Back to Journals » OncoTargets and Therapy » Volume 13
The Immunostimulative Effect and Mechanisms of a Novel Mouse Anti-Human PD-1 Monoclonal Antibody on Jurkat Lymphocytic Cells Cocultured with Hepatoma Cells
Authors Li Z, Li B, Li L, Wang G, Li Y, Fu R, Ming Y, Ni R, Wang J, Ye G, Chen J ![]()
Received 11 September 2020
Accepted for publication 5 November 2020
Published 27 November 2020 Volume 2020:13 Pages 12225—12241
DOI https://doi.org/10.2147/OTT.S281397
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Ziwei Li,1,* Bin Li,1,* Li Li,1 Guanying Wang,1 Yuanyuan Li,1 Ruoqiu Fu,1 Yue Ming,1 Rui Ni,1 Jiming Wang,2 George Ye,3 Jianhong Chen1
1Department of Pharmacy, Daping Hospital, Army Medical University, Chongqing 400042, People’s Republic of China; 2Center for Cancer Research, National Cancer Institute, Frederick, MD 21702, USA; 3Yes Biotech Laboratories Ltd, Mississauga, ON L5S 1V6, Canada
*These authors contributed equally to this work
Correspondence: Jianhong Chen
Department of Pharmacy, Daping Hospital, Army Medical University, 10# Changjiangzhilu, Chongqing 400042, People’s Republic of China
Tel +86 23 6875 7098
Fax +86 23 6889 8867
Email [email protected]
Background: Monoclonal antibodies (mAbs) that target the programmed cell death-1 (PD-1)/programmed death-ligand 1 (PD-L1) immune checkpoint have demonstrated substantial clinical benefit for a variety of solid tumors. However, their applications in patients with hepatocellular carcinoma (HCC) are reported with unclear molecular mechanisms. Here, we report a novel mouse anti-human PD-1 mAb that can reverse the immunosuppressive effect of HePG2 cells on Jurkat cells.
Materials and Methods: HepG2 liver cancer cells, which were induced to overexpress PD-L1 by IFN-γ, were co-cultured with PHA-activated Jurkat lymphocytic cells to investigate the immunostimulative effect and mechanisms of the 14 newly generated PD-1 mAbs. Multiple cellular and molecular biology experiments were performed in this study, such as CCK-8, ELISA, flow cytometry, immunofluorescence and Western blot.
Results: We found that mAb B1C4 significantly enhanced the tumor-killing cytokine secretion level by Jurkat cells in the co-culture system and increased the killing ability of Jurkat cells on HepG2 cells. Co-culture with HePG2 cells led to Jurkat cell cycle delay in S phase, and B1C4 promoted cell cycle progression from S to G2/M. Co-culture with HePG2 cells also caused apoptosis in Jurkat cells, which was inhibited by B1C4. B1C4 reversed the immunosuppression of Jurkat cells resulted from co-cultured with HePG2 cells through inhibiting PTEN and activating PI3K/AKT/mTOR signaling pathways.
Conclusion: Our study demonstrated that anti-PD-1 mAb B1C4 could inhibit the apoptosis of Jurkat cells induced by HePG2 hepatoma cells and reverse the immunosuppressive effect of HePG2 cells on Jurkat cells. The study provides a vital basis for applying PD-1 monoclonal antibodies in the treatment of HCC and provides antibody selection for the development of novel PD-1 mAb with blocking activity.
Keywords: hepatocellular carcinoma, monoclonal antibody, programmed cell death-1, PI3K/PTEN/AKT/mTOR signaling, T cell immunostimulation
Introduction
Programmed Death 1 (PD-1, CD279), as an immunosuppressive molecule of the B7-CD28 superfamily, is a transmembrane protein expressed on the surface of T cells. Two ligands that PD-1 has included PD-L1 (B7-H1) and PD-L2 (B7-DC). Tumor cells and tumor microenvironment inhibit the activation of T cells or induce T cell apoptosis by increasing PD-L1 expression and binding to PD-1 on the surface of tumor-specific CD8+ T cells, which in turn causes a decrease in the secretion of antitumor cytokines such as IFN-γ and IL-2.1,2 T cells are therefore unable to detect tumor cells and send signals to the immune system, allowing cancer cells to evade immune system clearance to limit the host’s immune response. PD-1/PD-L1 pathway inhibitor blocks the binding of PD-1 to PD-L1 to suppress the transmission of negative regulatory signals, which may restore T cell immune responses.3–5
PD-1/PD-L1 immune checkpoint inhibitors have been used in a variety of tumor immunotherapy,6–9 with problems of low response rates in a small number of cancer patients.10–13 Also, PD-1/PD-L1 treatment is often accompanied by direct or indirect toxicity, including fatigue, rash, itching, diarrhea, loss of appetite and nausea. Anti-PD-1 antibody immunotherapy may also cause mucosal mossy dermatitis in patients.14–16 In addition, in two cases of patients accepting anti-PD-1 antibody treatment, there was a severe skin toxicity reaction.17 Toxicity of anti-PD-1/PD-L1 antibody immunotherapy may compromise the efficacy of antibody in its clinical application. The mechanism of the action and toxicity of monoclonal antibody drugs is unclear, which needs further research to clarify. In the development and clinical application of anti-PD-1/PD-L1 antibodies, the toxic and side effects need to be fully considered to prevent adverse reactions. Therefore, the development of monoclonal antibodies (mAbs) with low side effects and high response rates in patients will maximize the use of antibodies in tumor immunotherapy.
In recent years, immunotherapy has made significant breakthroughs in the field of cancer treatment, eg, blocking PD-1/PD-L1 signal pathway with a monoclonal antibody against PD-1/PD-L1 has shown excellent antitumor efficacy in various solid tumors such as melanoma and nonsmall-cell lung cancer. Therefore, antibody therapy has become a focus in cancer in recent years.18–20 Studies have found that almost all types of human tumors express PD-L1, including melanoma, renal cell carcinoma, lung cancer, head and neck cancer, gastrointestinal cancer, bladder cancer, ovarian cancer, and hematological malignancies.21–24 Hepatocellular carcinoma (HCC) is a malignant tumor with a high fatality rate. The number of HCC cases in China accounts for about half of the total number of global cases, which provides a broad application prospect of immune checkpoint inhibitor for HCC.25 Nevertheless, PD-1/PD-L1 antibodies used in patients with hepatocellular carcinoma are reported with unclear molecular mechanisms. At present, whether PD-1 level could be used as a biomarker for hepatocellular carcinoma immunotherapy and prognosis evaluation is unclear, and whether the monoclonal antibody drugs blocking the PD-1/PD-L1 immune checkpoint can achieve good results in the treatment of HCC remains to be further investigated. Therefore, an in-depth study on the efficacy and mechanism of anti-PD-1 mAb therapy for HCC should be of great significance in the welfare of the HCC patients.
In previous studies, Sp2/0-Ag14 myeloma cells and spleen cells derived from BALB/c mice immunized with recombinant human PD-1/PDCD1 protein were fused for the production of novel antibodies. Fourteen mouse anti-human PD-1 mAbs were obtained in which the mAb 9E11 showed better antigen recognition specificity in HCC tissues. We investigated the clinical significance of PD-1 expression in HCC tissues from 77 cases with mAb 9E11 and found that the expression level of PD-1 in HCC tissues is closely related to the tumor size and differentiation degree.26 In this study, 14 mouse anti-human PD-1 mAbs were further studied to screen those with low cytotoxicity and better blocking activity on PD-1/PD-L1 immune checkpoints. Furthermore, the immunosuppressive effects of Jurkat lymphocytic cells caused by co-cultured with HePG2 liver cancer cells, and the immunostimulative mechanisms of the selected PD-1 mAb on Jurkat cells were evaluated.
Materials and Methods
Materials
RPMI-1640 medium, penicillin and streptomycin were purchased from Thermo Scientific; Fetal Bovine Serum (FBS) was purchased from Biological Industries (BI); 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), Phytohemagglutinin-P (PHA-P) were purchased from Sigma-Aldrich; Recombinant Human IFN-γ was purchased from Peprotech. Human IFN-γ ELISA Kit (Sensitivity: 4 pg/mL; Standard Curve Range: 4–500 pg/mL), Human IL-2 ELISA Kit (Sensitivity: 2 pg/mL; Standard Curve Range: 2–250 pg/mL), APC-conjugated Mouse IgG1 K Isotype Control and APC-conjugated anti-Human CD279 (PD-1) antibodies were purchased from eBioscience; Dimethyl Sulfoxide (DMSO), Propidium Iodide (PI), Phosphate Buffered Saline (PBS), Normal Mouse IgG, and Cell Counting Kit-8 (CCK-8) were purchased from Beyotime Biotechnology; Annexin V-FITC/PI Apoptosis Detection Kit was purchased from KeyGENBioTECH; Enhanced Chemiluminescence (ECL) was purchased from Millipore.
Preparation of mAbs
The mAbs were prepared by Yes Biotech Laboratories Ltd. Sp2/0-Ag14 myeloma cells and spleen cells derived from BALB/c mice immunized with recombinant human PD-1/PDCD1 protein were fused for the production of novel antibodies. The desired immunization effect was achieved after the serum antibody titers reached 1:107. PD-1 mAbs were generated and assigned the following names: 1C4, 1H8, 2C4, 2H7, 3E5, 5B2, 5H7, 7C9, 7E5, 7G12, 9A5, 9B4, 9E11 and B1C4. The heavy chains of 13 mAbs were IgG1, while clone 9A5 was observed to be IgG2a. All 14 mAbs were observed to contain κ light chains.
Cell Lines and Culture
Jurkat cells were obtained from ScienCell. HepG2 cells were obtained from American Type Culture Collection. They were incubated in complete RPMI-1640 medium (Thermo Fisher Scientific) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Biological Industries), 2 mM L-glutamine and penicillin-streptomycin solution (Thermo Fisher Scientific, 100 U/mL penicillin and 100 μg/mL streptomycin) at 37ºC with 5% CO2 and 95% air in a humidified atmosphere.
