tanaka N, Imai K, Okimoto K, et al. Development of novel sustained-release system, disintegration-controlled matrix tablet (DCMT) with solid dispersion granules of nilvadipine. J Control Release. 2005;108(2–3):386–395. doi:10.1016/j.jconrel.2005.08.024
56. Urbanetz NA. Stabilization of solid dispersions of nimodipine and polyethylene glycol 2000. Eur J Pharm Sci. 2006;28(1–2):67–76. doi:10.1016/j.ejps.2005.12.009
57. Vasconcelos T, Barrocas P, Lima R, Soares-da-Silva P. Development of an opicapone third generation solid dispersion. 2013. doi:10.13140/RG.2.1.1171.7604
58. Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today. 2007;12(23–24):1068–1075. doi:10.1016/j.drudis.2007.09.005
59. Budiman A, Hafidz NPM, Azzahra RSS, et al. Advancing the physicochemical properties and therapeutic potential of plant extracts through amorphous solid dispersion systems. Polymers. 2024;16(24):3489. doi:10.3390/polym16243489
60. He Y, Ho C. Amorphous solid dispersions: utilization and challenges in drug discovery and development. J Pharm Sci. 2015;104(10):3237–3258. doi:10.1002/jps.24541
61. Laitinen R, Löbmann K, Strachan CJ, Grohganz H, Rades T. Emerging trends in the stabilization of amorphous drugs. Int J Pharm. 2013;453(1):65–79. doi:10.1016/j.ijpharm.2012.04.066
62. Nielsen LH, Rades T, Müllertz A. Stabilisation of amorphous furosemide increases the oral drug bioavailability in rats. Int J Pharm. 2015;490(1–2):334–340. doi:10.1016/j.ijpharm.2015.05.063
63. Leuner C. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. doi:10.1016/S0939-6411(00)00076-X
64. Cln V, Park C, Lee BJ. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85(3):799–813. doi:10.1016/j.ejpb.2013.09.007
65. Zhang J, Han R, Chen W, et al. Analysis of the literature and patents on solid dispersions from 1980 to 2015. Molecules. 2018;23(7):1697. doi:10.3390/molecules23071697
66. Punčochová K, Heng JYY, Beránek J, Štěpánek F. Investigation of drug–polymer interaction in solid dispersions by vapour sorption methods. Int J Pharm. 2014;469(1):159–167. doi:10.1016/j.ijpharm.2014.04.048
67. Wilson VR, Lou X, Osterling DJ, et al. Amorphous solid dispersions of enzalutamide and novel polysaccharide derivatives: investigation of relationships between polymer structure and performance. Sci Rep. 2020;10(1):18535. doi:10.1038/s41598-020-75077-7
68. Budiman A, Aulifa DL. A comparative study of the pharmaceutical properties between amorphous drugs loaded-mesoporous silica and pure amorphous drugs prepared by solvent evaporation. Pharmaceuticals. 2022;15(6):730. doi:10.3390/ph15060730
69. Zhang J, Guo M, Luo M, Cai T. Advances in the development of amorphous solid dispersions: the role of polymeric carriers. Asian J Pharm Sci. 2023;18(4):100834. doi:10.1016/j.ajps.2023.100834
70. Li J, Wang Y, Yu D. Effects of additives on the physical stability and dissolution of polymeric amorphous solid dispersions: a review. AAPS Pharm Sci Tech. 2023;24(7):175. doi:10.1208/s12249-023-02622-8
71. Karagianni A, Kachrimanis K, Nikolakakis I. Co-Amorphous solid dispersions for solubility and absorption improvement of drugs: composition, preparation, characterization and formulations for oral delivery. Pharmaceutics. 2018;10(3):98. doi:10.3390/pharmaceutics10030098
