Back to Journals » The Application of Clinical Genetics » Volume 8
The genetics of Charcot–Marie–Tooth disease: current trends and future implications for diagnosis and management
Authors Hoyle JC, Isfort M, Roggenbuck J, Arnold WD
Received 21 May 2015
Accepted for publication 14 August 2015
Published 19 October 2015 Volume 2015:8 Pages 235—243
DOI https://doi.org/10.2147/TACG.S69969
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Martin Maurer
J Chad Hoyle,1 Michael C Isfort,1 Jennifer Roggenbuck,1,2 W David Arnold1,3,4
1Department of Neurology, Division of Neuromuscular Disorders, 2Department of Internal Medicine, Division of Human Genetics, 3Department of Physical Medicine and Rehabilitation, 4Department of Neuroscience, The Ohio State University Wexner Medical Center, Columbus, OH, USA
Abstract: Charcot–Marie–Tooth (CMT) disease is the most common hereditary polyneuropathy and is classically associated with an insidious onset of distal predominant motor and sensory loss, muscle wasting, and pes cavus. Other forms of hereditary neuropathy, including sensory predominant or motor predominant forms, are sometimes included in the general classification of CMT, but for the purpose of this review, we will focus primarily on the forms associated with both sensory and motor deficits. CMT has a great deal of genetic heterogeneity, leading to diagnostic considerations that are still rapidly evolving for this disorder. Clinical features, inheritance pattern, gene mutation frequencies, and electrodiagnostic features all are helpful in formulating targeted testing algorithms in practical clinical settings, but these still have shortcomings. Next-generation sequencing (NGS), combined with multigene testing panels, is increasing the sensitivity and efficiency of genetic testing and is quickly overtaking targeted testing strategies. Currently, multigene panel testing and NGS can be considered first-line in many circumstances, although obtaining initial targeted testing for the PMP22 duplication in CMT patients with demyelinating conduction velocities is still a reasonable strategy. As technology improves and cost continues to fall, targeted testing will be completely replaced by multigene NGS panels that can detect the full spectrum of CMT mutations. Nevertheless, clinical acumen is still necessary given the variants of uncertain significance encountered with NGS. Despite the current limitations, the genetic diagnosis of CMT is critical for accurate prognostication, genetic counseling, and in the future, specific targeted therapies. Although whole exome and whole genome sequencing strategies have the power to further elucidate the genetics of CMT, continued technological advances are needed.
Keywords: Charcot–Marie–Tooth disease, next-generation sequencing, neurogenetic testing, nerve conduction studies, neuropathy
Overview and clinical background
Charcot–Marie–Tooth (CMT) disease is a genetically and phenotypically heterogeneous group of disorders. Classically, CMT includes hereditary disorders associated with sensory and motor deficits of the peripheral nervous system, sometimes also referred to as hereditary motor and sensory neuropathy. Other variants, including hereditary sensory neuropathy (or hereditary sensory and autonomic neuropathy) and distal hereditary motor neuropathy (or distal spinal muscular atrophy), are also sometimes grouped under the general classification of CMT. These disorders are usually phenotypically distinct. Hereditary sensory neuropathy is usually sensory predominant and may be associated with autonomic dysfunction and skin ulcerations due to insensate skin.1,2 Conversely, distal hereditary motor neuropathy usually lacks or has minimal sensory involvement.3 In this review, our focus will be to provide an overview of the clinical presentation, laboratory workup, diagnosis, and management of the classic CMT phenotype of sensory and motor deficits.
The majority of patients with CMT have onset of symptoms in the first to second decade, although there is significant variability ranging from severe deficits in early childhood to only mild features in very late life.4 For instance, patients with the most common form, CMT1A, rarely require use of a wheelchair during their lifetime, but the most common axonal variant of CMT (CMT2A) has greater severity with most patients becoming nonambulatory at a young age, with 23 of 27 patients becoming nonambulatory before the age of 20 in one review of CMT2A.5–7 The symptoms and exam findings of CMT typically present insidiously and include distal predominant features of slowly progressive muscle weakness and wasting, most evident in the anterior leg and foot muscles, decreased reflexes, and vibratory sensory loss.4 Pes cavus (or high arched feet) and hammer toes are common, seen in approximately 70% of CMT patients.4 Scoliosis is less common but may occur in one-third to half of cases, usually with kyphoscoliosis.4,8–10 Hip dysplasia is another orthopedic complication that may occur.11 Although neuropathic pain is not a classic feature of CMT, moderate pain is noted by the majority of patients, and small nerve fiber function is frequently impaired in CMT1A patients.12–15 Soft tissue and joint-related pain are also significant sources of pain in patients with CMT.16 Sleep disturbances, including restless legs syndrome in 28% of patients, and muscle cramping may also be seen.17–19
Clinical presentation and examination
As is the case with hereditary neuromuscular conditions in general, the insidious onset and progression often make deficits less obvious to a patient subjectively compared with the examiner’s findings. For instance, overt vibratory sensation loss is often seen on examination in patients without sensory complaints. Similarly, profound distal leg weakness may not be noticed by patients until pointed out by a clinical examiner. Thus, historical clues should be sought by asking about functional activities at a younger age, such as whether the patient had difficulty playing sports, had frequent spraining of ankles, or had other functional limitations that in hindsight demonstrate an earlier age of onset than initially reported by the patient.20 This is a critical aspect of the clinical evaluation to ascertain the indolent onset of symptoms that are more suggestive of a slowly progressive hereditary etiology. Additionally, symptoms of symmetric significant distal weakness with polyneuropathy would not be expected to be encountered secondary to common causes of neuropathy such as glucose dysregulation or idiopathic axonal polyneuropathies, which are characteristically sensory predominant. In cases with notable distal motor involvement, acquired inflammatory etiologies, significant toxic exposures (such as lead or chemotherapeutic agents like vincristine) or certain vitamin deficiencies (such as thiamine, B12, or copper), and genetic causes become more relevant on the differential. Of those considerations, the slow onset and progression of symptoms, high arched feet and hammer toes, and family history of similar symptoms are then triggers to consider genetic causes at the top of the list.
