Back to Journals » Clinical Interventions in Aging » Volume 9
The genetics of Alzheimer's disease
Authors Bagyinszky E, Youn YC ![]() , An SSA
, An SSA ![]() , Kim S
, Kim S ![]()
Received 15 July 2013
Accepted for publication 9 August 2013
Published 1 April 2014 Volume 2014:9 Pages 535—551
DOI https://doi.org/10.2147/CIA.S51571
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Eva Bagyinszky,1 Young Chul Youn,2 Seong Soo A An,1,* SangYun Kim3,*
1Department of BioNano Technology Gachon University, Gyeonggi-do, 2Department of Neurology, Chung-Ang University College of Medicine, Seoul, 3Department of Neurology, Seoul National University Budang Hospital, Gyeonggi-do, South Korea
*These authors contributed equally to this work
Abstract: Alzheimer's disease (AD) is a complex and heterogeneous neurodegenerative disorder, classified as either early onset (under 65 years of age), or late onset (over 65 years of age). Three main genes are involved in early onset AD: amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). The apolipoprotein E (APOE) E4 allele has been found to be a main risk factor for late-onset Alzheimer's disease. Additionally, genome-wide association studies (GWASs) have identified several genes that might be potential risk factors for AD, including clusterin (CLU), complement receptor 1 (CR1), phosphatidylinositol binding clathrin assembly protein (PICALM), and sortilin-related receptor (SORL1). Recent studies have discovered additional novel genes that might be involved in late-onset AD, such as triggering receptor expressed on myeloid cells 2 (TREM2) and cluster of differentiation 33 (CD33). Identification of new AD-related genes is important for better understanding of the pathomechanisms leading to neurodegeneration. Since the differential diagnoses of neurodegenerative disorders are difficult, especially in the early stages, genetic testing is essential for diagnostic processes. Next-generation sequencing studies have been successfully used for detecting mutations, monitoring the epigenetic changes, and analyzing transcriptomes. These studies may be a promising approach toward understanding the complete genetic mechanisms of diverse genetic disorders such as AD.
Keywords: dementia, amyloid precursor protein, presenilin 1, presenilin 2, APOE, mutation, diagnosis, genetic testing
Introduction
Alzheimer’s disease (AD) is a complex and heterogeneous neurodegenerative disorder. Several genetic and environmental factors and gene interactions may be involved in the disease’s occurrence and progression.1 Experiments have been performed with mono- and dizygotic twins to estimate the role of genetics in AD, the environmental influences, and the disease heritability. Variation in age of onset, neuropathological patterns, and disease duration may be possible due to genetic–environmental interactions.2–4 AD can be categorized into two subtypes: early onset and late onset. As a polygenic disorder, several additional genes might be potential risk factors for AD. Many single-nucleotide polymorphisms (SNPs) have been identified and confirmed to be associated with AD. The majority of recent studies in the genetics of AD have focused on the identification of novel risk-factor genes and mutations.2,5,6
Early onset Alzheimer’s disease
Occurrence of familial Alzheimer’s disease (FAD) represents the minority (5%–10%) of all AD cases. Familial early onset Alzheimer’s disease (EOAD) can be characterized by the Mendelian inheritance pattern; however, EOAD patients have also been reported without any family history (termed “sporadic EOAD”). Three genes are considered the main risk factors for EOAD: amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2; Figure 1). Mutations in these genes might result in alteration of amyloid beta (Abeta) production (both Abeta 40 and Abeta 42), leading to apoptosis of the neurons and dementia.6–9 Figure 2 presents a timeline of AD onset according to age.5,10
 | Figure 1 The amyloid precursor protein (APP), presenilin (PSEN) 1, and PSEN2 genes involved in early onset Alzheimer’s disease (AD). |
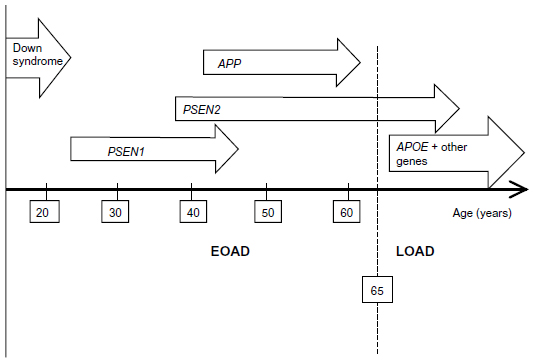 | Figure 2 The age onset of Alzheimer’s disease (AD), depending on the different involvement of genes. The symptoms of dementia can occur at an earlier age in Down syndrome patients than in AD patients without trisomy. |
The APP gene is located on chromosome 21. Triplication of chromosome 21 results in the triplication of the APP gene, which might enhance APP expression and Abeta accumulation. Down syndrome patients have been reported to develop AD pathology (deposition of senile plaques and neurofibrillary tangles) earlier than those without Down syndrome.11 These findings suggest that overexpression of APP might be related to AD pathology. The APP gene contains 19 exons for encoding the APP protein. The Abeta peptide is encoded by exons 16 and 17. Following transcription and alternative splicing, at least five isoforms of APP protein were identified, which contain the Abeta peptide sequence.12 However, APP seems to be a very rare risk factor for AD, as 21 and three mutations were described at exon 17 and 16, respectively. Most of the pathogenic APP mutations were located near the cleavage sites of alpha, beta, and gamma secretase enzymes, which suggests they might be involved in the onset of AD through altering the proteolysis of the Abeta peptide.13,14 N-terminal mutations in the Abeta sequence can affect the endosomal/lysosomal cleavage of Abeta, and might alter the beta secretase cleavages.12,15 Mutations near the cleavage site of alpha secretase (Glu693Lys, Glu693Gly, Glu693del, Asp694Asn) might change the processing of APP, in enhancing the proteolytic resistance of Abeta peptide.16,17 De Jonghe et al studied the APP mutations near the gamma secretase cleavage site.13 Missense mutations at codon 714-715 of APP decreased the secretion of Abeta 40, and the mutations at codon 716-717 increased the production and secretion of Abeta 42. This study suggests that gamma secretase cleavage might increase the ratio of Abeta 42 to Abeta 40.10–13,18
Linkage analyses (1996) identified two highly homologous genes – PSEN1 and PSEN2 – that might be involved in the onset of AD.19,20 The structures of PSEN1 and PSEN2 are similar, with a homology of 67%. Both of them contain 12 exons with ten coding exons (exons 3–12) for a protein of ~450 amino acids. Presenilin 1 (PS1) and presenilin 2 (PS2) proteins are transmembrane (TM) proteins with at least seven TM domains.19 The function of presenilins was first described by Wolfe et al, who proposed that two transmembrane aspartate (257 and 385) residues in PS1 are critical in gamma secretase activity.20 Most AD risk-factor mutations have been detected in PSEN1 (approximately 30%–70% of early onset FAD), which is located on chromosome 14. More than 180 mutations were found in PSEN1 in association with FAD, but they might be involved in sporadic AD or LOAD.14 Patients with PSEN1 mutations might develop AD symptoms in their 40s or early 50s, with a few cases occurring in persons in their late 30s and early 60s. Several missense mutations in PSEN1 can increase the production of Abeta 42 and 40. In an alternative mechanism, the levels of Abeta 42 and Abeta 40 might be increased and decreased, respectively.21
PSEN2, on chromosome 1, is another risk-factor gene for AD, especially EOAD among a very small European population. The most well-known group with dementia from PSEN2 mutation is families with Volga German ancestry. AD arising from PSEN2 mutations can be highly variable, and may occur between the ages of 40 and 75 years.5,21,22 The first PSEN2 mutation in AD patients was described in 1995.5,23–25 Patients with PSEN2 mutation have not been reported in Korea, the People’s Republic of China, or Japan, but silent mutations have been detected in Japan.26 A few PSEN2 mutations, such as Leu143His or Arg143His, have not been associated with any neurodegenerative phenotype.27 Two PSEN2 mutations, Arg62His and Arg71Trp, may be involved in breast cancer, although the pathomechanism is not clear.28 Table 1 summarizes all mutations described in APP, PSEN1, and PSEN2 genes that may be involved in AD progression.
