Back to Journals » The Application of Clinical Genetics » Volume 9
The genetics of uveal melanoma: current insights
Authors Helgadottir H, Hoiom V
Received 25 May 2016
Accepted for publication 11 July 2016
Published 6 September 2016 Volume 2016:9 Pages 147—155
DOI https://doi.org/10.2147/TACG.S69210
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Hildur Helgadottir,1,2 Veronica Höiom1
1Department of Oncology and Pathology, Karolinska Institutet, 2Department of Oncology, Karolinska University Hospital, Stockholm, Sweden
Abstract: Uveal melanoma (UM) is the most common malignant eye tumor in adults affecting ~7,000 individuals per year worldwide. UM is a rare subtype of melanoma with distinct clinical and molecular features as compared to other melanoma subtypes. UMs lack the most typical cutaneous melanoma-associated mutations (BRAF, NRAS, and NF1) and are instead characterized by a different set of genes with oncogenic or loss-of-function mutations. By next-generation sequencing efforts on UM tumors, several driver genes have been detected. The most frequent ones are BAP1, EIF1AX, GNA11, GNAQ, and SF3B1. In many cases, mutations in these genes appear in a mutually exclusive manner, have different risk of metastasis, and are consequently of prognostic importance. The majority of UM cases are sporadic but a few percentage of the cases occurs in families with an inherited predisposition for this malignancy. In recent years, germline mutations in the BAP1 gene have been found to segregate in an autosomal dominant pattern with numerous different cancer types including UM in cancer-prone families. This cancer syndrome has been denoted as the tumor predisposition syndrome.
Keywords: uveal melanoma, driver genes, oncogenes, tumor suppressor genes, familial cancer
Introduction
Uveal melanoma (UM) is the most common neoplasms of eye that develop in adults displaying a high propensity for metastasis. It is a rare subtype of melanoma, representing ~5% of all melanoma tumors. UM can appear in the choroid, ciliary body, or iris of the eye and is by far the most common ocular tumor in adults. Ocular melanomas can also rarely arise in melanocytes in the conjunctiva – melanoma of the conjunctiva accounts for ~2%–3% of all eye neoplasms. The incidence rate of UM ranges from 0.2 to 0.3 per million individuals in African/Asian populations to up to 6 per million individuals in white populations.1 The average age of diagnosis is ~60 years and it affects both sexes equally or slightly more frequently males as per some reports.2,3 It is more common among light-skinned individuals, but it can affect individuals from any ethnicity. Cutaneous melanoma (CM) and UM share some risk factors such as fair skin color, blue eyes, red/blond hair, and freckling/many nevi.4,5 Whereas the incidence of CM has been rising in many Caucasian populations, the incidence of UM has been stable over the years.6 Also in contrast to CM, the impact of ultraviolet (UV) light exposure is less clear for UM. UM is molecularly diverse from CM and shows a different pattern of driver mutations. Compared to CM that shows one of the highest mutational-load among different cancer types, UM displays a low mutational burden.7 It has, in many cases, a poor prognosis since about half of all patients develop metastatic disease, predominantly in the liver. By using gene expression profile (GEP) classification, UM can be stratified into two distinct molecular classes with a significant difference in prognosis.8 Class 1 tumors can be further divided into two subgroups (class 1A and 1B) and has in general a good prognosis and low metastatic risk, whereas class 2 tumors have high metastatic risk and thereby a worse prognosis. The risk of metastasis has been determined to be 2% for class 1A tumors, 21% for 1B tumors, and finally 72% for class 2.9 The different molecular classes are also associated with mutations in different UM driver genes.10 The most frequently mutated genes that are considered to be drivers in UM development and progression are BAP1, EIF1AX, GNA11, GNAQ, and SF3B1 (Table 1).10–15
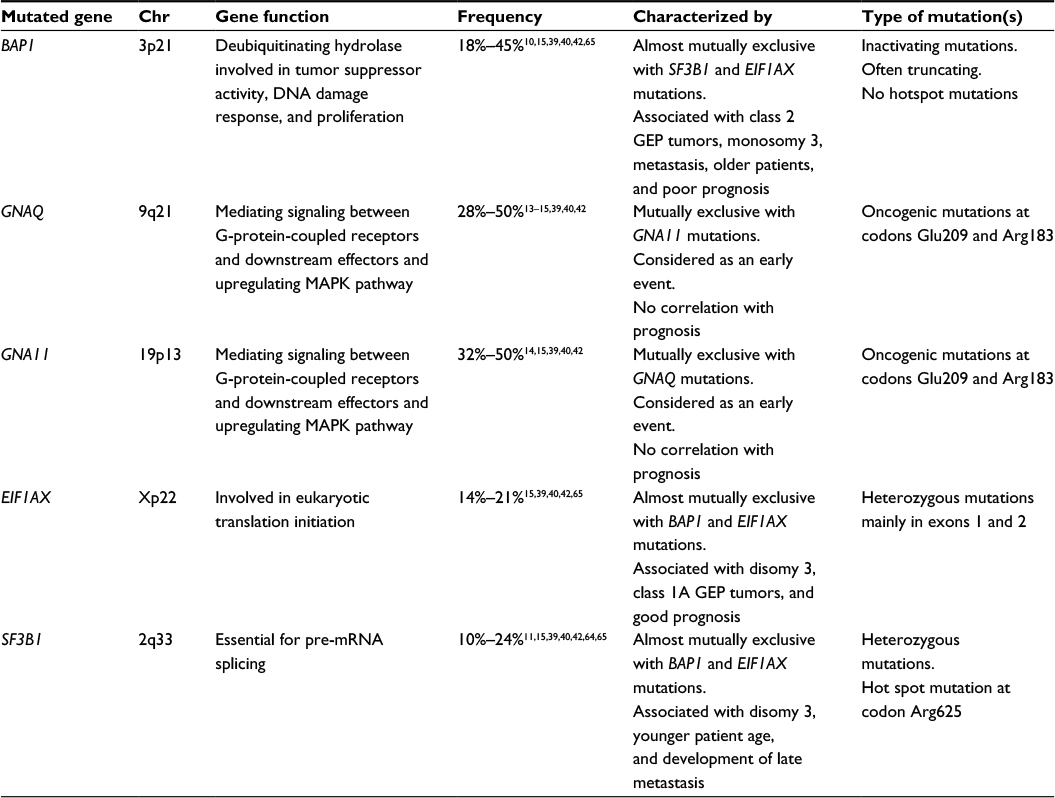 | Table 1 The most frequent driver mutated genes in uveal melanoma Abbreviations: Chr, chromosome; GEP, gene expression profile. |
Some UM cases occur in families with an inherited predisposition for UM. The only high-penetrance susceptibility gene for familial UM identified so far is BRCA1-associated protein 1 (BAP1). The frequency of predicted pathogenic germline BAP1 mutations observed in different cohorts of UM patients ranges from 1.6% to 3%, with mutations predominantly found in patients with a family history of UM.16–18 The cancer risk mediated by germline BAP1 mutations is inherited in an autosomal dominant pattern with incomplete penetrance. Cancer types associated with BAP1 germline mutations include UM, CM, mesothelioma, meningioma, renal cell cancer, basal cell carcinoma and melanocytic BAP1-mutated atypical intradermal tumors, and possibly additional cancer types.19 While carrying a germline mutation in the CDKN2A gene is the strongest known inherited risk factor for CM, such mutations do not seem to increase the risk of UM.20–22
