Back to Journals » The Application of Clinical Genetics » Volume 7
The genetics of Leigh syndrome and its implications for clinical practice and risk management
Received 8 July 2014
Accepted for publication 20 August 2014
Published 13 November 2014 Volume 2014:7 Pages 221—234
DOI https://doi.org/10.2147/TACG.S46176
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Martin Maurer
Ilene S Ruhoy, Russell P Saneto
Division of Pediatric Neurology, Seattle Children's Hospital/University of Washington, Seattle, WA, USA
Abstract: Leigh syndrome, also referred to as subacute necrotizing encephalomyelopathy, is a severe, early-onset neurodegenerative disorder that is relentlessly progressive and devastating to both the patient and the patient's family. Attributed to the ultimate failure of the mitochondrial respiratory chain, once it starts, the disease often results in the regression of both mental and motor skills, leading to disability and rapid progression to death. It is a mitochondrial disorder with both phenotypic and genetic heterogeneity. The cause of death is most often respiratory failure, but there are a whole host of complications, including refractory seizures, that may further complicate morbidity and mortality. The symptoms may develop slowly or with rapid progression, usually associated with age of onset. Although the disease is usually diagnosed within the first year of life, it is important to note that recent studies reveal phenotypic heterogeneity, with some patients having evidence of in utero presentation and others having adult-onset symptoms.
Keywords: mitochondrial disorder, neurodegeneration, multisystemic disease, oxidative phosphorylation, mitochondrial DNA, neuroimaging, seizures
Introduction
Denis Leigh, a British neuropathologist, first described Leigh syndrome in 1951 in a postmortem pathologic evaluation of a 7-month-old infant who presented with severe encephalopathy and respiratory difficulties.1 However, it has only been during the last 15 years that the knowledge of the myriad of genetic mutations and natural histories of Leigh syndrome have significantly increased our understanding of this mitochondrial disorder. Leigh syndrome is relatively common, with an incidence of 1 in 40,000 births, although certain populations have higher incidences. Because of two founder gene mutations found in two ethnic populations, one mitochondrial DNA (mtDNA)-encoded mutation leading to the disease occurs in 1 in 2,000 births in an isolated population near Quebec, Canada, and another nuclear-encoded mutation with an estimated homozygote frequency of 1:2,500 occurs in the Faroese population.2,3 There is no known predilection for sex or race. The preschool incidence of Leigh syndrome was 1 in 32,000 in one population study within Northern Europe.4
The onset of symptoms is typically seen at between 3 and 12 months of age, but there is a wide range of disease onset. Onset can begin in utero with oligohydramnios and intrauterine growth restriction, but some cases do not become clinically observable until the second or third decade of life.5 When onset takes place during the first year, parental complaints of loss of head control and other motor milestones and floppiness prompt the initial medical investigation. Cognitive deterioration may appear insidious or may be rapid, usually after a bout of fever or onset of seizures. Nutritional troubles resulting from the decreased ability to feed may precede or exacerbate the underlying encephalopathy. Decreased body and head growth become apparent.
When the disease starts in the second or third year of life, the first complaints are usually difficulty in walking, dystonia, dysarthria, or intellectual regression. Patients are brought to medical attention most often for failure to thrive related to dysphagia and gastrointestinal distress. It is during this first examination that diffuse hypotonia is commonly noted. Later in the course, there are often complications with dystonic posturing not related to seizures, encephalopathy, and ataxia. These symptoms make it difficult to move purposefully and both interfere with proper motor development and result in significant delays and regressions. Decreased strength of respiratory muscles creates difficult-to-manage respiratory complications of illness. Initially intermittent, the respiratory symptoms ultimately become more consistent; hence the high incidence of death by respiratory failure.
Survival is variable, with most patients dying within a few years of diagnosis. In a large cohort of patients with Leigh syndrome, the median age of death was 2.4 years (range, 1 month–21 years).5 In this study, the survival probability was about 0.4 if a patient lived to 20 years of age, suggesting there is variability in survival. This likely is underscored by the genetic mutation type and the frequency of environmental stressors such as illness. Relapses with disease exacerbations are commonly a result of infections, usually with fever.5–7 Poor survival has been associated with age of disease onset, genetically identified disease, epileptic seizures, brainstem lesion on neuroimaging, failure to gain weight, and intensive care admission.5
Epilepsy is a common clinical finding that replicates overall disease clinical and genetic heterogeneity. In a recent review, seizures were seen in about 39% of patients with Leigh syndrome from all causes.5 In another large review focusing only on nuclear-encoded genes, a frequency of 21% of Leigh syndrome patients were described as having seizures.7 Twenty-six percent of patients with pyruvate dehydrogenase (PDH) deficiency were noted to have seizures, but the review did not divide these patients into groups depending on whether they had Leigh syndrome or not, although only 27% of the total population had frank Leigh syndrome.8 The phenotypic expression of seizures in Leigh syndrome represents a genetic heterogeneous population, with identical genetic mutations giving rise to seizures in some patients and not in others. Phenotypically, the same epilepsy syndrome can be found in a variety of genetic mutations, both nuclear- and mtDNA-encoded.7,9 Seizure types range from infantile spasms to minor focal seizures.8–11 It has been our experience that up to 75% of Leigh syndrome patients with seizures remain medically intractable compared with other reports that suggest that only 30% are medically intractable (Saneto, unpublished data, 2014). In fact, the presence of seizures is a risk factor for early patient death and likely represents a more severe spectrum of cerebral involvement of the disorder.5 A complete understanding of this heterogeneity has not been attained.
Pathologically, Leigh syndrome is uniformly characterized by multifocal spongiform degeneration diffusely throughout the brain, including the basal ganglia, thalamus, cerebellum, brain stem, spinal cord, and optic nerves. Characteristic neuropathology includes spongiform lesions with relative preservation of neurons, associated with demyelination and gliosis.12 These lesions, often accompanied by demyelination, are seen bilaterally and symmetrically throughout the brain but have a particular predilection for the brainstem.13 Most of the symptoms seen on physical examination can be localized to regions of the brain and brainstem that correspond to magnetic resonance imaging (MRI) changes. This correlated correspondence among neurological exam, MRI, and pathological findings has been useful in replacing postmortem pathological analysis in diagnostic schemes.
Overall, the clinical picture of those afflicted by Leigh syndrome largely represents the areas of the brain involved, but there are characteristic, albeit variable, clinical manifestations (Table 1). Because of the often involved brainstem, cerebellum, and basal ganglia in the majority of cases, the disease expresses itself not only with intermittent respiratory abnormalities but also with progressive oculomotor and other cranial nerve palsies, abnormal thermoregulation, ataxia, a variety of involuntary movements, and optic atrophy. Encephalopathy is manifested by motor delay, mental retardation, progressive cognitive decline, seizures, and feeding difficulties.
 | Table 1 Clinical manifestations seen with Leigh syndrome patients |
Leigh syndrome: genetics
Leigh syndrome is an extremely genetically heterogeneous mitochondrial disorder. Newly identified nuclear genetic causes are increasing, largely as a result of the use of next-generation and whole-exome sequencing.14–16 Nuclear DNA mutations are inherited in a Mendelian fashion, with autosomal recessive and X-linked inheritance seen as the etiology of Leigh syndrome. Identification of causative mutations can be a cumbersome task, as the investigator is confronted by two separate genomes, both nuclear and mitochondrial.17 The identification of pathological mutations in the mtDNA can be unclear due to mitochondrial genetics and physiology. As a result of multiple mtDNA molecules in individual mitochondrion, mutations can be heteroplasmic, meaning that mutated mtDNA and normal mtDNA coexist within individual mitochondrion. Identical DNA genomes, either normal or mutated, within the mitochondrion are called homoplasmic. Both situations can give rise to Leigh syndrome.18 However, the precise genetic cause of multiple cases of Leigh syndrome remains unknown.19
As of 2013, Leigh syndrome can be caused by mutations in more than 35 different genes (Table 2) of both nuclear and mitochondria origin, involving all five respiratory chain complexes.20 There are 13 mtDNA-encoded structural proteins of complex I, III, IV, and V, whereas the other approximately 77 structural subunits are nuclear-encoded proteins. The electron donors of coenzyme Q10 and cytochrome c are also nuclear-encoded proteins. Mutations in any of these structural components of the respiratory chain can induce Leigh syndrome. Mutations in other genes involved in mitochondrial function, the tricarboxylic acid (TCA) cycle, and coenzyme Q10 have also been found as an etiology of Leigh syndrome.3,21–23 Depending on the mutated genome, Leigh syndrome can be inherited as a maternally inherited mitochondrial trait (mtDNA-encoded), as an autosomal recessive trait resulting from mutations in nuclear genes encoding mitochondrial respiratory chain complex subunits, complex assembly proteins, coenzyme Q10, mitochondrial targeted tRNA synthetases synthetases, and X-linked genes involved in PDHA1 and complex I assembly factor NDUFA1 (nuclear-encoded). Although most patients with Leigh syndrome have a mutation in nuclear DNA, about 25% have a mutation in mtDNA.