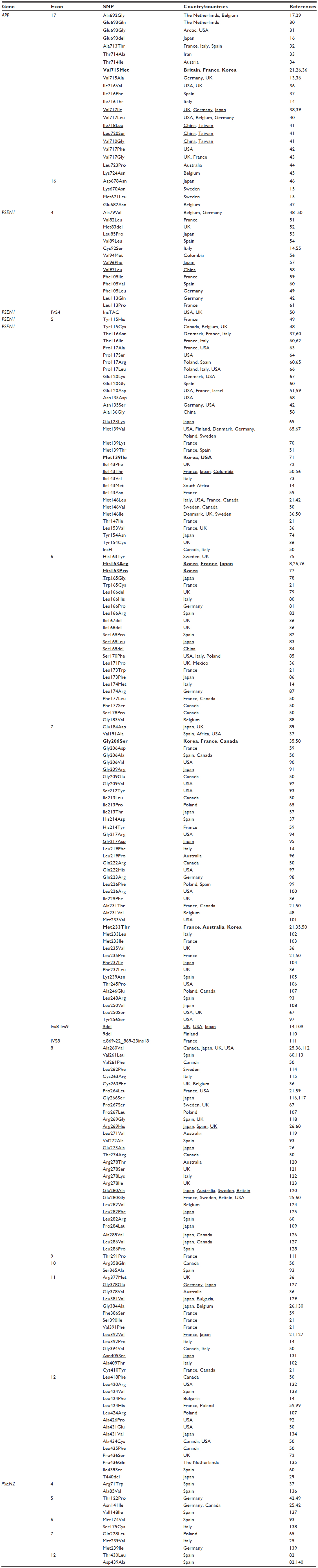 | Table 1 The known Alzheimer’s disease risk-factor mutations in APP and PSEN1–2 |
Late-onset Alzheimer’s disease
In late-onset Alzheimer’s disease (LOAD), several genes have been described as potential risk factors, but nongenetic factors may also be involved in the disease’s progression (Figure 3).9 The APOE gene, located on chromosome 19, is an important genetic risk factor for LOAD, and its importance has been validated from population studies. Apolipoprotein E (ApoE) protein is the major cholesterol carrier in the brain, which can be involved in neuronal maintenance and repair. ApoE binds to several receptors on the cell surface, which are involved in lipid delivery and transport, glucose metabolism, neuronal signaling, and mitochondrial function. Normally, ApoE binds to Abeta peptide and play a role in its clearance.141
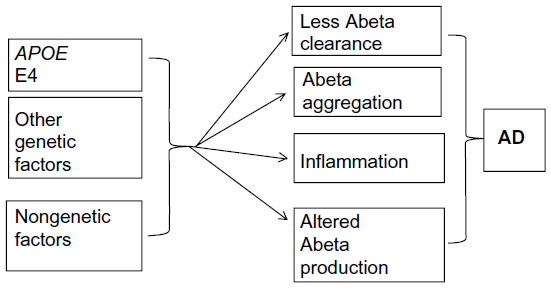 | Figure 3 Factors involved in late-onset Alzheimer’s disease (AD). |
Two polymorphic sites, located at codon 112 and 158, have been described in the human APOE gene. At least three main variations of the APOE gene have been identified, called “E2,” “E3,” and “E4” alleles. E3 was defined as a normal allele with Cys at codon 112 and Arg at codon 158. Two other APOE alleles have been described, the E2 and E4 alleles, which carry Arg158Cys and Cys112Arg polymorphisms, respectively.142,143 Six different genotypes can be distinguished with the following combinations: homozygous – E4/E4, E3/E3, and E2/E2 – and heterozygous – E2/E3, E2/E4, and E3/E4 (Table 2). E3 is the most common variant (77%), while E2 (8%) and E4 (15%) alleles have been detected less frequently. Higher frequencies of the E4 allele have been found among AD patients, and increased risk of AD can be found in patients with both homo- and heterozygous alleles.141 The pathogenic nature of the E4 allele might be associated with the structural change of ApoE protein. ApoE protein has two major functional domains: a 22 kDa N-terminal and a 10 kDa C-terminal domain, connected by a hinge region. The E4 allele can promote domain interactions through the altered orientation of Arg61 in the N-terminal domain. Arg112 can interact with the Glu255 in the C-terminal domain, resulting in structural changes to ApoE protein, neuronal death, and neurodegeneration. Mouse experiments revealed that the mutation of Arg61 to Thr, or of Glu255 to Ala, may reduce the domain interactions.144–148 Figure 4 shows the differences between the E3 and E4 alleles.
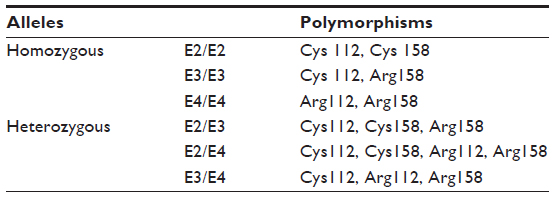 | Table 2 The six genotypes of the apolipoprotein E (APOE) gene |
 | Figure 4 The difference between apolipoprotein E (APOE) protein E3 allele (A) and APOE E4 allele (B). The pathomechanism of the APOE E4 allele could be based on the interaction between Arg112 and Glu255. |
The prevalence of the E2 allele has been found to be significantly lower in individuals with dementia.148 E2 allele was suggested to be protective against AD.145 Further, APOE E2 and E3 may participate in neuronal maintenance and repair.145 A Korean study detected significant correlation between the APOE E4 allele and AD.149 Genotyping analysis was performed in a group of AD patients and healthy individuals (controls). The allele and genotype frequency were compared using chi-square and Fisher’s exact tests. The frequency of the APOE E4 allele in the EOAD and LOAD groups was significantly higher than in the control group. However, the study failed to find any difference in the E2 allele between AD patients and controls. These findings suggest that the E2 allele might not play a protective role against AD in Korea.149
Genome-wide association studies (GWASs) have identified novel genes that might be associated with LOAD. Recently, SNP arrays have been developed and used for the analysis of several genes and SNPs. GWASs have been successfully applied to complex polygenic disorders, such as diabetes and macular degeneration.150,151 Several papers have been published on the association between AD and different genes or alleles. Bertram et al have created a publicly available, constantly updated, database summarizing the potential genes that may be related to AD (http://www.alzgene.org).152 Systematic meta-analyses were performed for each polymorphism with all genotype data described for them. At least three case-control samples were tested. This database collected all potential genes that may be involved in AD onset, thus is a powerful tool to further the understanding of AD genetics. Additionally, it may be considered a model for tracking gene candidates in other polygenic disorders.152,153
Clusterin (CLU) is a major inflammatory-related apolipoprotein (Apolipoprotein J; ApoJ) that is expressed in all mammalian tissues. Clusterin may play a protective role against apoptosis, cell damage, or oxidative stress. Clusterin expression has been found to be upregulated in the brains of AD patients.154 Animal models have suggested it might be secreted with soluble Abeta. Clusterin can act as a molecular chaperon, which might prevent Abeta oligomerization and fibrillization.151 GWASs have determined a strong association between CLU mutations (located on chromosome 8) and LOAD. Additionally, a significant association has been found between the APOE E4 allele and CLU mutations.154,155
The complement receptor 1 (CR1) gene, located on chromosome 1, encodes the receptor for C3b complement protein. CR1 and C3b can be involved in Abeta clearance and in the prevention of Abeta aggregation. Risk-factor mutations for LOAD have been found in CR1 (rs6656401 and rs3818361).155 The functional role of CR1 mutations in AD pathogenesis is not determined yet, and further studies are needed to find out the effect in Abeta deposition.155,156
Phosphatidylinositol binding clathrin assembly protein (PICALM or CALM), located on chromosome 11, may be a putative LOAD risk-factor gene. PICALM can play a role in APP endocytosis and Abeta generation. Additionally, its overexpression may increase Abeta cleavage and aggregation.157 Harold et al found strong association between two polymorphisms in PICALM and LOAD. Rs561655 is located within a transcription factor-binding site, and a silent mutation, rs592297, may be involved in the alternative splicing.158 Other SNPs in PICALM have also been suggested to be involved in LOAD, such as rs3851179 and rs541458.158
Sortilin-related receptor (SORL1) on chromosome 11q23-24 may be involved in Abeta recycling. The under-expression of SORL1 can increase Abeta generation. Intronic polymorphisms, located near the 3′ end of the SORL1 coding region, might be associated with AD.159,160
A poly-T repeat (rs10524523) was identified in exon 6 of the translocase of outer mitochondrial membrane 40 homolog (TOMM40; chromosome 19) gene that can be associated with an earlier age of onset of LOAD in patients with APOE E3/E3 and E3/E4 alleles. Cruchaga et al suggested that TOMM40 and other mitochondrial enzymes might be involved in the onset of LOAD.161
