")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 15
The Impact of Pharmacogenetics on Pharmacokinetics and Pharmacodynamics in Neonates and Infants: A Systematic Review
Authors Yalçin N, Flint RB, van Schaik RHN, Simons SHP, Allegaert K
Received 11 May 2022
Accepted for publication 14 June 2022
Published 30 June 2022 Volume 2022:15 Pages 675—696
DOI https://doi.org/10.2147/PGPM.S350205
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Nadir Yalçin,1,2 Robert B Flint,2,3 Ron HN van Schaik,3,4 Sinno HP Simons,3 Karel Allegaert2,5– 7
1Department of Clinical Pharmacy, Faculty of Pharmacy, Hacettepe University, Ankara, Turkey; 2Department of Hospital Pharmacy, Erasmus MC, Rotterdam, the Netherlands; 3Division of Neonatology, Department of Pediatrics, Erasmus MC, Rotterdam, the Netherlands; 4Department of Clinical Chemistry, Erasmus MC, Rotterdam, the Netherlands; 5Department of Pharmaceutical and Pharmacological Sciences, KU Leuven, Leuven, Belgium; 6Department of Development and Regeneration, KU Leuven, Leuven, Belgium; 7Child and Youth Institute, KU Leuven, Leuven, Belgium
Correspondence: Karel Allegaert, Neonatal Intensive Care Unit, UZ Leuven, Herestraat 49, Leuven, 3000, Belgium, Tel +32-016-342020, Fax +32-016-343209, Email [email protected]
Abstract: In neonates, pharmacogenetics has an additional layer of complexity. This is because in addition to genetic variability in genes that code for proteins relevant to clinical pharmacology, there are rapidly maturational changes in these proteins. Consequently, pharmacotherapy in neonates has unique challenges. To provide a contemporary overview on pharmacogenetics in neonates, we conducted a systematic review to identify, describe and quantify the impact of pharmacogenetics on pharmacokinetics and -dynamics in neonates and infants (PROSPERO, CRD42022302029). The search was performed in Medline, Embase, Web of Science and Cochrane, and was extended by a PubMed search on the ‘top 100 Medicines’ (medicine + newborn/infant + pharmacogen*) prescribed to neonates. Following study selection (including data in infants, PGx related) and quality assessment (Newcastle–Ottawa scale, Joanna Briggs Institute tool), 55/789 records were retained. Retained records relate to metabolizing enzymes involved in phase I [cytochrome P450 (CYP1A2, CYP2A6, CYP2B6, CYP2C8/C9/C18, CYP2C19, CYP2D6, CYP3A5, CYP2E1)], phase II [glutathione-S-transferases, N-acetyl transferases, UDP-glucuronosyl-transferase], transporters [ATP-binding cassette transporters, organic cation transporters], or receptor/post-receptor mechanisms [opioid related receptor and post-receptor mechanisms, tumor necrosis factor, mitogen-activated protein kinase 8, vitamin binding protein diplotypes, corticotrophin-releasing hormone receptor-1, nuclear receptor subfamily-1, vitamin K epoxide reductase complex-1, and angiotensin converting enzyme variants]. Based on the available overview, we conclude that the majority of reported pharmacogenetic studies explore and extrapolate observations already described in older populations. Researchers commonly try to quantify the impact of these polymorphisms in small datasets of neonates or infants. In a next step, pharmacogenetic studies in neonatal life should go beyond confirmation of these associations and explore the impact of pharmacogenetics as a covariate limited to maturation of neonatal life (ie, fetal malformations, breastfeeding or clinical syndromes). The challenge is to identify the specific factors, genetic and non-genetic, that contribute to the best benefit/risk balance.
Keywords: developmental pharmacology, infant, ontogeny, child development, genetic variation
Introduction
At the beginning of the second half of the 20th century, Friedrich Vogel developed the concept that genes play an important role in determining drug response and coined the term pharmacogenetics (PGx).1 Subsequently, PGx became a powerful tool to understand variability in drug exposure, efficacy, tolerability, or toxicity. When applied to clinical care, this holds the promise to be integrated in individualization and precision medicine.2,3 In neonates, PGx has an additional complexity next to genetic variability (ie, polymorphisms) in the genes that code for proteins responsible for drug pharmacokinetics (PK) (like metabolism, transporter) or pharmacodynamics (PD) (like receptor, target enzyme). This is because there are rapidly evolving, ontogenic changes in expression of these same proteins or processes related to maturation of renal, cardiac, intestinal function, etc.4 Maturation (like receptor expression, receptor activity, cellular metabolism, enzyme activity) hereby displays collinearity with age and weight, be it with a lot of diversity in maturational patterns.5,6
To a certain extent, this subpopulation displays a specific “phenoconversion” setting, with a maturational, age-dependent mismatch between genotype and phenotype.7 Using the same approach, PG holds the promise in related fields of neonatal medicine, like forensic medicine and sudden infant death syndrome.8
PK describes the relationship of concentration over time, PD describes the relationship between a concentration and (adverse) effects.9 In total about 18,000 variants in 231 pharmacogenes have been determined, the majority of which are rare variants, some of which may be deleterious regarding gene function with significant consequences in PK and PD.10 This genetic variability can affect drug absorption, distribution, metabolism, elimination, toxicity (ADMET), and effectiveness.
There are many illustrations on the integration of PGx and age-related maturation (ie ontogeny) to improve the prediction of phenotypic drug ADMET [like cytochrome P450 (CYP) C219, CYP 2D6 or N-acetyl transferase-2 (NAT2), UDP-glucuronosyltransferase 2B7 (UGT2B7), mu-opioid receptor (Opioid OPRM1), Catechol-O-methyltransferase (COMT), or P-glycoprotein (ABCB1)].11,12 This phenotypic variability is further aggravated by interfering disease characteristics (like renal or liver failure, sepsis, growth restriction) or interventions (like co-medication, extracorporeal membrane oxygenation, whole-body cooling).
As we are not aware of a recent systematic review on this topic and to provide a contemporary overview on the current knowledge on PGx in neonatal pharmacology, we performed a systematic review to identify, describe and quantify the impact of PGx on PK and PD in neonates and infants.
Materials and Methods
This review was performed based on the PRISMA (Preferred Reporting Items for Systematic Reviews and MetaAnalysis) guideline.13 The protocol was registered in PROSPERO on February 6th, 2022, an international prospective register of systematic reviews (CRD42022302029, review ongoing).14
Search Strategy
Potentially relevant publications published between January 1st, 1946 and January 1st, 2022 were identified by searching the following databases: Medline ALL (1946–present), Embase (1971–present), Web of Science Core Collection (1975–present), and Cochrane Central Register of Controlled Trials (1992–present). In addition, reference lists of relevant reviews were screened for any potentially relevant individual studies (backward snowballing). The search strategy, terms or subject headings (and related concepts, depending on the database) are provided in Supplementary File 1. Furthermore, a search was performed based on the “Top 100 medications prescribed in the NICU” (800,000 cases, 2010–2018), as recently reported by Stark et al.15 The names of each drug were searched with the keywords “newborn or infant” and “pharmacogen*” in the PubMed database until February 22nd, 2022.
Study Selection and Data Extraction
Search results were downloaded into EndNote X9, duplicates were removed automatically and manually. One reviewer (N.Y.) verified the removal of duplicates. Two reviewers (N.Y. and K.A.) subsequently screened the reports independently by title and abstract, and then by full-text for potentially eligible studies. Discrepancies were discussed with a third reviewer (R.F.) until consensus was reached. The following data were extracted when reported in the papers retained: first author, year, country, study aim, setting, study design, study population, sample size (male/female), ethnicity or nationality, postnatal or gestational age, and impact of PGx on PK/PD in these studies. Only clinical trials and case reports were included in this systematic review. Due to a significant degree of heterogeneity between PGx and PK/PD measures, a meta-analysis was not deemed suitable.
Inclusion and Exclusion Criteria
The inclusion criteria were formulated using the PICOS (Participant, Intervention, Comparison, Outcome, and Study) design.13 Studies were included if they met the following criteria:
Participants: the study population involved only patients <1 year, including neonates (day 1–28). For studies with a mixed population of participants, the studies were included for only infants according to the age (minimum or categorical values) of the patients. Studies in children and adults were excluded.
Intervention: since the majority of PGx findings are cohort (retrospective or prospective) studies, clinician-oriented intervention studies are not available. However, this may vary depending on the study design involved. For studies with a mixed population with ontogenetic and/or phenotypic study design, outcomes relating to the analysis of PGx and PK/PD findings were included if data could be extracted.
Comparator and outcome: data on impact of PGx variability on PK/PD parameters were included, with or without a comparator group.
Study design: Any clinical trial and case report published in English was considered for inclusion, regardless of study design.
The following types of studies were excluded: (a) papers that rather report on pre-assessment before PG implementation in children; (b) studies containing only ontogenic/phenotypic data, without PG data, or (c) non-primary clinical publications, such as conference abstracts, editorials, reviews, letters, and animal studies.