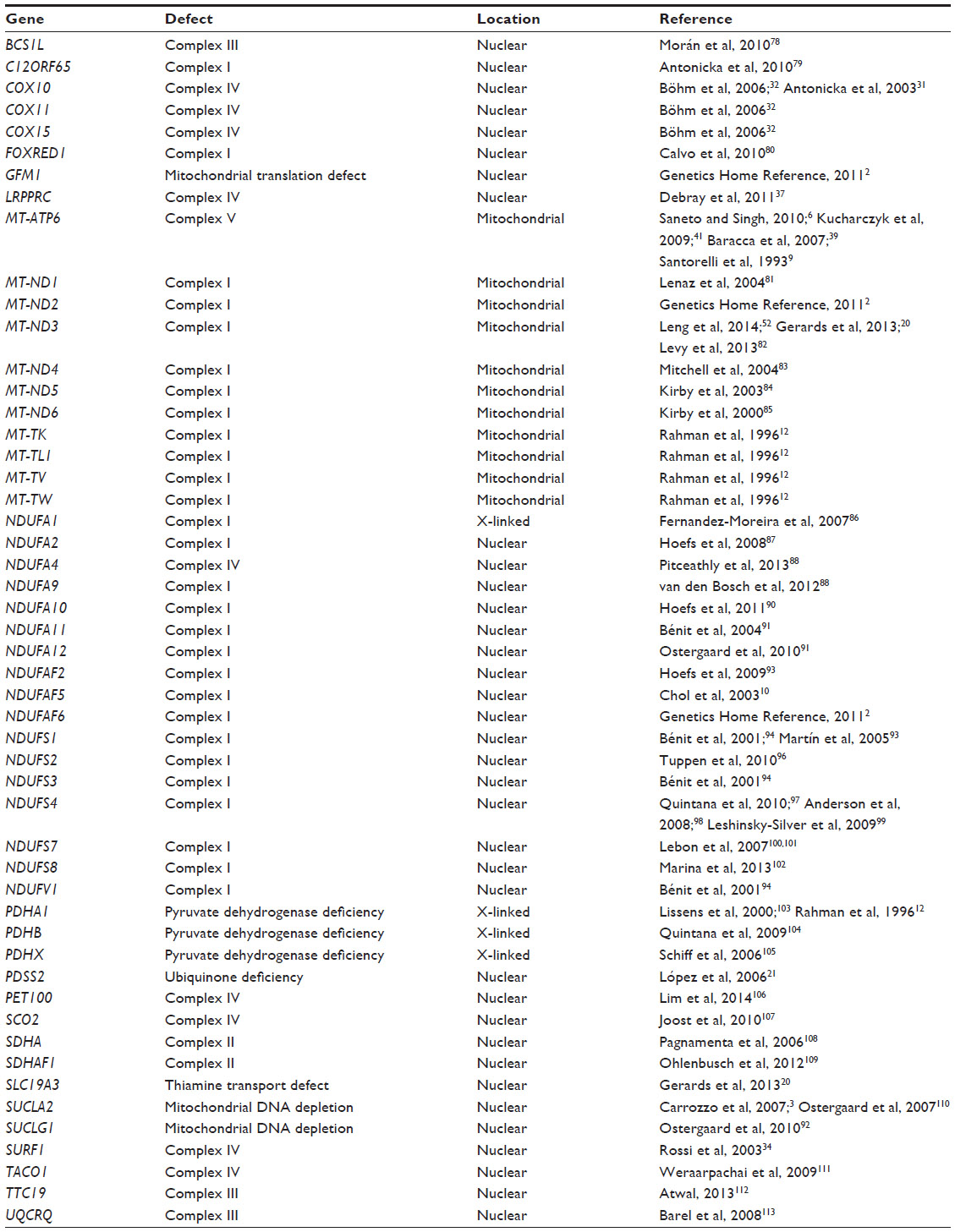 | Table 2 Genes known to be associated with Leigh syndrome |
Most pathological gene mutations are ultimately involved in the process of energy production in the mitochondria. Mitochondrial adenosine triphosphate (ATP) production is coupled to the oxidation of electron carriers nicotinamide adenine dinucleotide + hydrogen (NADH) and flavin adenine dinucleotide (FADH2).24 Five protein complexes make up the respiratory chain that are involved in oxidative phosphorylation (OXPHOS). Complex I, complex II, complex III, and complex IV make up the oxidative portion, and complex V makes up the phosphorylation segment of the mitochondrial respiratory chain. Complex V is also known as ATP synthase, as it uses the energy from the electrochemical proton gradient across the inner membrane produced by the reactions of complexes I–IV that reduce molecular oxygen to water. Complex V then uses this energy to catalyze the synthesis of ATP from adenosine diphosphate and inorganic phosphate.25 NADH and FADH2, formed via the TCA cycle and fatty acid oxidation, respectively, are used as electron donors to reduce oxygen molecules. Mutations associated with the respiratory chain complexes either reduce or eliminate their activity, impairing mitochondrial energy production, which in turn ultimately leads to cellular and organ dysfunction.26 Furthermore, the production of intracellular reactive oxygen species by both the mitochondria and the surrounding cells produces a proinflammatory environment that contributes to much of the tissue injury and organ impairment.27
Mitochondrial genetics imparts special inheritance patterns. Maternal inheritance imparts the disease mutation to all children of an affected woman. Multiple mtDNA per mitochondrion and multiple mitochondria per cell can lead to heteroplasmy, the combination of normal and abnormal mtDNA in the same cell. To date, most mitochondrial diseases resulting from mtDNA are either a result of a high degree of heteroplasmy or homozygous mtDNA mutations.26 Heteroplasmy creates one important characteristic feature of mtDNA-encoded disease: There is an association of the severity of the clinical manifestations of the disease with the extent to which mutant mtDNA exists in ratio to wild-type mtDNA in each cell. Intertissue variability likely occurs, and prospective studies could be considered and may help define organ specificity.
Nuclear-encoded mutations can be inherited in a Mendelian fashion. Genes encoding the proteins involved in the structural components of the respiratory chain complexes, the electron carrier of coenzyme Q10, assembly proteins of the various complexes and super complexes, protein transporters, and modifiers of mt tRNAs, can give rise to Leigh syndrome. Enzymes involved in the TCA cycle can also be involved in this syndrome. This wide range of components of mitochondrial pathophysiology produces a spectrum of disease.
Whether a result of nuclear- or mtDNA-encoded mutation, ultimately it is impaired OXPHOS that leads to a critical nadir of cellular energy and subsequent cell death. The high-energy demand tissues are most vulnerable to this process and, in part, are responsible for the clinical features of Leigh syndrome. It is not surprising that although a wide range of clinical symptoms is found, most patients present similarly with neurological symptoms of brainstem and/or basal ganglia dysfunction, seizures, hypotonia, periodic disease exacerbation with illness, and progressive neurological decline. Four main groups of defects appear to be responsible for the vast majority of cases.