Somatic alterations
Risk of metastatic disease and UM-associated survival is strongly correlated with the molecular subtype.8 Poor prognosis and high risk of metastatic disease are often accompanied with loss of chromosome 3 in the tumor, while tumors with intact chromosome 3 correlates with good prognosis and rarely leads to disseminated disease.23 The metastatic rate for tumors with partial loss of chromosome 3 has shown a great variation depending on the study (ranging from 0% to 48%).24 However, later studies indicate that partial monosomy of chromosome 3 often associates with a good prognosis.24,25 Other frequent chromosomal aberrations in UM include gain of chromosome 8q which, similar to the loss of chromosome 3, associates with decreased survival, both independently but in particular in combination with chromosome 3 monosomy.23 Loss of chromosome 1 or parts of this chromosome is also frequent aberration, affecting ~25% of all tumors. Gain of chromosome 6p and loss of 6q has been detected in about one-third of the tumors, often in the same tumor.26 This abnormality is usually associated with better patient survival, possibly because it rarely occurs in tumors with monosomy of chromosome 3.27 Inactivation of CDKN2A may be part of UM pathogenesis, either through methylation of the CDKN2A promotor region or through loss of chromosome 9p or a smaller region surrounding the 9p21, harboring the CDKN2A locus. Both promotor methylation and chromosomal loss affect up to one-third of the tumors each.28–30
Other important pathways often altered in UM, as in many cancer types, are the retinoblastoma (Rb) and p53 pathways.31–35 Mutations in the genes encoding for these proteins, RB1 and TP53, are infrequent in UM tumors suggesting other ways of inactivation. Cyclin D1 overexpression or CDKN2A promotor methylation are two plausible explanations for hyperphosphorylation and inactivation of the Rb-protein, while inactivation of p53 may be caused by MDM2 overexpression.30,33,36 Also, constitutively activation of the PI3K/AKT pathway and inactivation of the tumor suppressor PTEN (mainly by LOH of the PTEN locus) are common events in UM tumors.37,38
However, overall the extent of genomic instability and chromosomal aberrations is relatively low in UM tumors compared to many other cancer types such as in CM. Also the mutational load in UM tumors is low, and the mean mutation rate of UM tumors has been determined to be around 0.5 per Mb sequence, both concerning genomic and protein coding regions.39 In UM tumors, several frequent driver mutations have been described, none of them being described as key drivers in other melanoma subtypes. The most commonly mutated genes are BAP1, EIF1AX, GNA11, GNAQ, and SF3B1. In addition, there are numerous other genes with rare mutations.15,39 The list of rarely mutated genes will most probably increase with time due to ongoing and future sequencing studies. BAP1, EIF1AX, and SF3B1 often occur in a mutually exclusive manner as do GNA11 and GNAQ. Some of these driver mutations have also been shown to be of importance for the prognosis since they mediate a variable risk of metastatic disease. BAP1 is associated with monosomy 3, poor prognosis, and class 2 GEP tumors, while EIF1AX is associated with class 1 GEP tumors and good prognosis. SF3B1 has been associated with younger patient age and good prognosis.40
BRCA1-associated protein 1
Loss of chromosome 3 was for a long time the best predictor for metastatic disease in UM patients. Later, the identification of different GEPs, which led to the development of the GEP classification, has improved the prognostic accuracy. The class 2 tumors that are aggressive with high metastatic potential were found to be accompanied by loss of chromosome 3. Using next-generation sequencing, it was discovered that a vast majority of the class 2 tumors carried a mutation in the BAP1 gene, mapped to chromosome 3p21.1, while very few of the class 1 tumors harbored a mutation in this gene. Thus, inactivating hemizygous mutations of BAP1 leads to protein inactivation and loss of BAP1 expression.10 This implicate BAP1 to function as a tumor suppressor gene, with loss of one copy of chromosome 3 and mutation in the other allele, fulfilling the Knudsen two hits hypothesis definition of a tumor suppressor gene. Indeed, BAP1 has previously been shown to display tumor suppressor capacity by binding to the BRCA1 protein and thereby enhancing BRCA1-mediated tumor suppression.41 BAP1 mutations strongly correlate with metastatic disease in UM; over 80% of metastasizing UM has been