Risk of Bias and Quality Assessment
The risk of bias and quality assessment for each paper was assessed using the Newcastle–Ottawa Scale (NOS)16 for non-randomized (case-control, cohort, and randomized controlled trials (RCTs) respectively) studies and the Joanna Briggs Institute (JBI) Critical Appraisal Tools for case reports. The risk of bias and quality assessment was primarily performed by N.Y., in consultation with K.A. and R.F. According to the NOS, a study can be awarded a maximum of one star for each numbered item within the “Selection” (4 items) and Exposure (3 items) categories. A maximum of two stars can be given for Comparability (1 item).16 In the JBI checklist, a case report can be evaluated with 8 bias items, each of which answered as “Yes”, “No”, “Unclear” or “Not/Applicable”. The overall appraisal is completed with one of the options: “Include”, “Exclude” or “Seek further info”.17
Results
We summarized the numbers of records screened, assessed and retained following identification via databases or registries, or by other methods (backward snowballing and top 100 list of medications prescribed in the NICU) in Figure 1 and Table 1. The flow diagram provides information on reasons for exclusion, and the extent of (dis)agreement. In total 55 reports were retained, published from 1998 onwards (2 reports before 2000, 10 between 2000–2009, 38 between 2010–2019, and already 5 reports from 2020 onwards), with a diversity of countries involved (based on the corresponding author).
 |  |  |  |  |  | 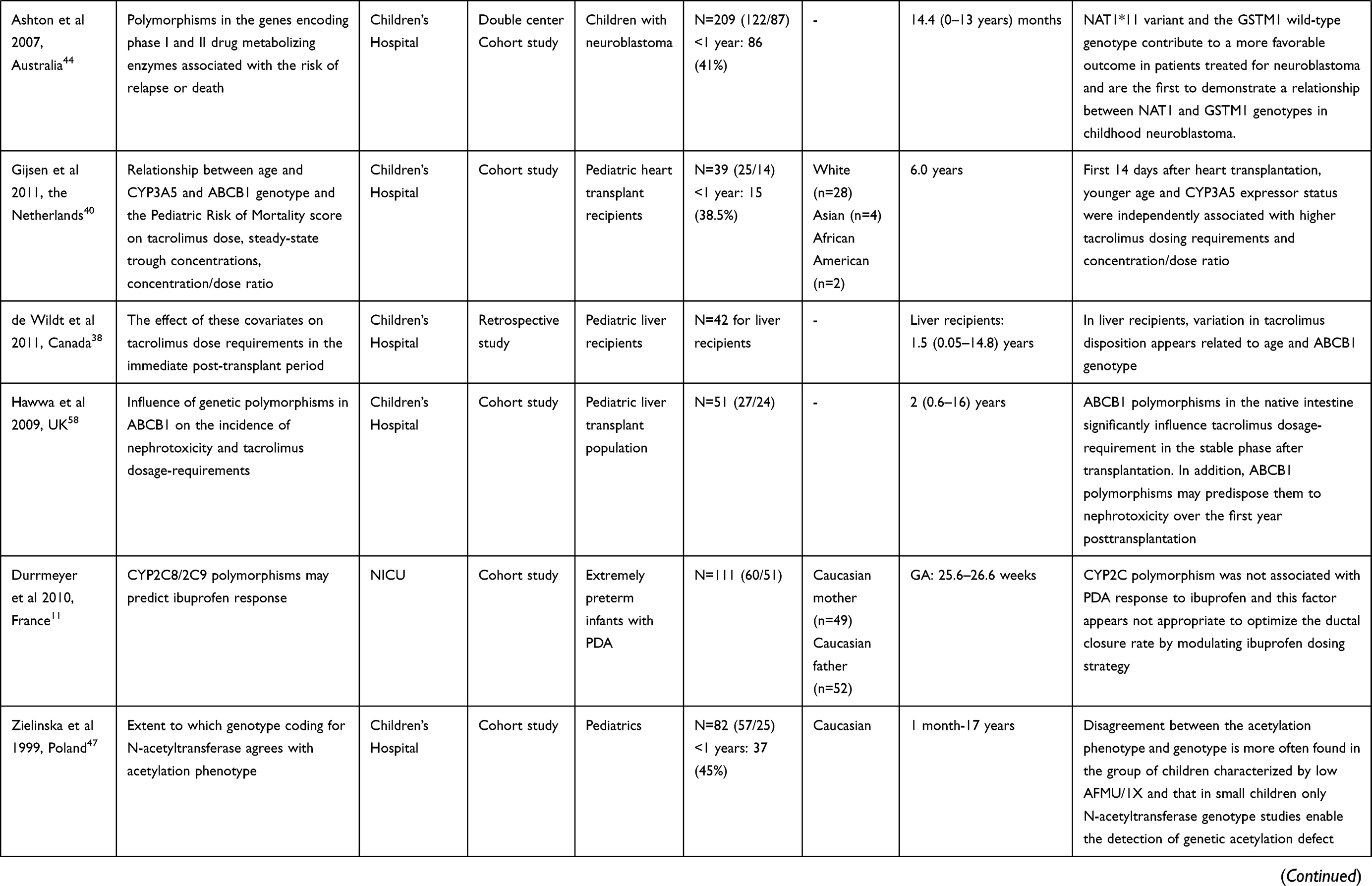 |
Table 1 Description of Studies About Impact of Pharmacogenetics on Pharmacokinetics and -Dynamics in Neonates and/or Infants |
 |
Figure 1 PRISMA flow diagram showing process of study selection for inclusion in systematic review. Notes: PRISMA figure adapted from Moher D, Liberati A, Altman D, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. Journal of clinical epidemiology. 2009;62(10). Creative Commons.13 |
Quality assessment was based on the NOS tool in 34 cohort studies (Table S1), 11 case-control studies (Table S2) and 6 randomized controlled trials (Table S3), and by the JBI critical appraisal tool for 4 case reports (Table S4). Because of the relevant number of reports retained, we post hoc decided to subdivide these findings by phase I related, phase II related, transporter related PG reports, or receptor and post receptor PGx related reports.
Phase I Related Pharmacogenetics
CYP1A2
In a dataset of 99 Chinese preterm neonates (gestational age (GA) 25–33 weeks, 201 caffeine concentrations), body weight, postmenstrual age (PMA) and serum creatinine affected caffeine clearance, while CYP1A2 polymorphism was not identified as independent covariate.18
CYP2A6
In a cohort of 270 children (2–72 months old) that had to undergo cardiac surgery, dexmedetomidine (3 µg/kg, intranasal) was administered for pre-sedation. In all cases, dexmedetomidine was quantified in blood samples collected one hour after administration. Comparing GG (185/257) versus T carriers (either TT or TG, 72/257) cases (CYP2A6 rs835309 genotypes), the mean concentration was 0.456 versus 0.397 ng/mL, so that T carriers had a 13% lower concentration at one hour, reflecting faster clearance (p=0.025).19 Unfortunately, no sub-analysis in infants, nor any analysis on maturational covariates were reported in this bio-analytical paper.19
CYP2B6
In a dataset of 25 young children (range 4–23 months), of whom 14 were infants (n = 4, ≤6 months, n = 10, 7–12 months), the median cyclophosphamide clearance was 46.6 (range 9.4–153) mL/min/m2. Despite this marked inter-patient variability, no covariates (age, weight, CYP2B6 or CYP2C19) were identified, with only a trend for lower clearance in CYP2B6 *1/*6 (n = 7) versus *1/*1. The authors suggested that the absence of a significant difference was due to the small number.20
CYP2C8/C9/C18
As non-steroidal anti-inflammatory drugs (NSAIDs) are commonly used in preterm neonates to induce closure of a patent ductus arteriosus (PDA), data on the impact of CYP2C8/2C9 polymorphisms on ibuprofen and indomethacin have been reported, all with focus on pharmacodynamic outcome (ie, the need for surgical closure). Durrmeyer et al reported on the response to ibuprofen in a cohort of 86 extremely preterm neonates with a hemodynamically significant PDA. While a higher gestational age (GA) and non-Caucasian ethnicity were associated with an ibuprofen response, CYP2C polymorphisms (CYP2C8*3, CYP2C9*2, CYP2C8*3) were not.11 On indomethacin, we retrieved two papers.21 Besides other factors (GA, surfactant use), the presence of CYP2C9*2 (adjusted OR, 3.74) was associated with indomethacin treatment failure (defined by the need for surgical ligation, 35% of cases) in a 3-center cohort study on 144 preterm (GA 22–32 weeks) cases.21 This paper confirmed a previously published case-control (52 non-responders versus 96 responders, <32 weeks) analysis. Besides GA, these authors identified an association between CYP2C9 rs2153628 (increased odds of response 1.92) or rs1799853, and DA response.22
We also retrieved a case report of phenytoin toxicity (lethargy) in a two-month-old Thai infant, associated with a CYP2C9 *1/*3 (slow metabolizer) and a CYP2C19 *1/*1 (normal metabolizer) genotype.23
Related to coumarins, we retrieved 3 datasets reporting on CYP2C9 and 2C18 polymorphisms in children, be it that the number of infants included in these studies was limited. Using an adult PK/PD model bridging effort to children for warfarin, the INR response was predicted reasonable well in 64 children (age range 0.05–18.9 years, weight range 3.4–94 kg), be it with a tendency to overpredict the international normalized ratio (INR), or underpredict dose in children <2 years. It was hereby extrapolated from adult findings that CYP2C9 polymorphisms explained up to a 4.2-fold (CYP2C9 *3/*3 vs *1/*1) difference in warfarin maintenance dose.24 Another cohort of 123 children (only 2 infants included) body surface area (BSA) and indication explained 45% of the variability in acenocoumarol dose requirement (INR targeting), while Fontan circulation was another covariate (4.3 to 3 mg/kg, 1.4 fold difference). Specific for CYP2C9*2/CYP2C9*3, the median maintenance dose displayed a 1.35 fold difference (median observed dose, 1.21 versus 1.88 mg/day), for CYP2C18 (GG versus AG) this was 1.15 fold (median observed dose, 1.62 to 1.85 mg/day).25 Finally, in a dataset with 118 cases (age range 3 months to 18 years, 19 cases <3 years) on acenocoumarol dosing, CYP2C9 (*1/*1, *1/*x, or *x/*x:) to a certain extent (2%) showed a 1.88 fold difference in weekly median dose requirement of warfarin, ie 24.8, 22.2, or 13.13 mg/week, respectively.26
CYP2C19
In the previously mentioned (related to CYP2B6) cyclophosphamide study in young children, CYP2C19 polymorphisms (*1/*1, *1/*2) were neither relevant covariates.20 Other observations relate to phenobarbital and proton pump inhibitors (pantoprazole, omeprazole) PK.27–29 Body weight and age determined phenobarbital clearance in a cohort of 52 neonates and young infants (age range, 8 days - 6 months), while CYP2C19 (*1/*1, *1/*2, *2/*2 or *2/*3) polymorphisms were not, in contrast to similar studies in adults.27 As part of a population PK study with oral pantoprazole (granules) in preterm infants and neonates, the mean area under the plasma concentration versus time curve (AUC) was substantially higher in 2 (2/40) cases (median PMA 37 weeks, range 33–44) with a CYP2C19 poor metabolizer profile. In these poor metabolizers (*2/*2), the AUC was 10.8 and 27 µg.h/mL (1.9- and 4.8-fold increase) respectively, while the median AUC value was 5.63 (range 1.3–27) µg.h/mL.28 Along the same line, Zhao et al quantified the impact of CYP2C19 polymorphisms (poor, intermediate to extensive metabolizer) on the formation clearance of omeprazole to 5-hydroxy-omeprazole.29 Compared to the extensive/ultrarapid metabolizer (*1/*1, *1/*17, *17/*17) genotype, clearance to 5-hydroxy-omeprazole is at the 44.9th and 12.5th percentile for the intermediate (*1/*2 or *2/*17) and poor metabolizer (*2/*2), respectively.