Bridging Integrator 1 (BIN1; chromosome 2) is a tumor suppressor gene that can be involved with protein for vesicle trafficking. Mutations in BIN1 may be associated with autosomal recessive centronuclear myopathy. Caenorhabditis elegans experiments have suggested that BIN1 protein might have a role in trafficking APP, ApoE proteins, and Abeta through the endolysosomal pathways, thus BIN1 mutations may be a putative risk factor for LOAD.162
The low-density lipoprotein receptor-related protein 6 (LRP6) gene on chromosome 12 is expressed as a co-receptor for Wnt signaling. Defects in Wnt signaling have been validated as risk factors for neurodegenerative disorders such as schizophrenia, autism, and AD. Wnt signaling proteins, such as beta-catenin or glycogen synthase kinase 3 beta, can form complexes with presenilins, which suggests they might play an important role in Abeta processing and neurotoxicity. Genetic linkage studies have suggested an association between LOAD and chromosome 12. Polymorphisms in LRP6 might result in abnormalities in plasma ApoE catabolism and in Wnt signaling.163
The cadherin-associated protein alpha 3 (CTNNA3) gene located on chromosome 10 encodes alpha-T catenin, which can be involved in AD pathogenesis by binding to beta-catenin and interacting with PS1. Miyashita et al identified seven putative LOAD risk-factor polymorphisms located at intron 9 of CTNNA.164 Polymorphisms in CTNNA3 have shown significant association with LOAD in female patients, who carried the APOE E3 allele, but not the E4.164,165
Growth factor receptor-bound protein 2-associated-binding protein 2 (GAB2) molecules are intracellular docking or scaffolding molecules. GAB2 can be involved in several signal transduction processes, associated with cell growth, survival, differentiation, and apoptosis. GAB2 might play a role in the suppression of Tau phosphorylation and in neurofibrillary tangles (NFTs) formation. Reiman et al detected six SNPs in GAB2 (chromosome 11) which might be associated with LOAD.166 Interaction was found between GAB2 haplotypes and the APOE E4 allele.166–168
Dynamin-binding protein (DNMBP) or Tuba protein plays a role in the transport of dynamin to the actin regulatory proteins. A Belgian study found a significant association between two SNPs (rs3740057 and rs10883421) in the 3′ region of the DNMBP (chromosome 10) gene and LOAD.169
The A disintegrin and metalloproteinase domain-containing protein 10 (ADAM10; chromosome 15) gene encodes the major brain alpha secretase. Alpha secretase cleavage can prevent Abeta formation and aggregation, and increase Abeta clearance. In vitro and in vivo studies have shown that two mutations (Gln171Gly and Arg181Gly) in the pre-domain region of ADAM10 may be associated with AD.170
ATP-binding cassette transporter A7 (ABCA7), located on chromosome 19, is a recently discovered potential risk factor for AD. ABCA7 protein, which is highly homologous to ABCA1, may be involved in the synthesis and transport of high-density lipoprotein cholesterol and generate phospholipid and cholesterol efflux from the cells. It can also play a key role in sterol homeostasis and in the host defense system.171,172 The two variants (rs3752246 and rs3764650) in ABCA7 have been suggested to be associated with LOAD.171 Rs3764650 is located in intron 13, and rs3752246 is a missense mutation in exon 32 (Gly1527 Ala).171 Recent findings have revealed an additional SNP (rs115550680) that might be involved in LOAD in African-Americans. Since ABCA7 plays a role in the lipid metabolism as well as in APP transport, mutations in ABCA7 gene might be involved in LOAD.173
Recent GWASs have revealed that triggering receptor expressed on myeloid cells 2 (TREM2), located on chromosome 6 can be involved in AD, especially in LOAD. TREM2 is a member of immunoglobulin family, and it contains a single variable domain. TREM2 is located on the membrane of several immune cells, such as macrophages and dendritic cells. Its main ligand is DNA clamp loader is Replication Factor C-activating protein of 12 kilodaltons (DAP12), which can be involved in downstream signaling. Functions of TREM2 protein can include the clearance of apoptotic cells and immunosupression.174 In an Icelandic population, a rare variant (Arg47His) has been suggested to increase the risk of impairment in inflammation, leading to LOAD.175 Other variants located in exon 2 have been shown higher percentage in AD patients, such as Glu33X or Asp87 Asn. AD, associated with TREM2 can be associated with chronic brain inflammation with aberrations in microglial phagocytosis or inflammatory pathways.176
Cluster of differentiation 33 (CD33; chromosome 19) is a 67 kDa transmembrane glycoprotein that is expressed on the surface of myeloid progenitor cells, mature monocytes, and macrophages. It can function as a lectin, a carbohydrate-binding protein, which inhibits cellular activity. The CD33 locus is related to altered monocyte function, which suggests it can be involved in innate immunology, leading to AD progression. Rs3865444 can be associated with elevated CD33 expression, leading to cognitive decline and AD. Mutations in CD33 can be associated with disturbances in myeloid function and amyloid pathology, thus may be involved in the progression of early AD.177
Methods of detecting mutation
PCR-based methods can be performed for monitoring the mutations in the AD risk factor genes (Figure 5).178 Genomic DNA can be extracted from total blood, buffy coat (white blood cells), bone marrow, or cell cultures, using a specific extraction kit. DNA should be amplified by specific primers, designed for the AD risk-factor genes such as APP, PSEN1, PSEN2, and APOE.6–8,22,26 Several mutation detection methods have been developed, such as restriction fragment length polymorphism (RFLP), single-strand conformation polymorphism (SSCP), denaturing gradient gel electrophoresis (DGGE), temperature gradient gel electrophoresis (TGGE), and heteroduplex analysis. RFLP is based on the recognition of a specific cleavage site and can be used for genetic mapping and linkage analysis. To identify the polymorphisms in the PCR products, the amplicons should be sequenced.178
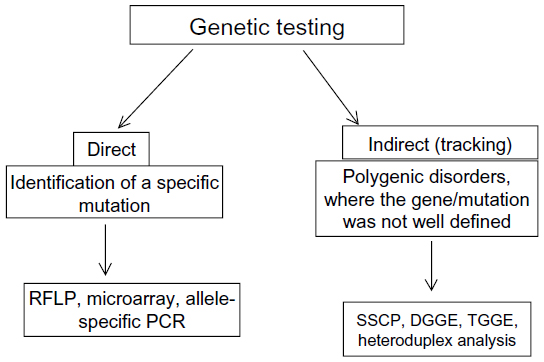 | Figure 5 Polymerase chain reaction (PCR)-based genetic methods. |
Methods based on the conformational changes of single-stranded DNA
DGGE is a rapid, commonly used method for mutation detection. The technology is based on the mobility of double-stranded DNA in polyacrylamide gel containing linearly increasing concentrations of denaturing chemicals.179,180 SSCP is a simple PCR-based mutation detection method. The mobility of double-stranded PCR fragments depends on the size of the DNA, since the polymorphisms might result in the altered mobility of single-stranded DNA by changing its conformation (Figure 6). The PCR products should be denatured by heat and formamide, followed by neutral polyacrylamide gel electrophoresis.181,182
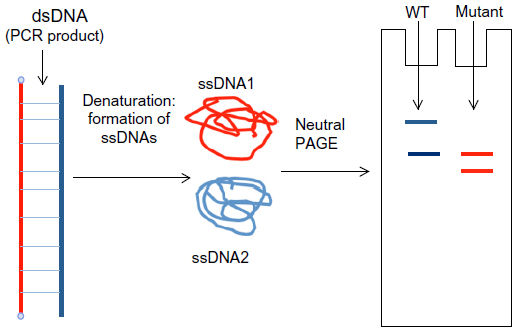 | Figure 6 The single-strand conformation polymorphism process. After denaturation of the polymerase chain reaction (PCR) product, the conformation of single-stranded DNA (ssDNA) could be different, resulting in altered mobility in polyacrylamide gel. |
Heteroduplex analysis with Surveyor® Nuclease