72. Pandi P, Bulusu R, Kommineni N, Khan W, Singh M. Amorphous solid dispersions: an update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int J Pharm. 2020;586:119560. doi:10.1016/j.ijpharm.2020.119560
73. Ma X, Williams RO. Characterization of amorphous solid dispersions: an update. J Drug Deliv Sci Technol. 2019;50:113–124. doi:10.1016/j.jddst.2019.01.017
74. Newman A, Knipp G, Zografi G. Assessing the performance of amorphous solid dispersions. J Pharm Sci. 2012;101(4):1355–1377. doi:10.1002/jps.23031
75. Newman A, Nagapudi K, Wenslow R. Amorphous solid dispersions: a robust platform to address bioavailability challenges. Ther Deliv. 2015;6(2):247–261. doi:10.4155/tde.14.101
76. Qu B, Wu H, Ding Z, et al. Amorphous solid dispersion formulation of pimobendan for bioavailability enhancement: a comprehensive study on miscibility, interactions, and in vitro dissolution behavior. J Drug Deliv Sci Technol. 2025;108:106884. doi:10.1016/j.jddst.2025.106884
77. Safna Hussan KP, Babu TD, Thayyil MS, Sreeshma TS, Archana A. Physicochemical characterization and biological evaluation of amorphous solid dispersions of an anticancerous drug: erlotinib HCl. Sci Rep. 2025;15(1):23787. doi:10.1038/s41598-025-07692-1
78. Bhujbal SV, Mitra B, Jain U, et al. Pharmaceutical amorphous solid dispersion: a review of manufacturing strategies. Acta Pharm Sin B. 2021;11(8):2505–2536. doi:10.1016/j.apsb.2021.05.014
79. Hussan KPS, Govindaraj G, Correia NT, Shinyashiki N, Thayyil MS, Babu TD. Molecular dynamics and interactions in amorphous solid dispersion of Erlotinib HCl for improved cancer therapy. J Mol Struct. 2025;1336:142014. doi:10.1016/j.molstruc.2025.142014
80. Anane-Adjei AB, Jacobs E, Nash SC, et al. Amorphous solid dispersions: utilization and challenges in preclinical drug development within AstraZeneca. Int J Pharm. 2022;614:121387. doi:10.1016/j.ijpharm.2021.121387
81. Baghel S, Cathcart H, O’Reilly NJ. Polymeric amorphous solid dispersions: a review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J Pharm Sci. 2016;105(9):2527–2544. doi:10.1016/j.xphs.2015.10.008
82. Qian K, Stella L, Jones DS, Andrews GP, Du H, Tian Y. Drug-Rich phases induced by amorphous solid dispersion: arbitrary or intentional goal in oral drug delivery? Pharmaceutics. 2021;13(6):889. doi:10.3390/pharmaceutics13060889
83. Suys EJA, Chalmers DK, Pouton CW, Porter CJH. Polymeric precipitation inhibitors promote fenofibrate supersaturation and enhance drug absorption from a type IV lipid-based formulation. Mol Pharm. 2018;15(6):2355–2371. doi:10.1021/acs.molpharmaceut.8b00206
84. Feng D, Peng T, Huang Z, et al. Polymer–Surfactant system based amorphous solid dispersion: precipitation inhibition and bioavailability enhancement of itraconazole. Pharmaceutics. 2018;10(2):53. doi:10.3390/pharmaceutics10020053
85. Chen T, Li Q, Ai G, et al. Enhancing hepatoprotective action: oxyberberine amorphous solid dispersion system targeting TLR4. Sci Rep. 2024;14(1):14924. doi:10.1038/s41598-024-65190-2
86. Wang L, Wu W, Wang L, Wang L, Zhao X. Highly water-soluble solid dispersions of honokiol: preparation, solubility, and bioavailability studies and anti-tumor activity evaluation. Pharmaceutics. 2019;11(11):573. doi:10.3390/pharmaceutics11110573