Inheritance patterns and specific phenotypic features
Most types of CMT are inherited in an autosomal dominant manner, but X-linked and autosomal recessive forms also occur (Table 1). X-linked forms are distinguished by a lack of male-to-male transmission on pedigree analysis and have intermediate-to-demyelinating range conduction velocities (see “Diagnostic evaluation” section for conduction velocity segregation of subtypes). Autosomal recessive inheritance is less common, but should be considered in consanguineous families or those with multiple affected siblings and asymptomatic parents, or in simplex cases. Autosomal recessive forms are frequently characterized by a more severe phenotype with an earlier onset of symptoms. Importantly, de novo mutations may occur (10% of CMT1A cases as an example), and there are many forms of CMT that may show reduced penetrance.21
 | Table 1 Classification and genetics of CMT disease |
Specific phenotypic features may be suggestive of a particular type of CMT. Examples of this include tonic pupils in MPZ-related neuropathies; white matter lesions with transient focal symptoms or stroke-like presentations in X-linked CMT (GJB1); or vocal cord/diaphragm involvement in CMT2C (TRPV4).22–28 Additionally, optic atrophy in MFN2 and deafness in MPZ, PMP22, and GJB1 may be characteristic features.29–32 Generally speaking, however, CMT clinical presentations are remarkably similar, and there is not enough phenotypic discriminatory value to rely on clinical clues alone for diagnosis of genetic type. Additionally, within a particular type of CMT, there can be significant variation in clinical involvement and expression even with the same mutation.33
Hereditary neuropathy with liability to pressure palsies is a relatively common form of hereditary neuropathy which most commonly presents clinically with painless, recurrent focal neuropathies at sites of compression but may also present with a more generalized CMT-like presentation.34,35 Other rare hereditary causes of neuropathy also need to be considered in the differential diagnosis of CMT, but typically these have distinguishing features. Leukodystrophies (autosomal recessive or X-linked recessive) are associated with confluent white matter changes on magnetic resonance imaging of the brain. Familial amyloid neuropathy (autosomal dominant) may be associated with cardiomyopathy, autonomic dysfunction, neuropathic pain, superimposed carpal tunnel syndrome, or nephropathy. Fabry disease (X-linked, though females carriers may be symptomatic) should be considered in the context of a history of periodic pain crises in the limbs (acroparesthesias), angiokeratomas on the skin, unexplained renal disease, or unexplained stroke. Refsum disease (autosomal recessive) may be associated with retinitis pigmentosa, deafness, ataxia, and icthyosis (scaly skin). Tangier disease is distinguished by the presence of enlarged orange tonsils, a low HDL, or a syrinx-like pattern of sensory loss. Mitochondrial disorders may also present with polyneuropathy as part of a particular syndrome, such as neuropathy ataxia retinitis pigmentosa. Alternatively, a multisystem presentation with features such as diabetes, myopathy, ptosis, external ophthalmoplegia, sensorineural deafness, optic atrophy, pigmentary retinopathy, and short stature would also suggest a mitochondrial etiology (Table 2).
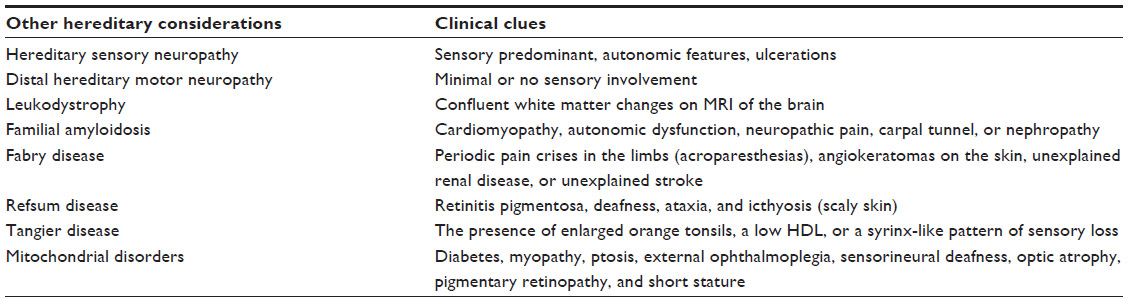 | Table 2 Other hereditary causes of neuropathy |
Diagnostic evaluation
Electrodiagnostic studies (EDx) are valuable clinical diagnostic tools for CMT. EDx can be used to confirm a clinically suspected sensorimotor polyneuropathy and exclude other possible mimicking conditions such as hereditary motor neuropathy or distal myopathy. Motor conduction velocities on nerve conduction study are particularly helpful to classify sensorimotor polyneuropathies into demyelinating versus axonal pathophysiological subtypes. A conduction velocity in an upper limb motor nerve in an adult of 38 m/s is utilized as the cutoff for differentiation of axonal (≥38 m/s) versus demyelinating (<38 m/s) disease.4 CMT1 designates demyelinating disease and CMT2, axonal. X-linked CMT typically has intermediate (defined as >35 and up to 45 m/s) to demyelinating range conduction velocity values.36 Similar to clinical features, EDx findings in autosomal recessive forms may often have more severe involvement.37