PDH complex deficiency
Leigh syndrome can be caused by mutations in genes that serve to form the PDH complex (PDHC). Many patients with PDHC deficiency have Leigh syndrome with classic imaging findings. Biochemical defects can involve any of the three subunits of the enzyme complex of the activation of the complex by PDH phosphatase and can lead to lower PDH activity and the limitation of pyruvate entering the respiratory chain. This produces a severe reduction of the TCA cycle activity. Deficiencies of the X-linked E1n subunit of the PDHC, the most common etiology of Leigh syndrome, have been well-documented.8 Leigh syndrome has also been associated with the E3 component of PDHC, which, if disease is present, is autosomal recessively inherited. Patients with PDHC deficiency have an earlier onset, usually between 3 and 6 months, with males usually having more severe disease. Females can also express the disease because of skewed X-inactivation.8 Interestingly, although more males died than females, females who had PDH deficiencies died earlier than the males.8 Clinical symptoms of PDHC deficiency can range from Leigh syndrome to intermediate ataxia or progressive neurodegenerative disorder. The reason for this range of phenotypes is not well understood.28
Isolated respiratory chain complexes
Complex II is the smallest complex of the respiratory chain, and all four subunits (A, B, C, and D) are nuclear-encoded. Mutations in complex II are relatively rare, as are documented causes of mitochondrial disorders in general. Complex II is located on the inner membrane. It functions to oxidize FADH2, transferring electrons to ubiquinone in the electron transport chain. These reactions are based on the functions of catalytic subunits A and B. More than ten different autosomal recessive pathogenic mutations in A have been described that cause mitochondrial disorders. Some patients described with complex II deficiency had the clinical phenotype and cerebral MRI findings consistent with Leigh syndrome, whereas others had other findings and were not considered to have Leigh syndrome.29,30 The heterogeneity of disease expression is not completely understood.
Cytochrome oxidase deficiency
The most common cause of nuclear-encoded mutation Leigh syndrome is disruption of complex IV or cytochrome oxidase (COX). There are 13 subunits, three of which are encoded by mtDNA and the other 10 are nuclear encoded.31 The assembly process of complex IV is not fully known, but mutations in these genes, and in particular SURF1, also can produce Leigh syndrome.32 This complex is involved in the last step of electron transfer in the oxidative process of ATP production. Mutations leading to the disruption of complex IV are the most common cause of Leigh syndrome in the United Kingdom.33 These patients have a slightly later age of onset than those with PDHC deficiency, usually between 8 and 12 months.
The most frequently mutated gene in COX-deficient Leigh syndrome is the nuclear-encoded SURF1 gene, located on chromosome 9q34 and important in the assembly and maintenance of COX activity. Eighty-three different mutations have been reported in 57 patients with approximately 80% truncating mutations.32 Truncating mutations in the SURF1 gene are subsequently degraded, resulting in the absence of functional SURF1 protein. The loss of this protein reduces the formation of normal COX complexes, which impairs mitochondrial energy production. Most patients with SURF1 mutations affecting COX have MRI abnormalities involving brainstem and subthalamic nuclei.34,35 However, there are some exceptions.36 The reasons for this heterogeneity in neuroimaging while retaining a homogeneous phenotypic presentation in Leigh syndrome are not well understood.
A particular unique subset of COX-deficient Leigh syndrome is French-Canadian Leigh disease (Saguenay-Lac-St-Jean cytochrome c oxidase deficiency [LRPPRC]). LRPPRC is clinically distinct, with acute fatal acidotic crises on a backdrop of chronic moderate developmental delay and hyperlactatemia. The Leigh syndrome of LRPPRC differs from that of SURF1-related COX deficiency. LRPPRC has a different spectrum of associated abnormalities, with acidotic crises being particularly suggestive of LRPPRC-related Leigh syndrome. LRPPRC encodes leucine-rich pentatricopeptide repeat-containing protein, which may regulate the stability and handling of mature mitochondrial mRNA. Even among p. A354V homozygotes, pronounced differences in survival and severity occur, showing that other genetic and/or environmental factors can influence outcome.37 It is unclear why the regulation of mRNA stability and handling alters COX activity more than the other respiratory chain complexes.
Mitochondrial ATPase 6 gene
Mutations of this gene result in the most common maternally inherited Leigh syndrome (MILS), the most widespread mtDNA mutation in Leigh syndrome. ATP 6 is part of complex V that alters the function of the proton channel and results in a loss of ATP-synthetic activity.38 When individuals have more than 90% heteroplasmy of mtDNA in their cells, they are classified as having MILS, and a rapidly progressive course is characterized by bilateral subacute lesions of the basal ganglia, which may extend to the brainstem, leading to the death of the patient.39 The ATP synthase enzyme is a multisubunit complex with a molecular mass of approximately 550,000 Da. It has both a hydrophobic domain and a hydrophilic ATPase. ATP synthase defects may be of either mitochondrial or nuclear genetic origin, as the biogenesis of the mitochondrial ATP synthase has 14 subunits that are nuclear-encoded in addition to two mtDNA-encoded proteins.40,41
A nucleotide change (T>G and T>C) at the same location in the mtDNA, 8993, affecting the ATP 6 subunit of the ATPase is also associated with neuropathy, ataxia, and retinitis pigmentosa (NARP), as well as MILS. Whereas MILS occurs when mutation load is greater than 90%, NARP is seen with percentages around 50%–60%.42 This difference in heteroplasmic load is a prime example of mitochondrial genetics, with low heteroplasmy versus a higher ratio of heteroplasmy causing different phenotypes.43 In 2007, Baracca et al tried to explain the difference in pathogenicity of these two diseases resulting from mutations of the same position of the ATP 6 gene of mtDNA and concluded that reactive oxygen species overproduction is likely a major pathogenic mechanism contributing to NARP versus MILS phenotypes.39
The degree of heteroplasmy producing NARP versus Leigh syndrome and producing varying levels of reactive oxygen species and degree of ATP production are likely not the full answer to phenotype. For example, a family at our institution demonstrated this continuum: A 45-year-old man presented with intellectual impairment, retinitis pigmentosa, cerebellar ataxia, peripheral neuropathy, pes cavus with foot drop, and a negative family history. MRI revealed cerebellar atrophy. He was ultimately diagnosed with NARP after targeted mutation analysis revealed more than a 95% of mtDNA with m.8993 T>C mutation. The percentage of heteroplasmy should have produced the Leigh syndrome phenotype. A pedigree traced to the maternal grandmother and subsequent testing demonstrated 50% of mtDNA with 8993 mutation in the maternal grandmother, who had a daughter, the patient’s mother, with 66% and a son, the patient’s uncle, with 80%. The grandmother and mother were clinically normal, but the uncle was affected. The patient also had a deceased brother with more than 95% of mtDNA with m.8993 T>C mutation. These data suggest that degree of ATP production via ATPase activity and amount of oxygen radical species produced cannot be the total etiology of the phenotypic requirement of Leigh syndrome versus NARP. Interestingly, retinitis pigmentosa is a major sign that is not found in Leigh syndrome as a result of nuclear-encoded mutations. Furthermore, there are no ragged red fibers. Five percent to 10% of Leigh syndrome cases have been attributed to mutations in the MT-ATP6 gene.
With the NARP m.8993 T>G mutation, it is possible to have NARP in one generation but then Leigh syndrome, NARP, and/or no clinical symptoms in another generation. This is an example of random segregation and “bottleneck effects” during oocyte development. MtDNA mutations are randomly segregated to the forming oocytes during postfertilization of the female fetus.44,45 Heteroplasmy and type of missense mutation in m. 8993 play an important role in age of disease onset. Leigh syndrome has infantile onset, whereas NARP usually has childhood or adult onset. Mitochondrial genetics and the degree of ATP production and oxidative reactive species as well as other undefined etiologies likely differentiate between onset and type of disorder. Interestingly, no other mutation in the mtDNA genome gives rise to NARP, although the reasons for this are not clear. A clear description of reproducible intertissue variability of mutation also has never been described.