found to carry a mutation in this gene.10 The frequency of BAP1 mutations in primary UM has been estimated to be approximately 30%–40%.39,42 Most of the BAP1 mutations are truncating variants or missense variants affecting the ubiquitin carboxyl-terminal hydrolase domain. In some cases, BAP1 is not altered by a sequence mutation but by hemizygous deletion of one or more exons. Such alterations may be missed by traditional Sanger sequencing because of the presence of normal DNA in the sample. Thus, in some cases, immunohistochemistry (IHC) might be a better choice of detection, if tissue samples are available. Loss of BAP1 expression using IHC has strongly been correlated with risk of metastasis, BAP1 mutation status, and loss of chromosome 3 and has, therefore, been proposed as a valid prognostic test.43,44 BAP1 is also frequently mutated in other tumor types, including cholangiocarcinoma, renal cell carcinoma, mesothelioma, and bladder cancer (www.cbioportal.org). Several of these cancer types are part of the hereditary cancer syndrome known as tumor predisposition syndrome that is characterized by germline mutations of BAP1 in patients belonging to cancer-prone families (discussed in detail in section Inherited susceptibility). BAP1 encodes a deubiquitinating hydrolase with multiple cellular functions, except tumor suppressor activity, such as regulation of chromatin dynamics, DNA damage response, cell cycle regulation, and cell growth. For example, BAP1 is involved in the polycomb multiprotein repressor complex that is critical for transcriptional silencing of target genes by removing ubiquitin molecules from histone H2A. As a consequence of this functional loss, an accumulation of monoubiquitinated histone H2A has been revealed, which in turn was found to cause a more dedifferentiated phenotype.45 BAP1 seems to be involved in other important cellular functions as well, for example, in cell proliferation by deubiquitinating the cell cycle regulator host factor 1.
Eukaryotic translation initiation factor 1A, X-linked
EIF1AX, located at chromosome Xp22, was identified as a UM driver gene by whole-exome sequencing.15 Approximately 14%–20% of all UM carries a mutation in this gene, with most mutations found in exons 1 and 2.15,39,42 EIF1AX mutations usually occur in nonmetastatic cases, are associated with class 1 GEP tumors and good prognosis, and are inversely associated with metastasis.40,46 EIF1AX mutations are usually mutually exclusive with BAP1 mutations and to a large extent also to SF3B1 mutations. As expected most EIF1AX mutations are identified in tumors with disomy 3 (48%) and rarely occur in monosomy 3 tumors (3%).15 In contrast to, for example, BAP1 mutations, which mainly are truncating and loss-of-function variants, the majority of the EIF1AX mutations are heterozygous nonsynonymous variants, or in some cases splicing variants, leading to deletions of one or two amino acids. Thus, in most cases, the core protein remains unchanged. EIF1AX, located on the X-chromosome, encodes the eukaryotic translation initiation factor 1A (eIF1A). This factor is essential in the initiation phase of translation of eukaryotic cells by the transfer of methionyl initiator tRNA to the small (40S) ribosomal unit.47 This stabilizes the formation of the ribosome around the AUG start codon, which enables translation. EIF1AX mutations are usually seen as heterozygous mutations in the tumor-DNA, suggesting that EIF1AX serves as a dominant acting oncogene. However, it has been reported that UM tumors carrying an EIF1AX mutation only express the mutant allele, which indicates that EIF1AX also may function in a recessive manner.15 Mutations in this gene have also been described in other cancers such as thyroid and ovarian cancers and in the rare melanocytic neoplasm primary leptomeningeal melanocytic neoplasms (LMNs). LMNs are also prevalent for mutations in GNAQ, GNA11 and SF3B1.48–50
Guanine nucleotide-binding protein subunit alpha-Qandguanine nucleotide-binding protein subunit alpha-11
GNAQ encodes the alpha subunit (Gαq) and GNA11 the alpha subunit 11 (Gα11), both being guanine nucleotide–binding proteins belonging to the heterotrimeric protein family, which are of importance in transmembrane signaling systems. The alpha subunits serve as a switch between the G-proteins active state – when bound to guanosine triphosphate (GTP) – and the inactive state – when GTP is hydrolyzed to guanosine diphosphate.51,52 Activating mutations in GNAQ/GNA11 were the first described driver mutations in UMs. GNAQ and GNA11 mutations occur in a mutual exclusive pattern and are exclusively found in codon 209 and in some cases in codon 183. Mutations at these positions lead to a constitutive activation of the Gαq and Gα11 subunits by abolishing their intrinsic GTPase activity, thereby preventing the return to an inactive state. In total, ~85% of