CYP2D6
For CYP2D6 polymorphism and its related CYP2D6 activity score, observations on dextromethorphan (oral) and tramadol (parenteral) have been reported.30–32 Dextromethorphan O- and N-demethylation in the first year of life was quantified, based on paired urine metabolic ratio’s following a single oral (0.3 mg/kg) dose at 0.5, 1, 2, 4, 6 and 12 months in 193 term born infants. There was a strong correlation between CYP2D6 genotype (CYP2D6 activity score) and O-demethylation (r=−0.638), with concordance from 2 weeks postnatal age onwards. The mean log urinary dextromethorphan/dextrorphan (DM/DX) metabolic ratio for a given CYP2D6 activity score (0, 0.5, 1, 2, >2) was −0.05, −1.29, −1.64, −2.05, or −2.21 respectively.30 In 86 neonates (median PMA 39 weeks, range 25–62) exposed to continuous intravenous tramadol, the plasma and urine log tramadol/0-demethyl tramadol (M/M1) ratio were determined (R2 0.59 and 0.64 respective) by PMA and the CYP2D6 activity score in a multiple regression model in 86 neonates (median PMA 39 weeks, range 25–62). Median urine log M/M1 values for a CYP2D6 activity score of 0, 1, 2 or 3 were 2.3, 1, 0.7 and 0.2, median plasma log M/M1 values for the same score (1, 2 or 3) were 1, 0.5 and 0.35 in this cohort. Tramadol clearance to M1 displayed a CYP2D6 activity score, with an additional maturational pattern. Clearance to M1 had a large between subject variability (111%). Size and PMA were the major contributors (52.7%), while CYP2D6 activity score contributed 6.4%.31,32
Two case reports on the impact of CYP2D6 polymorphism on pharmacotherapy related to lopinavir/ritonavir and dihydrocodeine were also retrieved.33,34 The first case relates to pharmacogenomic adaptation of antiretroviral therapy (lopinavir failure) in a 6 month old African infant with a CYP2D6 ultrarapid metabolizer profile.33 The second paper described two children, of whom one was a neonate (1 month, 3 kg) with a CYP2D6*1/*10-*36 genotype. The newborn was exposed to dihydrocodeine-containing cough mixture (1 mg q12h dihydrocodeine phosphate, for 2 days), and subsequently displayed acute respiratory depression, followed by ventilation for 3 days.34
CYP3A5
Related to tacrolimus PK, we retrieved 6 reports (5 hepatic and 1 cohort on cardiac transplants). However, the number of infants in these observations were rather limited, so these data should be interpreted with caution in this specific population. Fukudo et al reported on a population PK study in 130 (including PGx in 65/130) pediatric transplant recipients (age 0.1–15 years) shortly after (<50 days) transplantation. CYP3A5*1 carrier donor liver was associated with a 2-fold higher oral clearance (CL/F) compared to CYP3A5*3/*3.35 Chen et al also studied personalized tacrolimus dosing requirements by CYP3A5 recipient and donor polymorphisms in 90 children (median 10, range 4–120 months). The recipients with an expressor genotype required more time to achieve the tacrolimus therapeutic range during the induction phase, and needed more upward dose during late induction and the maintenance phases compared with those with CYP3A5 *3/*3. Donor CYP3A5 genotypes impacted the tacrolimus trough concentrations after liver transplantation.36 Uesugi assessed the effect of intestinal CYP3A5 on post-liver transplant tacrolimus through levels in a population of 204 transplanted patients (age range 0.25–70 years). The tacrolimus concentration/dose (C/D) ratio was lower in recipients with the CYP3A5*1/*1 and *1/*3 genotype (CYP3A5 expressors) compared to the CYP3A5*3/*3 genotype (non-expressors). Amongst the combination of CYP3A5 genotypes between the graft liver and native intestines, CYP3A5 expressors in both graft liver and native intestine had the lowest C/D tacrolimus ratio (1.7–2.6 fold difference between *1/*1 and *3/*3, respectively) in the first 35 days after liver transplant. This indicates that intestinal CYP3A5, as well as hepatic CYP3A5 affect the first-pass effect of oral tacrolimus.37
With focus on the immediate (14 days) post-transplant period in kidney (48, no infants) and liver (42, including infants) recipients, de Wildt et al observed no impact of CYP3A5 expression in the hepatic transplant cases (age range 0.05–14.8 years) in liver recipients (in contrast to kidney transplants).38 Xue et al reported-on liver recipient and donor data in 64 cases (median age 10 months, range 5–60), and analysed the impact of CYP3A5 (CYP3A5, *1 expression allele) polymorphisms on tacrolimus PK and infectious complications. The authors hereby confirmed the contribution of CYP3A5 expression (38%) to the C/D ratio, and the graft liver was a key determinant. Expressors (recipient + donor) showed a significant higher incidence of infectious complications (Odd ratio 3.86).39 Finally, Gijsen et al reported on 39 cardiac transplant cases (median age 6 years, 15 infants) where CYP3A5 expressors (CYP3A5 *1/*1 or *1/*3) needed higher doses (+ 133%, 0.14 vs 0.06 mg/kg/12 h). Both age and CYP3A5 affected dosing, although this has not been separately analyzed in infants.40
Multiple CYPs
Sridharan et al evaluated the association between urinary acetaminophen metabolites (glucuronides, sulphated metabolites) and serum acetaminophen concentrations, and a set of key CYP enzyme (CYP1A2, CYP2D6, CYP2E1, CYP3A4, and CYP3A5) polymorphisms in cohort preterm neonates (GA range 23–34.5 weeks) exposed to acetaminophen because of a symptomatic PDA. Postnatal age was associated with and increased formation of urinary metabolites and decreased serum acetaminophen concentrations, while none of the CYP enzyme polymorphisms contributed to acetaminophen disposition.41
The relationship between genetic variations in relevant drug disposition genes and nevirapine PK parameters were explored in 53 Ghanaian children (age 3–35 months, or <10 kg) living with HIV eligible to initiate nevirapine-based antiretroviral therapy, with nevirapine minimum concentration (Cmin), AUC0–12h and CL/F as outcome variables. On CYPs, CYP2B6, CYP2A6 and CYP3A5 polymorphisms were hereby explored. CYP2B6 rs3745274 (reduced clearance) turned out to be one of the predictors of these outcome variables, although the multivariate (age, sex, co-medication, tuberculosis co-infection, and PGx) models only explained a limited portion of the variability (29–37%).42
Phase II Related Pharmacogenetics
Glutathione-S-Transferases (GST)
Ansari et al reported on the PK of busulfan used for myelo-ablation in 28 children (age range 0.4–19.8 years, weight range 6.2–84 kg) who underwent hematopoietic stem cell transplantation (HSCT).43 In this cohort, GSTA1, GSTM1 and GSTP1 polymorphisms were explored as covariates of first-dose intravenous busulfan (age range 3–12 months, median dose 0.8 mg/kg). In a multivariate regression analysis, dose, age and GSTM1 genotype were the best predictors of the first-dose PK variability. Furthermore, GSTM1-null patients received a lower cumulative busulfan dose. In contrast, GSTA1 or GSTP1 gene variants were not predicting. As this review focusses on the findings in infants (n = 4, 3 GSTM1-Null cases, 1 non-Null), differences in AUC or cumulative dose were not (yet) observed. Besides some information on PK, Ashton et al assessed the influence of polymorphisms of GSTM1 and N-acetyl transferase (NAT, see below) on outcome of children (86/209 < 1 year) with neuroblastoma.44 These authors hereby documented that the GSTM1 wild type and NAT1*11 was associated with a more favorable outcome in these patients, while GSTM1 null were more likely to relapse or die (hazard ratio 1.6, after adjusting).