Co-Culture System
HepG2 cells were cultured in 6-well plates (2×105 cells/well) and stimulated with 10 ng/mL IFN-γ (PeproTech) after adherence for 24 h to overexpress PD-L1. Jurkat cells were activated with 2 μg/mL phytohemagglutinin (PHA, Sigma) for 48 h. The activated Jurkat cells (2×106 cells/well) were added to HepG2 cells at an E/T ratio (effective cells to target cells) of 10:1 and co-cultured for 48 h.
PD-L1 Overexpression in HePG2 Cells by IFN-γ Stimulation
HepG2 cells were collected in the logarithmic growth phase. 2×105 cells per well were seeded into 6-well plates. The serum-containing medium with different concentrations of IFN-γ (5, 10, 20 ng/mL) was added into each well. Control wells were added with 2 mL serum-containing medium without IFN-γ. Cells were incubated for 24 h and the total protein was extracted. The expression level of PD-L1 protein in HepG2 cells was detected by Western bolt to determine the optimal stimulation conditions by IFN-γ. The proteins were separated by 10% SDS-polyacrylamide gels and transferred onto PVDF membranes (Millipore). The membranes were blocked with 6% non-fat milk at 37ºC for 1 h and then incubated overnight with anti-PD-L1 antibody (1:500, Thermo Fisher, #17-5983-41) at 4ºC. Blots were incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:5000, ZSGB-BIO, ZB-2305) as the secondary antibody at 37ºC for 1 h, and then detected with Immobilon Western Chemiluminescent HRP Substrate (Millipore). Protein levels were quantified by densitometry using an AlphaEaseFC 4.0 software and then normalized against vehicle control (100%). β-Actin (Santa Cruz, sc-47778) was used as an internal control.
Activation of Jurkat Cells by PHA
The cytotoxicity of PHA on Jurkat cells was detected by CCK-8 (Beyotime, C0038). Jurkat cells were collected in the logarithmic growth phase. Approximately 5×104 cells were seeded onto 96-well plate and treated with increasing concentrations of PHA (0.5, 1, 2 and 5 μg/mL) for 48 h. Control cells were treated with an equal volume of culture medium without PHA. CCK-8 solution (20 μL) was then added to the medium. After incubation for 2 h, the amount of orange formazan dye generated was determined by measuring the absorbance at 450 nm using a microplate reader (Bio-Rad). The viability of treated cells was compared with control cells (100%). Jurkat cells at 2×106 per well were seeded into a 6-well plate. After incubation for 48 h, morphology under the microscope of Jurkat cells was observed and the cell culture supernatants were collected by centrifugation and filtered through a 0.22-μm steriflip (Millipore). Interleukin-2 (IL-2) concentrations were quantified using a Human IL-2 ELISA Kit (eBioscience) according to the manufacturer’s directions. Based on the analyses, the results were synthesized to determine the optimal stimulation conditions for PHA on Jurkat cells.
Analysis of PD-1 Expression on Jurkat Cells
The expression of PD-1 on the surface of Jurkat cells was detected by flow cytometry after incubated with HePG2 cells for a different time. Approximately 2×105 HePG2 cells were seeded onto a 6-well plate and treated with 10 ng/mL IFN-γ after adherence for 24 h to overexpress PD-L1. Jurkat cells were activated with 2 μg/mL PHA for 48 h. Added the activated Jurkat cells (2×106 cells/well) to HepG2 cells at an E/T ratio (effective cells to target cells) of 10:1. Jurkat cells were collected after co-culture for 12 h, 24 h, 48 h and 72 h. The cells were washed twice with PBS and resuspended in 100 μL PBS. The cell density was adjusted to 1×106 cells/vial. After incubation with 5 μL APC-conjugated anti-PD-1 antibodies (eBioscience) for 1 h at 4°C, the cells were washed with PBS and fixed in 2% paraformaldehyde. Cell surface expression of PD-1 on different co-culture conditions was detected by a NovoCyte flow cytometer (ACEA Biosciences) to determine the optimal experimental conditions for the co-culture system.
Screening of Newly Developed PD-1 mAb for Blocking PD-1/PD-L1 Immune Checkpoints
The HepG2 killing ability of Jurkat cells was detected by MTT assay. A co-culture system of adherent HepG2 and suspension Jurkat cells was established. Approximately 5×103 HePG2 cells were seeded onto a 96-well plate and treated with 10 ng/mL IFN-γ for 24 h. Activated Jurkat cells (5×104 cells/well) were added into HepG2 cells at an E/T ratio of 10:1. Fourteen newly generated anti-PD-1 antibodies (10 μg/mL) were added into the co-culture system for 48 h to block the interaction of PD-1 and PD-L1. Mouse IgG was used as a negative control. After co-culture for 48 h, suspended Jurkat cells were isolated and 20 μL of MTT solution (5 mg/mL in PBS) were added to HepG2 cells for another 4 h, and then bleached with DMSO. The cytolytic effect of Jurkat cells on HepG2 cells was analyzed by a microplate reader (Bio-Rad) at 490 nm. Cell viability was compared with blank control cells (100%). Cell culture supernatants were collected and filtered through a 0.22-μm Steriflip (Millipore). Levels of IL-2 in the supernatants were measured by ELISA (eBioscience) following the manufacturer’s instructions. Jurkat cells activated by 2 μg/mL PHA for 48 h served as a positive control.
Effects of mAb 2H7 and B1C4 on Jurkat Cell Activity and Cytokine Secretion
In order to obtain PD-1 mAb with better blocking activity and lower cytotoxicity, CCK-8 method was used to study the chronergy and quantitative efficiency of mAb 2H7 and B1C4 on Jurkat cells. Approximately 5×104 cells were seeded onto a 96-well plate and treated with increasing concentrations of mAb 2H7 and B1C4 (5, 10, 20 and 40 μg/mL). Control cells were treated with an equal volume of culture medium. After incubated for 24, 48 and 72 h, 20 μL Cell Counting Kit-8 solution was added to the medium. After incubation for two more hours, the amount of orange formazan dye generated was determined by measuring the absorbance at 450 nm using a microplate reader (Bio-Rad). Based on CCK-8 results, mAb 2H7 (10 μg/mL) and B1C4 (5 μg/mL) were added into the co-culture system and incubated for 48 h. Levels of IL-2 and IFN-γ in the supernatants were measured by ELISA (eBioscience) based on the manufacturer’s instructions. Mouse IgG was used as a negative control. The cell co-culture system without adding any stimulants was used as the blank control. Jurkat cells activated by 2 μg/mL PHA for 48 h served as the positive control.
Cell Cycle Analysis
The co-culture system was established according to the above methods. Jurkat cells activated by 2 μg/mL PHA for 48 h were used as control. Changes in the cell cycle were investigated after co-culture for 24 and 48 h and then treated with blocking PD-1 mAb B1C4 (5 μg/mL) for 48 h. Suspended Jurkat cells were collected after co-culture and fixed with 75% ethanol at 4°C overnight. The proportion of cells in each stage of the cell cycle was quantitatively estimated by propidium iodide (PI) staining and detected with a NovoCyte flow cytometer (ACEA Biosciences).
Apoptosis Assay
The cell co-culture system was established according to the above methods. Jurkat cells without activation of PHA were used as control. Changes in apoptosis level were investigated in Jurkat cells activated by 2 μg/mL PHA for 48 h, co-cultured for 48 h and co-cultured for 48 h with PD-1 mAb B1C4 (5 μg/mL) added. The effect of co-culture system and PD-1 mAb B1C4 on apoptosis of Jurkat cells was measured. Morphological characteristics of chromatin indicating cell apoptosis were observed by DAPI staining under a confocal microscope (Leica). The early and late apoptosis cells were quantitatively estimated by Annexin V-FITC/PI double staining from the NovoCyte flow cytometer (ACEA Biosciences) using an Annexin V-FITC/PI apoptosis detection kit (KeyGEN BioTECH).
Immunofluorescence Staining
The cell co-culture system was established according to the above methods. Jurkat cells activated by 2 μg/mL PHA for 48 h were used as control. On the basis of the cell co-culture system, 5 μg/mL mAb B1C4 were added to study the effects of PD-1 mAb B1C4 on apoptosis-related protein expression in Jurkat cells. After co-culture, suspended Jurkat cells were collected and fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 for 20 min, and blocked with normal goat serum for 30 min to block non-specific protein–protein interactions. The anti-PTEN (1:500, Abcam, ab170941), anti-phospho-Akt (Thr308) (1:1000, Cell Signaling Technology, #2965), anti-Akt (1:1000, Abcam, ab200195), anti-phospho-mTOR (1:200, Cell Signaling Technology, #5536), anti-mTOR (1:100, Abcam, ab2732), anti-cleaved caspase-3 (1:100, Abcam, ab32042), anti-cleaved caspase-9 (1:500, Cell Signaling Technology, #52873), and anti-Bax (1:100, Abcam, ab199677) were then incubated with the cells overnight at 4°C. The secondary antibody Alexa Fluor 488-conjugated (green) goat Anti-Rabbit IgG (1:100, ZSGB-BIO, ZF-0511) was incubated with the cells for 1 h at 37°C. After a final wash, the nuclei were counterstained with DAPI (4ʹ,-6-diamidino-2-phenylindole, Beyotime, C1005) in 1×PBS for 5 min before imaging. Cells were visualized with a confocal microscope (Leica).
Western Blot Analysis
Cell grouping was the same as above. After co-culture, suspended Jurkat cells were collected and analyzed for protein on 10% or 12% SDS-polyacrylamide gels (SDS-PAGE). Proteins were transferred onto PVDF membranes (Millipore). The membranes were blocked with 6% skim milk at 37ºC for 1 h and then incubated overnight with corresponding antibodies at 4ºC. The following antibodies were used: anti-PTEN (1:500, Abcam, ab170941), anti-phospho-Akt (Thr308) (1:500, Cell Signaling Technology, #2965), anti-Akt (1:500, Abcam, ab200195), anti-phospho-mTOR (1:500, Cell Signaling Technology, #5536), anti-mTOR (1:500, Abcam, ab2732), anti-cleaved caspase-3 (1:500, Abcam, ab32042), anti-cleaved caspase-9 (1:500, Cell Signaling Technology, #52873), anti-Bax (1:500, Abcam, ab199677) and anti-β-Actin (1:1000, Santa Cruz, sc-47778). The membranes were then incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit (1:2000, ZSGB-BIO, ZB-2301) or anti-mouse IgG (1:2000, ZSGB-BIO, ZB-2305) as the secondary antibody at 37ºC for 1 h. Specific proteins were visualized with Immobilon Western Chemiluminescent HRP Substrate (Millipore). Protein levels were quantified via densitometry using AlphaEaseFC 4.0 software and then normalized against control (100%). β-Actin was used as an internal control.