all UMs carry a mutation in either of these genes. Both GNAQ and GNA11 have been found to upregulate the MAP kinase pathway when constitutively activated in a similar fashion as BRAF and NRAS mutations. In CM activating mutations in BRAF are a very common event, whereas UM rarely carries any mutation in BRAF.53–55 The activation of the MAPK pathway in the absence of BRAF/NRAS mutations in UMs was at first unforeseen until the identification of GNAQ and later GNA11 mutations that had the same effect as the V600EBRAF mutation. Interestingly, BRAF mutations have been seen in up to nearly half of all iris melanomas.56 This could be explained by the iris being more anterior and therefore more exposed to UV radiations than the ciliary body or the choroid. Cell lines with a GNAQ Q209L mutation have also been found to be highly sensitive to mitogen-activated protein kinase (MEK) inhibition.13 Mutations in these genes have not been associated with the two different molecular classes of UM tumors. In addition, GNAQ/GNA11 mutations have not been reported to be of prognostic value and they occur at similar frequencies in metastatic and nonmetastatic lesions. Furthermore, they have not been linked to patient outcomes. Taken all this data into consideration is supportive of GNAQ/GNA11 being early events.12 The hotspot mutations in GNAQ or GNA11 are also commonly found in benign nevi such as blue nevi.14,57 Actually, GNAQ Q209 was most frequently found in blue nevi, observed in 55% of the lesions, whereas 45% of the primary UMs and 22% of the metastatic UM, respectively, carry this mutation.14 Inverse relationship was seen for the GNA11 Q209 mutation where metastatic lesions showed the highest number (56%) followed by primary UM tumors (32%) and lastly blue nevi (6%).14 Mutations affecting codon R183 are less frequent, present in 2% of the blue nevi and 5% of primary UM tumors, GNAQ and GNA11 mutations combined.14
Splicing factor 3b subunit 1
SF3B1 located at chromosome 2 is another driver gene identified by whole-exome sequencing of UM tumors. SF3B1 is essential in pre-mRNA splicing by encoding the unit of the splicing factor 3b protein complex that is a critical part of both major (U2-like) and minor (U12-like) spliceosomes.58 The spliceosomes are part of the splicing machinery, which bind to the intron near the branchpoint.59,60 SF3B1 has recently also been designated as a factor involved in DNA-damage repair.61Missense mutations in specific regions of the SF3B1gene have been found to alter the splicing of many target genes.62,63 These mutations predominantly alter codon Arg625 in exon 14 of the SF3B1 gene and have been identified in UMs with a reported mutation rate between 10% and 21%.11,15,64 Some studies report an association between SF3B1 mutations and good prognosis, lower age at diagnosis (a favorable prognostic factor), and tumors with disomy 3.11,64 However, in a study with longer follow-up time, tumors with disomy 3 and a SF3B1 mutation showed significant worse prognosis and development of late metastasis compared to wild-type tumors. In patients with a SF3B1 mutation, most metastasis occurred more than 5 years after diagnosis (median 8.2 years, range 23–145 months).65
In a study by Martin et al,15 29% of the tumors with disomy 3 carried a heterozygous mutation in SF3B1 compared to only 3% in tumors with monosomy 3. Furthermore, in tumors with partial monosomy 3, preferentially with loss of 3q and retention of 3p, 54% were found to carry Arg625 mutation in SF3B1.15 In hematological and lymphoid malignancies, mutational hotspots have been detected in specific codons, for example, codon 700, coding for the HEAT repeats (HD) 4–9.62,66,67 Resequencing of the exons encoding for this region in UM tumors revealed a mutation rate of 15% in UM tumors (n=66).15
SF3B1 mutations often occur in tumors that express the oncogene PRAME.68 Expression of PRAME has been found to be associated with class 1 tumors with an intermediate risk of metastasis, suggesting that there is a risk class of tumors lying between the high-risk tumors characterized by BAP1 mutations and low-risk tumors frequently harboring EIF1AX mutations.
Other rare mutations/alterations in UM tumors
By whole-genome and whole exome sequencing of UM tumors, a recurrent gain-of-function mutation in the phospholipase C, beta 4 (PLCB4), gene was identified.39 This was the only gene with a recurrent mutation (two out of 28 samples) that was found above the known driver genes in UM (BAP1, EIF1AX, GNA11, GNAQ, and SF3B1). The mutation (c.G1888T, p.D630Y) lies in the Y-domain of the highly conserved catalytic core of PLCB4 and was predicted to be deleterious/probably damaging using the prediction tools SIFT and PolyPhen. Interestingly, the PLCB4 protein is a downstream target of GNA11/GNAQ and the p.D630Y PLCB4 mutation was mutually exclusive with mutations in GNA11 and GNAQ.