Surveyor Nuclease (Transgenomic, Inc, Omaha, NE, USA) is a plant (celery) endonuclease that cleaves double-stranded DNA at mismatch sites, including SNPs, insertions, and deletions. A novel PCR-based mutation detection method has been developed by Transgenomic. The process has four main steps: 1) amplification of target DNAs from patients and healthy controls; 2) hybridization of normal DNA with the DNA of the patient; 3) digestion of homo- and heteroduplexes by Surveyor Nuclease; and finally, 4) separation of cleavage products by standard gel electrophoresis or high-pressure liquid chromatography (Figure 7). This method may be promising in molecular diagnosis, and it has been successfully used for the identification of genetic-based disorders.183–185
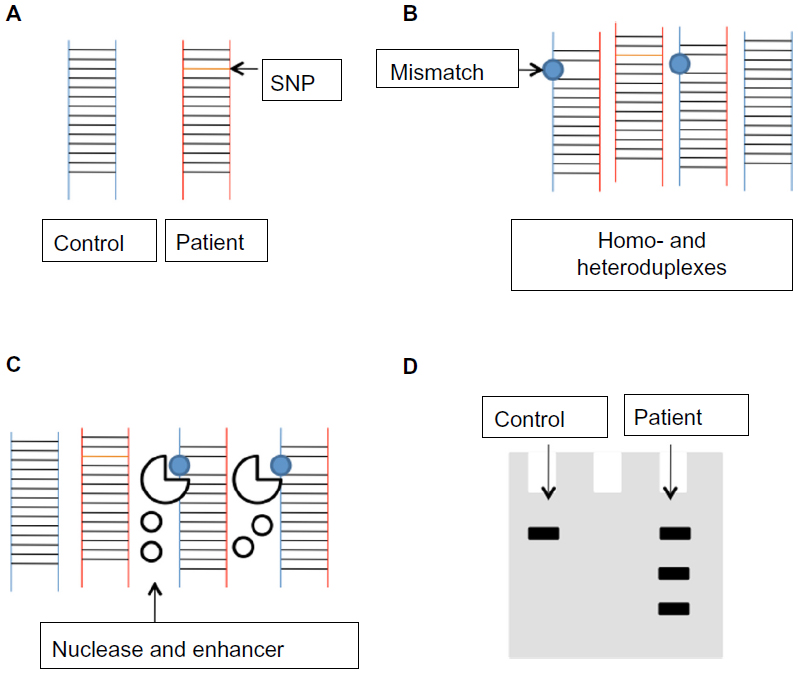 | Figure 7 The basic steps of genotyping with Surveyor® Nuclease (Transgenomic, Inc, Omaha, NE, USA). After mixing the polymerase chain reaction amplicons of healthy control and patient (A), hybridization should be performed, resulting in homo- and heteroduplex formation (B). Treatment with Surveyor Nuclease cleaves the DNA at the mismatch site (C). Cleavage products can be separated by electrophoresis (D). |
APOE genotyping
Allele-specific, multiplex PCR has been developed for APOE genotyping, with common and specific inner primers for polymorphism detection at codons 112 and 158. The agarose electrophoresis pattern can show the homozygous and heterozygous genotypes of E2, E3, and E4 alleles.186 Various kits have been designed for APOE PCR genotyping. One of the most frequently used kits is the LightCycler® ApoE Mutation Kit by Roche Diagnostics (Basel, Switzerland).187 PCR-RFLP is a widely used, simple and fast method for APOE genotyping. The genomic DNA should be amplified with specific primers, followed by HhaI digestion. The samples can be separated in 8% polyacrylamide (PAGE) gel, and visualized with fluorescent dye.188
Future insights into AD genetics: from GWASs to next-generation sequencing (NGS)
Since AD is a genetically heterogeneous disorder, GWASs have been performed for identification of novel disease risk-factor loci. Several genes and mutations have been tested to find association with disease-related phenotypes, such as changes in biomarker levels and/or neuropathology.189 Sanger sequencing is a widely used technology, but it has limitations in terms of cost, speed, and efficacy. High-throughput or NGS technologies are recent hot topics in genomic research of animals and humans. NGS technologies included sequencing by synthesis, ligation, or hybridization; single-molecule sequencing; nanopore sequencing; and colony sequencing. NGS technologies provide fast and cost-effective sequencing strategies that can be used in various genetic applications; for example, in high-throughput mutation detection, small RNA detection, or the monitoring of epigenetic changes. The most well-known NGS technologies have been developed by Illumina (and Solexa, Inc, purchased by Illumina in 2007; San Diego, CA, USA), Helicos BioSciences (Cambridge, MA, USA), ABI/SOLiD, and 454 Life Sciences (a subsidiary of Roche; Branford, CT, USA) and use a single-molecule template for mutation detection with cloning-free approaches.190,191
Jin et al performed pooled DNA sequencing with APP, PSEN1, PSEN2, progranulin (PGRN) and microtubule-associated Tau protein (MAPT) genes that was applied in a large population for monitoring rare human-specific mutations.192 Samples were collected from selected groups of patients and pooled in complex mixtures with negative control samples (validated as wild-type alleles). The mixes were then sequenced by NGS analyzers. The sequencing data were mapped back to the sample and to the control as reference. The pooled sequencing analysis detected PGRN and MAPT mutations in patients with clinically diagnosed AD. These findings show that the clinical phenotype of amnesic frontotemporal dementia and that of AD may be similar, and the overlapping symptoms can result in difficulties in the disease diagnosis. Complex genetic analysis might improve the diagnosis of neurodegenerative disorders.192,193
It has been suggested that the development of the human brain depends on the level of transcription. Alterations in transcription regulation are responsible for the unique gene expression patterns in the brain. Aging is the main risk factor for AD, but normal aging itself can result in only a low degree of neuronal loss. Alternative splicing and gene expression may be involved in AD pathogenesis. Microarrays are widely used for transcriptome analysis, but their accuracy might be limited because of mistakes in hybridization. Transcriptome studies have been performed in animals, various cell lines, cells derived from AD patients, and in postmortem brain tissues. Twine et al performed a whole-transcriptome analysis in different regions of an AD brain.194 Illumina RNA-Seq analysis was used for whole-transcriptome profiling. This study provided a possible insight into the changes in gene expressions and alternative splicing. NGS can produce digital signals directly from the complementary DNA, decrease the risk for false-positive data, and correspond to the existing genomic sequence.194,195
Conclusion
AD is the most common form of senile dementia, but it can sometimes be difficult to distinguish heterogeneous neurodegenerative disorders, such as frontotemporal dementia, dementia with Lewy Bodies, Parkinson’s disease, and Creutzfeldt–Jakob disease.5 AD is a complex disorder, so several genes on different chromosomes could be involved in its onset. Finding the potential genes involved in AD progression is an essential step in molecular diagnosis. Genetic testing should be important to understand the mechanisms and pathways leading to neurodegeneration and disease symptoms. It is believed that disease-modifying therapies are more likely to be effective in the earlier stages of AD, especially before the clinical symptoms appear. Genetic testing in the family members of patients should also be important to predict the risk for disease onset in the future. Using disease markers with genetic testing together may provide more effective disease diagnosis. In addition, the discovery of novel genes may provide more information on AD-related pathways.9,25,196,197
Genetic analysis can improve the differential diagnosis of neurodegenerative dementias. Standard Sanger sequencing is still a widely used technology, but can be costly and time consuming. NGS technologies offer a faster, less expensive approach, not only for mutation detection but also for transcriptome analysis or epigenetics.198 Several loci have been identified that might be involved in both familial and sporadic forms of neurodegenerative disorders. Understanding the complete genetic mechanisms of AD can provide additional information about the pathological mechanisms of neurodegeneration. GWASs and NGS studies may improve the prevention and treatment of AD.199
Acknowledgments
This work was supported by the GRRC program of Gyeonggi province (GRRC Gachon 2013-B04, Development of Microfluidic Chip for diagnosis of disease).
Disclosure
The authors declare no conflicts of interest in this work.