Effect of the Dual PI3K/mTOR Inhibitor on Jurkat Cells
In order to investigate the influence of the dual PI3K/mTOR inhibitor (NVP-BEZ235, MedChemExpress, HY-50673) on co-cultured HePG2 and Jurkat cells, the cytotoxicity was detected by the MTT method and CCK-8, respectively. HePG2 cells (5×103) and Jurkat cells (5×104) were seeded onto 96-well plates and treated with increasing concentrations of NVP-BEZ235 (0, 125, 250, 500 and 1000 nM) for 24, 48 and 72 h, respectively. Control cells were treated with an equal volume of culture medium without NVP-BEZ235. Cell viability was compared with control cells (100%). After that, the cell co-culture system was established according to the methods described above and served as a blank control group. mAb B1C4 (5 μg/mL) was used as the positive control. mAb B1C4 in combination with NVP-BEZ235 was added into the co-culture system for 48 h to investigate the effect of the dual PI3K/mTOR inhibitor on the expression of phospho-Akt and phospho-mTOR. The killing effect of Jurkat cells on HepG2 cells was analyzed by MTT.
Statistical Analysis
Results were expressed as the mean ± SD. Statistical analysis was performed by one-way analysis of variance (ANOVA). P<0.05 was considered statistically significant. All analyses were performed using IBM SPSS Statistics 23.0.
Results
IFN-γ Induced the Overexpression of PD-L1 on the Surface of HepG2 Cells
As illustrated in Figure 1A, the level of PD-L1 expression on the surface of HepG2 cells was significantly increased after 24 h of treatment with IFN-γ (P<0.01). With the increasing of IFN-γ concentrations, PD-L1 expression on the surface of HepG2 cells increased progressively. The expression of PD-L1 achieved a maximum of 241.99% (P<0.01) with IFN-γ at 10 ng/mL. However, PD-L1 expression decreased when IFN-γ reached 20 ng/mL. IFN-γ at 10 ng/mL was then used in subsequent experiments to induce the overexpression of PD-L1 on the surface of HepG2 cells.27
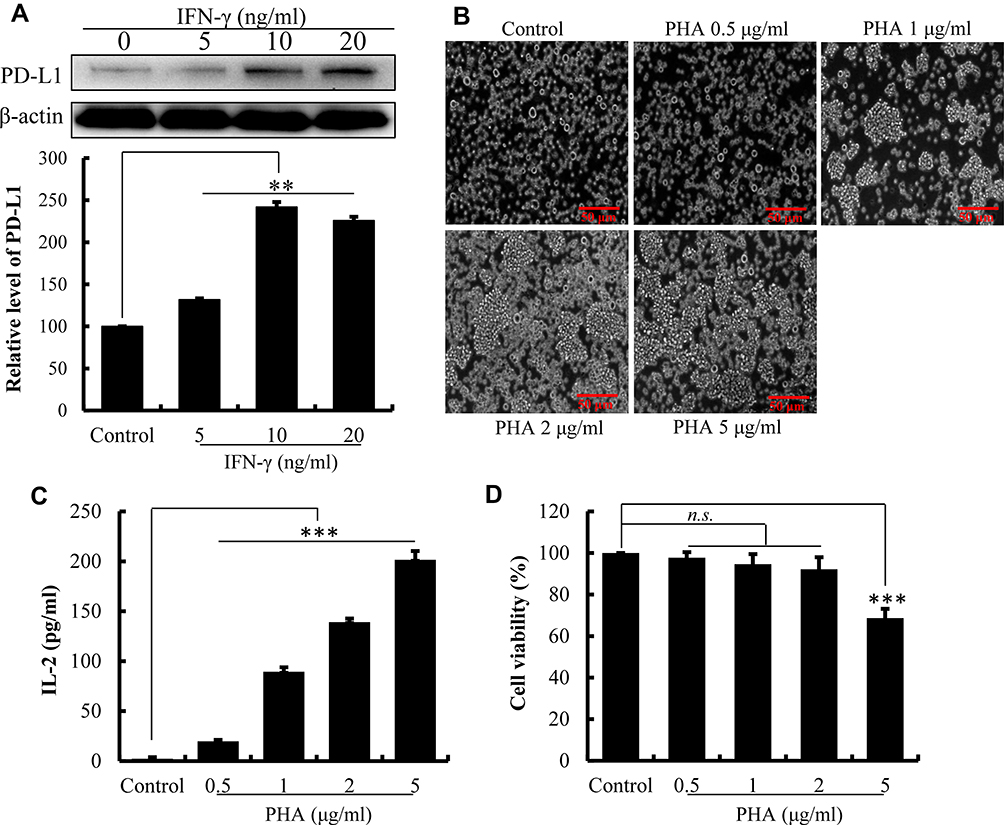 |
Figure 1 Establishment of the co-culture system of Jurkat lymphocytic cell and HePG2 liver cancer cell. (A) The effect of IFN-γ on the expression of PD-L1 by HepG2 cell determined by Western blot. Densitometric values analyzed using the AlphaEaseFC 4.0 software program. β-Actin served as an internal control. Protein expression was normalized against the control. (B) The effect of PHA on morphology of Jurkat cell observed using an inverted microscope. (C) The cytotoxicity of PHA on Jurkat cells detected by Cell Counting-Kit 8. Cell viability was compared with control cells (100%). (D) PHA promotes the secretion of IL-2 by Jurkat cells. The data are presented as the mean ± S.D. (n=3), **p<0.01, ***p<0.001 vs control. |
PHA-Activated Jurkat Cells
The results in Figure 1B demonstrate that PHA promoted the Jurkat cells clump together to accelerate their proliferation. Moreover, PHA promoted the release of IL-2 by Jurkat cell. As the concentration of PHA increased, the secretion level of IL-2 cytokines significantly increased (Figure 1C, P<0.01). Indicated that the PHA could promote the activation of Jurkat cells through promoting the cell proliferation and cytokine secretion. However, excessive PHA was cytotoxic to Jurkat cells. When PHA concentration reached 5 μg/mL, the survival rate of Jurkat cells decreased compared to the control cell (Figure 1D, P<0.01). Therefore, the stimulating concentration of PHA was set at 2 μg/mL in the cell co-culture system.
Co-Culture with HePG2 Cells Induced PD-1 Overexpression on Jurkat Cells
PD-1 expression on Jurkat cells was determined by FACS analysis after co-culture with HepG2 cells. Activated Jurkat cells were also cultured alone as controls. As illustrated in Figure 2, the expression of PD-1 was induced on Jurkat cells after co-culture with HepG2 cells (P<0.001). With the increasing co-culture time, the expression level of PD-1 on the surface of Jurkat cells also increased, with a reduction after 72 h. Given that the prolonged co-culture may lead to changes in cell viability or stability, the time limit for cell co-culture was set at 48 h.
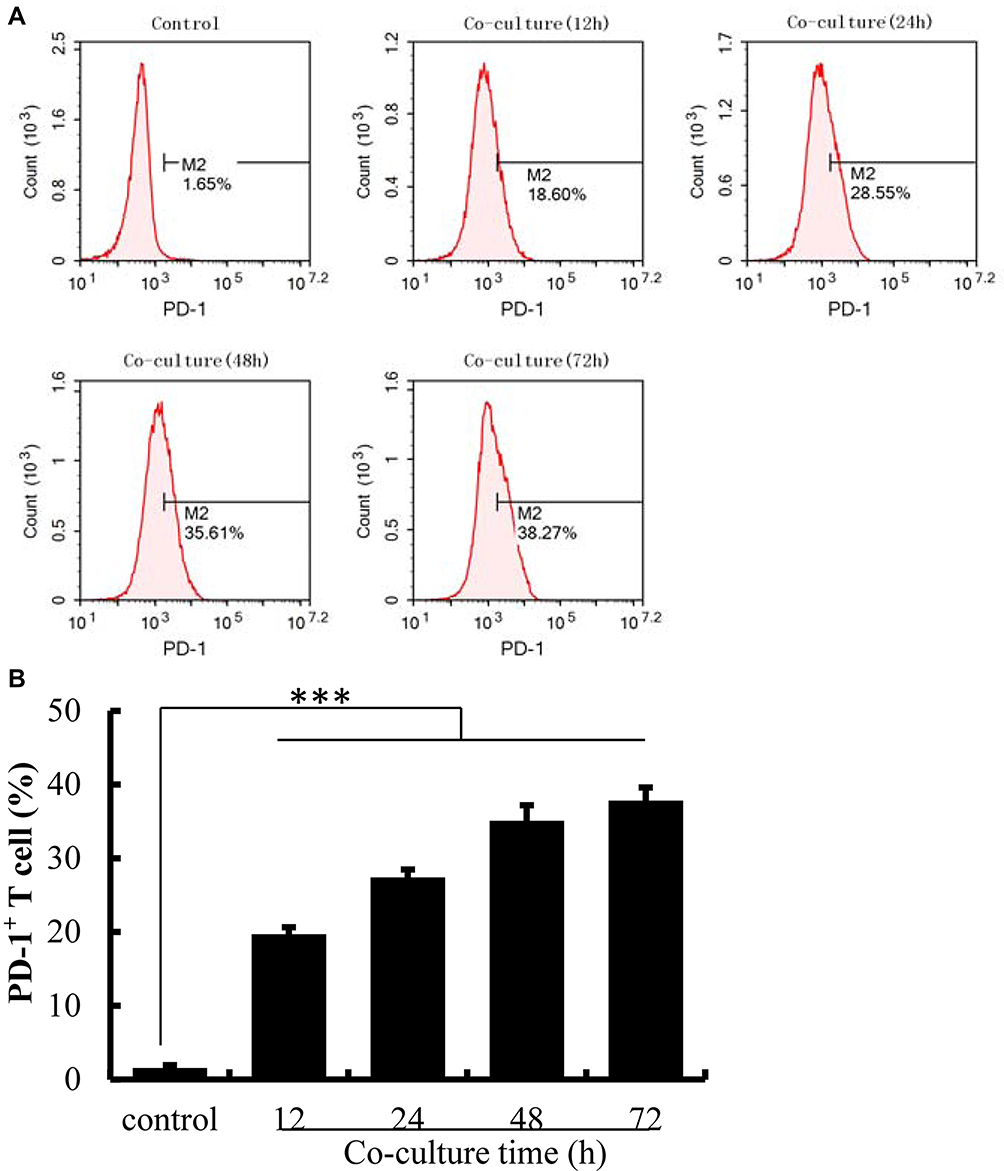 |
Figure 2 The expression of PD-1 was up-regulated on Jurkat cells after co-culture with HePG2 cells. (A) The expression of PD-1 on Jurkat cells detected by flow cytometry. (B) The percentage of PD-1+ T cells analyzed using IBM SPSS Statistics 23.0. Jurkat cells activated by PHA were cultured alone as controls. The data are presented as the mean ± S.D. (n=3), ***p<0.001 vs control. |
mAb 2H7 and B1C4 Showed Better Blocking Activity for PD-1/PD-L1 Interaction
As illustrated in Figure 3A, the survival rate of HepG2 cells in the co-culture system was significantly decreased (P<0.01) after cultured with mAb 2H7, B1C4, 5B2 and 5H7, indicating that the tumor-killing ability of Jurkat cells was enhanced by adding four antibodies into co-culture. This suggests that the above mAbs exhibit blocking activity on PD-1/PD-L1 signal pathway, with a more potent effect show by mAb 2H7 and B1C4 (P<0.001). The levels of IL-2 in the supernatant were also increased compared with the control cell (Figure 3B, P<0.001). Hence, mAb 2H7 and B1C4 were selected for further experiments.