In CM, recurrent mutations in the core promotor of the telomerase reverse transcriptase (TERT) gene are common. TERT is part of the telomerase enzyme and maintains the telomere ends by adding the telomere repeat TTAGGG. Deregulation of telomerase and aberrant expression of TERT have been found in several different cancer forms such as thyroid and bladder cancers (www.cbioportal.org). Approximately 70% of CM tumors have been reported to carry any of the two mutual exclusive recurrent mutations in the TERT promotor, both being consistent with the typical UV-damage signature. These mutations affect the expression levels of TERT by creating a novel binding site for the transcription factor E-twenty-six.69,70 A germline TERT promotor mutation with the same functional effect as the described somatic mutations has been found to segregate with high penetrance in two large melanoma-prone families in two separate studies.69,71 In UM tumors, TERT promotor mutations are very rare.72 In a study by Dono et al,42 one out of 50 patients carried one of the previously described TERT promotor mutations.42 Here, the promotor mutation was observed in combination with mutations in GNA11 and EIF1AX as well as two normal copies of chromosome 3. TERT promotor mutation thus seems to be infrequent in UM; however, an elevated level of TERT expression has been observed in a subset of UM tumors with wild-type TERT promotors42 suggesting that telomere maintenance might be an important factor also in UM etiology, although with less impact than for CM.
Inherited susceptibility
UM predominantly occurs in a sporadic fashion, with approximately only 1% of the cases considered to be hereditary cases. However, patients with an inherited predisposition are likely to be more common than initially believed due to the discovery of UM being part of cancer syndromes. Between 2% and 5% of the UM cases have, therefore, been proposed to be familial cases.73 The first report of a germline mutation in BAP1 was published in 2010.10 One year later, two independent groups described inactivating germline mutations segregating in cancer-prone families, mainly characterized by distinct melanocytic neoplasms and mesothelioma in combination with UM and other cancers.74,75 Several other studies have, thereafter, described the link between familial UM and BAP1 germline mutations.76,77 The neoplasms associated with BAP1 germline mutations, also called the tumor predisposition syndrome, have been expanded to include CM, renal cell carcinoma, meningioma, and basal cell carcinoma.76,78–83 Additional cancer types are continuously being linked to this syndrome. In a review by Rai et al,19 it was reported that 56 out of 57 families with a reported BAP1 germline mutation had one or more family members diagnosed with any of the main cancer types associated with this cancer syndrome (UM, CM, mesothelioma, or renal cell carcinoma).19 Still, only a subset of the families with an inherited predisposition for UM carries a germline mutation in BAP1 suggesting the presence of other, yet to be identified, high penetrance susceptibility genes. Several attempts identifying such genes have been done through large-scale sequencing approaches (ie, whole exome- and whole-genome sequencing) with little success so far, indicating that these genes are very rare and collaborations between research groups are needed. Another plausible explanation for the absence of additional UM susceptibility genes could be the presence of phenocopies in families that often comprise two relatives affected by UM, and where no clear aggregation of cancer cases is seen in the pedigree. Whether there is a polygenic component behind the inherited susceptibility for UM, as has been suggested for CM, is unclear. To date, no low to intermediate risk genes have been associated with UM.
Clinical implications
About half of all UM patients will subsequently suffer from metastatic disease, by hematogenous spread mainly affecting the liver. The survival for these patients is poor since UM is resistant to standard treatments with chemotherapy. Novel targeted therapies and immunotherapies have revolutionized the treatment of metastatic CM during the past years. In contrast, activating BRAF mutations are very rare in UM and, consequently, BRAF inhibitory treatment is not applicable. In CM, treatment with antibodies against immune checkpoint molecules has improved both disease-free survival and overall survival. A high burden of coding mutations has been associated with a better response to immune checkpoint inhibitors, such as CTLA-4.84,85 A high mutational load has also been correlated to a greater repertoire of neoantigens, which is associated with a better treatment response. Thus, the low burden of mutations found in UM might, therefore, be an explanation for the lower response to immunotherapies as compared to other melanoma subtypes.7 Due to the recent advances in sequencing technologies, multiple driver genes for UM have been discovered that hopefully will improve the understanding of the carcinogenesis behind this neoplasm and subsequently lead to prolonged survival of patients suffering from metastatic UM. The identification of driver genes has led to the identification of novel treatment targets and several clinical trials are ongoing investigating these targets in UM therapy.
Targeting mutated GNQ/GNA11 directly is difficult because of the molecular nature of the mutations causing an inactivation of intrinsic GTPase within the cell. However, for several downstream molecules of GNAQ/GNA11, targeted therapies have become available. These include mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK) that is shown to be upregulated in GNAQ/GNA11 mutated tumors.86,87 Inhibition of MEK has actually been found to decrease the proliferation of UM tumors both in vivo and in vitro.88,89 Furthermore, a clinical Phase II trial has shown a prolonged progression-free survival of nearly 9 weeks when treating patients with the MEK inhibitor selumetinib compared to chemotherapy (temozolamide).90 However, in another Phase II trial, there was no significant effect on overall survival when treating with semurafenib compared to chemotherapy, although there was a modest increase in response rate and progression-free survival.91 Other putative downstream targets of GNAQ/GNA11 mutated tumors are protein kinas C and molecules of the protein kinase B (AKT)/mammalian target of rapamycin pathway.92–94
Also BAP1 mutations are difficult to target directly because of their recessive nature. However, the effects of the mutations are possible to target by the use of histone deacetylase (HDAC) inhibitors in tumors with loss of BAP1 function. The absence of functional BAP1 protein leads to hyperubiquitination of H2A in the cells.45 The use of HDAC inhibitors can reverse this phenotype, thereby causing a shift from aggressive, dedifferentiated class-2 UM cells to more differentiated and less aggressive cells.45 HDAC inhibitors have also been suggested as adjuvant treatment in high-risk patients.95
In familial cancer combining clinical and genetic information can be used to improve prognostic estimates and to improve strategies for early diagnosis. By genetic testing of cancer-prone families, the clinical outcome can in many situations be improved, by detecting precursor lesions and tumors at an early stage in members of mutation-positive families. Often, however, this is not straightforward, as in familial melanoma, where a low frequency of mutations in high penetrance genes is seen and risk estimates for mutation carriers have not been well established at this point. Genetic testing is often only recommended when the result is of importance in the management of the patient and where there is a possibility of improving the clinical outcome. However, in families exhibiting the phenotype specific for BAP1 tumor predisposition syndrome, genetic testing should be offered. Additional research will be of importance to elucidate the penetrance and risk of developing different types of cancer in mutation carriers. Identifying the susceptibility factor in cancer-prone families will be of importance for choice of surveillance programs and follow-up of the patient and their relatives. For families with a high cancer burden but without mutation in any known high predisposing gene, next-generation sequencing will be the natural choice to search for novel susceptibility genes. This will subsequently increase the knowledge about genetic susceptibility and may in the future be the basis for improved early detection and prevention of UM as well as lead to the development of new targeted treatments.