Features of demyelination on nerve conduction study in patients with CMT are usually uniform and symmetric without conduction blocks or temporal dispersion.38 Asymmetric involvement, temporal dispersion, and conduction block typically imply acquired diseases like chronic immune demyelinating polyneuropathy. This is confounded, though, by the observation that EDx findings mimicking acquired demyelinating disease may be seen occasionally, particularly in certain CMT subtypes, such as CMTX, CMT1B, CMT1C, and CMT4J.22,39–41 Additionally, CMT4J may also mimic the clinical presentation of acquired demyelination.39 Cerebrospinal fluid (CSF) analysis is frequently used in the evaluation of possible autoimmune forms of neuropathy, but CSF findings usually do not differentiate this well. In CMT1, the CSF protein is typically elevated, and in a quarter of cases is above 100 (mg per 100 mL).42 Peripheral nerve ultrasound usually reveals diffuse nerve enlargement in demyelinating forms of CMT, whereas acquired immune demyelinating polyneuropathies tend to have more patchy and multifocal enlargement of nerves.43–45 Informative genetic testing is particularly helpful in these cases to ultimately clarify the diagnosis and to avoid unnecessary, potentially toxic immunosuppressive treatments. Of note, hereditary neuropathy with liability to pressure palsies has characteristic electrophysiological features of not only focal neuropathies at entrapment sites, but also a background of disproportionately slow distal motor latencies and sensory conduction velocities with rather normal motor conduction velocities that are not measured across common sites of nerve compression.46
Genetic testing for CMT first became available after the discovery of the PMP22 duplication in 1991.47 This duplication remains the most common genetic etiology accounting for the majority of CMT1 cases and close to 40% of all CMT cases.36,48,49 The three next most common genetic forms of CMT are those caused by mutations in GJB1, MPZ, and MFN2, accounting for the majority of the additional yield of current genetic testing, even though there are more than 50 other known genetic forms of CMT (Table 3).48 With conventional testing methods, it has been estimated that approximately 60% of CMT cases can be genetically confirmed with higher rates of genetic confirmation for CMT1.36,48,49 The yield is lower for CMT2, where, in approximately one-third of cases, a molecular diagnosis is obtained.48,49
 | Table 3 Relative frequency of known mutations (Inherited Neuropathy Consortium 2014) |
Given the high cost of genetic testing historically using Sanger technology, algorithms were developed to help prioritize the most likely candidate gene(s) for testing.36 These algorithms take advantage of the small number of high-yield genes to test, along with contextualizing those gene associations with other differentiating features such as the clinical, EDx, and inheritance patterns to guide testing toward the highest yield genetic testing strategy (Table 4).
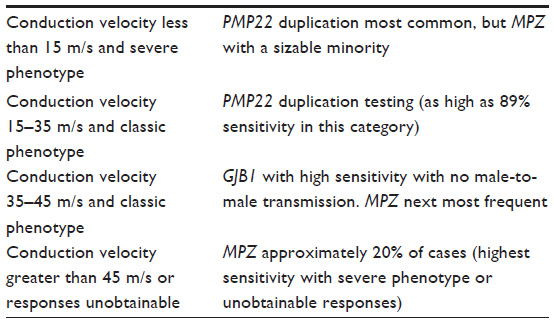 | Table 4 Targeted testing strategies |
Diagnostic algorithms for conventional targeted genetic testing can provide a reasonable bypass of low-yield testing despite the significant genetic heterogeneity of CMT. However, newer technology is offering cost-effective, higher yield alternatives. Next-generation sequencing (NGS) offers the advantage of sequencing vast amounts of genetic material in parallel and is transforming the approach to the genetic diagnosis of CMT.50 This strategy can be applied to a set of genes (panels), all protein-coding genetic material (the exome), or all genetic material (the genome). Of these options, targeted panel testing is currently the least expensive, has the highest per-gene technical performance, and offers the most straightforward result interpretation with fewer variants of unknown significance. Mutations in novel genes cannot be identified with panel testing, but for clinical testing programs, panel testing is rapidly gaining favor. In our opinion, NGS panel testing for CMT can now be reasonably considered as the first line for clinical testing, with the exception of initially testing for the PMP22 duplication in CMT1. NGS technology does not directly ascertain the presence of pathogenic duplications or deletions, but indirect methods such as relative read depth are increasingly being applied to identify duplications and deletions from NGS data.51 This means that a single NGS panel test has the potential to ascertain all known major mutation types, including PMP22 duplications and deletions. Until this technology fully matures, conventional methods to validate the presence of an abnormality of PMP22 should be obtained, whether that be an independent test or as an additional component of the panel testing.