N-Acetyl Transferase
We retrieved 5 different reports on the exploration of NAT-related polymorphisms in neonates and infants. PK findings were explored by Keller et al (isoniazide, NAT-2), Schaaf et al (isoniazide, NAT-2), and Zielinska et al (caffeine, NAT-2), PD finding by Ashton et al (neuroblastoma, see above, and Zielinska et al (trimethoprim-sulfamethoxazole toxicity).45–48 Related to isoniazide PK, Keller et al used the metabolic ratio (MR, acetyl-isoniazide/isoniazide) in blood, and hereby described a positive correlation with age (r = 0.53, age range 4 months to 17 years, 25/88 cases were 4–23 months).45 The slow genotype (no alleles) had a much lower MR compared to the rapid (two alleles) genotype (0.5 versus 1.15). Schaaf et al estimated the first order elimination rate constant (k) and AUC in 64 children (18 cases < 2 years). The k value was dependent on the age, as well as on NAT-2 allele frequency (SS = 0.254; FS = 0.51; FF = 0653 h−1). Finally, Zhu et al quantified the pharmacogenetics of NAT2 enzyme maturation in perinatal HIV exposed infants receiving isoniazide, to describe the genotype-dependent enzyme maturation processes for the NAT2 enzyme. Consecutive plasma concentration-time measurements of isoniazide from 151 infants (starting at 3–4 months of age) receiving isoniazide 10 to 20 mg/kg/day orally during the course of the 24-month study were incorporated in a population analysis along with NAT2 genotype, body weight, age, and sex.49 Apparent clearance (70 kg body weight-normalized) for fast and intermediate acetylators increasing from 14.25 L/h and 10.88 L/h at 3 months of age to 22.84 L/h and 15.58 L/h at 24 months of age, while slow acetylators displayed no changes over age (7.35 L/h).49
UDP-Glucuronosyl-Transferase (UGT)
For UGT-related polymorphisms, we retrieved data on acetaminophen (UGT1A9) and morphine (UGT2B7).50,51 Linakis re-explored a previously reported parent-metabolite (glucuronidated, sulfated, and oxidative metabolites) PK model following iv acetaminophen in 33 neonates (PNA range 1–26 days). Integration of a specific UGT1A9 gene promoter region covariate (rs3832043, - 118 > ins T, T9 > T10) significantly improved model fit (reduced between-subject variability in formation clearance of acetaminophen-glucuronide). UGT1A9 T10 was associated with a 42% reduction in clearance to acetaminophen-glucuronide.50
Morphine plasma concentrations in 17 preterm neonates (range 25–32 weeks GA) collected 20 min post intubation (morphine for rapid sequence intubation) were associated with postnatal age (range 0.14–16 days) and UGT2B7 −900G>A, while the morphine-3 and morphine-6-glucuronide/morphine ratio was higher compared to −900G/G type (GG, GA, and AA, MG3/M and MG6/M: 15.3 and 4.6-fold difference).52
Transporter Related PG Reports
ATP-Binding Cassette Transporters
In an exploratory study on the impact of ABCB1 and ABCC2 polymorphisms on cyclosporine PK (intravenous and oral, pre-transplant) in children (104 cases, age 0.36–16.3 years, 31 infants), Fanta et al observed an age-dependent influence of ABCB1 polymorphisms.53 BCB1 polymorphisms affected bio-availability (c.2677GG, GT or TT, 0.64, 0.52 and 0.41, 1.5-fold difference). However, this observation only to a limited extent explained the inter-patient variability (3.2-fold variability in oral bioavailability, prehepatic extraction ratio 3.7-fold, hepatic extraction 2.4-fold), and specific in infants (31/104 cases), no PG related effects were observed.
Hill et al explored the impact of ABCB1 PGx on actinomycin PK in 117 sampled children (age range 0.3–19.8 years, 9 infants). Body weight was the dominant covariate, while pharmacogenetic variation in candidate drug transporter genes - identified from preclinical studies – had no significant impact on intravenous actinomycin disposition.54
With focus on neonatal abstinence syndrome (NAS) cases, Wachman et al explored the association of ABCB1 gene polymorphisms with the length of hospital stay (LOS) and the need for pharmacological treatment in 86 NAS newborns, but this association was not confirmed.55 Similarly, a signal on the contribution of ABCB1 polymorphisms on nevirapine PK variability (cf supra) was not observed.42 Hronova reported on the effect of ABCB1 polymorphisms in a group of 30 neonates and 18 children exposed to midazolam or sufentanil on withdrawal syndrome characteristics. Treatment duration and cumulative doses for midazolam and/or sufentanil were associated with a higher risk to develop withdrawal syndrome: While ABCB1 polymorphisms affected the dose, there was no effect on the likelihood of withdrawal syndrome.56 We also retained one case report, describing transient bilateral hydronephrosis and obstruction related acute kidney injury secondary to bladder sphincter spasm, solved with bladder catheterization and stopping morphine exposure in a preterm (GA 34 weeks) newborn.57 In further PGx screening, the patient turned out to be homozygous for C3435T polymorphism (ABCB1 gene), associated with impaired P-glycoprotein activity. As P-glycoprotein is also expressed in urothelial cells, the authors suggested that this polymorphism contributed to opioid-induced urinary retention.
In a dataset of 105 childhood acute lymphoblastic leukemia (ALL, 7 infants), Yang et al explored associations with 17 genetic polymorphisms in 13 targets (Taiwan ALL-93 protocol), and observed three polymorphic alleles in the multi-drug resistance 1 (MDR1) ABCB1 gene. Furthermore, they found that homozygotic MDR1 2677GG, 3435CC, and 2677G-3435C genotypes were highly associated with a significant reduction in event free survival (Hazard ratios were 6.8, 21.7, and 6.8) in those patients treated by the standard risk (SR) protocol.
Related to tacrolimus PK, we retrieved 5 reports (4 hepatic, with 1 also including data in renal transplant cases, 1 cardiac) on ABCB1 polymorphisms or haplotypes. Chen et al studied personalized tacrolimus dosing requirements by recipient and donor polymorphisms in 90 liver transplant children (median 10 months, range 4–120), including ABCB1. No effects of ABCB1 polymorphisms on dosing requirement were observed.36 In contrast, Fukudo et al reported on a population PK study in 130 (including PGx in 65/130) pediatric liver transplant recipients (age range 0.1–15 years) shortly after (<50 days) transplantation, and observed an effect of ABCB1 mRNA expression on CL/F (+15–20%), much smaller than the 2-fold impact of CYP3A5 polymorphisms.35 Hawwa et al assessed the tacrolimus dose and nephrotoxicity after liver transplant in 51 pediatric cases (recipient PGx, range 0.6–16 years). Trough levels in TTT haplotype and G2677>T and C3435>T were correlated to higher exposure, with a different C/D ratio from 3 years after transplant onwards (like G2677->T and C3435->T 62 versus 39, and 60 versus 33 ng/mL/mg/kg.58 Reduction in estimated glomerular filtration rate (eGFR) was more common and more pronounced in T-T-T haplotype carriers and G2677->T or C1236->T.58 With focus on the immediate (14 days) post-transplant period in kidney (48, no infants) and liver (42, including infants) recipients, de Wildt et al observed in liver recipients (age range 0.05–14.8 years) that homozygous cases (T-T-T haplotype) needed higher doses (+136%, 0.26 versus 0.11 mg/kg/12h).38 In pediatric liver recipients, but not renal transplant recipients, variation in tacrolimus disposition appeared related to age and ABCB1 genotype shortly after transplantation. Using a similar approach in 39 cardiac transplant cases, Gijsen et al documented that age and CYP3A5 expression affected tacrolimus dosing, but not ABCB1 polymorphisms or disease severity (PRISM = Pediatric Risk of Mortality).