References
Ertekin-Taner N. Genetics of Alzheimer’s disease: a centennial review. Neurol Clin. 2007;25(12):611–617. | |
Reitz C, Mayeux R. Use of genetic variation as biomarkers for Alzheimer’s disease. Ann N Y Acad Sci. 2009;1180:75–96. | |
Brickell KL, Leverenz JB, Steinbart EJ, et al. Clinicopathological concordance and discordance in three monozygotic twin pairs with familial Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2007;78(10):1050–1055. | |
Meyer JM, Breitner JC. Multiple threshold model for the onset of Alzheimer’s disease in the NAS-NRC twin panel. Am J Med Genet. 1998;81(1):92–97. | |
Bird DT. Genetic aspects of Alzheimer disease. Genet Med. 2008;10(4):231–239. | |
Sorbi S, Forleo P, Tedde A, et al. Genetic risk factors in familial Alzheimer’s disease. Mech Ageing Dev. 2001;122(16):1951–1960. | |
Schellenberg GD, Anderson L, O’dahl S, et al. APP717, APP693, and PRIP gene mutations are rare in Alzheimer disease. Am J Med Genet. 1991;49(3):511–517. | |
Tanzi RE, Vaula G, Romano DM, et al. Assessment of amyloid beta-protein precursor gene mutations in a large set of familial and sporadic Alzheimer disease cases. Am J Med Genet. 1992;51(2):273–282. | |
Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Investig. 2005;115(6):1449–1457. | |
Rademakers R, Cruts M, Van Broeckhoven C. Genetics of early-onset Alzheimer dementia. ScientificWorldJournal. 2003;3:497–519. | |
Prasher VP, Farrer MJ, Kessling AM, et al. Molecular mapping of Alzheimer-type dementia in Down’s syndrome. Ann Neurolol. 1998;43(3):380–383. | |
König G, Mönning U, Czech C, et al. Identification and differential expression of a novel alternative splice isoform of the beta A4 amyloid precursor protein (APP) mRNA in leukocytes and brain microglial cells. J Biol Chem. 1992;267(15):10804–10809. | |
De Jonghe C, Esselens C, Kumar-Singh S, et al. Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum Mol Genet. 2001;10(16):1665–1671. | |
Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33(9):1340–1344. | |
Mullan M, Crawford F, Axelman K, et al. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1(5):345–347. | |
Tomiyama T, Nagata T, Shimada H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol. 2008;63(3):377–387. | |
Roks G, Van Harskamp F, De Koning I, et al. Presentation of amyloidosis in carriers of the codon 692 mutation in the amyloid precursor protein gene (APP692). Brain. 2000;123(Pt 10):2130–2140. | |
Golaz J, Charnay Y, Vallet P, Bouras C. [Alzheimer’s disease and Down’s syndrome. Some recent etiopathogenic data.] Encephale. 1991;17(1):29–31. French [with English abstract]. | |
Cruts M, Hendriks L, Van Broeckhoven C. The presenilin genes: a new gene family involved in Alzheimer disease pathology. Hum Mol Genet. 1996;5 Spec No:1449–1455. | |
Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398(6727):513–517. | |
Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–670. | |
Sleegers K, Roks G, Theuns J, et al. Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain. 2004;127(7):1641–1649. | |
Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer Disease. J Geriatr Psychiatry Neurol. 2010;23(4):213–227. | |
Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. | |
Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376(6543):775–778. | |
Kamimura K, Tanahashi H, Yamanaka H, Takahashi K, Asada T, Tabira T. Familial Alzheimer’s disease genes in Japanese. J Neurol Sci. 1998;160(1):76–81. | |
Jayadev S, Leverenz JB, Steinbart E, et al. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133(4):1143–1154. | |
To MD, Gokgoz N, Doyle TG, et al. Functional characterization of novel presenilin-2 variants identified in human breast cancers. Oncogene. 2008;25(25):3557–3564. | |
Ishikawa A, Piao YS, Miyashita A, et al. A mutant PSEN1 causes dementia with Lewy bodies and variant Alzheimer’s disease. Ann Neurol. 2005;57(3):429–434. | |
Kumar-Singh S, Cras P, Wang R, et al. Dense-core senile plaques in the Flemish variant of Alzheimer’s disease are vasocentric. Am J Pathol. 2002;161(2):507–520. | |
Kamino K, Orr HT, Payami H, et al. Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am J Hum Genet. 1992;51(5):998–1014. | |
Giaccone G, Rossi G, Morbin M, Tagliavini F, Bugiani O. A713T mutation of the APP gene in an Italian family with Alzheimer disease and severe congophilic angiopathy. Neurobiol Aging. 2002;23(Suppl 1): S320. | |
Pasalar P, Najmabadi H, Noorian AR, et al. An Iranian family with Alzheimer’s disease caused by a novel APP mutation (Thr714Ala). Neurology. 2002;58(10):1574–1575. | |
De Jonghe C, Kumar-Singh S, Cruts M, et al. Unusual Aβ amyloid deposition in Alzheimer’s disease due to an APP T714I mutation at the γ42-secretase site. Neurobiol Aging. 2000;21(Suppl 1):S200. | |
Park HK, Na DL, Lee JH, Kim JW, Ki CS. Identification of PSEN1 and APP gene mutations in Korean patients with early-onset Alzheimer’s disease. J Korean Med Sci. 2008;23(2):213–217. | |
Janssen JC, Beck JA, Campbell AT, et al. Early onset familial Alzheimer’s disease: Mutation frequency in 31 families. Neurology. 2003;60(20):235–239. | |
Guerreiro RJ, Baquero M, Blesa R, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2008;31(5):725–731. | |
Yoshioka K, Miki T, Katsuya T, Ogihara T, Sakaki Y. The 717Val–Ile substitution in amyloid precursor protein is associated with familial Alzheimer’s disease regardless of ethnic groups. Biochem Biophys Res Commun. 1991;178(3):1141–1146. | |
Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. | |
Murrell JR, Hake AM, Quaid KA, Farlow MR, Ghetti B. Early-onset Alzheimer disease caused by a new mutation (V717L) in the amyloid precursor protein gene. Arch Neurol. 2000;57(6):885–887. | |
Thajeb P, Wang P, Chien CL, Harrigan R. Novel polymorphisms of the amyloid precursor protein (APP) gene in Chinese/Taiwanese patients with Alzheimer’s disease. J Clin Neurosci. 2009;16(2):259–263. | |
Finckh U, Kuschel C, Anagnostouli M, et al. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics. 2005;6(2):85–89. | |
Chartier-Harlin MC, Crawford F, Houlden H, et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353(6347):844–846. | |
Kwok JB, Li QX, Hallupp M, et al. Novel Leu723Pro amyloid precursor protein mutation increases amyloid beta42(43) peptide levels and induces apoptosis. Ann Neurol. 2000;47(2):249–253. | |
Theuns J, Marjaux E, Vandenbulcke M, et al. Alzheimer dementia caused by a novel mutation located in the APP C-terminal intracytosolic fragment. Hum Mutat. 2006;27(9):888–896. | |
Wakutani Y, Watanabe K, Adachi Y, et al. Novel amyloid precursor protein gene missense mutation (D678N) in probable familial Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75(7):1039–1042. | |
Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease: an update. Ann Medicine. 2008;40(8):562–583. | |
Cruts M, van Duijn CM, Backhovens H, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998;7(1):43–51. | |
Finckh U, Müller-Thomsen T, Mann U, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet. 2000;66(1):110–117. | |
Rogaeva EA, Fafel KC, Song YQ, et al. Screening for PS1 mutations in a referral-based series of AD cases: 21 novel mutations. Neurology. 2001;57(4):621–625. | |
Campion D, Flaman JM, Brice A, et al. Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Hum Mol Genet. 1995;4(12):2373–2377. | |
Houlden H, Baker M, McGowan E, et al. Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurol. 2000;48(5):806–808. | |