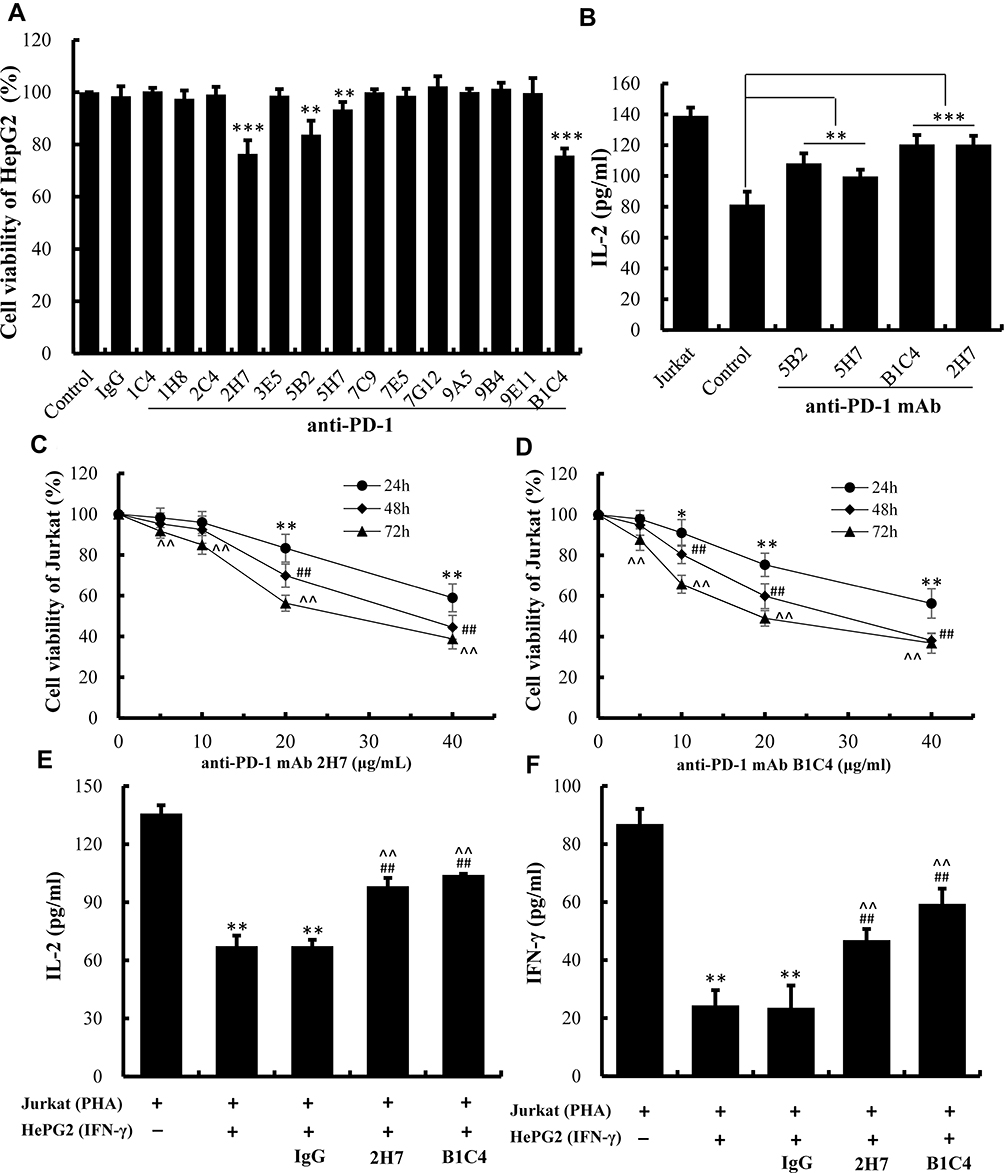 |
Figure 3 B1C4 was screened out from the 14 newly developed mAbs with better blocking activity for PD-1/PD-L1 interaction. (A) Killing ability of Jurkat cells on HepG2 cells detected by MTT assay after treatment with 14 anti-PD-1 mAbs. (B) Secretion of IL-2 by Jurkat cells measured by ELISA. Co-culture system of HepG2 and Jurkat cells were used as controls. The data are presented as the mean ±S.D. (n=3), **p<0.01, ***p<0.001 vs control. (C) The viability of Jurkat cells after treatment with mAb 2H7. (D) The viability of Jurkat cells treated with mAb B1C4. The data are presented as the mean ±S.D. (n=3). *p<0.05, **p<0.01 vs 0 μg/mL (24 h); ##p<0.01 vs 0 μg/mL (48 h); ^^p<0.01 vs 0 μg/mL (72 h). (E) The influence of mAb B1C4 and 2H7 on IL-2 secretion by Jurkat cells after co-culture with HePG2 cells. (F) Influence of mAb B1C4 and 2H7 on IFN-γ secretion in Jurkat cells after co-cultured with HePG2 cells. The data are presented as the mean ± S.D. (n=3). **p<0.01 vs Jurkat; ##p<0.01 vs Jukat+HepG2; ^^p<0.01 vs Jukat+HepG2+IgG. |
Function Stimulation of T Cells by mAb B1C4
The chronergy and quantitative efficiency of mAb 2H7 and B1C4 on Jurkat cells were detected using CCK-8 assay (Figure 3C and D). As the concentrations of the mAbs increased or the incubation time of the cells was prolonged, mAb 2H7 and B1C4 showed a certain degree of cytotoxicity to Jurkat cells. mAb 2H7 showed no cytotoxicity at the concentration of 10 μg/mL within 48 h of culture. Moderate cytotoxicity was observed as the concentration of mAb 2H7 increased to 20 μg/mL or the incubation time was prolonged to 72 h (P<0.01). mAb B1C4 showed no cytotoxicity at the concentration of 5 μg/mL within 48 h of incubation. Cytotoxicity was shown as the concentration of mAb B1C4 increased to 10 μg/mL (P<0.05) or the incubation time was prolonged to 72 h (P<0.01).
Based on these results, mAb 2H7 (10 μg/mL) and B1C4 (5 μg/mL) were added into the co-culture system for 48 h, respectively, to further investigate the effects of the two blocking PD-1 mAbs on cytokine secretion by Jurkat cells. As shown in Figure 3E and F, the levels of IL-2 and IFN-γ released by PHA-activated Jurkat cells were 132.85±4.29 pg/mL and 86.92±5.20 pg/mL, respectively, which were significantly decreased after co-culture with HePG2 cells for 48 h (P<0.01). There were no significant changes compared with the co-culture group with a control antibody (mouse IgG). Compared with the co-culture group and the mouse IgG group, the levels of IL-2 and IFN-γ were significantly increased after added mAb 2H7 and B1C4 into the co-culture (P<0.01), where B1C4 showed a better effect. The results showed that mAb B1C4 more effectively reverses the immunosuppressive status of Jurkat cells in the co-culture system according to promote the secretion of cytokines IL-2 and IFN-γ in Jurkat cells.
mAb B1C4 Improved the Cell Cycle Delay of Jurkat Cells Caused by Co-Cultured with HePG2 Cells
We then used flow cytometry to perform cell cycle analysis. Figure 4 shows the cell cycle distribution of Jurkat cells activated by 2 μg/mL PHA for 48 h was 44.73%±1.76% in G1/G0 phase, 28.61%±1.19% in S phase, and 25.35%±1.35% in G2/M phase. After co-culture with HepG2 cells for 24 and 48 h, the percentage of Jurkat cells decreased in G1/G0 phase and G2/M phase (P<0.05), but significantly increased in S phase (P<0.01). In addition to mAb B1C4 into the co-culture system for 48 h, the percentage of Jurkat cells showed no significant changes in G1/G0 phase but significantly decreased in S phase and increased in G2/M phase (Figure 4B, P<0.01). These results indicate that co-culture with HePG2 cells leads to Jurkat cell cycle delay in S phase. Anti-PD-1 mAb B1C4 promotes cell cycle progression from S to G2/M. Therefore, co-culture with HePG2 cells may suppress the immune function of Jurkat cells by delaying the cell cycle in S phase. B1C4 promotes the progression of cell cycle to restore the function of Jurkat cells.