We reviewed costs based on the currently available commercial genetic testing for CMT in the USA. There is wide variability in cost of molecular testing. For some commercial labs, the price of panel testing is much higher than individual gene testing, while other commercial labs have a “fixed price” cost that is the same for either full panel or individual gene testing. Additionally, this full panel testing can be commercially obtained with PMP22 validation testing included along with the NGS. Comparing across several options, the least expensive PMP22 duplication test option was approximately 45% of the cost of the least expensive available CMT full panel. The cost for other single gene tests (other than the PMP22 duplication testing) was not any less expensive than the cost of performing an NGS multigene panel based on current cost information. Taking GJB1 as an example, single gene testing has a high sensitivity (upwards of 78%) in pedigrees with no male-to-male transmission, a classic CMT phenotype, and intermediate conduction velocities on EDx testing.36 Nevertheless, considering that a multigene NGS panel can be obtained for the same cost, initial targeted testing is not advantageous. The next two most common genetic forms of CMT (MPZ- and MFN2-related) have lower yield with targeted testing and are also not individually less expensive than performing full panel NGS testing.36,48
If one assumes that all CMT1 patients with a negative PMP22 duplication on initial screening will go on to get panel testing as a second step, then we can evaluate the practicality of initial targeted testing of the PMP22 duplication versus including it as part of an initial multigene panel screen. Gess et al52 reported that 51% of a German CMT1 cohort tested positive for a PMP22 duplication, and an even lower percentage was reported in a Norwegian cohort by Ostern et al.53 Given that targeted initial testing for the PMP22 duplication is, again, approximately 45% of the cost of full panel testing, sensitivities of initial targeted testing of 51% or less suggest that even in CMT1 patients, there might be rather similar overall cost for that cohort in seeking initial PMP22 targeted testing versus starting with multipanel gene testing. On the other hand, a greater than 70% sensitivity for PMP22 duplication was noted by both Murphy et al49 in a UK population and Saporta et al36 in the USA. They found that up to 89% of CMT1 patients with a classic phenotype and a conduction velocity between 15 and 35 m/s tested positive for the PMP22 duplication.36 Taking into account these data, we believe it is still reasonable to proceed with targeted testing for the PMP22 duplication in CMT1 phenotypes (particularly the classic clinical cohort with conduction velocities between 15 and 35 m/s) initially. However, additional time cost for repeat testing in patients who test negative is another factor to consider. Continued cost reductions in price and continued technological advances of NGS panel testing (such as further validation of utilizing relative read depth of NGS data to identify PMP22 duplications and deletions) will likely further support multigene panel testing as the first line diagnostic strategy for CMT in general in the near future.
Whole exome or genome sequencing can be considered if panel testing is negative to further increase the yield of diagnosis, but clinical caution is necessary.54–57 Result interpretation and disclosure may be complicated by the identification of variants of uncertain significance, incidental findings, and other scenarios such as identification of misattributed parentage. Due to the complexity of this testing, whole exome and whole genome testing is best performed in consultation with a tertiary referral to an inherited neuropathy clinic that includes a genetic counselor or medical geneticist. A significant limitation of whole exome and genome testing currently is that it does not detect small tandem repeats, other copy number variations, mutations in regulatory sequences distant from a gene, epigenetic changes, or mutations in mitochondrial DNA. As such, this technology does not yet have the capability to provide genetic diagnosis in all cases of CMT.58 With large multigene panels, whole exome, and whole genome testing for CMT, it is critical that clinicians are able to determine the relevance of molecular findings in the setting of the patient’s clinical phenotype. Additionally, the scope and complexity of test results mean that pre- and posttest genetic counseling is increasingly critical.
Management
Treatment for patients with CMT is essentially supportive at this point. Ankle foot orthoses can improve walking economy in patients with significant ankle dorsiflexion weakness.59–63 Thumb opposition splints have been shown to improve hand function in those with more severe intrinsic hand muscle weakness.64 Scoliosis typically does not require treatment, but referral to an orthopedic surgeon may be required in significant cases.8 There is very limited data on stabilization surgeries for foot deformities foot drop, and altered mechanics around the ankle, and invasive procedures should be considered with caution.65–68 Nevertheless, surgical intervention may be very helpful in select cases when performed by experienced surgeons. Although data is limited, exercise appears to be a safe therapeutic intervention. There have been variable reports on whether overwork weakness exists in CMT, but the majority of data argue against any detrimental effects and resistance training appears to correlate with some benefit.69–74
The genetic defects and associated pathophysiology of various forms of CMT may increase a patient’s susceptibility to toxicity in the context of concomitant illness or toxin exposure.6,75–77 Logically, neurotoxic medications should be avoided whenever possible in CMT patients.78 Rapid worsening and clinical decompensation of CMT may occur following exposure to toxic medications, and a prime example is vincristine in the setting of CMT1A which can lead to profound worsening, but exacerbation may also be noted in other forms of CMT.79–84
There are no established disease-modifying therapies to date. High doses of ascorbic acid demonstrated positive animal model data by downregulating PMP22 production, but human trials were disappointing and did not demonstrate clinically significant improvements.85–90 Animal studies also have shown some promise with progesterone antagonism to decrease PMP22 production.91,92 Also, curcumin in CMT1B improved Schwann cell differentiation and alleviated endoplasmic reticulum stress as part of reducing the activation of the unfolded protein response.93,94 Histone deacetylase (HDAC) 6 inhibition demonstrated positive results in an animal model of CMT2F (HSPB1) with an increase of α-tubulin acetylation correcting axonal transport defects.95 An exploratory human clinical study has shown some recent promise for a combination of baclofen, naltrexone, and sorbitol (PXT3003), showing early evidence for consistent improvement beyond stabilization.96 This study, built on preclinical experiments, shows pleiotropic mechanisms for downregulating PMP22.97 Research has most commonly focused on altering mechanisms unique to an individual genetic subtype of CMT (with CMT1A garnering the most research). This underscores the importance of continued improvement in genetic diagnostic strategies to provide personalized medicine for individual genetic subtypes and patients with CMT. Some avenues for more generalized disease modification are being sought after, as well. Neurotrophin 3 was tested both on animals and in a small human pilot study.98,99 This neurotrophic growth factor showed trends for improved axonal regeneration and needs further follow-up. Additionally, there are theories that multiple subtypes of CMT2 have shared endpoint pathology, such as with axonal transport. HDAC inhibitors could then potentially translate to treatment of multiple subtypes.100 Regardless, the future with better genetic diagnostic options and understanding will lead to multiple new avenues for potential treatments.