Complex I deficiency
Abnormal respiratory chain enzyme activity is commonly found within complex I (NADH:ubiquinone oxidoreductase). It is the first and largest enzyme complex of the mitochondrial respiratory chain and OXPHOS system and plays critical roles in transferring electrons from NADH to ubiquinone and redox-driven proton translocation to maintain the electrochemical gradient across the inner mitochondrial membrane.46 It consists of 45 subunits: seven subunits are encoded by mtDNA, and 38 subunits are from nuclear DNA. In addition, there are multiple assembly proteins involved in functional holoenzyme activity. There are at least ten assembly proteins that are of nuclear DNA origin that have associated disease.47 However, there are many more punitive candidate assembly factor genes involved in complex I assembly, suggesting that more nuclear-encoded genes remain to be discovered.7,47 Leigh syndrome can result from mtDNA and nuclear-encoded gene mutations associated with isolated complex I defects. In one large review study, 84% of patients with complex I dysfunction had complex I enzymatic activity changes as a result of nuclear-encoded mutations.7
Deficiency of complex I is the most commonly identified defect in childhood-onset mitochondrial disease and accounts for approximately a third of all cases of OXPHOS disorders.48 There are seven mtDNA-encoded subunits of complex I that are part of the highly conserved core of complex I. However, age of onset may differ by whether mtDNA- or nuclear-encoded defects are the etiology. Leigh syndrome induced by mtDNA-encoded gene mutations at age of onset may be related to a specific mutation. In three large cohort studies (n=188) of neonatal-onset mitochondrial disease, only three neonates had Leigh syndrome, and all three had the condition as a result of m.8993 T>G mutation.49–51 However, in infants and children, all of the mtDNA-encoded subunits of complex I (ND1, ND2, ND3, ND4, ND4l, ND5, and ND6) have been associated with Leigh syndrome.26,52–54 There have been at least 26 distinct mtDNA-encoded mutations giving rise to Leigh syndrome.55 Phenotypical expressions may vary, with some patients with identical mtDNA-encoded mutations having either Leigh syndrome or another type of nonsyndromic mitochondrial disease. Heteroplasmy plays a role in phenotypic variation, but there must be other reasons for this variation in the phenotype, as similar percentages of heteroplasmy can give rise to distinct phenotypes.
There are 38 nuclear-encoded complex I subunits, of which seven form the highly conserved core subunits, NDUFS1–NDUFS3, NDUFS7, NDUFS8, and NDUFV1 and NDUFV2.56 Leigh syndrome has been described in each of these proteins.55 Associated subunits of complex I, NDUFS4, NDUFS7, and NDUFS8 and NDUFA1, NDUFA2, NDUFA9, and NDUFA10, have been linked to Leigh syndrome.7,57,58 Assembly proteins have also been described, including NDUFAF1, NDUFAF4, ACAD9, NUBPL, C20orf7, C8orf38, and FOXRED1.7 As more refinements in gene discovery methodology occur, it is likely that more nuclear-encoded complex I proteins will be found.
Leigh syndrome patients with complex I deficiency typically have normal early development but present in late infancy or early childhood with progressive neurological abnormalities consistent with Leigh syndrome. Presentation before 6 months usually includes symptoms of hypotonia, feeding problems, vomiting, encephalopathy, epilepsy, and eye movement disturbances, whereas after 6 months, presentation was psychomotor retardation, pyramidal signs and symptoms, dystonia, ataxia, epilepsy, failure to thrive, vomiting, and optic atrophy.7 Stepwise neurodevelopmental regression may follow intercurrent illnesses. Although there may be some recovery noted, usually the child never returns to baseline before the illness. Nuclear-encoded complex I deficiency was associated with brainstem involvement, optic atrophy, and basal ganglia involvement on MRI scans.7
Other mutations
Various mutations in mtDNA-encoded tRNA genes have been described as causing Leigh syndrome. Mutations include those in m.8344 A>G tRNA Lys, m.1644 G>T tRNA Val, m.3537 Tins, m.3243 A>G tRNA Leu, m.10410 T>A tRNA Arg, and m.5523 T>G tRNA Trp.59–64 Other mtDNA-encoded mutations have been described in ribosomal proteins, such as m.896 A>G 12S rRNA and m.270 A>G 16S rRNA.63
There are other nuclear-encoded genes involved in Leigh syndrome, although they are relatively uncommon. In their study, Gerards et al found a homozygous nonsense mutation in the thiamine transporter gene SLC19A3 (c.20 C>A).20 This mutation resulted in a premature stop codon and, subsequently, decreased thiamine uptake by components of the respiratory complex of the mitochondria. The phenotype was consistent with known clinical presentations and the disease course of Leigh syndrome.20
Carrozzo et al characterized a group of patients with SUCLA2 gene mutations in whom clinical presentation and MRI findings were consistent with Leigh syndrome.3 There were many patients from the Faroe Islands having a common ancestor, suggesting a founder effect for this mutation. Presenting signs and symptoms included early-onset hypotonia, psychomotor delay and regression, progressive dystonic/athetoid movements, and sensorineural deafness. The SUCLA2 gene encodes the beta-subunit of the adenosine diphosphate-forming succinyl-CoA synthetase, a mitochondrial matrix enzyme that catalyzes the reversible synthesis of succinyl-CoA from succinate and CoA. The reverse reaction occurs in the TCA cycle, whereas the forward reaction may produce succinyl-CoA for the activation of ketone and heme synthesis. Methylmalonic aciduria results from buildup of succinyl CoA and increased conversion to methylmalonyl CoA. It is not entirely clear how a mutation in the SUCLA2 gene causes mtDNA dysfunction. Elpeleg et al theorized an interaction between succinyl-CoA synthetase and nucleoside diphosphate kinase, both of which are crucial for the maintenance of ribonucleotides, and deoxyribonucleotides.22 Indeed, mtDNA depletion has been found in some patients.22
Coenzyme Q10 is a lipophilic molecule that functions as an electron carrier involved in the transfer of electrons from complex I and complex II to complex III, as well as a potent antioxidant. Recently, patients with mutations in the synthetic pathway of coenzyme Q10 synthesis, decaprenyl diphosphate synthase subunit 2, have a multisystem disorder that includes Leigh syndrome signs and symptoms together with nephrotic syndrome and focal seizures.21,23
Diagnosis