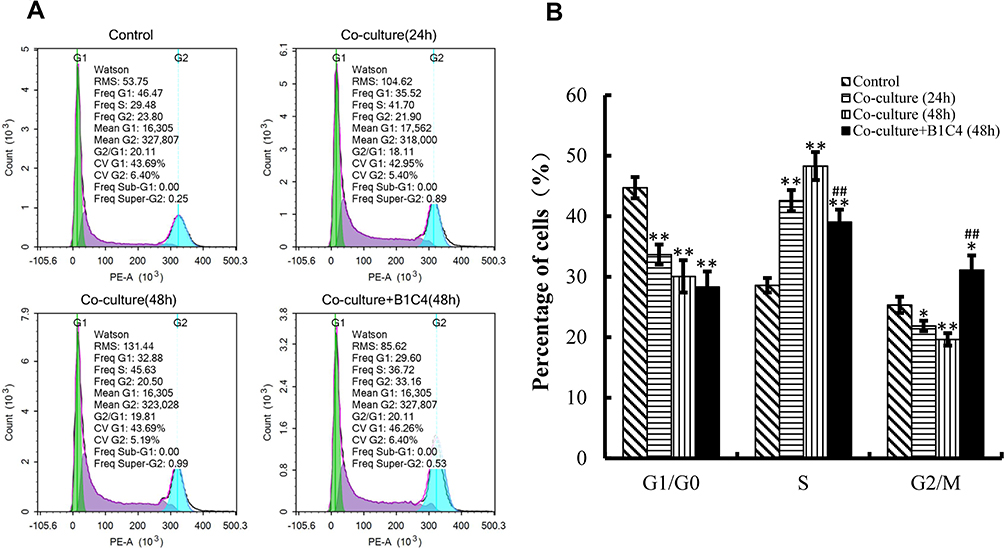 |
Figure 4 mAb B1C4 improved cycle delay of Jurkat cells caused by co-culture with HePG2 cells. (A) The percentage of Jurkat in each phase of the cell cycle was estimated using a flow cytometer. (B) The percentage of Jurkat in each phase analyzed using IBM SPSS Statistics 23.0. Jurkat cells activated by 2 μg/mL PHA for 48 h were used as control. Data are presented as the means ± S.D. (n=3). *p<0.05, **p<0.01 vs control; ##p<0.01 vs the co-culture group for 48 h. |
mAb B1C4 Inhibited the Apoptosis of Jurkat Cells Induced by Co-Culture with HePG2 Cells
We examined changes in the morphological characteristics of chromatin indicative of apoptosis. Homogeneous chromatin morphology was observed in Jurkat cells cultured alone (inactivated and activated). After co-culture with HePG2 cells, Jurkat cells exhibited morphological features of apoptosis, such as chromatin condensation and marginalization or nuclei fragmentation. Cell apoptosis in co-culture system was improved after incubation with mAb B1C4 (Figure 5A). The results in Figure 5B and C show that, compared with Jurkat cells without PHA treatment (3.56%±0.60%), the percentage of apoptotic cells was increased to 9.32%±0.62% after activated by PHA (2 μg/mL). When PHA-activated (2 μg/mL, 48 h) Jurkat cells were co-cultured with IFN-γ pretreated (10 ng/mL, 24 h) HepG2 cells for 48 h, the total apoptosis rate of Jurkat cells was increased to 33.52%±0.97% (P<0.01). When the cell co-culture system was incubated with mAb B1C4 for 48 h, the total apoptosis rate of Jurkat cells was decreased to 25.85%±1.24% (P<0.01). The results indicate that co-culture with hepatoma cells induces the apoptosis of Jurkat cells, resulting in immune function suppression. B1C4 inhibits the apoptosis of Jurkat cells in the co-culture system to promote the immune function recovery.
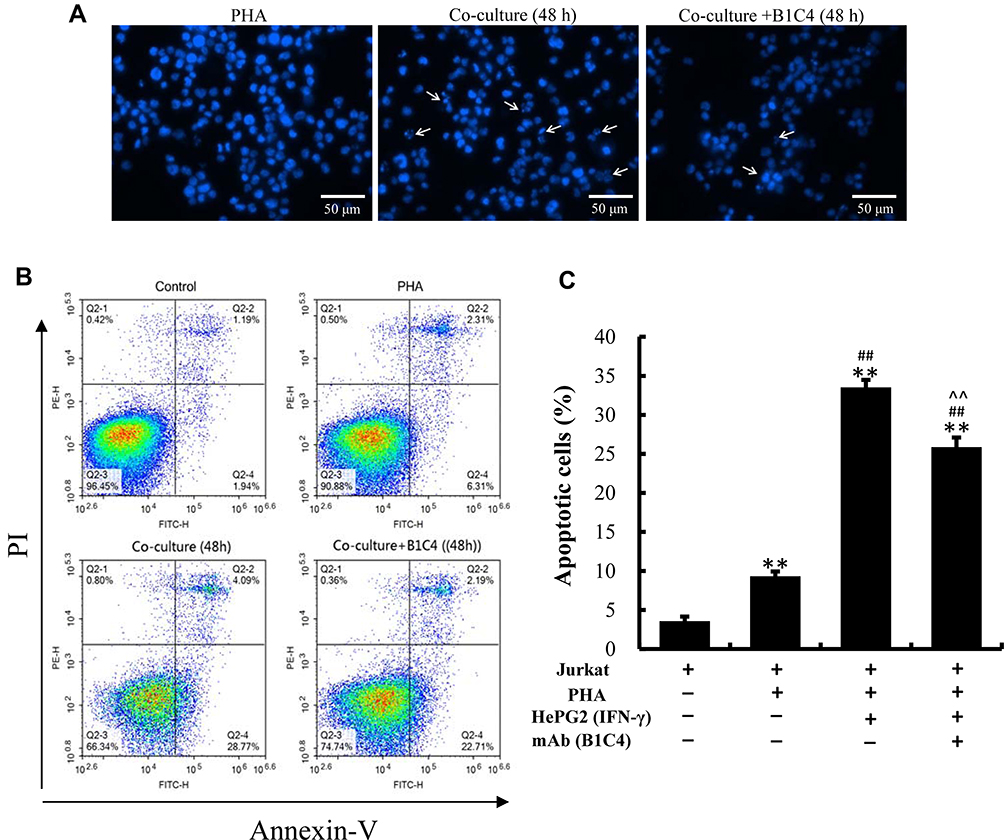 |
Figure 5 mAb B1C4 inhibited the apoptosis of Jurkat cells induced by co-culture with HePG2 cells. (A) Jurkat cells stained with DAPI for cell nuclei and observed under laser-scanning confocal microscopy. White arrows indicate condensed chromatin, marginalization or nuclei fragmented into apoptotic bodies. (B, C) Apoptosis of Jurkat cells estimated using a flow cytometer. Jurkat cells without activation by PHA were used as controls. The bar graphs show the percentage of apoptotic cells for each of the indicated conditions. The data are presented as the mean ± S.D. (n=3), **p<0.01 vs control; ##p<0.01 vs the PHA-activated Jurkat cells; ^^p<0.01 vs Jurkat (PHA)+HepG2 (IFN-γ). |
mAb B1C4 Reversed the Immunosuppression of Jurkat Cells Caused by Co-Cultured with HePG2 Cells Through Inhibiting PTEN and Activating PI3K/AKT/mTOR Signaling Pathway
Since phosphatases and tensin homolog (PTEN) is an essential tumor suppressor gene that encodes a phosphatase that antagonizes PI3K/AKT/mTOR anti-apoptotic pathway,28,29 we next examined whether mAb B1C4 inhibit the apoptosis of Jurkat cells through PTEN. As shown in Figure 6, PTEN protein level in Jurkat cells was significantly increased after co-culture with HePG2 cells. However, PTEN was decreased after treatment with mAb B1C4. In contrast, p-AKT and p-mTOR were significantly increased after treatment with mAb B1C4, which inhibited Bax, cleaved caspase-9 and the downstream effector cleaved caspase-3 leading to apoptosis of Jurkat cells induced by co-cultured with hepatoma cells. Western blot further confirmed the decrease of PTEN in Jurkat cells treated by mAb B1C4 (Figure 7). Figure 8A and B showed that cytotoxicity on both HePG2 and Jurkat cells appeared as the concentration of NVP-BEZ235 increased to 500 nM (P<0.01). NVP-BEZ235 showed no cytotoxicity on HePG2 cells at the concentration of 250 nM within 48 h of incubation, but with cytotoxicity for Jurkat cells within 24 h incubation (P<0.05). Therefore, 125 nM NVP-BEZ235 together with 5 μg/mL mAb B1C4 were added into the co-culture system for 48 h in the following studies. Results in Figure 8C show that compared with mAb B1C4 added alone, the expression of p-AKT (P<0.01) and p-mTOR (P<0.05) was significantly decreased in the presence of NVP-BEZ235. The killing ability of Jurkat cells on HePG2 which was promoted by mAb B1C4 was also inhibited by NVP-BEZ235 (Figure 8D). These results indicate that activation of PI3K/AKT/mTOR signaling pathway by mAb B1C4 was blocked by the dual PI3K/mTOR inhibitor. These results suggest that mAb B1C4 inhibits PTEN and activate PI3K/AKT/mTOR signaling pathway in Jurkat cells, which may inhibit the apoptosis of Jurkat cells induced by PD-L1 expressed on hepatoma cells (Figure 9).