Conclusion
CMT is a disorder with a significant amount of genetic heterogeneity. Improvements are being made in the ability to provide cost-effective large panel testing through NGS. This technology is changing diagnostic algorithms and paradigms for molecular testing. Further genetic technological advances are still needed to diagnose the remaining unknown mutations in this disorder. Disease-modifying therapies specific to the varied underlying genetic abnormalities and pathophysiology of CMT will be the next crucial step forward for clinical impact.
Acknowledgment
WDA receives funding from a career development award through NIH-NICHD (5K12HD001097-17).
Disclosure
The authors report no conflicts of interest in this work.
References
Mroczek M, Kabzinska D, Kochanski A. Molecular pathogenesis, experimental therapy and genetic counseling in hereditary sensory neuropathies. Acta Neurobiol Exp (Wars). 2015;75(2):126–143. | |
Rotthier A, Baets J, Timmerman V, Janssens K. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat Rev Neurol. 2012;8(2):73–85. | |
Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatr. 2012;83(1):6–14. | |
Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103(2):259–280. | |
Feely SM, Laura M, Siskind CE, et al. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology. 2011;76(20):1690–1696. | |
Sheth S, Francies K, Siskind CE, Feely SM, Lewis RA, Shy ME. Diabetes mellitus exacerbates motor and sensory impairment in CMT1A. J Peripher Nerv Syst. 2008;13(4):299–304. | |
Verhamme C, van Schaik IN, Koelman JH, de Haan RJ, de Visser M. The natural history of Charcot-Marie-Tooth type 1A in adults: a 5-year follow-up study. Brain. 2009;132(Pt 12):3252–3262. | |
Karol LA, Elerson E. Scoliosis in patients with Charcot-Marie-Tooth disease. J Bone Joint Surg Am. 2007;89(7):1504–1510. | |
Horacek O, Mazanec R, Morris CE, Kobesova A. Spinal deformities in hereditary motor and sensory neuropathy: a retrospective qualitative, quantitative, genotypical, and familial analysis of 175 patients. Spine. 2007;32(22):2502–2508. | |
Walker JL, Nelson KR, Stevens DB, Lubicky JP, Ogden JA, VandenBrink KD. Spinal deformity in Charcot-Marie-Tooth disease. Spine. 1994;19(9):1044–1047. | |
Novais EN, Bixby SD, Rennick J, Carry PM, Kim YJ, Millis MB. Hip dysplasia is more severe in Charcot-Marie-Tooth disease than in developmental dysplasia of the hip. Clin Orthop Relat Res. 2014;472(2):665–673. | |
Carter GT, Jensen MP, Galer BS, et al. Neuropathic pain in Charcot-Marie-Tooth disease. Arch Phys Med Rehabil. 1998;79(12):1560–1564. | |
Ramchandren S, Jaiswal M, Feldman E, Shy M. Effect of pain in pediatric inherited neuropathies. Neurology. 2014;82(9):793–797. | |
Ribiere C, Bernardin M, Sacconi S, et al. Pain assessment in Charcot-Marie-Tooth (CMT) disease. Ann Phys Rehabil Med. 2012;55(3):160–173. | |
Zambelis T. Small fiber neuropathy in Charcot-Marie-Tooth disease. Acta Neurol Belg. 2009;109(4):294–297. | |
Jeong NY, Shin YH, Jung J. Neuropathic pain in hereditary peripheral neuropathy. J Exerc Rehabil. 2013;9(4):397–399. | |
Boentert M, Knop K, Schuhmacher C, Gess B, Okegwo A, Young P. Sleep disorders in Charcot-Marie-Tooth disease type 1. J Neurol Neurosurg Psychiatr. 2014;85(3):319–325. | |
Luigetti M, Del Grande A, Testani E, et al. Restless leg syndrome in different types of demyelinating neuropathies: a single-center pilot study. J Clin Sleep Med. 2013;9(9):945–949. | |
Johnson NE, Sowden J, Dilek N, et al. Prospective study of muscle cramps in Charcot-Marie-Tooth disease. Muscle Nerve. 2015; 51(4):485–488. | |
Arnold WD, Flanigan KM. A practical approach to molecular diagnostic testing in neuromuscular diseases. Phys Med Rehabil Clin N Am. 2012;23(3):589–608. | |
Blair IP, Nash J, Gordon MJ, Nicholson GA. Prevalence and origin of de novo duplications in Charcot-Marie-Tooth disease type 1A: first report of a de novo duplication with a maternal origin. Am J Hum Genet. 1996;58(3):472–476. | |
Murphy SM, Laura M, Blake J, Polke J, Bremner F, Reilly MM. Conduction block and tonic pupils in Charcot-Marie-Tooth disease caused by a myelin protein zero p.Ile112Thr mutation. Neuromuscul Disord. 2011;21(3):223–226. | |
Anand G, Maheshwari N, Roberts D, et al. X-linked hereditary motor sensory neuropathy (type 1) presenting with a stroke-like episode. Dev Med Child Neurol. 2010;52(7):677–679. | |
Sagnelli A, Piscosquito G, Chiapparini L, et al. X-linked Charcot-Marie-Tooth type 1: stroke-like presentation of a novel GJB1 mutation. J Peripher Nerv Syst. 2014;19(2):183–186. | |