87. Newman A. Pharmaceutical Amorphous Solid Dispersions. Wiley; 2015.
88. Dannenfelser R, Yalkowsky SH. Database for aqueous solubility of nonelectrolytes. Bioinformatics. 1989;5(3):235–236. doi:10.1093/bioinformatics/5.3.235
89. Kumar A. Solid dispersion- strategy to enhance solubility and dissolution of poorly water soluble drugs. Univer J Pharmaceut Res. 2017;2(5):54–59. doi:10.22270/ujpr.v2i5.RW4
90. Sakure K, Kumari L, Badwaik H. Development and evaluation of solid dispersion based rapid disintegrating tablets of poorly water-soluble anti-diabetic drug. J Drug Deliv Sci Technol. 2020;60:101942. doi:10.1016/j.jddst.2020.101942
91. Maggi L, Canobbio A, Bruni G, Musitelli G, Conte U. Improvement of the dissolution behavior of gliclazide, a slightly soluble drug, using solid dispersions. J Drug Deliv Sci Technol. 2015;26:17–23. doi:10.1016/j.jddst.2015.01.002
92. Surti N, Mahajan AN, Patel D, Patel A, Surti Z. Spray dried solid dispersion of repaglinide using hypromellose acetate succinate: in vitro and in vivo characterization. Drug Dev Ind Pharm. 2020;46(10):1622–1631. doi:10.1080/03639045.2020.1812631
93. Qushawy M, Nasr A, Swidan S, Mortagi Y. Development and characterization of glimepiride novel solid nanodispersion for improving its oral bioavailability. Sci Pharm. 2020;88(4):52. doi:10.3390/scipharm88040052
94. Reginald-Opara JN, Attama A, Ofokansi K, Umeyor C, Kenechukwu F. Molecular interaction between glimepiride and Soluplus ® -PEG 4000 hybrid based solid dispersions: characterisation and anti-diabetic studies. Int J Pharm. 2015;496(2):741–750. doi:10.1016/j.ijpharm.2015.11.007
95. Petkov V, Vinarov Z, Tcholakova S. Mechanisms of dissolution and crystallization of amorphous glibenclamide. Int J Pharm. 2024;666:124820. doi:10.1016/j.ijpharm.2024.124820
96. Mansour HF, Aly FU. In vitro evaluation and in vivo performance of lyophilized gliclazide. Drug Dev Ind Pharm. 2015;41(4):650–657. doi:10.3109/03639045.2014.891131
97. Mohamed MS, Abdelhafez WA, Zayed G, Samy AM. In vitro and in vivo characterization of fast dissolving tablets containing gliquidone–pluronic solid dispersion. Drug Dev Ind Pharm. 2019;45(12):1973–1981. doi:10.1080/03639045.2019.1689993
98. Chauhan B, Shimpi S, Paradkar A. Preparation and evaluation of glibenclamide-polyglycolized glycerides solid dispersions with silicon dioxide by spray drying technique. Eur J Pharm Sci. 2005;26(2):219–230. doi:10.1016/j.ejps.2005.06.005
99. Biswal S, Sahoo J, Murthy PN, Giradkar RP, Avari JG. Enhancement of dissolution rate of gliclazide using solid dispersions with polyethylene glycol 6000. AAPS Pharm Sci Tech. 2008;9(2):563–570. doi:10.1208/s12249-008-9079-z
100. Pahovnik D, Reven S, Grdadolnik J, Borštnar R, Mavri J, Žagar E. Determination of the interaction between glimepiride and hyperbranched polymers in solid dispersions. J Pharm Sci. 2011;100(11):4700–4709. doi:10.1002/jps.22662
101. Li H, Ma L, Li X, et al. A simple and effective method to improve bioavailability of glimepiride by utilizing hydrotropy technique. Eur J Pharm Sci. 2015;77:154–160. doi:10.1016/j.ejps.2015.06.016
102. Pisay M, Navti PD, Rao V, Koteshwara KB, Mutalik S. Investigation of drug-polymer miscibility and design of ternary solid dispersions for oral bioavailability enhancement by Hot Melt Extrusion. J Drug Deliv Sci Technol. 2023;90:105107. doi:10.1016/j.jddst.2023.105107
103. Adibkia K, Ghajar S, Osouli-Bostanabad K, Balaei N, Emami S, Barzegar-Jalali M. Novel gliclazide electrosprayed nano-solid dispersions: physicochemical characterization and dissolution evaluation. Adv Pharm Bull. 2019;9(2):231–240. doi:10.15171/apb.2019.026
104. Yin LF, Huang SJ, Zhu CL, et al. In vitro and in vivo studies on a novel solid dispersion of repaglinide using polyvinylpyrrolidone as the carrier. Drug Dev Ind Pharm. 2012;38(11):1371–1380. doi:10.3109/03639045.2011.652635
105. Censi R, Gigliobianco MR, Dubbini A, Malaj L, Di Martino P. New nanometric solid dispersions of glibenclamide in neusilin® UFL2. AAPS Pharm Sci Tech. 2016;17(5):1204–1212. doi:10.1208/s12249-015-0457-z
106. Guan J, Han J, Zhang D, et al. Increased dissolution rate and oral bioavailability of hydrophobic drug glyburide tablets produced using supercritical CO2 silica dispersion technology. Eur J Pharm Biopharm. 2014;86(3):376–382. doi:10.1016/j.ejpb.2013.10.008
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Nanometerizing Taxifolin Into Selenized Liposomes to Ameliorate Its Hypoglycemic Effect by Optimizing Drug Release and Bioavailability
Qi C, Xing H, Ding N, Feng W, Wu Y, Zhang X, Yu Y
International Journal of Nanomedicine 2025, 20:2225-2240
Published Date: 21 February 2025