In view of the inconsistent nature of the disease and the notable absence of a specific biochemical or molecular defect,65 definitive diagnoses has relied on demonstration of the typical pattern of brain lesions on MRI or autopsy and clinical findings.12 Although affected areas appear hypodense as a result of tissue necrosis, on computed tomography scan, this test is much less reliable. Primary striatal involvement on MRI can suggest Leigh syndrome. However, neuroimaging findings can also vary depending on the particular genetic and/or biochemical defect. Other mitochondrial conditions such as PDH deficiency and complex V deficiency without Leigh syndrome, as well as nonmitochondrial diseases such as carbon monoxide intoxication, infantile bilateral striatal necrosis, Wilson disease, and juvenile Huntington’s chorea, can demonstrate basal ganglia changes. Biochemical findings, clinical history, and physical examination can usually distinguish these disorders from Leigh syndrome. White matter lesions in the cerebral hemispheres may be so prominent as to mimic a leukodystrophy. Therefore, to be reasonably certain of the diagnosis of Leigh syndrome, strict criteria have been proposed on the basis of the combination of typical MRI findings (Figures 1 and 2), biochemical abnormalities such as the demonstration of a increased level of lactate in the cerebrospinal fluid, and a clinical picture that is consistent with the disease.12
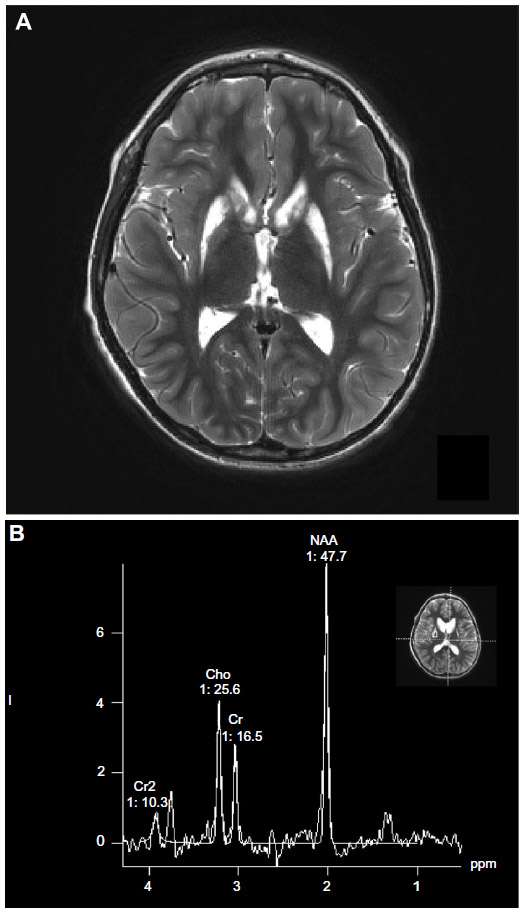 | Figure 1 Axial MRI scan (A) and MRS spectrum (B) of a 9-year old boy with Leigh syndrome due to a mutation in the mtDNA |
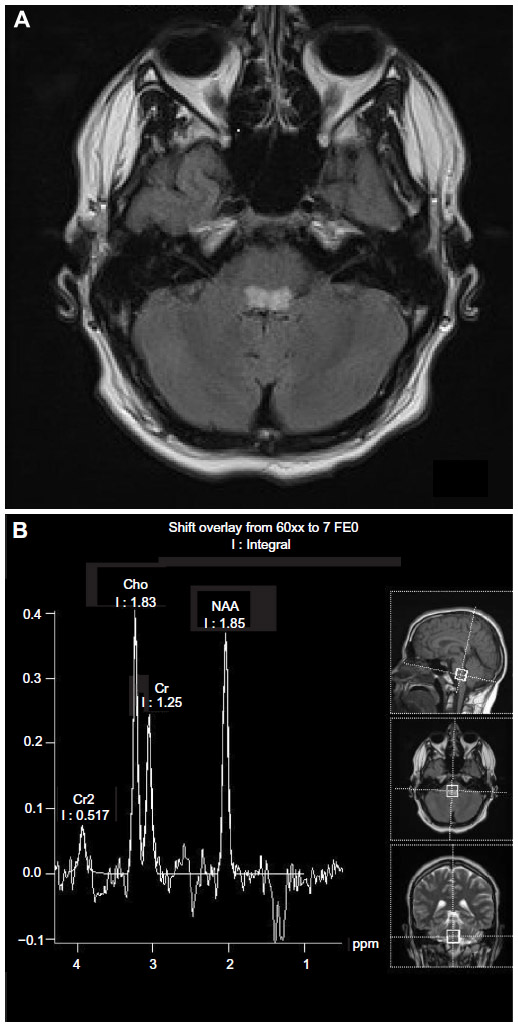 | Figure 2 |
Other neuroimaging findings can provide clues to the diagnosis. Diffusion characteristics and proton magnetic resonance spectroscopy (MRS) characteristics vary depending on the acuity of the lesion (Figures 1 and 2).66 In the acute phase of injury, when mitochondrial function is acutely impaired and ATP production is reduced, water motion will be reduced; this is seen as a hyperintense signal on diffusion weighted image acquisition. If no permanent damage ensues, diffusion may return to normal tissue intensity. However, if significant astroglial response or necrosis ensues in the injured area seen as hypointensity, diffusion or hyperintensity will be increased in the surrounding tissue. MRS has shown a decreased N-acetylaspartate peak and an elevated lactate peak, with lactate elevation most pronounced in areas with acute mitochondrial dysfunction (with consequent anaerobic glycolysis). The decrease in N-acetylaspartate suggests neuronal and/or axonal loss, evolving necrosis resulting from neuronal death, whereas the elevation of the lactate doublet peak is a sign of dysfunctional OXPHOS.
Individual causes of Leigh syndrome can demonstrate variable MRI findings. PDH deficiency typically has classic findings described in Leigh syndrome with T1 and T2 prolongation or hyperintensity of the corpus striatum (caudate and putamen) in addition to delayed myelination. MRS can show large lactate doublet and small N-acetylaspartate peaks in affected regions.66,67 Patients with COX deficiency with SURF1 mutations can have characteristic T1 and T2 prolongations in the subthalamic nuclei, medulla, inferior cerebellar peduncles, inferior olives, and nucleus of the solitary tract in the medulla and central tegmental tract and reticular formation in the dorsal pons and periaqueductal gray matter of the mesencephalon.33–35 However, even within patients having SURF1 mutations, there is variability in brain findings on MRI.33 Lactate peak is not seen on proton MRS in these patients, likely stemming from quiescence of the disease, at which time lactate is likely not present.6 Cerebral dysgenesis can be seen, especially in PDH deficiency, with E1 alpha subunit deficiency. MILS arising from mutations of the ATPase 6 gene (complex V) is less homogeneous than the SURF1 group. However, some cases do demonstrate involvement of the anterior putamen, dorsal mesencephalon, and dorsal pons.36,68 The etiology of the heterogeneity of MRI findings within and between specific genetic etiologies is not clear.
Biochemical confirmation is most often seen in alterations of enzyme activity of one or a combination of the respiratory chain complexes. As described in further detail later, the respiratory chain is responsible for the synthesis of ATP. Muscle biopsy is the most common diagnostic tool for assay material.69 However, there are situations in which other tissue testing can be used for respiratory chain analysis, such as cultured fibroblasts.7,47 Other biochemical modalities such as blue native gel electrophoresis and ultrastructural analysis of muscle can give clues to diagnosis, as can abnormal histochemical staining findings of COX deficiency, succinate dehydrogenase deficiency, and/or Gomori trichrome.7,47,70 Structural changes in mitochondria may or may not be seen. Enzymology is not diagnostic in many instances of Leigh syndrome, but alterations in respiratory chain complex activity can be compromised by reductions in amounts resulting from nuclear-encoded assembly factor activity or other factors in respiratory chain function, such as decreased mitochondrial protein synthesis resulting from decreased formulation of Met-tRNA.7,14,36,47
At this time, diagnosis is based on the clinical scenario of a young child with developmental delay/regressions and, often, seizures. MRI findings often reveal bilateral, often symmetrical, necrotic lesions extending from the thalamus to the brainstem and the posterior columns of the spinal cord.5,6 Lactate and pyruvate are less reliably elevated in the blood than they are in the cerebrospinal fluid. Cerebrospinal fluid values of lactate and pyruvate are important measurements for diagnosis. Elevated blood lactate levels are likely a result of respiratory chain dysfunction but are not highly specific.71 If lactic acidosis is present because OXPHOS defects are present, corresponding elevations in plasma alanine are commonly seen. Renal tubular acidosis (Fanconi syndrome) may be present. Of note, significant elevated levels of ammonia, decreased blood glucose, and organic aciduria are usually not present.
As a result of the lack of a pathognomonic finding, a set of clinical, biochemical, and neuroimaging findings have been developed. Rahman et al12 established diagnostic criteria including progressive neurological disease with motor and intellectual developmental retardation/stagnation, usually regression; signs and symptoms of brainstem and/or basal ganglia disease; raised lactate levels in blood and/or cerebrospinal fluid; and characteristic symmetric necrotic lesions in the basal ganglia and/or brainstem.12 As variability with biochemical findings may be dependent on disease activity, criteria for Leigh syndrome should be reflective of an overall clinical picture of degeneration, mitochondrial dysfunction, and neuroimaging findings.72 Recently, Baertling et al offered a pathway for the diagnosis of Leigh syndrome.72 This pathway offers further diagnostic guidance with genetic testing and tissue histology in addition to the history, examination, laboratory values, and MRI findings within the diagnostic criteria of Rahman et al.12 It is important to note, however, that as genetic testing improves, the field is moving toward a genetic diagnosis being part of the criteria for Leigh syndrome, but we feel that until genetic testing becomes more sensitive and specific, it should not be included as a “required” criteria.