Ataka S, Tomiyama T, Takuma H, et al. A novel presenilin-1 mutation (Leu85Pro) in early-onset Alzheimer disease with spastic paraparesis. Arch Neurol. 2004;61(11):1773–1776. | |
Queralt R, Ezquerra M, Lleó A, et al. A novel mutation (V89L) in the presenilin 1 gene in a family with early onset Alzheimer’s disease and marked behavioural disturbances. J Neurol Neurosurg Psychiatry. 2002;72(2):266–269. | |
Tedde A, Nacmias B, Ciantelli M, et al. Identification of new presenilin gene mutations in early-onset familial Alzheimer disease. Arch Neurol. 2003;60(11):1541–1544. | |
Arango D, Cruts M, Torres O, et al. Systematic genetic study of Alzheimer disease in Latin America: mutation frequencies of the amyloid beta precursor protein and presenilin genes in Colombia. Am J Med Genet. 2001;103(2):138–143. | |
Kamino K, Sato S, Sakaki Y, et al. Three different mutations of presenilin 1 gene in early-onset Alzheimer’s disease families. Neurosci Lett, 1997;208(3):195–198. | |
Fang B, Jia L, Jia J. Chinese Presenilin-1 V97L mutation enhanced Abeta42 levels in SH-SY5Y neuroblastoma cells. Neurosci Lett. 2006;406(1–2):33–37. | |
Raux G, Guyant-Maréchal L, Martin C, et al. Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: an update. J Med Genet. 2005;42(10):793–795. | |
Gómez-Tortosa E, Barquero S, Barón M, et al. Clinical-genetic correlations in familial Alzheimer’s disease caused by presenilin 1 mutations. J Alzheimers Dis. 2010;19(3):873–888. | |
Raux G, Gantier R, Thomas-Anterion C, et al. Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology. 2000;55(10):1577–1579. | |
La Bella V, Liguori M, Cittadella R, et al. A novel mutation (Thr116Ile) in the presenilin 1 gene in a patient with early-onset Alzheimer’s disease. Eur J Neurol. 2004;11(8):521–524. | |
Anheim M, Hannequin D, Boulay C, Martin C, Campion D, Tranchant C. Ataxic variant of Alzheimer’s disease caused by Pro117Ala PSEN1 mutation. J Neurol Neurosurg Psychiatry. 2007;78(12):1414–1415. | |
Dowjat WK, Kuchna I, Wisniewski T, Wegiel J. A novel highly pathogenic Alzheimer presenilin-1 mutation in codon 117 (Pro117Ser): Comparison of clinical, neuropathological and cell culture phenotypes of Pro117Leu and Pro117Ser mutations. J Alzheimers Dis. 2004;6(1):31–43. | |
Zekanowski C, Styczynska M, PepŁonska B, et al. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer’s disease in Poland. Exp Neurol. 2003;184(2):991–996. | |
Wisniewski T, Dowjat WK, Buxbaum JD, et al. A novel Polish presenilin-1 mutation (P117L) is associated with familial Alzheimer’s disease and leads to death as early as the age of 28 years. Neuroreport. 1998;9(2):217–221. | |
Hutton H, Busfield F, Wragg M, et al. Complete analysis of the presenilin 1 gene in early onset Alzheimer’s disease. Neuroreport. 1996;7(3):801–805. | |
Crook R, Ellis R, Shanks M, et al. Early-onset Alzheimer’s disease with a presenilin-1 mutation at the site corresponding to the Volga German presenilin-2 mutation. Ann Neurol. 1997;42(1):124–128. | |
Yasuda M, Maeda K, Hashimoto M, et al. A pedigree with a novel presenilin 1 mutation at a residue that is not conserved in presenilin 2. Arch Neurol. 1999;56(1):65–69. | |
Dumanchin C, Brice A, Campion D, et al. De novo presenilin 1 mutations are rare in clinically sporadic, early onset Alzheimer’s disease cases. French Alzheimer’s Disease Study Group. J Med Genet. 1998;35(8):672–673. | |
Kim HJ, Kim HY, Ki CS, Kim SH. Presenilin 1 gene mutation (M139I) in a patient with an early-onset Alzheimer’s disease: clinical characteristics and genetic identification. Neurol Sci. 2010;31(8):781–783. | |
Palmer MS, Beck JA, Campbell TA, et al. Pathogenic presenilin 1 mutations (P436S and I143F) in early-onset Alzheimer’s disease in the UK. Hum Mutat. 1999;13(3):256. | |
Gallo M, Marcello N, Curcio SA, et al. A novel pathogenic PSEN1 mutation in a family with Alzheimer’s disease: phenotypical and neuropathological features. J Alzheimers Dis. 2011;25(3):425–431. | |
Hattori S, Sakuma K, Wakutani YS, et al. A novel presenilin 1 mutation (Y154N) in a patient with early onset Alzheimer’s disease with spastic paraparesis. Neurosci Lett. 2004;368(3):319–322. | |
Axelman K, Basun H, Lannfelt L. Wide range of disease onset in a family with Alzheimer disease and a His163Tyr mutation in the presenilin-1 gene. Arch Neurol. 1998;55(5):698–702. | |
Hong KS, Kim SP, Na DL, et al. Clinical and genetic analysis of a pedigree of a thirty-six-year-old familial Alzheimer’s disease patient. Biol Psychiatry. 1997;42(12):1172–1176. | |
Kim J, Bagyinszky E, Chang YH, et al. A novel PSEN1 H163P mutation in a patient with early-onset Alzheimer’s disease: clinical, neuroimaging, and neuropathological findings. Neurosci Lett. 2012;530(2):109–114. | |
Higuchi S, Yoshino A, Matsui T, et al. A novel PS1 Mutation (W165G) in a Japanese family with early-onset Alzheimer’s disease. Alzheimer Reports. 2000;3(4):227–231. | |
Knight WD, Kennedy J, Mead S, et al. A novel presenilin 1 deletion (p.L166del) associated with early onset familial Alzheimer’s disease. Eur J Neurol. 2007;14(7):829–831. | |
Pantieri R, Pardini M, Cecconi M, et al. A novel presenilin 1 L166H mutation in a pseudo-sporadic case of early-onset Alzheimer’s disease. Neurol Sci. 2005;26(5):349–350. | |
Moehlmann T, Winkler E, Xia X, et al. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci U S A. 2002;99(12):8025–8030. | |
Lleó A, Blesa R, Queralt R, et al. Frequency of mutations in the presenilin and amyloid precursor protein genes in early-onset Alzheimer disease in Spain. Arch Neurol. 2002;59(11):1759–1763. | |
Takao M, Ghetti B, Murrell JR, et al. Ectopic white matter neurons, a developmental abnormality that may be caused by the PSEN1 S169L mutation in a case of familial AD with myoclonus and seizures. J Neuropathol Exp Neurol. 2001;60(12):1137–1152. | |
Guo J, Wei J, Liao S, Wang L, Jiang H, Tang B. A novel presenilin 1 mutation (Ser169del) in a Chinese family with early-onset Alzheimer’s disease. Neurosci Lett. 2010;468(1):34–37. | |
Piccini A, Zanusso G, Borghi R, et al. Association of a presenilin 1 S170F mutation with a novel Alzheimer disease molecular phenotype. Arch Neurol. 2007;64(5):738–745. | |
Kasuga K, Ohno T, Ishihara T, et al. Depression and psychiatric symptoms preceding onset of dementia in a family with early-onset Alzheimer disease with a novel PSEN1 mutation. J Neurol. 2009;256(8):1351–1353. | |
Klünemann HH, Rogaeva E, Neumann M, et al. Novel PS1 mutation in a Bavarian kindred with familial Alzheimer disease. Alzheimer Dis Assoc Disord. 2004;18(4):256–258. | |
Dermaut B, Kumar-Singh S, Engelborghs S, et al. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann Neurol. 2004;55(5):617–626. | |
Yasuda M, Maeda K, Ikejiri Y, Kawamata T, Kuroda S, Tanaka C. A novel missense mutation in the presenilin-1 gene in a familial Alzheimer’s disease pedigree with abundant amyloid angiopathy. Neurosci Lett. 1997;232(1):29–32. | |
Goldman JS, Reed B, Gearhart R, Kramer JH, Miller BL. Very early-onset familial Alzheimer’s disease: a novel presenilin 1 mutation. Int J Geriatr Psychiatry. 2002;17(7):649–651. | |
Sugiyama N, Suzuki K, Matsumura T, et al. A novel missense mutation (G209R) in exon 8 of the presenilin 1 gene in a Japanese family with presenile familial Alzheimer’s disease. Hum Mutat. 1999;14(1):90. | |
Poorkaj P, Sharma V, Anderson L, et al. Missense mutations in the chromosome 14 familial Alzheimer’s disease presenilin 1 gene. Hum Mutat. 1998;11(3):216–222. | |