Conclusion
Five driver genes have so far been found to be frequently mutated in UM. Two of these, GNAQ and GNA11, are considered to occur early in carcinogenesis and to be of no prognostic relevance. Mutations occurring in the other driver genes are likely to have arisen later in the tumor development and thus are of importance for patient outcome. EIF1AX-mutated tumors show in general a strong correlation with class 1 GEP tumors and increased patient survival; BAP1 mutations, in contrast, associate with GEP class 2 tumors and poor survival; and SF3B1-mutated tumors seem to fall in between, which associate with late-onset metastatic disease. As being a complement to the GEP classification, mutation status of UM driver genes will hopefully increase the prognostic accuracy and be of help for deciding different treatment regimens, such as MEK inhibition therapy in GNAQ- and GNA11-mutated tumors. Other, probably more infrequent, mutated genes are continuously being detected, which will help us add more details to resolve the puzzle. UM also occurs in families with an inherited predisposition. The only high penetrance gene for hereditary UM identified so far is the BAP1 gene. Germline mutations in BAP1, mainly truncating mutations, have been found to segregate with reduced penetrance in families with many different cancer diagnoses, including but not exclusively to UM, CM, mesothelioma, and renal cell carcinoma. The search for additional novel UM susceptibility genes in BAP1-mutation negative families are ongoing through large whole-exome and genome sequencing. Knowledge on highly segregating penetrant mutations in affected families is of great importance for the management and surveillance of the patients and their relatives and will hopefully have positive impact on prevention and early diagnosis in the future.
Disclosure
The authors report no conflicts of interest in this work.
References
Kivela T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009;93(9):1129–1131. | ||
Nathan P, Cohen V, Coupland S, et al. Uveal melanoma UK national guidelines. Eur J Cancer. 2015;51(16):2404–2412. | ||
Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;18(1):75–84, viii. | ||
Vajdic CM, Kricker A, Giblin M, et al. Eye color and cutaneous nevi predict risk of ocular melanoma in Australia. Int J Cancer. 2001;92(6):906–912. | ||
Weis E, Shah CP, Lajous M, Shields JA, Shields CL. The association between host susceptibility factors and uveal melanoma: a meta-analysis. Arch Ophthalmol. 2006;124(1):54–60. | ||
Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118(9):1881–1885. | ||
Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44(9):1006–1014. | ||
Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64(20):7205–7209. | ||
Field MG, Harbour JW. Recent developments in prognostic and predictive testing in uveal melanoma. Curr Opin Ophthalmol. 2014;25(3):234–239. | ||
Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330(6009):1410–1413. | ||
Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45(2):133–135. | ||
Onken MD, Worley LA, Long MD, et al. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49(12):5230–5234. | ||
Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. | ||
Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363(23):2191–2199. | ||
Martin M, Masshofer L, Temming P, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45(8):933–936. | ||
Aoude LG, Vajdic CM, Kricker A, Armstrong B, Hayward NK. Prevalence of germline BAP1 mutation in a population-based sample of uveal melanoma cases. Pigment Cell Melanoma Res. 2013;26(2):278–279. | ||
Gupta MP, Lane AM, DeAngelis MM, et al. Clinical characteristics of uveal melanoma in patients with germline BAP1 mutations. JAMA Ophthalmol. 2015;133(8):881–887. | ||
Turunen JA, Markkinen S, Wilska R, et al. BAP1 Germline mutations in Finnish patients with uveal melanoma. Ophthalmology. 2016;123(5):1112–1117. | ||
Rai K, Pilarski R, Cebulla CM, Abdel-Rahman MH. Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin Genet. 2016;89(3):285–294. | ||
Helgadottir H, Hoiom V, Jonsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51(8):545–552. | ||
Soufir N, Bressac-de Paillerets B, Desjardins L, et al. Individuals with presumably hereditary uveal melanoma do not harbour germline mutations in the coding regions of either the P16INK4A, P14ARF or cdk4 genes. Br J Cancer. 2000;82(4):818–822. | ||
Singh AD, Croce CM, Wary KK, et al. Familial uveal melanoma: absence of germline mutations involving the cyclin-dependent kinase-4 inhibitor gene (p16). Ophthalmic Genet. 1996;17(1):39–40. | ||
Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jockel KH, Becher R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347(9010):1222–1225. | ||
Abdel-Rahman MH, Christopher BN, Faramawi MF, et al. Frequency, molecular pathology and potential clinical significance of partial chromosome 3 aberrations in uveal melanoma. Mod Pathol. 2011;24(7):954–962. | ||