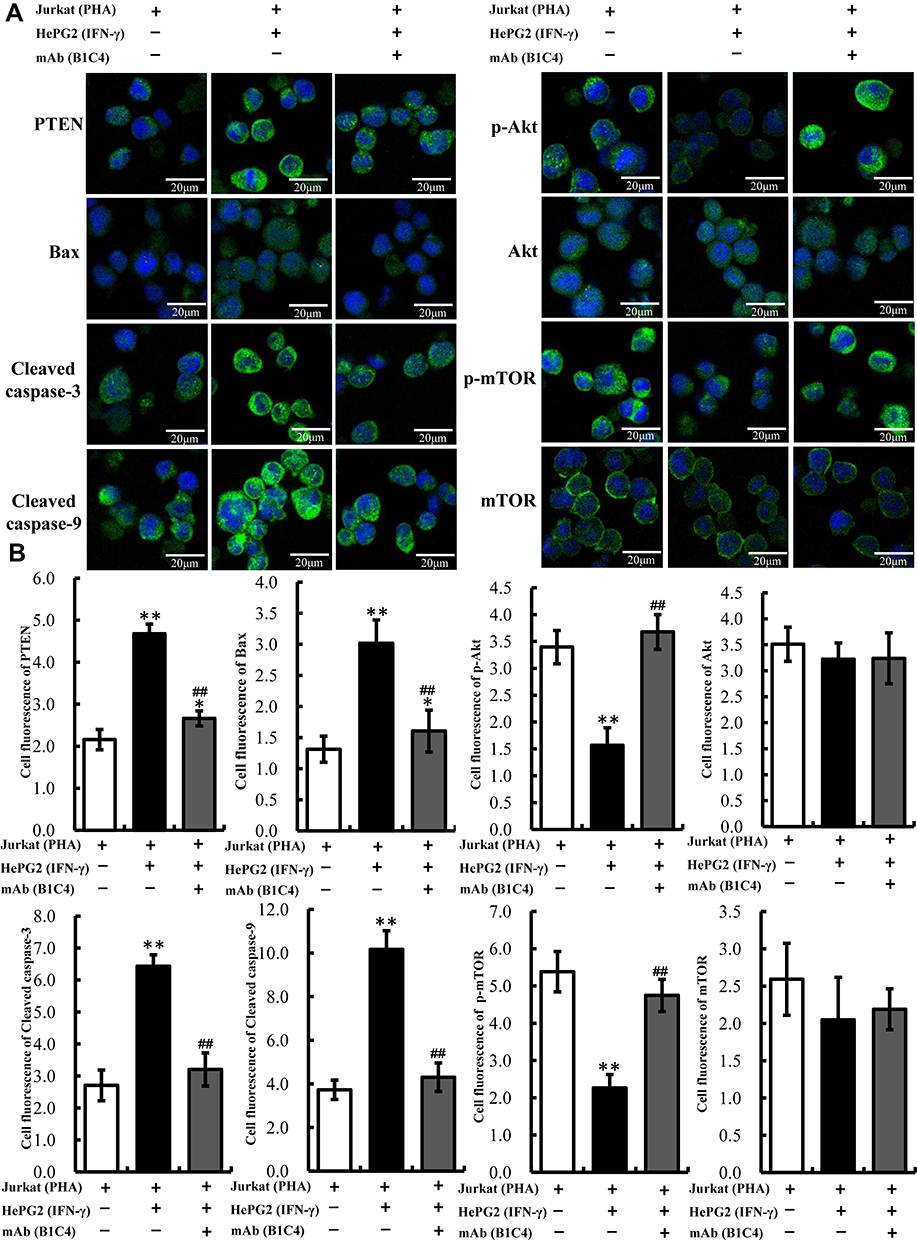 |
Figure 6 The expression of PTEN, p-Akt, Akt, p-mTOR, mTOR, Cleaved caspase-9, Cleaved caspase-3 and Bax in Jurkat cells determined by immunofluorescence staining. (A) Proteins stained by indirect fluorescence with secondary antibodies conjugated to Alexa Fluor-488 (green). Nuclei were counterstained with DAPI (blue). A representative image (n=3) is shown. Images were collected using a Leica TCS SP5 confocal imaging system. (B) The bar graphs show the fluorescence intensity of the proteins. The data are presented as the mean ± S.D. (n=10). *p<0.05, **p<0.01 vs Jurkat (PHA); ##p<0.01 vs Jurkat (PHA)+HepG2 (IFN-γ). |
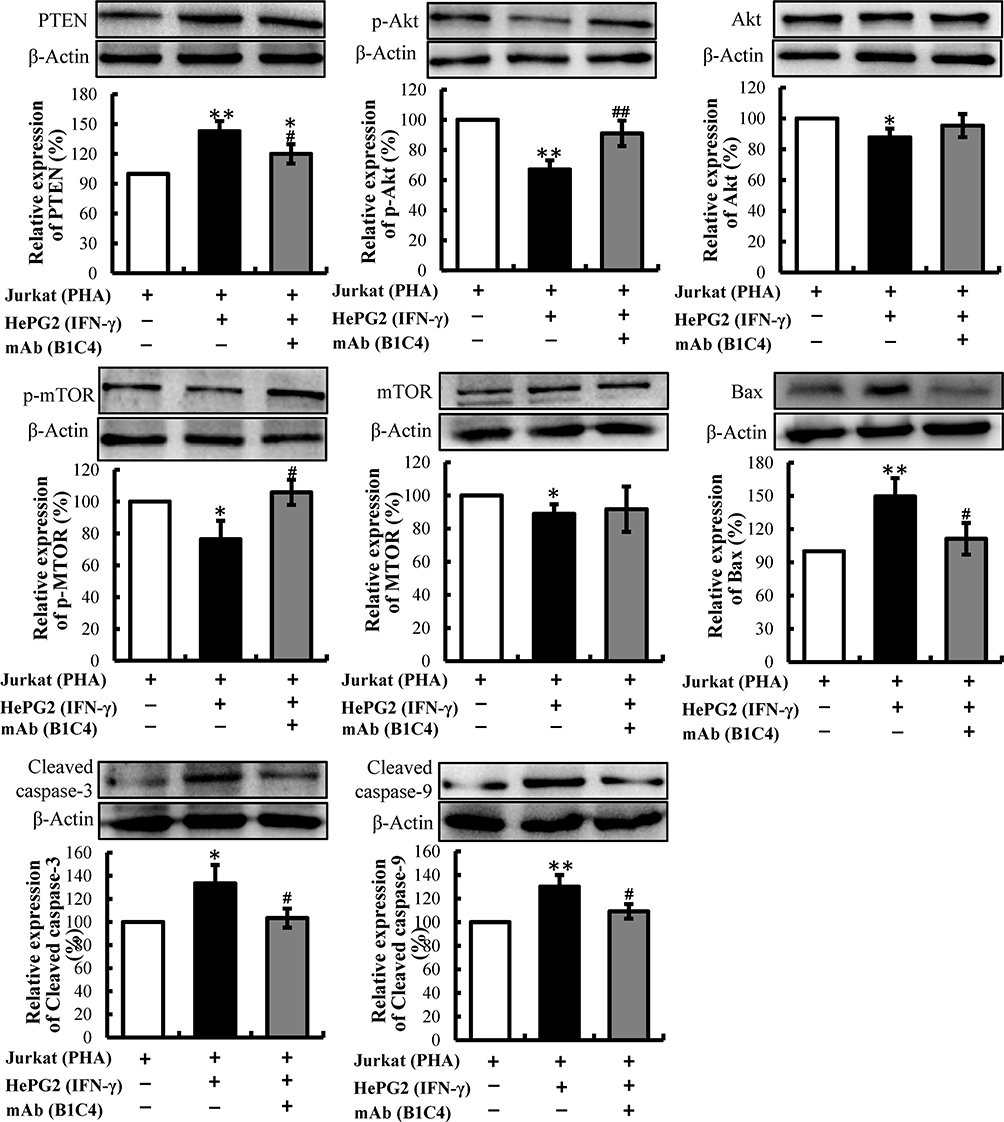 |
Figure 7 The expressions of PTEN, p-Akt, Akt, p-mTOR, mTOR, Bax, Cleaved caspase-3 and Cleaved caspase-9 in Jurkat cells determined by Western blot. Densitometric values were analyzed using the AlphaEaseFC 4.0 software program. β-Actin was used as an internal control. Jurkat cells activated by 2 μg/mL PHA for 48 h were used as controls and protein levels were normalized against the control. The data are presented as the means ± S.D. (n=3). *p<0.05, **p<0.01 vs Jurkat (PHA); #p< 0.05, ##p< 0.01 vs Jurkat (PHA)+HepG2 (IFN-γ). |
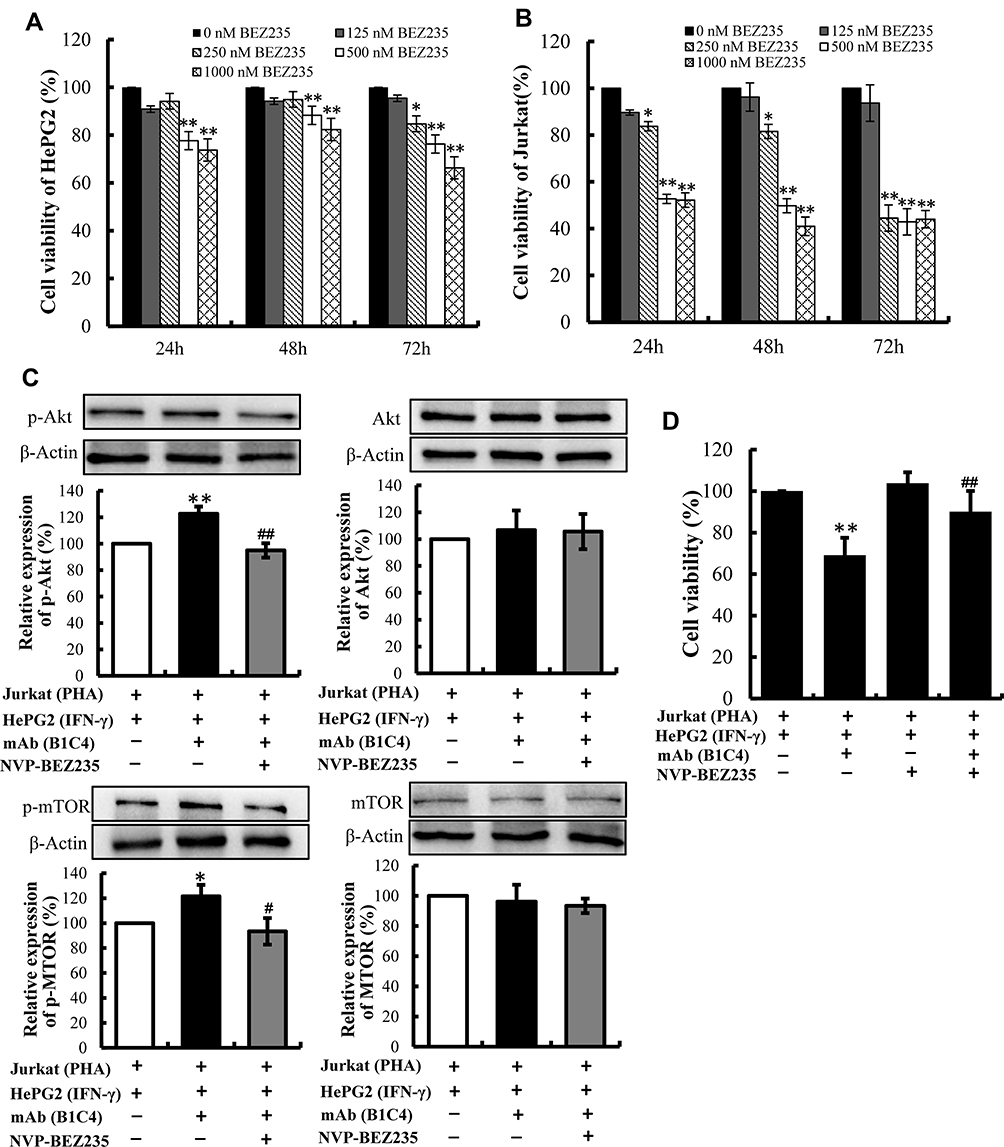 |
Figure 8 Activation of PI3K/AKT/mTOR signaling pathway by mAb B1C4 was blocked by the dual PI3K/mTOR inhibitor (NVP-BEZ235). (A, B) The viability of HePG2 and Jurkat cells after treatment with NVP-BEZ235. The data are presented as the mean ±S.D. (n=3). *p<0.05, **p<0.01 vs 0 nM NVP-BEZ235. (C) Influence of NVP-BEZ235 on the protein expression by Jurkat cells in co-culture system detected by Western blot. Densitometric values were analyzed using the AlphaEaseFC 4.0 software program. β-Actin was used as an internal control. Jurkat cells co-cultured with HePG2 cells for 48 h were used as controls and protein expression was normalized against the control. The data are presented as the means ± S.D. (n=3). *p<0.05, **p<0.01 vs Jurkat (PHA)+HepG2 (IFN-γ); #p<0.05 vs (PHA)+HepG2 (IFN-γ)+mAb (B1C4). (D) Influence of the dual PI3K/mTOR inhibitor (NVP-BEZ235) on killing ability of Jurkat cells in co-culture system detected by MTT assay. The data are presented as the means ± S.D. (n=3). **p<0.01 vs (PHA)+HepG2 (IFN-γ); ##p<0.01 vs Jurkat (PHA)+HepG2 (IFN-γ)+mAb (B1C4). |
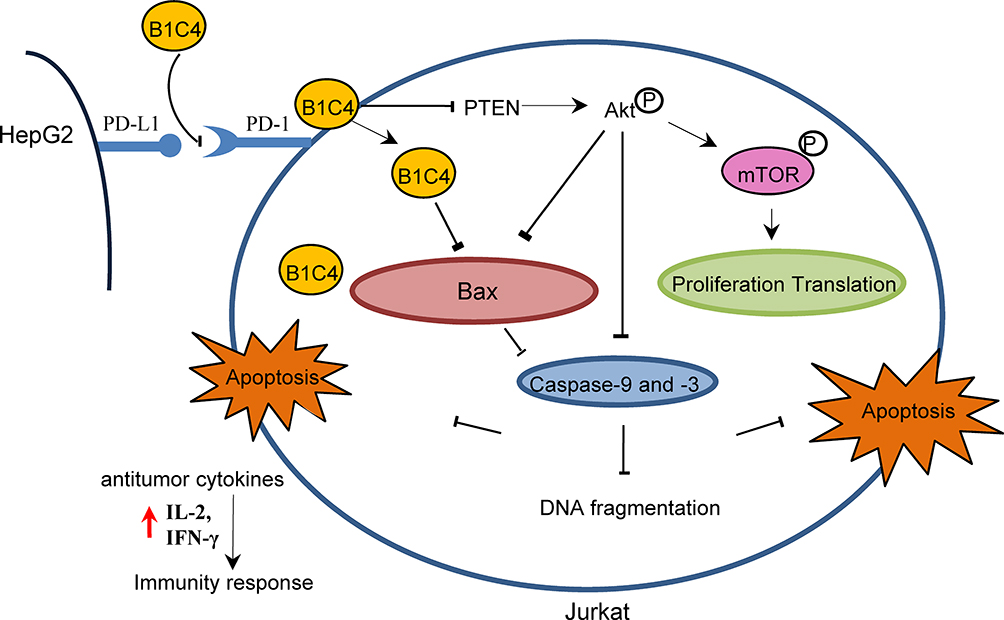 |
Figure 9 mAb B1C4 reversed the immunosuppression of Jurkat cells caused by co-culture with HePG2 cells through inhibiting PTEN and activating PI3K/AKT/mTOR signaling pathway. |
Discussion
PD-1 and PD-L1 interaction has been demonstrated to negatively regulate T-cell receptor signaling to further inhibit the cell activation and function, leading to inhibition of the immune response in cancer patients.30,31 PD-1 is selectively overexpressed on depleted effector T cells. In acute infection stage of LCMV (lymphocytic choriomeningitis virus), the expression level of PD-1 on the surface of effector T lymphocytes increased, and the expression of PD-1 on memory T cells decreased or no expression after the infection was restored.32,33 A CD4+ T cell line Jurkat cells were used as effector T cells co-cultured with hepatoma cells. FACS analysis showed that tumor cells induced PD-1 expression on T cells.34 In this study, Jurkat cells activated by PHA to promote the secretion of IL-2 and IFN-γ were used to simulate effector T cells. IFN-γ is a cytokine that induces PD-L1 in tumor cells and was therefore used to stimulate HepG2 cells to overexpress PD-L1.27 The two pretreated cells were co-cultured to mimic the tumor microenvironment where the immune activity of T cells was inhibited after interaction with hepatoma cells. The co-culture system was used to evaluate the blocking activity of 14 newly generated anti-human PD-1 mAbs. We found that co-cultured with hepatoma cells can up-regulate the expression of PD-1 on Jurkat cells. The levels of cytokines including IL-2 and INF-γ in culture supernatant and the killing ability of T cells for hepatoma cells were significantly elevated in culture with mAb B1C4. This suggests that the immune reaction by blocking PD-1/PD-L1 pathway with anti-PD-1 mAb B1C4 restores the function of Jurkat T cells.