Ringman JM, Gylys KH, Medina LD, et al. Biochemical, neuropathological, and neuroimaging characteristics of early-onset Alzheimer’s disease due to a novel PSEN1 mutation. Neurosci Lett. 2011;487(3):287–292. | |
Norton JB, Cairns NJ, Chakraverty S, et al. Presenilin1 G217R mutation linked to Alzheimer disease with cotton wool plaques. Neurology. 2009;73(6):480–482. | |
Takao M, Ghetti B, Hayakawa I, et al. A novel mutation (G217D) in the Presenilin 1 gene ( PSEN1) in a Japanese family: presenile dementia and parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta Neuropathol. 2002;104(2):155–170. | |
Smith MJ, Gardner RJ, Knight MA, et al. Early-onset Alzheimer’s disease caused by a novel mutation at codon 219 of the presenilin-1 gene. Neuroreport. 1999;10(3):503–507. | |
Miklossy J, Taddei K, Suva D, et al. Two novel presenilin-1 mutations (Y256S and Q222H) are associated with early-onset Alzheimer’s disease. Neurobiol Aging. 2003;24(5):655–662. | |
Uttner I, Kirchheiner J, Tumani H, et al. A novel presenilin1 mutation (Q223R) associated with early onset Alzheimer’s disease, dysarthria and spastic paraparesis and decreased Abeta levels in CSF. Eur J Neurol. 2010;17(4):631–633. | |
Zekanowski C, Golan MP, Krzyśko KA, et al. Two novel presenilin 1 gene mutations connected with frontotemporal dementia-like clinical phenotype: genetic and bioinformatic assessment. Exp Neurol. 2006;200(1):82–88. | |
Coleman P, Kurlan R, Crook R, Werner J, Hardy J. A new presenilin Alzheimer’s disease case confirms the helical alignment of pathogenic mutations in transmembrane domain 5. Neurosci Lett. 2004;364(3):139–140. | |
Houlden H, Crook R, Dolan RJ, McLaughlin J, Revesz T, Hardy J. A novel presenilin mutation (M233V) causing very early onset Alzheimer’s disease with Lewy bodies. Neurosci Lett. 2001;313(1–2):93–95. | |
Aldudo J, Bullido MJ, Valdivieso F. DGGE method for the mutational analysis of the coding and proximal promoter regions of the Alzheimer’s disease presenilin-1 gene: two novel mutations. Hum Mutat. 19991;14(5):433–439. | |
Portet F, Dauvilliers Y, Campion D, et al. Very early onset AD with a de novo mutation in the presenilin 1 gene (Met 233 Leu). Neurology. 2003;61(8):1136–1137. | |
Sodeyama N, Iwata T, Ishikawa K, et al. Very early onset Alzheimer’s disease with spastic paraparesis associated with a novel presenilin 1 mutation (Phe237Ile). J Neurol Neurosurg Psychiatry. 2001;71(4):556–557. | |
Lladó A, Fortea J, Ojea T, et al. A novel PSEN1 mutation (K239N) associated with Alzheimer’s disease with wide range age of onset and slow progression. Eur J Neurol. 2010;17(7):994–996. | |
Edwards-Lee T, Wen J, Bell J, Hardy J, Chung J, Momeni P. A presenilin-1 mutation (T245P) in transmembrane domain 6 causes early onset Alzheimer’s disease. Neurosci Lett. 2006;398(3):251–252. | |
Kowalska A, Wender M, Florczak J, et al. Molecular genetics of Alzheimer’s disease: presenilin 1 gene analysis in a cohort of patients from the Poznań region. J Appl Genet. 2003;44(2):231–234. | |
Furuya H, Yasuda M, Terasawa K, et al. A novel mutation (L250V) in the presenilin 1 gene in a Japanese familial Alzheimer’s disease with myoclonus and generalized convulsion. J Neurol Sci. 2003;209(1–2):75–77. | |
Tabira T, Chui DH, Nakayama H, Kuroda S, Shibuya M. Alzheimer’s disease with spastic paresis and cotton wool type plaques. J Neurosci Res. 2002;70(3):367–372. | |
Verkkoniemi A, Somer M, Rinne OJ, et al. Variant Alzheimer’s disease with spastic paraparesis: clinical characterization. Neurology. 2000;54(5):1103–1109. | |
Dumanchin C, Tournier I, Martin C, et al. Biological effects of four PSEN1 gene mutations causing Alzheimer disease with spastic paraparesis and cotton wool plaques. Hum Mutat. 2006;27(10):1063. | |
Ikeda M, Sharma V, Sumi SM, et al. The clinical phenotype of two missense mutations in the presenilin I gene in Japanese patients. Ann Neurol. 1996;40(6):912–917. | |
Jiménez Caballero PE, Lladó A, de Diego Boguna C, Martin Correa E, Serviá Candela M, Marsal Alonso C. A novel presenilin 1 mutation (V261L) associated with presenile Alzheimer’s disease and spastic paraparesis. Eur J Neurol. 2008;15(9):991–994. | |
Forsell C, Froelich S, Axelman K, et al. A novel pathogenic mutation (Leu262Phe) found in the presenilin 1 gene in early-onset Alzheimer’s disease. Neurosci Lett. 1997;234(1):3–6. | |
Wasco W, Pettingell WP, Jondro PD, et al. Familial Alzheimer’s chromosome 14 mutations. Nat Med. 1995;1(9):848. | |
Akatsu H, Yamagata H, Wake A, et al. The first autopsy case report of familial Alzheimer’s disease (AD) associated with a mutation at G266S in the presenilin 1 (PSEN1) gene. Alzheimers Dement. 2008; 4 Suppl 2:T578. | |
Matsubara-Tsutsui M, Yasuda M, Yamagata H, et al. Molecular evidence of presenilin 1 mutation in familial early onset dementia. Am J Med Genet. 2002;114(3):292–298. | |
Perez-Tur J, Croxton R, Wright K, et al. A further presenilin 1 mutation in the exon 8 cluster in familial Alzheimer’s disease. Neurodegeneration. 1996;5(3):207–212. | |
Kwok JB, Halliday GM, Brooks WS, et al. Presenilin-1 mutation L271V results in altered exon 8 splicing and Alzheimer’s disease with non-cored plaques and no neuritic dystrophy. J Biol Chem. 2003;278(9):6748–6754. | |
Kwok JB, Taddei K, Hallupp M, et al. Two novel (M233T and R278T) presenilin-1 mutations in early-onset Alzheimer’s disease pedigrees and preliminary evidence for association of presenilin-1 mutations with a novel phenotype. Neuroreport. 1997;8(6):1537–1542. | |
Raman A, Lin X, Suri M, Hewitt M, Constantinescu CS, Phillips MF. A presenilin 1 mutation (Arg278Ser) associated with early onset Alzheimer’s disease and spastic paraparesis. J Neurol Sci. 2007;260(1–2):78–82. | |
Assini A, Terreni L, Borghi R, et al. Pure spastic paraparesis associated with a novel presenilin 1 R278 K mutation. Neurology. 2003;60(1):150. | |
Godbolt AK, Beck JA, Collinge J, et al. A presenilin 1 R278I mutation presenting with language impairment. Neurology. 2004;63(9):1702–1704. | |
Dermaut B, Kumar-Singh S, De Jonghe C, et al. Cerebral amyloid angiopathy is a pathogenic lesion in Alzheimer’s disease due to a novel presenilin 1 mutation. Brain. 2001;124(12):2383–2392. | |
Hamaguchi T, Morinaga A, Tsukie T, Kuwano R, Yamada M. A novel presenilin 1 mutation (L282F) in familial Alzheimer’s disease. J Neurol. 2009;256(9):1575–1577. | |
Aoki M, Abe K, Oda N, et al. A presenilin-1 mutation in a Japanese family with Alzheimer’s disease and distinctive abnormalities on cranial MRI. Neurology. 1997;48(4):1118–1120. | |
Ikeuchi T, Kaneko H, Miyashita A, et al. Mutational analysis in early-onset familial dementia in the Japanese population. The role of PSEN1 and MAPT R406 W mutations. Dement Geriatr Cogn Disord. 2008;26(1):43–49. | |
Sánchez-Valle R, Lladó A, Ezquerra M, Rey MJ, Rami L, Molinuevo JL. A novel mutation in the PSEN1 gene (L286P) associated with familial early-onset dementia of Alzheimer type and lobar haematomas. Eur J Neurol. 2007;14(12):1409–1412. | |
Dintchov Traykov L, Mehrabian S, Van den Broeck M, et al. Novel PSEN1 mutation in a Bulgarian patient with very early-onset Alzheimer’s disease, spastic paraparesis, and extrapyramidal signs. Am J Alzheimers Dis Other Demen. 2009;24(5):404–407. | |
Cruts M, Backhovens H, Wang SY, et al. Molecular genetic analysis of familial early-onset Alzheimer’s disease linked to chromosome 14q24.3. Hum Mol Genet. 1995;4(12):2363–2371. | |