Thomas S, Putter C, Weber S, Bornfeld N, Lohmann DR, Zeschnigk M. Prognostic significance of chromosome 3 alterations determined by microsatellite analysis in uveal melanoma: a long-term follow-up study. Br J Cancer. 2012;106(6):1171–1176. | ||
Aalto Y, Eriksson L, Seregard S, Larsson O, Knuutila S. Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42(2):313–317. | ||
Ehlers JP, Worley L, Onken MD, Harbour JW. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin Cancer Res. 2008;14(1):115–122. | ||
Merbs SL, Sidransky D. Analysis of p16 (CDKN2/MTS-1/INK4A) alterations in primary sporadic uveal melanoma. Invest Ophthalmol Vis Sci. 1999;40(3):779–783. | ||
Ohta M, Berd D, Shimizu M, et al. Deletion mapping of chromosome region 9p21-p22 surrounding the CDKN2 locus in melanoma. Int J Cancer. 1996;65(6):762–767. | ||
van der Velden PA, Metzelaar-Blok JA, Bergman W, et al. Promoter hypermethylation: a common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001;61(13):5303–5306. | ||
Chana JS, Wilson GD, Cree IA, et al. c-myc, p53, and Bcl-2 expression and clinical outcome in uveal melanoma. Br J Ophthalmol. 1999;83(1):110–114. | ||
Scholes AG, Liloglou T, Maloney P, et al. Loss of heterozygosity on chromosomes 3, 9, 13, and 17, including the retinoblastoma locus, in uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42(11):2472–2477. | ||
Brantley MA Jr, Harbour JW. Deregulation of the Rb and p53 pathways in uveal melanoma. Am J Pathol. 2000;157(6):1795–1801. | ||
Brantley MA Jr, Harbour JW. Inactivation of retinoblastoma protein in uveal melanoma by phosphorylation of sites in the COOH-terminal region. Cancer Res. 2000;60(16):4320–4323. | ||
Sun Y, Tran BN, Worley LA, Delston RB, Harbour JW. Functional analysis of the p53 pathway in response to ionizing radiation in uveal melanoma. Invest Ophthalmol Vis Sci. 2005;46(5):1561–1564. | ||
Coupland SE, Anastassiou G, Stang A, et al. The prognostic value of cyclin D1, p53, and MDM2 protein expression in uveal melanoma. J Pathol. 2000;191(2):120–126. | ||
Saraiva VS, Caissie AL, Segal L, Edelstein C, Burnier MN Jr. Immunohistochemical expression of phospho-Akt in uveal melanoma. Melanoma Res. 2005;15(4):245–250. | ||
Abdel-Rahman MH, Yang Y, Zhou XP, Craig EL, Davidorf FH, Eng C. High frequency of submicroscopic hemizygous deletion is a major mechanism of loss of expression of PTEN in uveal melanoma. J Clin Oncol . 2006;24(2):288–295. | ||
Johansson P, Aoude LG, Wadt K, et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016;7(4):4624–4631. | ||
Decatur CL, Ong E, Garg N, et al. Driver mutations in uveal melanoma: associations With gene expression profile and patient outcomes. JAMA Ophthalmol. 2016;134(7):728–733. | ||
Jensen DE, Proctor M, Marquis ST, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16(9):1097–1112. | ||
Dono M, Angelini G, Cecconi M, et al. Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in uveal melanoma: detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. Br J Cancer. 2014;110(4):1058–1065. | ||
Koopmans AE, Verdijk RM, Brouwer RW, et al. Clinical significance of immunohistochemistry for detection of BAP1 mutations in uveal melanoma. Mod Pathol. 2014;27(10):1321–1330. | ||
van de Nes JA, Nelles J, Kreis S, et al. Comparing the prognostic value of BAP1 mutation pattern, chromosome 3 status, and BAP1 immunohistochemistry in uveal melanoma. Am J Surg Pathol. 2016;40(6):796–805. | ||
Landreville S, Agapova OA, Matatall KA, et al. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin Cancer Res. 2012;18(2):408–416. | ||
Ewens KG, Kanetsky PA, Richards-Yutz J, et al. Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Invest Ophthalmol Vis Sci. 2014;55(8):5160–5167. | ||
Chaudhuri J, Si K, Maitra U. Function of eukaryotic translation initiation factor 1A (eIF1A) (formerly called eIF-4C) in initiation of protein synthesis. J Biol Chem. 1997;272(12):7883–7891. | ||
Hunter SM, Anglesio MS, Ryland GL, et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget. 2015;6(35):37663–37677. | ||
Kunstman JW, Juhlin CC, Goh G, et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet. 2015;24(8):2318–2329. | ||
Kusters-Vandevelde HV, Creytens D, van Engen-van Grunsven AC, et al. SF3B1 and EIF1AX mutations occur in primary leptomeningeal melanocytic neoplasms; yet another similarity to uveal melanomas. Acta Neuropathol Commun. 2016;4:5. | ||
Markby DW, Onrust R, Bourne HR. Separate GTP binding and GTPase activating domains of a G alpha subunit. Science. 1993;262(5141):1895–1901. | ||
Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340(6236):692–696. | ||
Malaponte G, Libra M, Gangemi P, et al. Detection of BRAF gene mutation in primary choroidal melanoma tissue. Cancer Biol Ther. 2006;5(2):225–227. | ||
Rimoldi D, Salvi S, Lienard D, et al. Lack of BRAF mutations in uveal melanoma. Cancer Res. 2003;63(18):5712–5715. | ||
Cruz F 3rd, Rubin BP, Wilson D, et al. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Res. 2003;63(18):5761–5766. | ||