Roberts et al reported on a population PK study of oral topotecan in infants and young children with brain tumors. Based on PK observations in 61 cases (age range 0.48–4.59 years, 20/61 cases <2 years). After including BSA in the V/F and CL/F as a power model centered on the population median, the ABCG2 rs4148157 allele had a significant role in the Ka value. Patients homozygous or heterozygous for G>A demonstrated a Ka value 2-fold higher than GG, complemented with a 2-fold higher maximal concentration as well.59
Related to ABCC3 polymorphisms (Multidrug Resistance Protein 3), Hahn et al observed a decreasing trend on morphine clearance with MRP3 efflux transporter (rs4793665) genotypes (CC>CT>TT) (morphine clearance, standardized to 70 kg, 31.3, vs 26.39 vs 20.76 L/h, 1.5-fold difference) in a subgroup of OCT1 wild-type cases.60
Organic Cation Transporters
Matic et al reported that the plasma log M/M1 ratio in 50 preterm neonates and infants (27–54 weeks PMA) exposed to intravenous tramadol, was also affected by the OCT1 allele frequency, in addition to the previously mentioned CYP2D6 activity score and age, with a 1.14-fold difference between cases with ≥2 functional gene copies (n=27/50) or with <2 (n = 23/50).31,32,61 Using a physiologically-based pharmacokinetic modelling effort in 85 neonates and young infants (PMA range 24.9–57.3 weeks), the impact of age, OCT1 ontogeny, and OCT1 allele frequency (loss of function) on morphine disposition were explored based on in vivo morphine observations. Comparing the wild, to the hetero- or homo-haplotype, there were not yet differences in morphine clearance in the 24–34 weeks group, while these differences appeared (about 2-fold differences) at 34–40 (19.3, 15.5 and 10.9) and 40–58 weeks (38.7, 30 and 20 L/h, standardized to 70 kg).62
Organic Anion Transporter Protein 1B1 (OATP1B1, SLCO1B1 genotype) polymorphisms were also explored in the previously mentioned cyclosporine cohort ((intravenous and oral, pre-transplant) in children (104 cases, age range 0.36–16.3 years, 31 infants), but this analysis remained negative.53
Receptor and Post Receptor Mechanism Reports
Opioid Related Receptor and Post Receptor Mechanisms
The relevance of the KCNJ6-1250G.A (rs6517442, c.-1787G.A, G-protein-activated inwardly rectifying potassium channel 2) and the COMT c.472G.A (rs4680, Val158Met) single-nucleotide polymorphisms were studied in preterm infants needing intubation and randomized to a premedication strategy including remifentanil (n = 17) or morphine (n = 17) by Elens et al.63 The authors hereby documented that KCNJ6-1250A (time to response to pain therapy, delay to reach a pain free score, AA vs AG vs GG: 182, 109 and 60 minutes, 3-fold difference) and COMT 158 Val alleles (Val/val, vs Val/Met vs Met/Met: 285, 137 and 63 minutes, 4.5-fold difference) were associated with diminished opioid-induced pain relief in preterm neonates. Within the same theme, Matic et al assessed the need for rescue morphine within a randomized controlled trial in preterm (n = 64, placebo cases) neonates that required mechanical ventilation. The authors hereby aimed to determine whether single nucleotide polymorphisms (SNPs) of OPRM1 118A>G (asn40asp), COMT 472G>A (val158met) and beta-arrestin-2 (G-protein coupled receptor, ARRB2) 8622C>T were associated with morphine rescue. For OPRM1 and COMT separately, the expected risk for rescue morphine or morphine dose was not significantly increased. However, the combined OPRM1/COMT “high-risk” genotype had a higher likelihood for the need for rescue (OR: 5.12).52
With focus on neonatal abstinence syndrome (NAS) cases, Wachman et al explored the association of OPRM1 and COMT genes (ABCB1 discussed higher) with LOS and the need for pharmacological treatment in 86 NAS newborns.55 OPRM1 (118A>G AG/GG shorter LOS (−8.5 days versus average 22.3 days, −38%) compared to AA genotype and were less likely to receive treatment (48% vs 72%). COMT AG/GG shorter LOS (−10.8 days versus average LOS 22.3 days, −48%) and less “advanced, ≥2 drugs” treatment (18% versus 56%) compared to AA genotype.55
In a subsequent study of the same researchers, the association COMT genes with NAS severity was confirmed in a replication cohort (n = 113).55 Furthermore, infants with the PNOC (prepronociceptin, ligand of OPRM) rs4732636 A allele had a reduced need for pharmacological treatment (adjusted OR 2).55 In the study of Hronova et al, the impact of COMT and human pregnane X receptor (PXR) (besides ABCB1, cf higher) on sufentanil and midazolam dosing were also explored, and observed that these polymorphisms affected dosing, but not the depth of analgo-sedation or withdrawal syndrome.56
Tumor Necrosis Factor (TNF) and Mitogen-Activated Protein Kinase 8 (MAPK8)
The relevance of tumor necrosis factor (TNF) or mitogen-activated protein kinase 8 (MAPK8) polymorphisms on the immune response to HBV vaccination (at birth, 1 and 6 months) in 753 infants of HBsAg positive, HBeAg negative mothers has been explored. Formula feeding and MAPK8 polymorphisms (rs17780725, AA versus GA or GG genotype), but not TNF polymorphisms, were associated a “low responder” profile (antibody titers, 5.8% of the cases) after hepatitis B vaccination (OR = 2.9) at 7 months.64
Vitamin D Binding Protein SNPs
Vitamin D protein (6 different diplotypes) genotype affects 25 OH-vitamin D concentration and efficiency of D3 supplementation (either 10 or 30 µg/day) in infants. Major allele homozygote haplotypes and diplotype 1 (1S/1S, 1F/1S and 1F/1F) result in higher 25 OH-vitamin D concentrations compared to diplotype 2 (1S/2, 1 F/S, and 2/2) with a mean effects size from −4.4 nmol/l to −10.9 nmol/l at 24 months (median 25OH vitamin D 114 and 118 nmol, so 4% and 9% lower), for Haplo3SNP and Haplo4SNP the effect size was −12.6 to −33.7 nmol/L (so 11 and 29% lower).65
Corticotrophin-Releasing Hormone Receptor-1 (CRHR1) Polymorphisms
77/126 cases included in the TOLSURF trial (RCT on late surfactant to prevent bronchopulmonary dysplasia in ventilated preterms, receiving inhaled nitric oxide), and exposed to dexamethasone or hydrocortisone, a panel of candidate SNP associations were tested, with the change in respiratory severity score as dependent variable. Using this explorative approach, genetic variation in CRHR1 polymorphism was associated with differences in absolute decrease in respiratory severity score (RSS) at day 7 (Rs7225082, each T allele is associated with a smaller decrease in RSS at day 7 (RSS change: −44, −50 or −31% for GG, GT and TT respectively, 1.6-fold variability).66
Nuclear Receptor Subfamily 1
The previously mentioned nevirapine PK study also explored the relationship between nuclear receptor subfamily 1, group I, member 2 and member 3 (NR1I2, NR113) polymorphisms in the same cohort. In the multivariate model to explore the between individual PK findings, age, sex, co-medication, tuberculosis co-infection and PGx markers were retained. In addition to CYP2B6 rs3745274, NR1I2 rs6785049 was also retained in the model (GA versus GG). Combining the individual observations in CYP2B6 and NR1I2 polymorphism, a 3-fold variability in nevirapine AUC0-12h was described.42
Vitamin K Epoxide Reductase Complex 1 (VKORC1) Polymorphism
We retrieved 4 datasets reporting on VKORC1 polymorphisms (3/4 also reported on CYP2C9 polymorphism), but we would like to restress that the pooled number of infants is limited. Using an adult PK/PD model bridging effort to children for warfarin, the INR response was predicted reasonably well in 64 children (age and weight range, 0.05–18.9 years and 3.4–94 kg), be it with a tendency to overpredict INR (or underpredict dose) in children <2 years. It was hereby extrapolated from adult findings that VKORC1 (A/A versus G/G) polymorphisms explained up to a 2.1-fold difference in warfarin maintenance dose.24 In 48 pediatric cases (age range 0.42–19.25 years), age and VKORC1 genotype were major factors for the weight normalized warfarin dose and INR. The anticoagulant effect of warfarin in patients with the VKORC1 1173CT or 1173CC genotype was 52.3% of that in patients with the 1173TT genotype. However, only 3/48 cases were <1 year.67 In a cohort of 123 children (only 2 infants included), VKORC1 polymorphisms (GG, to AA, 2.52 versus 1 mg/day, 1.3-fold difference) affected dosing.25 In a multivariate model, BSA and indication explained 45% of the variability in acenocoumarol dose requirement (INR targeting), adding VKORC1 (GG, AG, AA=5, 3.99, 2.7 mg/day, 1.3-fold difference), CYP2C9 (*2 or *3, CC/AA, CC/CA, CT/AA, TT/AA = 2.6-fold) and CYP2C18 (GG, AG or AA = 1.33-fold) resulted in 61.8% explained.25 Finally, in a dataset with 118 cases (age range 3 months to 18 years, 19 cases <3 years) on acenocoumarol, VKORC1 polymorphisms contributed to the variability in dosing.26 VKORC1 (GG, GA to AA: 31, 22, 9.7 mg/week) displayed a 3.2-fold difference in weekly dosing. In a multiple regression model, height (48.1%), target INR (4.4%), CYP2C9 (2%), and VKORC1 (18.2%) resulted in an explained variability of 69.7%.26
Angiotensin Converting Enzyme (ACE) Variants
Chen et al studied personalized tacrolimus dosing requirements by recipient and donor polymorphisms in 90 liver transplant children (age range 4–120 months), including CYP3A5 (positive), ABCB1. (negative), but also ACE variants were explored. However, no effects of ACE variants on dosing requirements were observed.36
Discussion
Retained records relate to phase I [CYP1A2, CYP2A6, CYP2B6, CYP2C8/C9/C18, CYP2C19, CYP2D6, CYP3A5, CYP2E1], phase II [GST, NAT, UGT], transporters [ABC transporters, OTC], or receptor/post-receptor mechanisms [OPRM-1 and post-receptor mechanisms, TNF, MAPK-8, vitamin binding protein diplotypes, CRHR-1, NR subfamily 1, VKORC1, or ACE enzymes variants].