A remitting course with acute exacerbations is common. Episodes of decompensation, usually associated with illnesses, result in developmental regression with an exacerbation of symptoms such as abnormal tone and ataxia. In the majority of patients, the disease presents in the first year of life, most frequently before the age of 6 months, and death often occurs within 2 years of onset. Sofou et al proposed onset at 2 years of age as the cutoff for late-onset Leigh syndrome.5 Clearly, however, the diagnosis of Leigh syndrome in adults questions the validity of such a chronologic spectrum. It appears prognosis is, in part, related to the mutation identified and to heterogeneity of the phenotypic spectrum. SURF1-deficient Leigh syndrome was found to have a more favorable survival outcome compared with disease associated with complex I deficiency or LRPPRC mutations.33 Predictors of poorer prognosis include disease onset at age younger than 6 months, failure to thrive, brainstem lesions of neuroimaging, epileptic seizures that are difficult to treat, and history of intensive care hospitalization. However, all of the factors involved remain to be elucidated.
There is no specific treatment for Leigh syndrome, and generally specific therapy for mitochondrial disorders in child is not widely available. In spite of making considerable progress in our understanding of the molecular mechanisms underlying mitochondrial disease, the available therapeutic approaches are extremely limited but are still a very hot topic for current research goals. The aim of symptomatic treatment is to manage symptoms and to improve the energy state by increasing and optimizing ATP production and lowering lactate levels. This is accomplished by the elimination of noxious metabolics and the supply of oxygen radical scavengers. Antioxidants are a commonly employed mechanism in treatment, and there is currently no clear evidence in support of regular use of these drugs. EPI-743 has been suggested to improve clinical outcomes in cases of genetically confirmed Leigh syndrome in one small and uncontrolled study.73 Of note, EPI-743 also arrested disease progression and reversed vision loss in all but one of the five consecutively treated patients with LHON.74 EPI-743 is an analogue of coenzyme Q10 and is thought to help increase intracellular glutathione, which is reduced in mitochondrial disorders.73 Encouraging studies suggest new medications might be helpful in Leigh syndrome, but well-controlled prospective studies are needed.
Supplementation with B vitamins, such as thiamine, which is a cofactor of PDH, has had no lasting effect, despite reports of neurological improvement with riboflavin treatment.75,76 The beneficial effects of a ketogenic diet in a patient with PDHC deficiency have been reported.77 Response to therapy has been monitored by a combination of MR imaging and MRS.
Symptomatic treatments including antiseizure medication to control seizures are an important part of treatment. Quality of life can improve significantly if seizures can be controlled without further adverse effects. Physiotherapy and pharmacotherapy for neuromuscular concerns, appropriate feeding methods to prevent malnutrition and aspiration, and aggressive treatment of fever and infections are further important treatment parameters to hopefully improve quality of life and decrease stress on both the patient and the family. Cardiology, pulmonology, and ophthalmology evaluations are important, as these organ manifestations would be important to identify early on.
Considering the known genetic defects in the causation of mitochondrial disorders in general, and Leigh syndrome in particular, genetic-based therapeutic strategies are inarguably an important area of treatment available to patients. Although they are certainly in their infancy, there are several experimental strategies currently under research and development. Examples include complementation through allotropic expression of protein and manipulation of heteroplasmy25 by, for example, inhibiting the replication of mutant mtDNA through the sequence-specific binding of peptide nucleic acids. The long-term effectiveness of gene-based therapy has yet to be proven.
Conclusion
Leigh syndrome is a severe, progressive, multisystem, metabolic neurodegenerative disorder with distinct neuroradiological and neuropathological changes but prominent clinical and genetic heterogeneity. Leigh syndrome should be considered in all cases of early infantile progressive encephalopathy of unknown origin, especially when there is an associated nutritional disorder. There is currently limited knowledge regarding correlation phenotype and biochemical markers with the location of the mutant protein within the assembly, function, or structure of each complex of the mitochondrial respiratory chain. Indeed, this quest is complicated because a single family harboring the same mutation can have variable presentations and life expectancy. Although patients with mutations in core subunits have a significantly poorer prognosis than patients with mutations in noncore subunits, the variability in prognosis is extremely high.7
With current genetic diagnostic technology, genetic mutations in both nuclear and mitochondrial genomes are identified at an increasing rate. Randomized controlled studies are still scarce. Successful treatments are even more elusive, and experimental therapeutic interventions should be aggressively considered and attempted.
Acknowledgments
The authors thank the families and patients who have added to their learning and understanding of Leigh syndrome.
Disclosure
RPS has received grants from the National Institutes of Health (7RO1GM75184, 1RC1NS070232-01, 1U54NS078059-01) and Edison Pharmaceuticals, and has also received funds from the Mitochondrial Research Guild at Seattle Children’s Hospital. The authors have no conflicts of interest in this work.
References
Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14(3):216–221. | |
Genetics Home Reference. Leigh syndrome. Available from: http://ghr.nlm.nih.gov/condition/leigh-syndrome; 2011. Accessed: Jun 2014. | |
Carrozzo R, Dionisi-Vici C, Steuerwald U, et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007;130(Pt 3):862–874. | |
Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA anbormalities. Ann Neurol. 2001;49(3):377–383. | |
Sofou K, De Coo IFM, Isohanni P, et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis. 2014;9(1):52. | |
Saneto RP, Singh KK. Illness-induced exacerbation of Leigh syndrome in a patient with the MTATP6 mutation, m. 9185 T>C. Mitochondrion. 2010;10(5):567–572. | |
Koene S, Rodenburg RJ, van der Knaap MS, et al. Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: what we learned from 130 cases. J Inherit Metab Dis. 2012;35(5):737–747. | |
Patel KP, O’Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012;105(1):34–43. | |
Santorelli FM, Shanske S, Macaya A, DeVivo DC, DiMauro S. The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh’s syndrome. Ann Neurol. 1993;34(6):827–834. | |
Chol M, Lebon S, Bénit P, et al. The mitochondrial DNA G13513A MELAS mutation in the NADH dehydrogenase 5 gene is a frequent cause of Leigh-like syndrome with isolated complex I deficiency. J Med Genet. 2003;40(3):188–191. | |
Tsuji M, Kuroki S, Maeda H, et al. Leigh syndrome associated with West syndrome. Brain Dev. 2003;25(4):245–250. | |
Rahman S, Blok RB, Dahl HHM, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39(3):343–351. | |
Cavanagh JB, Harding BN. Pathogenic factors underlying the lesions in Leigh’s disease. Tissue responses to cellular energy deprivation and their clinico-pathological consequences. Brain. 1994;117(Pt 6):1357–1376. | |