Henriquez F, Janssen C, Kemp EG, Roberts F. The T1799A BRAF mutation is present in iris melanoma. Invest Ophthalmol Vis Sci. 2007;48(11):4897–4900. | ||
Emley A, Nguyen LP, Yang S, Mahalingam M. Somatic mutations in GNAQ in amelanotic/hypomelanotic blue nevi. Hum Pathol. 2011;42(1):136–140. | ||
Golas MM, Sander B, Will CL, Luhrmann R, Stark H. Molecular architecture of the multiprotein splicing factor SF3b. Science. 2003;300(5621):980–984. | ||
Corrionero A, Minana B, Valcarcel J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 2011;25(5):445–459. | ||
Folco EG, Coil KE, Reed R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 2011;25(5):440–444. | ||
Te Raa GD, Derks IA, Navrkalova V, et al. The impact of SF3B1 mutations in CLL on the DNA-damage response. Leukemia. 2015;29(5):1133–1142. | ||
Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. | ||
Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44(1):47–52. | ||
Furney SJ, Pedersen M, Gentien D, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3(10):1122–1129. | ||
Yavuzyigitoglu S, Koopmans AE, Verdijk RM, et al. Uveal melanomas with SF3B1 mutations: a distinct subclass associated with late-onset metastases. Ophthalmology. 2016;123(5):1118–1128. | ||
Rossi D, Bruscaggin A, Spina V, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118(26):6904–6908. | ||
Hahn CN, Scott HS. Spliceosome mutations in hematopoietic malignancies. Nat Genet. 2011;44(1):9–10. | ||
Field MG, Decatur CL, Kurtenbach S, et al. PRAME as an independent biomarker for metastasis in uveal melanoma. Clin Cancer Res. 2016;22(5):1234–1242. | ||
Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339(6122):959–961. | ||
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–959. | ||
Harland M, Petljak M, Robles-Espinoza CD, et al. Germline TERT promoter mutations are rare in familial melanoma. Fam Cancer. 2016;15(1):139–144. | ||
Griewank KG, Murali R, Schilling B, et al. TERT promoter mutations in ocular melanoma distinguish between conjunctival and uveal tumours. Br J Cancer. 2013;109(2):497–501. | ||
Harbour JW. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res. 2012;25(2):171–181. | ||
Testa JR, Cheung M, Pei J, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43(10):1022–1025. | ||
Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43(10):1018–1021. | ||
Aoude LG, Wadt K, Bojesen A, et al. A BAP1 mutation in a Danish family predisposes to uveal melanoma and other cancers. PLoS One. 2013;8(8):e72144. | ||
Hoiom V, Edsgard D, Helgadottir H, et al. Hereditary uveal melanoma: a report of a germline mutation in BAP1. Genes Chromosomes Cancer. 2013;52(4):378–384. | ||
Abdel-Rahman MH, Pilarski R, Cebulla CM, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48(12):856–859. | ||
Njauw CN, Kim I, Piris A, et al. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS One. 2012;7(4):e35295. | ||
Wadt KA, Aoude LG, Johansson P, et al. A recurrent germline BAP1 mutation and extension of the BAP1 tumor predisposition spectrum to include basal cell carcinoma. Clin Genet. 2015;88(3):267–272. | ||
Wadt K, Choi J, Chung JY, et al. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res. 2012;25(6):815–818. | ||
Popova T, Hebert L, Jacquemin V, et al. Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet. 2013;92(6):974–980. | ||
de la Fouchardiere A, Cabaret O, Savin L, et al. Germline BAP1 mutations predispose also to multiple basal cell carcinomas. Clin Genet. 2015;88(3):273–277. | ||
Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. | ||
Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. | ||
Weber A, Hengge UR, Urbanik D, et al. Absence of mutations of the BRAF gene and constitutive activation of extracellular-regulated kinase in malignant melanomas of the uvea. Lab Invest. 2003;83(12):1771–1776. | ||
Zuidervaart W, van Nieuwpoort F, Stark M, et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br J Cancer. 2005;92(11):2032–2038. | ||
von Euw E, Atefi M, Attar N, et al. Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol Cancer. 2012;11:22. | ||
Mahipal A, Tijani L, Chan K, Laudadio M, Mastrangelo MJ, Sato T. A pilot study of sunitinib malate in patients with metastatic uveal melanoma. Melanoma Res. 2012;22(6):440–446. | ||
Selumetinib shows promise in metastatic uveal melanoma. Cancer Discov. 2013;3(7):OF8. | ||
Carvajal RD, Sosman JA, Quevedo JF, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311(23):2397–2405. | ||
Patel M, Smyth E, Chapman PB, et al. Therapeutic implications of the emerging molecular biology of uveal melanoma. Clin Cancer Res. 2011;17(8):2087–2100. | ||
Khalili JS, Yu X, Wang J, et al. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res. 2012;18(16):4345–4355. | ||
Abdel-Wahab O, Adli M, LaFave LM, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180–193. | ||
Harbour JW, Chao DL. A molecular revolution in uveal melanoma: implications for patient care and targeted therapy. Ophthalmology. 2014;121(6):1281–1288. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.