Based on the available overview, we conclude that the majority of reported pharmacogenetic studies explored and extrapolated observations previously described in older populations. The researchers hereby commonly quantified the impact of these polymorphisms in neonates or infants, integrated in the ontogenic process. The between study comparisons are further complicated by a diversity in methods and polymorphisms sets explored, and rather small datasets in neonates or infants.
In this way, “appearance” of PGx signals similar as reported in adults suggests that the phenotypic activity or expression in neonates or infants already sufficiently reflects “adult level of activity” and “phenoconversion”: integrated developmental pharmacogenetics are the key. To further illustrate this, we can compare the observations on CYP1A polymorphisms (caffeine) with CYP2D6 ontogeny (dextromethorphan, tramadol).18,30–32 For caffeine, CYP1A2 polymorphisms are not yet relevant in the metabolic elimination of methylxanthines in neonates, in contrast to observations on the relevance of CYP1A2 polymorphisms in adults.68 This can simply be explained by the fact that CYP1A2 activity is almost absent in the first months of life, and caffeine elimination is almost exclusively renal.69,70 The same holds true for acetaminophen metabolism. Postnatal age was the main determinant of urinary metabolites and the serum acetaminophen concentrations, while none of the CYP enzyme polymorphisms contributed to acetaminophen disposition.41 This is to a large extent anticipated, as only CYP2E1 is involved in acetaminophen metabolism, and this iso-enzyme displays a slow ontogeny pattern, only reaching the adult level of activity after infancy.69 In contrast, CYP2D6 ontogeny occurs already in early infancy, so that it is anticipated that the CYP2D6 activity score will already contribute to the variability, to remain relevant throughout human life.71 The same is true for eg transporters, as OCT1 allele frequency already contributes to plasma concentrations of M1. This informs us that OCT1 is indeed already sufficiently well expressed in neonates.61 Additional information on the ontogeny pattern has been explored, based on a physiologically-based pharmacokinetic morphine modelling effort in 85 neonates and young infants (PMA 24.9–57.3 weeks). Comparing the wild, to the hetero- or homo-haplotype, there were not yet differences in morphine clearance in the 24–34 weeks group, while these differences appeared (about 2-fold differences) at 34–40 (19.3, 15.5 and 10.9) and 40–58 weeks (38.7, 30 and 20 L/h, standardized to 70 kg).62 In a next step, PGx studies in neonatal life should go beyond confirmation of these associations and explore the impact of PGx as a covariate limited to maturation of perinatal life (ie fetal malformations, breastfeeding or clinical syndromes following placental transfer, or iso-enzymes that are more active in perinatal life, like CYP3A7).72 Applying population PK analyses hereby strengthens the analysis when based on a relatively small number of observations, and can be integrated with similar datasets in older populations, as illustrated by the tramadol example.70
Conclusion
We conducted a systematic review to identify, describe and quantify the impact of PGx on PK and PD in neonates and infants. We conclude that the majority of reported PGx studies were reported in the past 20 years. These studies commonly extrapolate observations already described in older populations, to subsequently quantify the impact of these polymorphisms in neonates or infants. There is still uncertainty on the power and extent of observations needed in neonates to truly confirm or reject these already described observations in older populations.
In a next step, PGx studies in perinatal life should go beyond confirmation of these associations and explore the impact of PGx as a covariate limited to perinatal life or early infancy. The challenge is to identify the specific factors, genetic and non-genetic, that contribute to the best benefit/risk balance with the need for big datasets/populations.
Acknowledgments
The authors wish to thank Wichor Bramer from the Erasmus MC Medical Library for developing and updating the search strategies. Also, we would like to thank The Scientific and Technological Research Council of Turkey (TÜBİTAK) for contributing to N.Y., to obtain a scholarship to research at Erasmus Medical Center and thus to collaborate with co-authors (ref no: 53325897-115.02-109267).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Roden DM, McLeod HL, Relling MV, et al. Pharmacogenomics. Lancet. 2019;394(10197):521–532. doi:10.1016/S0140-6736(19)31276-0
2. Gregornik D, Salyakina D, Brown M, Roiko S, Ramos K. Pediatric pharmacogenomics: challenges and opportunities: on behalf of the Sanford Children’s Genomic Medicine Consortium. Pharmacogenomics J. 2021;21(1):8–19. doi:10.1038/s41397-020-00181-w
3. Lewis T, Leeder JS. Pharmacogenomics and implementation of precision therapeutics in the neonatal ICU: a new frontier? Pharmacogenomics. 2018;19(16):1231–1233. doi:10.2217/pgs-2018-0132
4. Lewis T. Neonatal pharmacogenetics. In: Benitz WE, Smith PB, editors. Infectious Disease and Pharmacology. Amsterdam: Elsevier; 2019:141–153.
5. De Cock RFW, Piana C, Krekels EHJ, Danhof M, Allegaert K, Knibbe CAJ. The role of population PK-PD modelling in paediatric clinical research. Eur J Clin Pharmacol. 2011;67:S5–S16. doi:10.1007/s00228-009-0782-9
6. Smits A, Kulo A, de Hoon JN, Allegaert K. Pharmacokinetics of drugs in neonates: pattern recognition beyond compound specific observations. Curr Pharm Design. 2012;18(21):3119–3146. doi:10.2174/1381612811209023119
7. Shah RR, Smith RL. Addressing phenoconversion: the Achilles’ heel of personalized medicine. Br J Clin Pharmacol. 2015;79(2):222–240. doi:10.1111/bcp.12441
8. Di Nunno N, Esposito M, Argo A, Salerno M, Sessa F. Pharmacogenetics and forensic toxicology: a new step towards a multidisciplinary approach. Toxics. 2021;9(11):292. doi:10.3390/toxics9110292
9. Allegaert K, van den Anker JN. Clinical pharmacology in neonates: small size, huge variability. Neonatology. 2014;105(4):344–349. doi:10.1159/000360648
10. Patrinos GP. Sketching the prevalence of pharmacogenomic biomarkers among populations for clinical pharmacogenomics. Eur J Human Genet. 2020;28(1):1–3. doi:10.1038/s41431-019-0499-x
11. Durrmeyer X, Hovhannisyan S, Medard Y, et al. Are cytochrome P450 CYP2C8 and CYP2C9 polymorphisms associated with ibuprofen response in very preterm infants? PLoS One. 2010;5(8):e12329. doi:10.1371/journal.pone.0012329
12. Sistonen J, Madadi P, Ross CJ, et al. Prediction of codeine toxicity in infants and their mothers using a novel combination of maternal genetic markers. Clin Pharmacol Ther. 2012;91(4):692–699. doi:10.1038/clpt.2011.280
13. Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535. doi:10.1136/bmj.b2535
14. Yalcin N, Flint R, Koch B, van Schaik RH, Simons S, Allegaert A. A systematic review on the until currently reported observations on the impact of pharmacogenetics on pharmacokinetics or pharmacodynamics in neonates. PROSPERO 2022 CRD42022302029. Available from: https://www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42022302029.