PD-L1 expressed on tumor cells interact with PD-1 on effector T lymphocytes that induces a series of immunosuppressive responses, allowing tumor cells to evade immune clearance.35 Studies have found that PD-1 protein expressed in T lymphocytes blocks the progression of their cycles, thus inhibiting T cell proliferation.36 The expression of PD-1 on CD8+ T lymphocytes in HCC patients has an adverse regulative effect on cell activation and proliferation.37 PD-L1 expressed on tumor cells contributed to tumor aggressiveness and postoperative recurrence in HCC patients.38 In addition, the interaction between PD-1 and PD-L1 may be involved in inducing T lymphocyte apoptosis.39 However, the molecular mechanisms about how PD-1/PD-L1 pathway operates between T cells and tumor cells in HCC patients are rarely reported. In this study, we found that co-culture with HePG2 cells enhanced the expression of PD-1 on Jurkat cells that result in cell cycle delay in S phase. mAb B1C4 reduces the S phase delay and promotes the transition of Jurkat cells from S phase to G2/M phase. Also, the apoptosis of Jurkat cells induced by co-culture with hepatoma cells is inhibited by mAb B1C4. These results indicate the potential of mAb B1C4 in the treatment of liver cancer. But recent studies have found that the liver tumors not only compromise intrahepatic immunity but also affect distant antitumor immunity and may contribute to the reduced efficacy of systemic anti–PD-1 treatment seen in the clinical setting. Liver involvement in patients with cancer could significantly compromise systemic antitumor immunity even at distant sites and may provide a mechanism of checkpoint inhibitor immunotherapies (CPIs) resistance in the setting of liver metastasis. Treg-targeting combination immunotherapy strategies restored the systemic antitumor immunity and CPI responsiveness.40 In future research, we need to further investigate the therapeutic effect of PD-1 mAb B1C4 on liver cancer.
The PI3K/AKT signal pathway plays an essential role in regulating cell proliferation and apoptosis. External factors stimulating PI3K promote the phosphorylation of downstream AKT protein, which activates or inhibits downstream apoptosis-related proteins, thus regulating the proliferation, differentiation, apoptosis, and migration of cells.41,42 PTEN is a primary negative regulator of Akt signaling, and the loss of PTEN function has been associated with many human cancers.43,44 PTEN antagonizes the effect of PI3K, inhibits the phosphorylation of AKT, ie, inhibits the activation of PI3K/AKT signaling pathway which is involved in anti-apoptosis effect.45 Our study showed co-culture with HePG2 cells induces the upregulation of PTEN protein in Jurkat cells, thus inhibiting the activation of PI3K/AKT signal pathway and leading to apoptosis of the cells. Monoclonal antibody B1C4, however, inhibits the expression of PTEN, activates the PI3K/AKT signaling pathway to increase the expression of downstream anti-apoptotic protein p-mTOR, and reduces the expression of proapoptotic proteins Bax, cleaved caspase-9, and cleaved caspase-3, thus inhibiting the apoptosis of Jurkat cells. The ATP-competitive PI3K and mTOR dual inhibitor NVP-BEZ235 counteract the effect of mAb B1C4 in the promotion of p-Akt and p-mTOR. However, we found that NVP-BEZ235 had no significant effect on downstream apoptosis-related molecules, ie, Bax, cleaved caspase-9, and cleaved caspase-3. The reason may be that the downstream signaling pathway of PI3K is more complex, beyond the involvement of mTOR, Bax, caspase-3, and caspase-9 proteins in the pathway. In addition, some studies have reported the mutation of PTEN in Jurkat cells. The PTEN deficiency in Jurkat T cells causes constitutive activation and premature priming of T-cell receptor (TCR) signaling pathways. Although the PTEN-deficient cells were also hyperresponsive to TCR stimulation, signaling models based on results from Jurkat T-cell lines may underestimate the role of PI3K in TCR signaling. Moreover, considering the complexity of the tumor microenvironment and tumor heterogeneity, the extraction of T lymphocytes from human peripheral blood can better simulate the tumor microenvironment.46 Therefore, the whole process of PD-1 monoclonal antibody B1C4 regulating Jurkat cell apoptosis by activating the PI3K/AKT signaling pathway remains further investigation with a better model of cell disease.