McKinney JL, De Los Reyes EC, Lo WD, Flanigan KM. Recurrent central nervous system white matter changes in Charcot-Marie-tooth type X disease. Muscle Nerve. 2014;49(3):451–454. | |
Echaniz-Laguna A, Dubourg O, Carlier P, et al. Phenotypic spectrum and incidence of TRPV4 mutations in patients with inherited axonal neuropathy. Neurology. 2014;82(21):1919–1926. | |
Landoure G, Sullivan JM, Johnson JO, et al. Exome sequencing identifies a novel TRPV4 mutation in a CMT2C family. Neurology. 2012;79(2):192–194. | |
Santoro L, Manganelli F, Di Maio L, et al. Charcot-Marie-Tooth disease type 2C: a distinct genetic entity. Clinical and molecular characterization of the first European family. Neuromuscul Disord. 2002;12(4):399–404. | |
Bombelli F, Stojkovic T, Dubourg O, et al. Charcot-Marie-Tooth disease type 2A: from typical to rare phenotypic and genotypic features. JAMA Neurol. 2014;71(8):1036–1042. | |
De Jonghe P, Timmerman V, Ceuterick C, et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain. 1999;122(Pt 2):281–290. | |
Takashima H, Nakagawa M, Umehara F, et al. Gap junction protein beta 1 (GJB1) mutations and central nervous system symptoms in X-linked Charcot-Marie-Tooth disease. Acta Neurol Scand. 2003;107(1):31–37. | |
Huehne K, Benes V, Thiel C, et al. Novel mutations in the Charcot-Marie-Tooth disease genes PMP22, MPZ, and GJB1. Hum Mutat. 2003;21(1):100. | |
Mathis S, Corcia P, Tazir M, et al. Peripheral myelin protein 22 gene duplication with atypical presentations: a new example of the wide spectrum of Charcot-Marie-Tooth 1A disease. Neuromuscul Disord. 2014;24(6):524–528. | |
Meier C, Moll C. Hereditary neuropathy with liability to pressure palsies. Report of two families and review of the literature. J Neurol. 1982;228(2):73–95. | |
Pareyson D, Scaioli V, Taroni F, et al. Phenotypic heterogeneity in hereditary neuropathy with liability to pressure palsies associated with chromosome 17p11.2–12 deletion. Neurology. 1996;46(4):1133–1137. | |
Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol. 2011;69(1):22–33. | |
Tazir M, Bellatache M, Nouioua S, Vallat JM. Autosomal recessive Charcot-Marie-Tooth disease: from genes to phenotypes. J Peripher Nerv Syst. 2013;18(2):113–129. | |
Kaku DA, Parry GJ, Malamut R, Lupski JR, Garcia CA. Uniform slowing of conduction velocities in Charcot-Marie-Tooth polyneuropathy type 1. Neurology. 1993;43(12):2664–2667. | |
Cottenie E, Menezes MP, Rossor AM, et al. Rapidly progressive asymmetrical weakness in Charcot-Marie-Tooth disease type 4J resembles chronic inflammatory demyelinating polyneuropathy. Neuromuscul Disord. 2013;23(5):399–403. | |
Michell AW, Laura M, Blake J, et al. GJB1 gene mutations in suspected inflammatory demyelinating neuropathies not responding to treatment. J Neurol Neurosurg Psychiatr. 2009;80(6):699–700. | |
Gerding WM, Koetting J, Epplen JT, Neusch C. Hereditary motor and sensory neuropathy caused by a novel mutation in LITAF. Neuromuscul Disord. 2009;19(10):701–703. | |
Bouche P, Gherardi R, Cathala HP, Lhermitte F, Castaigne P. Peroneal muscular atrophy. Part 1. Clinical and electrophysiological study. J Neurol Sci. 1983;61(3):389–399. | |
Zaidman CM, Harms MB, Pestronk A. Ultrasound of inherited vs acquired demyelinating polyneuropathies. J Neurol. 2013;260(12):3115–3121. | |
Schreiber S, Oldag A, Kornblum C, et al. Sonography of the median nerve in CMT1A, CMT2A, CMTX, and HNPP. Muscle Nerve. 2013; 47(3):385–395. | |
Sugimoto T, Ochi K, Hosomi N, et al. Ultrasonographic nerve enlargement of the median and ulnar nerves and the cervical nerve roots in patients with demyelinating Charcot-Marie-Tooth disease: distinction from patients with chronic inflammatory demyelinating polyneuropathy. J Neurol. 2013;260(10):2580–2587. | |
Andersson PB, Yuen E, Parko K, So YT. Electrodiagnostic features of hereditary neuropathy with liability to pressure palsies. Neurology. 2000;54(1):40–44. | |
Roa BB, Garcia CA, Lupski JR. Charcot-Marie-Tooth disease type 1A: molecular mechanisms of gene dosage and point mutation underlying a common inherited peripheral neuropathy. Int J Neurol. 1991;25–26: 97–107. | |
Fridman V, Bundy B, Reilly MM, et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry. 2015;86(8):873–878. | |
Murphy SM, Laura M, Fawcett K, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatr. 2012;83(7):706–710. | |
Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol. 2013;9(10):562–571. | |
Ankala A, da Silva C, Gualandi F, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol. 2015;77(2):206–214. | |
Gess B, Schirmacher A, Boentert M, Young P. Charcot-Marie-Tooth disease: frequency of genetic subtypes in a German neuromuscular center population. Neuromuscul Disord. 2013;23(8):647–651. | |
Ostern R, Fagerheim T, Hjellnes H, Nygard B, Mellgren SI, Nilssen O. Diagnostic laboratory testing for Charcot Marie Tooth disease (CMT): the spectrum of gene defects in Norwegian patients with CMT and its implications for future genetic test strategies. BMC Med Genet. 2013;14:94. | |
Choi BO, Koo SK, Park MH, et al. Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth disease. Hum Mutat. 2012;33(11):1610–1615. | |
Daud D, Griffin H, Douroudis K, et al. Whole exome sequencing and the clinician: we need clinical skills and functional validation in variant filtering. J Neurol. 2015;262(7):1673–1677. | |
Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362(13):1181–1191. | |
Menezes MP, Waddell L, Lenk GM, et al. Whole exome sequencing identifies three recessive FIG4 mutations in an apparently dominant pedigree with Charcot-Marie-Tooth disease. Neuromuscul Disord. 2014;24(8):666–670. | |
Hoyer H, Braathen GJ, Busk OL, et al. Genetic diagnosis of Charcot-Marie-Tooth disease in a population by next-generation sequencing. Biomed Res Int. 2014;2014:210401. | |
Menotti F, Laudani L, Damiani A, Mignogna T, Macaluso A. An anterior ankle-foot orthosis improves walking economy in Charcot-Marie-Tooth type 1A patients. Prosthet Orthot Int. 2014;38(5):387–392. | |
Dufek JS, Neumann ES, Hawkins MC, O’Toole B. Functional and dynamic response characteristics of a custom composite ankle foot orthosis for Charcot-Marie-Tooth patients. Gait Posture. 2014;39(1):308–313. | |
Ramdharry GM, Day BL, Reilly MM, Marsden JF. Foot drop splints improve proximal as well as distal leg control during gait in Charcot-Marie-Tooth disease. Muscle Nerve. 2012;46(4):512–519. | |
Phillips MF, Robertson Z, Killen B, White B. A pilot study of a crossover trial with randomized use of ankle-foot orthoses for people with Charcot-Marie-Tooth disease. Clin Rehabil. 2012;26(6):534–544. | |
Guillebastre B, Calmels P, Rougier PR. Assessment of appropriate ankle-foot orthoses models for patients with Charcot-Marie-Tooth disease. Am J Phys Med Rehabil. 2011;90(8):619–627. | |
Videler A, Eijffinger E, Nollet F, Beelen A. A thumb opposition splint to improve manual dexterity and upper-limb functioning in Charcot-Marie-Tooth disease. J Rehabil Med. 2012;44(3):249–253. | |
Wetmore RS, Drennan JC. Long-term results of triple arthrodesis in Charcot-Marie-Tooth disease. J Bone Joint Surg Am. 1989;71(3):417–422. | |
Ortiz C, Wagner E. Tendon transfers in cavovarus foot. Foot Ankle Clin. 2014;19(1):49–58. | |
Boffeli TJ, Tabatt JA. Minimally invasive early operative treatment of progressive foot and ankle deformity associated with Charcot-Marie-Tooth disease. J Foot Ankle Surg. 2015;54(4):701–708. | |
Dreher T, Wolf SI, Heitzmann D, Fremd C, Klotz MC, Wenz W. Tibialis posterior tendon transfer corrects the foot drop component of cavovarus foot deformity in Charcot-Marie-Tooth disease. J Bone Joint Surg Am. 2014;96(6):456–462. | |
Piscosquito G, Reilly MM, Schenone A, et al. Is overwork weakness relevant in Charcot-Marie-Tooth disease? J Neurol Neurosurg Psychiatr. 2014;85(12):1354–1358. | |
Chetlin RD, Gutmann L, Tarnopolsky M, Ullrich IH, Yeater RA. Resistance training effectiveness in patients with Charcot-Marie-Tooth disease: recommendations for exercise prescription. Arch Phys Med Rehabil. 2004;85(8):1217–1223. | |
Burns J, Raymond J, Ouvrier R. Feasibility of foot and ankle strength training in childhood Charcot-Marie-Tooth disease. Neuromuscul Disord. 2009;19(12):818–821. | |
Vinci P, Esposito C, Perelli SL, Antenor JA, Thomas FP. Overwork weakness in Charcot-Marie-Tooth disease. Arch Phys Med Rehabil. 2003;84(6):825–827. | |
Lindeman E, Leffers P, Spaans F, et al. Strength training in patients with myotonic dystrophy and hereditary motor and sensory neuropathy: a randomized clinical trial. Arch Phys Med Rehabil. 1995;76(7):612–620. | |
Maggi G, Monti Bragadin M, Padua L, et al. Outcome measures and rehabilitation treatment in patients affected by Charcot-Marie-Tooth neuropathy: a pilot study. Am J Phys Med Rehabil. 2011;90(8):628–637. | |