15. Stark A, Smith PB, Hornik CP, et al. Medication use in the neonatal intensive care unit and changes from 2010 to 2018. J Pediatr. 2022;240:
16. Wells GA, Shea B, O’Connell D, et al. The Newcastle–Ottawa Scale (NOS) for assessing the quality if nonrandomized studies in meta-analyses; 2009. Available from: http://www.ohrica/programs/clinical_epidemiology/oxfordasp.
17. The Joanna Briggs Institute critical appraisal tools for use in JBI systematic reviews checklist for case reports. Available from: https://jbi.global/critical-appraisal-tools.
18. Gao XB, Zheng Y, Yang F, et al. Developmental population pharmacokinetics of caffeine in Chinese premature infants with apnoea of prematurity: a post-marketing study to support paediatric labelling in China. Br J Clin Pharmacol. 2021;87(3):1155–1164. doi:10.1111/bcp.14483
19. Guan Y, Li B, Wei W, et al. Quantitative ultra-high-performance liquid chromatography-tandem mass spectrometry for determination of dexmedetomidine in pediatric plasma samples: correlation with genetic polymorphisms. Biomed Chromatogr. 2019;33(12):e4683. doi:10.1002/bmc.4683
20. Barnett S, Errington J, Sludden J, et al. Pharmacokinetics and pharmacogenetics of cyclophosphamide in a neonate and infant childhood cancer patient population. Pharmaceuticals. 2021;14:272. doi:10.3390/ph14030272
21. Rooney SR, Shelton EL, Aka I, et al. CYP2C9*2 is associated with indomethacin treatment failure for patent ductus arteriosus. Pharmacogenomics. 2019;20(13):939–946. doi:10.2217/pgs-2019-0079
22. Smith CJ, Ryckman KK, Bahr TM, Dagle JM. Polymorphisms in CYP2C9 are associated with response to indomethacin among neonates with patent ductus arteriosus. Pediatr Res. 2017;82(5):776–780. doi:10.1038/pr.2017.145
23. Veeravigrom M, Jaroonvanichkul V, Netbaramee W, Phaisarn P, Uyathanarat T. Phenytoin toxicity in two-month-old Thai infant with CYP2C9 gene polymorphism - A case report. Brain Dev. 2016;38(1):136–138. doi:10.1016/j.braindev.2015.05.001
24. Hamberg AK, Friberg LE, Hanseus K, et al. Warfarin dose prediction in children using pharmacometric bridging–comparison with published pharmacogenetic dosing algorithms. Eur J Clin Pharmacol. 2013;69(6):1275–1283. doi:10.1007/s00228-012-1466-4
25. Maagdenberg H, Bierings MB, van Ommen CH, et al. The pediatric acenocoumarol dosing algorithm: the children anticoagulation and pharmacogenetics study. J Thromb Haemost. 2018;16(9):1732–1742. doi:10.1111/jth.14211
26. Moreau C, Bajolle F, Siguret V, et al. Vitamin K antagonists in children with heart disease: height and VKORC1 genotype are the main determinants of the warfarin dose requirement. Blood. 2012;119(3):861–867. doi:10.1182/blood-2011-07-365502
27. Lee SM, Chung JY, Lee YM, et al. Effects of cytochrome P450 (CYP)2C19 polymorphisms on pharmacokinetics of phenobarbital in neonates and infants with seizures. Arch Dis Child. 2012;97(6):569–572. doi:10.1136/archdischild-2011-300538
28. Ward RM, Tammara B, Sullivan SE, et al. Single-dose, multiple-dose, and population pharmacokinetics of pantoprazole in neonates and preterm infants with a clinical diagnosis of gastroesophageal reflux disease (GERD). Eur J Clin Pharmacol. 2010;66(6):555–561. doi:10.1007/s00228-010-0811-8
29. Zhao W, Leroux S, Biran V, Jacqz-Aigrain E. Developmental pharmacogenetics of CYP2C19 in neonates and young infants: omeprazole as a probe drug. Br J Clin Pharmacol. 2018;84(5):997–1005. doi:10.1111/bcp.13526
30. Blake MJ, Gaedigk A, Pearce RE, et al. Ontogeny of dextromethorphan O- and N-demethylation in the first year of life. Clin Pharmacol Ther. 2007;81(4):510–516. doi:10.1038/sj.clpt.6100101
31. Allegaert K, van Schaik RH, Vermeersch S, et al. Postmenstrual age and CYP2D6 polymorphisms determine tramadol o-demethylation in critically ill neonates and infants. Pediatr Res. 2008;63(6):674–679. doi:10.1203/PDR.0b013e31816ff712
32. Allegaert K, de Hoon J, Naulaers G, Van De Velde M. Neonatal clinical pharmacology: recent observations of relevance for anaesthesiologists. Acta Anaesthesiol Belg. 2008;59(4):283–288.
33. Gorny M, Röhm S, Läer S, Morali N, Niehues T. Pharmacogenomic adaptation of antiretroviral therapy: overcoming the failure of lopinavir in an African infant with CYP2D6 ultrarapid metabolism. Eur J Clin Pharmacol. 2010;66(1):107–108. doi:10.1007/s00228-009-0753-1
34. Shimizu M, Kondo T, Fukuoka T, Tanaka T, Yamazaki H. Dihydrocodeine overdoses in a neonate and in a 14-year-old girl who were both genotyped as cytochrome P450 2D6*1/*10-*36: comparing developmental ages and drug monitoring data with the results of pharmacokinetic modeling. Ther Drug Monit. 2018;40(2):162–165. doi:10.1097/FTD.0000000000000482
35. Fukudo M, Yano I, Masuda S, et al. Population pharmacokinetic and pharmacogenomic analysis of tacrolimus in pediatric living-donor liver transplant recipients. Clin Pharmacol Ther. 2006;80(4):331–345. doi:10.1016/j.clpt.2006.06.008
36. Chen YK, Han LZ, Xue F, et al. Personalized tacrolimus dose requirement by CYP3A5 but not ABCB1 or ACE genotyping in both recipient and donor after pediatric liver transplantation. PLoS One. 2014;9(10). doi:10.1371/journal.pone.0109464
37. Uesugi M, Masuda S, Katsura T, Oike F, Takada Y, Inui K. Effect of intestinal CYP3A5 on postoperative tacrolimus trough levels in living-donor liver transplant recipients. Pharmacogenet Genomics. 2006;16(2):119–127. doi:10.1097/01.fpc.0000184953.31324.e4
38. de Wildt SN, van Schaik RH, Soldin OP, et al. The interactions of age, genetics, and disease severity on tacrolimus dosing requirements after pediatric kidney and liver transplantation. Eur J Clin Pharmacol. 2011;67(12):1231–1241. doi:10.1007/s00228-011-1083-7
39. Xue F, Han L, Chen Y, et al. CYP3A5 genotypes affect tacrolimus pharmacokinetics and infectious complications in Chinese pediatric liver transplant patients. Pediatr Transplant. 2014;18(2):166–176. doi:10.1111/petr.12216
40. Gijsen V, Mital S, van Schaik RH, et al. Age and CYP3A5 genotype affect tacrolimus dosing requirements after transplant in pediatric heart recipients. J Heart Lung Transplant. 2011;30(12):1352–1359. doi:10.1016/j.healun.2011.08.001
41. Sridharan K, Al Jufairi M, Al Ansari E, et al. Evaluation of urinary Acetaminophen metabolites and its association with the genetic polymorphisms of the metabolising enzymes, and serum Acetaminophen concentrations in preterm neonates with patent ductus arteriosus. Xenobiotica. 2021;51:1335–1342. doi:10.1080/00498254.2021.1982070
42. Langaee T, Al-Shaer MH, Gong Y, et al. Pharmacogenetic predictors of nevirapine pharmacokinetics in Ghanaian children living with HIV with or without TB coinfection. Infect Genet Evol. 2021;92:104856. doi:10.1016/j.meegid.2021.104856
43. Ansari M, Lauzon-Joset JF, Vachon MF, et al. Influence of GST gene polymorphisms on busulfan pharmacokinetics in children. Bone Marrow Transplant. 2010;45(2):261–267. doi:10.1038/bmt.2009.143
44. Ashton LJ, Murray JE, Haber M, Marshall GM, Ashley DM, Norris MD. Polymorphisms in genes encoding drug metabolizing enzymes and their influence on the outcome of children with neuroblastoma. Pharmacogenet Genomics. 2007;17(9):709–717. doi:10.1097/FPC.0b013e3280e1cc92
45. Keller GA, Fabian L, Gomez M, Gonzalez CD, Diez RA, Girolamo GD. Age-distribution and genotype-phenotype correlation for N-acetyltransferase in Argentine children under isoniazid treatment. Int J Clin Pharmacol Ther. 2014;52(4):292–302. doi:10.5414/CP201957
46. Schaaf HS, Parkin DP, Seifart HI, et al. Isoniazid pharmacokinetics in children treated for respiratory tuberculosis. Arch Dis Child. 2005;90(6):614–618. doi:10.1136/adc.2004.052175