Haack TB, Gorza M, Danhauser K, et al. Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol Genet Metab. 2014;111(3):342–352. | |
Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4(118):118ra10. | |
Lieber DS, Calvo SE, Shanahan K, et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology. 2013;80(19):1762–1770. | |
DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9(8):429–444. | |
Bannwarth S, Procaccio V, Lebre AS, et al. Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J Med Genet. 2013;50(10):704–714. | |
Shrikhande DY, Kalakoti P, Syed MMA, Ahya K, Singh G. A rare mitochondrial disorder: Leigh syndrome – a case report. Ital J Pediatr. 2010;36(1):62–66. | |
Gerards M, Kamps R, van Oevelen J, et al. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain. 2013;136(Pt 3):882–890. | |
López LC, Schuelke M, Quinzii CM, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79(6):1125–1129. | |
Elpeleg O, Miller C, Hershkovitz E, et al. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76(6):1081–1086. | |
Van Maldergem L, Trijbels F, DiMauro S, et al. Coenzyme Q-responsive Leigh’s encephalopathy in two sisters. Ann Neurol. 2002;52(6):750–754. | |
Wong LJC. Mitochondrial syndromes with leukoencephalopathies. Semin Neurol. 2012;32(1):55–61. | |
Kucharczyk R, Rak M, di Rago JP. Biochemical consequences in yeast of the human mitochondrial DNA 8993T>C mutation in the ATPase6 gene found in NARP/MILS patients. Biochim Biophys Acta. 2009;1793(5):817–824. | |
Saneto RP, Sedensky MM. Mitochondrial disease in childhood: mtDNA encoded. Neurotherapeutics. 2013;10(2):199–211. | |
Morató L, Bertini E, Verrigni D, et al. Mitochondrial dysfunction in central nervous system white matter disorders. Glia. 2014;n/a. | |
Barnerias C, Saudubray JM, Touati G, et al. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol. 2010;52(2):e1–e9. | |
Jain-Ghai S, Cameron JM, Al Maawali A, et al. Complex II deficiency – a case report and review of the literature. Am J Med Genet A. 2013;161A(2):285–294. | |
Renkema GH, Wortmann SB, Smeets RJ, et al. SDHA mutations causing a multisystem mitochondrial disease: novel mutations and genetic overlap with hereditary tumors. Eur J Hum Genet. Epub April 30, 2014. | |
Antonicka H, Leary SC, Guercin G-H, et al. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet. 2003;12(20):2693–2702. | |
Böhm M, Pronicka E, Karczmarewicz E, et al. Retrospective, multicentric study of 180 children with cytochrome C oxidase deficiency. Pediatr Res. 2006;59(1):21–26. | |
Wedatilake Y, Brown RM, McFarland R, et al. SURF1 deficiency: a multi-centre natural history study. Orphanet J Rare Dis. 2013;8(1):96–110. | |
Rossi A, Biancheri R, Bruno C, et al. Leigh Syndrome with COX deficiency and SURF1 gene mutations: MR imaging findings. AJNR Am J Neuroradiol. 2003;24(6):1188–1191. | |
Farina L, Chiapparini L, Uziel G, Bugiani M, Zeviani M, Savoiardo M. MR findings in Leigh syndrome with COX deficiency and SURF-1 mutations. AJNR Am J Neuroradiol. 2002;23(7):1095–1100. | |
Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49(9):578–590. | |
Debray FG, Morin C, Janvier A, et al. LRPPRC mutations cause a phenotypically distinct form of Leigh syndrome with cytochrome c oxidase deficiency. J Med Genet. 2011;48(3):183–189. | |
Tatuch Y, Robinson BH. The mitochondrial DNA mutation at 8993 associated with NARP slows the rate of ATP synthesis in isolated lymphoblast mitochondria. Biochem Biophys Res Commun. 1993; 192(1):124–128. | |
Baracca A, Sgarbi G, Mattiazzi M, et al. Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993. Biochim Biophys Acta. 2007;1767(7):913–919. | |
Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. | |
Kucharczyk R, Zick M, Bietenhader M, et al. Mitochondrial ATP synthase disorders: molecular mechanisms and the quest for curative therapeutic approaches. Biochim Biophys Acta. 2009;1793(1):186–199. | |
Uziel G, Ghezzi D, Zeviani M. Infantile mitochondrial encephalopathy. Semin Fetal Neonatal Med. 2011;16(4):205–215. | |
Sgarbi G, Baracca A, Lenaz G, Valentino LM, Carelli V, Solaini G. Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA. Biochem J. 2006;395(3):493–500. | |
Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet. 1996;14(2):146–151. | |
Cao L, Shitara H, Sugimoto M, Hayashi J, Abe K, Yonekawa H. New evidence confirms that the mitochondrial bottleneck is generated without reduction of mitochondrial DNA content in early primordial germ cells of mice. PLoS Genet. 2009;5(12):e1000756. | |
Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem. 2006;281(43):32724–32727. | |
Hoefs SJG, Rodenburg RJ, Smeitink JA, van den Heuvel LP. Molecular base of biochemical complex I deficiency. Mitochondrion. 2012;12(5):520–532. | |
Loeffen JL, Smeitink JA, Trijbels JM, et al. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat. 2000;15(2):123–134. | |
Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003; 126(Pt 8):1905–1912. | |
García-Cazorla A, De Lonlay P, Nassogne MC, Rustin P, Touati G, Saudubray JM. Long-term follow-up of neonatal mitochondrial cytopathies: a study of 57 patients. Pediatrics. 2005;116(5):1170–1177. | |
Gibson K, Halliday JL, Kirby DM, Yaplito-Lee J, Thorburn DR, Boneh A. Mitochondrial oxidative phosphorylation disorders presenting in neonates: clinical manifestations and enzymatic and molecular diagnoses. Pediatrics. 2008;122(5):1003–1008. | |
Leng Y, Liu Y, Fang X, et al. The mitochondrial DNA 10197 G > A mutation causes MELAS/Leigh overlap syndrome presenting with acute auditory agnosia. Mitochondrial DNA. Epub April 8, 2014. | |
Negishi Y, Hattori A, Takeshita E, et al. Homoplasmy of a mitochondrial 3697G>A mutation causes Leigh syndrome. J Hum Genet. 2014;59(7):405–407. | |
Ma YY, Wu TF, Liu YP, et al. Genetic and biochemical findings in Chinese children with Leigh syndrome. J Clin Neurosci. 2013;20(11):1591–1594. | |
Finsterer J. Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol. 2008;39(4):223–235. | |
Brandt U. Energy converting NADH:quinone oxidoreductase (complex I). Annu Rev Biochem. 2006;75(1):69–92. | |
Loeffen J, Smeitink J, Triepels R, et al. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet. 1998;63(6):1598–1608. | |
Triepels RH, van den Heuvel LP, Loeffen JL, et al. Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann Neurol. 1999;45(6):787–790. | |
Howell N, Kubacka I, Smith R, Frerman F, Parks JK, Parker WD Jr. Association of the mitochondrial 8344 MERRF mutation with maternally inherited spinocerebellar degeneration and Leigh disease. Neurology. 1996;46(1):219–222. | |
Chalmers RM, Lamont PJ, Nelson I, et al. A mitochondrial DNA tRNA(Val) point mutation associated with adult-onset Leigh syndrome. Neurology. 1997;49(2):589–592. | |
Santorelli FM, Tanji K, Sano M, et al. Maternally inherited encephalopathy associated with a single-base insertion in the mitochondrial tRNATrp gene. Ann Neurol. 1997;42(2):256–260. | |
Piao YS, Tang GC, Yang H, Lu DH. Clinico-neuropathological study of a Chinese case of familial adult Leigh syndrome. Neuropathology. 2006;26(3):218–221. | |
Naess K, Freyer C, Bruhn H, et al. MtDNA mutations are a common cause of severe disease phenotypes in children with Leigh syndrome. Biochim Biophys Acta. 2009;1787(5):484–490. | |
Mkaouar-Rebai E, Chamkha I, Kammoun F, et al. Two new mutations in the MT-TW gene leading to the disruption of the secondary structure of the tRNA(Trp) in patients with Leigh syndrome. Mol Genet Metab. 2009;97(3):179–184. | |
Jiang YW, Qin J, Yuan Y, Qi Y, Wu XR. Neuropathologic and clinical features in eight Chinese patients with Leigh disease. J Child Neurol. 2002;17(6):450–452. | |
Saneto RP, Friedman SD, Shaw DW. Neuroimaging of mitochondrial disease. Mitochondrion. 2008;8(5–6):396–413. | |
Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R; Mitochondrial Medicine Society Clinical Directors Working Group; MMS Clinical Director’s Work Group. Practice patterns of mitochondrial disease physicians in North America. Part 1: diagnostic and clinical challenges. Mitochondrion. 2014;14(1):26–33. | |
Barkovich AJ, editor. Pediatric Neuroimaging. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:80–81. | |
Parikh S, Goldstein A, Koenig MK, et al; Clinical Director’s Work Group. Practice patterns of mitochondrial disease physicians in North America. Part 2: treatment, care and management. Mitochondrion. 2013;13(6):681–687. | |
Malfatti E, Bugiani M, Invernizzi F, et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain. 2007;130(Pt 7):1894–1904. | |
Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc. 2013;88(10):1127–1140. | |
Baertling F, Rodenburg RJ, Schaper J, et al. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry. 2014;85(3):257–265. | |
Martinelli D, Catteruccia M, Piemonte F, et al. EPI-743 reverses the progression of the pediatric mitochondrial disease--genetically defined Leigh Syndrome. Mol Genet Metab. 2012;107(3):383–388. | |
Sadun AA, Chicani CF, Ross-Cisneros FN, et al. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol. 2012;69(3):331–338. | |
Bar-Meir M, Elpeleg ON, Saada A. Effect of various agents on adenosine triphosphate synthesis in mitochondrial complex I deficiency. J Pediatr. 2001;139(6):868–870. | |
Pinard JM, Marsac C, Barkaoui E, et al. Syndrome de Leigh et leucodystrophie par déficit partiel en succinate déshydrogénase: régression sous riboflavine. [Leigh syndrome and leukodystrophy due to partial succinate dehydrogenase deficiency: regression with riboflavin]. Arch Pediatr. 1999;6(4):421–426. French. | |
Malojcic B, Brinar V, Poser C, Djakovic V. An adult case of Leigh disease. Clin Neurol Neurosurg. 2004;106(3):237–240. | |
Morán M, Marín-Buera L, Gil-Borlado MC, et al. Cellular pathophysiological consequences of BCS1L mutations in mitochondrial complex III enzyme deficiency. Hum Mutat. 2010;31(8):930–941. | |
Antonicka H, Ostergaard E, Sasarman F, et al. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet. 2010;87(1):115–122. | |
Calvo SE, Tucker EJ, Compton AG, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet. 2010;42(10):851–858. | |
Lenaz G, Baracca A, Carelli V, D’Aurelio M, Sgarbi G, Solaini G. Bioenergetics of mitochondrial diseases associated with mtDNA mutations. Biochim Biophys Acta. 2004;1658(1–2):89–94. | |
Levy RJ, Ríos PG, Akman HO, Sciacco M, Vivo DC, Dimauro S. Long Survival in Patients With Leigh Syndrome and the m.10191T>C Mutation in MT-ND3: A Case Report and Review of the Literature. J Child Neurol. Epub November 27, 2013. | |
Mitchell AL, Elson JL, Howell N, Taylor RW, Turnbull DM. Sequence variation in mitochondrial complex I genes: mutation or polymorphism? J Med Genet. 2006;43(2):175–179. | |
Kirby DM, Boneh A, Chow CW, et al. Low mutant load of mitochondrial DNA G13513A mutation can cause Leigh’s disease. Ann Neurol. 2003;54(4):473–478. | |
Kirby DM, Kahler SG, Freckmann ML, Reddihough D, Thorburn DR. Leigh disease caused by the mitochondrial DNA G14459A mutation in unrelated families. Ann Neurol. 2000;48(1):102–104. | |
Fernandez-Moreira D, Ugalde C, Smeets R, et al. X-linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann Neurol. 2007;61(1):73–83. | |
Hoefs SJ, Dieteren CE, Distelmaier F, et al. NDUFA2 complex I mutation leads to Leigh disease. Am J Hum Genet. 2008;82(6):1306–1315. | |
Pitceathly RD, Rahman S, Wedatilake Y, et al; UK10K Consortium. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013;3(6):1795–1805. | |
van den Bosch BJ, Gerards M, Sluiter W, et al. Defective NDUFA9 as a novel cause of neonatally fatal complex I disease. J Med Genet. 2012;49(1):10–15. | |
Hoefs SJ, van Spronsen FJ, Lenssen EW, et al. NDUFA10 mutations cause complex I deficiency in a patient with Leigh disease. Eur J Hum Genet. 2011;19(3):270–274. | |
Bénit P, Slama A, Cartault F, et al. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J Med Genet. 2004;41(1):14–17. | |
Ostergaard E, Schwartz M, Batbayli M, et al. A novel missense mutation in SUCLG1 associated with mitochondrial DNA depletion, encephalomyopathic form, with methylmalonic aciduria. Eur J Pediatr. 2010;169(2):201–205. | |
Hoefs SJ, Dieteren CE, Rodenburg RJ, et al. Baculovirus complementation restores a novel NDUFAF2 mutation causing complex I deficiency. Hum Mutat. 2009;30(7):E728–E736. | |
Bénit P, Chretien D, Kadhom N, et al. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet. 2001;68(6):1344–1352. | |
Martín MA, Blázquez A, Gutierrez-Solana LG, et al. Leigh syndrome associated with mitochondrial complex I deficiency due to a novel mutation in the NDUFS1 gene. Arch Neurol. 2005;62(4):659–661. | |
Tuppen HA, Hogan VE, He L, et al. The p. M292T NDUFS2 mutation causes complex I-deficient Leigh syndrome in multiple families. Brain. 2010;133(10):2952–2963. | |
Quintana A, Kruse SE, Kapur RP, Sanz E, Palmiter RD. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc Natl Acad Sci U S A. 2010;107(24):10996–11001. | |
Anderson SL, Chung WK, Frezzo J, et al. A novel mutation in NDUFS4 causes Leigh syndrome in an Ashkenazi Jewish family. J Inherit Metab Dis. 2008;31(S2)(Suppl 2):S461–S467. | |
Leshinsky-Silver E, Lebre AS, Minai L, et al. NDUFS4 mutations cause Leigh syndrome with predominant brainstem involvement. Mol Genet Metab. 2009;97(3):185–189. | |
Lebon S, Minai L, Chretien D, et al. A novel mutation of the NDUFS7 gene leads to activation of a cryptic exon and impaired assembly of mitochondrial complex I in a patient with Leigh syndrome. Mol Genet Metab. 2007;92(1–2):104–108. | |
Lebon S, Rodriguez D, Bridoux D, et al. A novel mutation in the human complex I NDUFS7 subunit associated with Leigh syndrome. Mol Genet Metab. 2007;90(4):379–382. | |
Marina AD, Schara U, Pyle A, et al. NDUFS8-related Complex I Deficiency Extends Phenotype from “PEO Plus” to Leigh Syndrome. JIMD Rep. 2013;10:17–22. | |
Lissens W, De Meirleir L, Seneca S, et al. Mutations in the X-linked pyruvate dehydrogenase (E1) alpha subunit gene (PDHA1) in patients with a pyruvate dehydrogenase complex deficiency. Hum Mutat. 2000;15(3):209–219. | |
Quintana E, Mayr JA, García Silva MT, et al. PDH E1β deficiency with novel mutations in two patients with Leigh syndrome. J Inherit Metab Dis. 2009;32(S1)(Suppl 1):S339–S343. | |
Schiff M, Miné M, Brivet M, et al. Leigh’s disease due to a new mutation in the PDHX gene. Ann Neurol. 2006;59(4):709–714. | |
Lim SC, Smith KR, Stroud DA, et al. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am J Hum Genet. 2014;94(2):209–222. | |
Joost K, Rodenburg R, Piirsoo A, van den Heuvel B, Zordania R, Ounap K. A novel mutation in the SCO2 gene in a neonate with early-onset cardioencephalomyopathy. Pediatr Neurol. 2010;42(3):227–230. | |
Pagnamenta AT, Hargreaves IP, Duncan AJ, et al. Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol Genet Metab. 2006;89(3):214–221. | |
Ohlenbusch A, Edvardson S, Skorpen J, et al. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J Rare Dis. 2012;7(1):69. | |
Ostergaard E, Hansen FJ, Sorensen N, et al. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130(Pt 3):853–861. | |
Weraarpachai W, Antonicka H, Sasarman F, et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat Genet. 2009;41(7):833–837. | |
Atwal PS. Mutations in the complex III assembly factor tetratricopeptide 19 gene TTC19 are a rare cause of Leigh syndrome. JIMD Reports. Berlin: Springer; 2013. | |
Barel O, Shorer Z, Flusser H, et al. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am J Hum Genet. 2008;82(5):1211–1216. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.