Fonkem E, Skordilis MA, Binkley EM, et al. Ethambutol toxicity exacerbating the phenotype of CMT2A2. Muscle Nerve. 2013;48(1):140–144. | |
Beutler AS, Kulkarni AA, Kanwar R, et al. Sequencing of Charcot-Marie-Tooth disease genes in a toxic polyneuropathy. Ann Neurol. 2014;76(5):727–737. | |
Ursino G, Alberti MA, Grandis M, et al. Influence of comorbidities on the phenotype of patients affected by Charcot-Marie-Tooth neuropathy type 1A. Neuromuscul Disord. 2013;23(11):902–906. | |
Weimer LH, Podwall D. Medication-induced exacerbation of neuropathy in Charcot Marie Tooth disease. J Neurol Sci. 2006;242(1–2):47–54. | |
Nakamura T, Hashiguchi A, Suzuki S, Uozumi K, Tokunaga S, Takashima H. Vincristine exacerbates asymptomatic Charcot-Marie-Tooth disease with a novel EGR2 mutation. Neurogenetics. 2012;13(1):77–82. | |
Hogan-Dann CM, Fellmeth WG, McGuire SA, Kiley VA. Polyneuropathy following vincristine therapy in two patients with Charcot-Marie-Tooth syndrome. JAMA. 1984;252(20):2862–2863. | |
Cil T, Altintas A, Tamam Y, Battaloglu E, Isikdogan A. Low dose vincristine-induced severe polyneuropathy in a Hodgkin lymphoma patient: a case report (vincristine-induced severe polyneuropathy). J Pediatr Hematol Oncol. 2009;31(10):787–789. | |
Porter CC, Carver AE, Albano EA. Vincristine induced peripheral neuropathy potentiated by voriconazole in a patient with previously undiagnosed CMT1X. Pediatr Blood Cancer. 2009;52(2):298–300. | |
Nishikawa T, Kawakami K, Kumamoto T, et al. Severe neurotoxicities in a case of Charcot-Marie-Tooth disease type 2 caused by vincristine for acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2008;30(7):519–521. | |
Moudgil SS, Riggs JE. Fulminant peripheral neuropathy with severe quadriparesis associated with vincristine therapy. Ann Pharmacother. 2000;34(10):1136–1138. | |
Lewis RA, McDermott MP, Herrmann DN, et al. High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: results of a randomized, double-masked, controlled trial. JAMA Neurol. 2013;70(8):981–987. | |
Micallef J, Attarian S, Dubourg O, et al. Effect of ascorbic acid in patients with Charcot-Marie-Tooth disease type 1A: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2009;8(12):1103–1110. | |
Pareyson D, Reilly MM, Schenone A, et al. Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): a double-blind randomised trial. Lancet Neurol. 2011;10(4):320–328. | |
Gess B, Rohr D, Young P. Ascorbic acid and sodium-dependent vitamin C transporters in the peripheral nervous system: from basic science to clinical trials. Antioxid Redox Signal. 2013;19(17):2105–2114. | |
Passage E, Norreel JC, Noack-Fraissignes P, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10(4):396–401. | |
Burns J, Ouvrier RA, Yiu EM, Ryan MM. Extended treatment of childhood Charcot-Marie-Tooth disease with high-dose ascorbic acid. J Peripher Nerv Syst. 2011;16(3):272–274. | |
Meyer zu Horste G, Prukop T, Liebetanz D, Mobius W, Nave KA, Sereda MW. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann Neurol. 2007;61(1):61–72. | |
Sereda MW, Meyer zu Horste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med. 2003;9(12):1533–1537. | |
Patzko A, Bai Y, Saporta MA, et al. Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice. Brain. 2012;135(Pt 12):3551–3566. | |
Bai Y, Patzko A, Shy ME. Unfolded protein response, treatment and CMT1B. Rare Dis. 2013;1:e24049. | |
d’Ydewalle C, Krishnan J, Chiheb DM, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17(8):968–974. | |
Attarian S, Vallat JM, Magy L, et al. An exploratory randomised double-blind and placebo-controlled phase 2 study of a combination of baclofen, naltrexone and sorbitol (PXT3003) in patients with Charcot-Marie-Tooth disease type 1A. Orphanet J Rare Dis. 2014;9:199. | |
Chumakov I, Milet A, Cholet N, et al. Polytherapy with a combination of three repurposed drugs (PXT3003) down-regulates Pmp22 over-expression and improves myelination, axonal and functional parameters in models of CMT1A neuropathy. Orphanet J Rare Dis. 2014;9:201. | |
Sahenk Z, Nagaraja HN, McCracken BS, et al. NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology. 2005;65(5):681–689. | |
Sahenk Z, Galloway G, Clark KR, et al. AAV1.NT-3 gene therapy for Charcot-Marie-Tooth neuropathy. Mol Ther. 2014;22(3):511–521. | |
d’Ydewalle C, Benoy V, Van Den Bosch L. Charcot-Marie-Tooth disease: emerging mechanisms and therapies. Int J Biochem Cell Biol. 2012;44(8):1299–1304. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.