47. Zielinska E, Bodalski J, Niewiarowski W, Bolanowski W, Matusiak I. Comparison of acetylation phenotype with genotype coding for N-acetyltransferase (NAT2) in children. Pediatr Res. 1999;45(3):403–408. doi:10.1203/00006450-199903000-00019
48. Zielińska E, Niewiarowski W, Bodalski J, et al. Genotyping of the arylamine N-acetyltransferase polymorphism in the prediction of idiosyncratic reactions to trimethoprim-sulfamethoxazole in infants. Pharm World Sci. 1998;20(3):123–130. doi:10.1023/A:1008664707825
49. Zhu R, Kiser JJ, Seifart HI, et al. The pharmacogenetics of NAT2 enzyme maturation in perinatally HIV exposed infants receiving isoniazid. J Clin Pharmacol. 2012;52(4):511–519. doi:10.1177/0091270011402826
50. Linakis MW, Cook SF, Kumar SS, et al. Polymorphic expression of UGT1A9 is associated with variable acetaminophen glucuronidation in neonates: a population pharmacokinetic and pharmacogenetic study. Clin Pharmacokinet. 2018;57(10):1325–1336. doi:10.1007/s40262-018-0634-9
51. Matic M, Norman E, Rane A, et al. Effect of UGT2B7-900G>A (−842G>A; rs7438135) on morphine glucuronidation in preterm newborns: results from a pilot cohort. Pharmacogenomics. 2014;15(12):1589–1597. doi:10.2217/pgs.14.115
52. Matic M, Simons SH, van Lingen RA, et al. Rescue morphine in mechanically ventilated newborns associated with combined OPRM1 and COMT genotype. Pharmacogenomics. 2014;15(10):1287–1295. doi:10.2217/pgs.14.100
53. Fanta S, Niemi M, Jonsson S, et al. Pharmacogenetics of cyclosporine in children suggests an age-dependent influence of ABCB1 polymorphisms. Pharmacogenet Genomics. 2008;18(2):77–90. doi:10.1097/FPC.0b013e3282f3ef72
54. Hill CR, Cole M, Errington J, Malik G, Boddy AV, Veal GJ. Characterisation of the clinical pharmacokinetics of actinomycin D and the influence of ABCB1 pharmacogenetic variation on actinomycin D disposition in children with cancer. Clin Pharmacokinet. 2014;53(8):741–751. doi:10.1007/s40262-014-0153-2
55. Wachman EM, Hayes MJ, Sherva R, et al. Association of maternal and infant variants in PNOC and COMT genes with neonatal abstinence syndrome severity. Am J Addict. 2017;26(1):42–49. doi:10.1111/ajad.12483
56. Hronová K, Pokorná P, Posch L, Slanař O. Sufentanil and midazolam dosing and pharmacogenetic factors in pediatric analgosedation and withdrawal syndrome. Physiol Res. 2016;65(4):S463–S472. doi:10.33549/physiolres.933519
57. Pogliani L, Mameli C, Cattaneo D, et al. Acute kidney injury in a preterm infant homozygous for the C3435T polymorphism in the ABCB1 gene given oral morphine. Clin Kidney J. 2012;5(5):431–433. doi:10.1093/ckj/sfs099
58. Hawwa AF, McKiernan PJ, Shields M, Millership JS, Collier PS, McElnay JC. Influence of ABCB1 polymorphisms and haplotypes on tacrolimus nephrotoxicity and dosage requirements in children with liver transplant. Br J Clin Pharmacol. 2009;68(3):413–421. doi:10.1111/j.1365-2125.2009.03461.x
59. Roberts JK, Birg AV, Lin T, et al. Population pharmacokinetics of oral topotecan in infants and very young children with brain tumors demonstrates a role of ABCG2 rs4148157 on the absorption rate constant. Drug Metab Dispos. 2016;44(7):1116–1122. doi:10.1124/dmd.115.068676
60. Hahn D, Fukuda T, Euteneuer JC, et al. Influence of MRP3 genetics and hepatic expression ontogeny for morphine disposition in neonatal and pediatric patients. J Clin Pharmacol. 2020;60(8):992–998. doi:10.1002/jcph.1592
61. Matic M, de Wildt SN, Elens L, et al. SLC22A1/OCT1 genotype affects O-desmethyltramadol exposure in newborn infants. Ther Drug Monit. 2016;38(4):487–492. doi:10.1097/FTD.0000000000000307
62. Hahn D, Emoto C, Euteneuer JC, Mizuno T, Vinks AA, Fukuda T. Influence of OCT1 ontogeny and genetic variation on morphine disposition in critically ill neonates: lessons from PBPK modeling and clinical study. Clin Pharmacol Ther. 2019;105(3):761–768. doi:10.1002/cpt.1249
63. Elens L, Norman E, Matic M, Rane A, Fellman V, van Schaik RHN. Genetic predisposition to poor opioid response in preterm infants: impact of KCNJ6 and COMT polymorphisms on pain relief after endotracheal intubation. Ther Drug Monit. 2016;38(4):525–533. doi:10.1097/FTD.0000000000000301
64. Cao MZ, Wu YH, Wen SM, et al. Mitogen-activated protein kinase eight polymorphisms are associated with immune responsiveness to HBV vaccinations in infants of HBsAg(+)/HBeAg(-) mothers. BMC Infect Dis. 2018;18(1). doi:10.1186/s12879-018-3166-x
65. Enlund-Cerullo M, Koljonen L, Holmlund-Suila E, et al. Genetic variation of the vitamin D binding protein affects vitamin D status and response to supplementation in infants. J Clin Endocrinol Metab. 2019;104(11):5483–5498. doi:10.1210/jc.2019-00630
66. Lewis T, Truog W, Norberg M, Ballard PL, Torgerson D, Group TS. Genetic variation in CRHR1 is associated with short-term respiratory response to corticosteroids in preterm infants at risk for bronchopulmonary dysplasia. Pediatr Res. 2019;85(5):625–633. doi:10.1038/s41390-018-0235-1
67. Kato Y, Ichida F, Saito K, et al. Effect of the VKORC1 genotype on warfarin dose requirements in Japanese pediatric patients. Drug Metab Pharmacokinet. 2011;26(3):295–299. doi:10.2133/dmpk.DMPK-10-NT-082
68. Nehlig A, Alexander SPH. Interindividual differences in caffeine metabolism and factors driving caffeine consumption. Pharmacol Rev. 2018;70(2):384–411. doi:10.1124/pr.117.014407
69. van Groen BD, Nicolai J, Kuik AC, et al. Ontogeny of hepatic transporters and drug-metabolizing enzymes in humans and in nonclinical species. Pharmacol Rev. 2021;73(2):597–678. doi:10.1124/pharmrev.120.000071
70. Aranda JV, Beharry KD. Pharmacokinetics, pharmacodynamics and metabolism of caffeine in newborns. Semin Fetal Neonatal Med. 2020;25(6):101183. doi:10.1016/j.siny.2020.101183
71. Allegaert K, Holford N, Anderson BJ, et al. Tramadol and o-desmethyl tramadol clearance maturation and disposition in humans: a pooled pharmacokinetic study. Clin Pharmacokinet. 2015;54(2):167–178. doi:10.1007/s40262-014-0191-9
72. Leeder JS. Developmental pharmacogenetics: a general paradigm for application to neonatal pharmacology and toxicology. Clin Pharmacol Ther. 2009;86(6):678–682. doi:10.1038/clpt.2009.195
73. Xue F, Han L, Chen Y, et al. CYP3A5 genotypes affect tacrolimus pharmacokinetics and infectious complications in Chinese pediatric liver transplant patients. Pediatr Transplant. 2014;18(2):166–76. PMID: 2443821. doi:10.1111/petr.12216
74. Yang YL, Lin DT, Chang SK. Pharmacogenomic variations in treatment protocols for childhood acute lymphoblastic leukemia. Pediatr Blood Cancer. 2010;54(2):206–11. doi:10.1002/pbc.22292
75. Yang TH, Chen YK, Xue F, et al. Influence of CYP3A5 genotypes on tacrolimus dose requirement: age and its pharmacological interaction with ABCB1 genetics in the Chinese paediatric liver transplantation. Int J Clin Pract Suppl. 2015;(183):53–62. PMID: 26176181. doi:10.1111/ijcp.12667
76. Zwaveling J, Press RR, Bredius RG, et al. Glutathione S-transferase polymorphisms are not associated with population pharmacokinetic parameters of busulfan in pediatric patients. Ther Drug Monit. 2008;30(4):504–10. PMID: 18641537. doi:10.1097/FTD.0b013e3181817428
77. Wachman EM, Hayes MJ, Brown MS, et al. Association of OPRM1 and COMT single-nucleotide polymorphisms with hospital length of stay and treatment of neonatal abstinence syndrome. JAMA. 2013;309(17):1821–7. PMID: 23632726 PMCID: PMC4432911. doi:10.1001/jama.2013.3411
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.