In conclusion, we established a cell-co-culture system of HePG2 liver cancer cell and Jurkat T lymphocyte. Killing ability of Jurkat cells on HepG2 cells and the secretion levels of IL-2 and IFN-γ in the supernatants were detected to screen out the blocking mAb B1C4 from the 14 newly generated mouse anti-human PD-1 mAbs. Furthermore, we investigate the effect and mechanism of the mAb B1C4 on reversing the immunosuppression of Jurkat cells. Our results demonstrate that mAb B1C4 enhances the killing ability of Jurkat cells on HePG2 cells by promoting the secretion of IL-2 and IFN-γ. Further, mAb B1C4 promotes Jurkat cell cycle progression from S to G2/M and inhibits cell apoptosis to restore the function suppressed by hepatoma cells. The molecular mechanism study demonstrates that mAb B1C4 reverses the immunosuppression of Jurkat cells resulted by co-culture with HePG2 cells by inhibiting PTEN and activating PI3K/AKT/mTOR signaling pathway. Our study provides a vital basis for applying PD-1 monoclonal antibodies on HCC, and provides antibody selection for the development of novel PD-1 monoclonal antibodies with blocking activity.
Funding
This study was supported by grants from the Key Project of Basic Science and Frontier Technology of Chongqing (cstc2015jcyjBX0018), Transformation Project of Scientific and Technological Achievements of Third Military Medical University (2015XZH19), Major New Drug Creation Project of China’s Ministry of Science and Technology (2018ZX09J18109-05).
Disclosure
The authors declare that they have no conflicts of interest concerning this article.
References
1. Li YS, Li FF, Jiang F, et al. A Mini-Review for Cancer Immunotherapy: molecular Understanding of PD-1/PD-L1 pathway & translational blockade of immune checkpoints. Int J Mol Sci. 2016;17(7):1151. doi:10.3390/ijms17071151
2. Mortara L, Balza E, Bruno A, et al. Anti-cancer therapies employing il-2 cytokine tumor targeting: contribution of innate, adaptive and immunosuppressive cells in the anti-tumor efficacy.Fron Immunol. 2018;9:2905. doi:10.3389/fimmu.2018.02905
3. Jr BD, Bakan CA, Mishra A, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116:2286–2294. doi:10.1182/blood-2010-02-271874
4. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;188:2148–2149. doi:10.1016/j.juro.2012.08.169
5. Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy–inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012;18:6580–6587. doi:10.1158/1078-0432.CCR-12-1362
6. Horn L, Herbst RS, Spigel D, et al. An Analysis of the Relationship of Clinical Activity to Baseline EGFR Status, PD-L1 Expression and Prior Treatment History in Patients with Non-Small Cell Lung Cancer (NSCLC) Following PD-L1 Blockade with MPDL3280A (Anti-PDL1). Amsterdam: IASLC 14th World Conference on Lung Cancer; 2012.
7. Zhang Y, Huang S, Gong D, et al. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell Mol Immunol. 2010;7:389–395. doi:10.1038/cmi.2010.28
8. Creelan BC. Update on immune checkpoint inhibitors in lung cancer. Cancer Control. 2014;21:80–89. doi:10.1177/107327481402100112
9. Gloria M, Sofia G, Massimo A, Giorgio V. Immune Checkpoint Inhibitors: A New Opportunity in the Treatment of Ovarian Cancer? Int J Mol Sci. 2016;17:1169. doi:10.3390/ijms17071169
10. Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–562. doi:10.1038/nature13904
11. Sharma P, Callahan MK, Bono P, et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, Phase 1/2 trial. Lancet Oncol. 2017;17:1590–1598. doi:10.1016/S1470-2045(16)30496-X
12. Stewart R, Morrow M, Hammond SA, et al. Identification and Characterization of MEDI4736, an Antagonistic Anti-PD-L1 Monoclonal Antibody. Cancer Immunol Res. 2015;3(9):1052–1062. doi:10.1158/2326-6066.CIR-14-0191
13. Massard C, Gordon MS, Sharma S, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti–Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol. 2016;34(26):3119–3125. doi:10.1200/JCO.2016.67.9761
14. West EE, Jin H-T, Rasheed A-U, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest. 2013;123(6):2604–2615. doi:10.1172/JCI67008
15. Strome SE, Dong H, Tamura H, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501–6505. doi:10.1097/00002820-200310000-00012
16. Maruyama D, Hatake K, Kinoshita T, et al. Multicenter Phase II study of nivolumab in Japanese patients with relapsed or refractory classical Hodgkin lymphoma. Cancer Science. 2017;108(5):1007–1012. doi:10.1111/cas.13230
17. Hodi FS, Sznol M, Kluger HM, et al. Long-term survival of ipilimumab-naive patients (pts) with advanced melanoma (mel) treated with nivolumab (anti-pd-1, bms-936558, ono-4538) in a Phase I trial. J Clin Oncol. 2014;25:374–393. doi:10.1200/jco.2014.32.15_suppl.9002
18. Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi:10.1038/nm730
19. Woo S-R, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. doi:10.1158/0008-5472.CAN-11-1620
20. Mangsbo SM, Sandin LC, Anger K, et al. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J Immunother. 2010;33(3):225–235. doi:10.1097/cji.0b013e3181c01fcb
21. Steidl C, Shah SP, Woolcock BW, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471(7338):377–381. doi:10.1038/nature09754
22. Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–3029. doi:10.1084/jem.20090847
23. Inman BA, Sebo TJ, Frigola X, et al. PD-L1 (B7-H1) expression by urothelial carcinoma of the bladder and BCG-induced granulomata. Cancer. 2007;109(8):1499–1505. doi:10.1002/cncr.22588
24. Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114(8):1537–1544. doi:10.1182/blood-2008-12-195792
25. Okudaira K, Hokari R, Tsuzuki Y, et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int J Oncol. 2009;35:741–749. doi:10.3892/ijo_00000387
26. Li ZW, Li B, Peng D, et al. Expression and clinical significance of PD-1 in hepatocellular carcinoma tissues detected by a novel mouse anti-human PD-1 monoclonal antibody. Int J Oncol. 2018;52:2079–2092. doi:10.3892/ijo.2018.4358
27. Coombs MRP, Harrison ME, Hoskin DW. Apigenin inhibits the inducible expression of programmed death ligand 1 by human and mouse mammary carcinoma cells. Cancer Lett. 2016;380(2):424–433. doi:10.1016/j.canlet.2016.06.023
28. Hopkins BD, Hodakoski C, Barrows D, et al. PTEN function: the long and the short of it. Trends Biochem Sci. 2014;39(4):183–190. doi:10.1016/j.tibs.2014.02.006
29. Lim HJ, Crowe P, Yang JL. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J Cancer Res Clin Oncol. 2015;141:671–689. doi:10.1007/s00432-014-1803-3
30. Xu-Monette ZY, Zhang M, Li J, et al. PD-1/PD-L1 Blockade: have We Found the Key to Unleash the Antitumor Immune Response? Front Immunol. 2017;4:1597. doi:10.3389/fimmu.2017.01597
31. Yang ZZ, Grote DM, Ziesmer SC, et al. PD-1 expression defines two distinct T-cell sub-populations in follicular lymphoma that differentially impact patient survival. Blood Cancer J. 2015;20:e281. doi:10.1038/bcj.2015.1
32. Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi:10.1038/nature04444
33. Li Q, Quan L, Lyu J, et al. Discovery of peptide inhibitors targeting human programmed death 1 (PD-1) receptor. Oncotarget. 2016;7:64967–64976. doi:10.18632/oncotarget.11274
34. Chen J, Wu XJ, Wang GQ. Hepatoma cells up-regulate expression of programmed cell death-1 on T cells. World J Gastroentero. 2008;14:6853–6857. doi:10.3748/wjg.14.6853
35. Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293–12297. doi:10.1073/pnas.192461099
36. Patsoukis N, Brown J, Petkova V, et al. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5:ra46–ra46. doi:10.1126/scisignal.2002796
37. Shi F, Shi M, Zeng Z, et al. PD-1 and PD-L1 upregulation promotes CD8+ T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer. 2011;128:887–896. doi:10.1002/ijc.25397
38. Gao Q, Wang XY, Qiu SJ, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15:971–979. doi:10.1158/1078-0432.CCR-08-1608
39. Chiku VM, Silva KL, de Almeida BF, et al. PD-1 function in apoptosis of T lymphocytes in canine visceral leishmaniasis. Immunobiology. 2016;221:879–888. doi:10.1016/j.imbio.2016.03.007
40. Lee JC, Mehdizadeh S, Smith J, et al. Regulatory T cell control of systemic immunity and immunotherapy response in liver metastasis. Sci Immunol. 2020;5:eaba0759. doi:10.1126/sciimmunol.aba0759
41. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi:10.1038/nrc839
42. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi:10.1038/nrg1879
43. Tekcham DS, Gupta S, Shrivastav BR, et al. Epigenetic Downregulation of PTEN in Gallbladder Cancer. J Gastrointest Cancer. 2017;48:110–116. doi:10.1007/s12029-017-9919-8
44. Wise HM, Hermida MA, Leslie NR. Prostate cancer, PI3K, PTEN and prognosis. Clin Sci. 2017;131:197–210. doi:10.1042/CS20160026
45. Fu X, Wen H, Jing L, et al. MicroRNA-155-5p promotes hepatocellular carcinoma progression by suppressing PTEN through the PI3K/Akt pathway. Cancer Sci. 2017;108:620–631. doi:10.1111/cas.13177
46. Shan X, Czar MJ, Bunnell SC, et al. Deficiency of PTEN in Jurkat T Cells Causes Constitutive Localization of Itk to the Plasma Membrane and Hyperresponsiveness to CD3 Stimulation. Mol Cell Biol. 2000;20:6945–6957. doi:10.1128/mcb.20.18.6945-6957.2000
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.