Yasuda M, Maeda S, Kawamata T, et al. Novel presenilin-1 mutation with widespread cortical amyloid deposition but limited cerebral amyloid angiopathy. J Neurol Neurosurg Psychiatry. 2000;68(2):220–223. | |
Shrimpton AE, Schelper RL, Linke RP, et al. A presenilin 1 mutation (L420R) in a family with early onset Alzheimer disease, seizures and cotton wool plaques, but not spastic paraparesis. Neuropathology. 2007;27(3):228–232. | |
Robles A, Sobrido MJ, García-Murias M, et al. Clinical picture of a patient with a novel PSEN1 mutation (L424V). Am J Alzheimers Dis Other Demen. 2009;24(1):40–45. | |
Matsushita S, Arai H, Okamura N, et al. Clinical and biomarker investigation of a patient with a novel presenilin-1 mutation (A431V) in the mild cognitive impairment stage of Alzheimer’s disease. Biol Psychiatry. 2002;52(9):907–910. | |
Beck JA, Poulter M, Campbell TA, et al. Somatic and germline mosaicism in sporadic early-onset Alzheimer’s disease. Hum Mol Genet. 2004;13(12):1219–1224. | |
Piscopo P, Marcon G, Piras MR, et al. A novel PSEN2 mutation associated with a peculiar phenotype. Neurology. 2008;70(17):1549–1554. | |
Lao JI, Beyer K, Fernández-Novoa L, Cacabelos R. A novel mutation in the predicted TM2 domain of the presenilin 2 gene in a Spanish patient with late-onset Alzheimer’s disease. Neurogenetics. 1998;1(4):293–296. | |
Piscopo P, Talarico G, Crestini A, et al. A novel mutation in the predicted TMIII domain of the PSEN2 gene in an Italian pedigree with atypical Alzheimer’s disease. J Alzheimers Dis. 2010;20(1):43–47. | |
Finckh U, Alberici A, Antoniazzi M, et al. Variable expression of familial Alzheimer disease associated with presenilin 2 mutation M239I. Neurology. 2000;54(10):2006–2008. | |
Lleó A, Blesa R, Gendre J, et al. A novel presenilin 2 gene mutation (D439A) in a patient with early-onset Alzheimer’s disease. Neurology. 2001;57(10):1926–1928. | |
Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10(5):333–344. | |
Green RC, Roberts JS, Cupples LA, et al; REVEAL Study Group. Disclosure of APOE genotype for risk of Alzheimer’s disease. N Engl J Med. 2009;361(3):245–254. | |
Rihn BH, Berrahmoune S, Jouma M, Chamaa S, Marcocci L, Le Faou A. APOE genotyping: comparison of three methods. Clin Exp Med. 2009;9(1):61–65. | |
Mahley RW, Huang Y. Alzheimer disease: multiple causes, multiple effects of apolipoprotein E4, and multiple therapeutic approaches. Ann Neurol. 2009;65(6):623–625. | |
Mahley RW, Huang Y. Apolipoprotein (apo) E4 and Alzheimer’s disease: unique conformational and biophysical properties of apoE4 can modulate neuropathology. Acta Neurol Scand Suppl. 2006;185:8–14. | |
Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103(15):5644–5651. | |
Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. | |
Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–118. | |
Kim KW, Jhoo JH, Lee KU, et al. Association between apolipoprotein E polymorphism and Alzheimer’s disease in Koreans. Neurosci Lett. 1999;277(3):145–148. | |
Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry. 2007;68(4):613–618. | |
Waring SC, Rosenberg RN. Genome-wide association studies in Alzheimer disease. Arch Neurol. 2008;65(3):329–334. | |
Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39(1):17–23. | |
Bettens K, Sleegers K, Van Broeckhoven C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013;12(1):92–104. | |
Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J. Apolipoprotein J (clusterin) and Alzheimer’s disease. Microsc Res Tech. 2000;50(4):305–315. | |
Lambert JC, Heath S, Even G, et al; European Alzheimer’s Disease Initiative Investigators, de Pancorbo MM, Lendon C, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–1099. | |
Crehan H, Holton P, Wray S, Pocock J, Guerreiro R, Hardy J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology. 2012;217(2):244–250. | |
Xiao Q, Gil SC, Yan P, et al. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J Biol Chem. 2012;287(25):21279–21289. | |
Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–1093. | |
Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39(2):168–177. | |
Meng Y, Lee JH, Cheng R, St George-Hyslop P, Mayeux R, Farrer LA. Association between SORL1 and Alzheimer’s disease in a genome-wide study. Neuroreport. 2007;18(17):1761–1764. | |
Cruchaga C, Nowotny P, Kauwe JS, et al; Alzheimer’s Disease Neuroimaging Initiative. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch Neurol. 2011;68(8):1013–1019. | |
Hu X, Pickering E, Liu YC, et al; Alzheimer’s Disease Neuroimaging Initiative. Meta-analysis for genome-wide association study identifies multiple variants at the BIN1 locus associated with late-onset Alzheimer’s disease. PLoS One. 2011;6(2):e16616. | |
De Ferrari GV, Papassotiropoulos A, Biechele T, et al. Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104(22):9434–9439. | |
Miyashita A, Arai H, Asada TA, et al. Japanese Genetic Study Consortium for Alzeheimer’s Disease. Genetic association of CTNNA3 with late-onset Alzheimer’s disease in females. Hum Mol Genet. 2007;16(23):2854–2869. | |
Ertekin-Taner N, Ronald J, Asahara H, et al. Fine mapping of the alpha-T catenin gene to a quantitative trait locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Hum Mol Genet. 2003;12(23):3133–3143. | |
Reiman EM, Webster JA, Myers AJ, et al. GAB2 alleles modify Alzheimer’s risk in APOE epsilon4 carriers. Neuron. 2007;54(5):713–720. | |
Pan XL, Ren RJ, Wang G, Tang HD, Chen SD. The Gab2 in signal transduction and its potential role in the pathogenesis of Alzheimer’s disease. Neurosci Bull. 2010;26(3):241–246. | |
Liang WS, Chen K, Lee W, et al. Association between GAB2 haplotype and higher glucose metabolism in Alzheimer’s disease-affected brain regions in cognitively normal APOEµ4 carriers. Neuroimage. 2011;54(3):1896–1902. | |
Bettens K, Brouwers N, Engelborghs S, et al. DNMBP is genetically associated with Alzheimer dementia in the Belgian population. Neurobiol Aging. 2009;30(12):2000–2009. | |
Kim M, Suh J, Romano D, et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum Mol Genet. 2009;18(20):3987–3996. | |
Holton P, Ryten M, Nalls M, et al. Initial assessment of the pathogenic mechanisms of the recently identified Alzheimer risk Loci. Ann Hum Genet. 2013;77(2):85–105. | |
Tanaka N, Abe-Dohmae S, Iwamoto N, Yokoyama S. Roles of ATP-binding cassette transporter A7 in cholesterol homeostasis and host defense system. J Atheroscler Thromb. 2011;18(4):274–281. | |
Reitz C, Jun G, Naj A, Rajbhandary R, et al; Alzheimer Disease Genetics Consortium. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ∈4, and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013;309(14):1483–1492. | |
Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol. 2006;7(12):1266–1273. | |
Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. | |
Guerreiro R, Wojtas A, Bras J, et al; Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. | |
Bradshaw EM, Chibnik LB, Keenan BT, et al; Alzheimer Disease Neuroimaging Initiative, Morris MC, Evans DA, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16(7):848–850. | |
NSW Government Centre for Genetics Testing. DNA Genetic Testing: Screening for Genetic Conditions and Genetic Susceptibility. Fact Sheet 21. Sydney: NSW Government Centre for Genetics Testing; nd. Available from: http://www.genetics.edu.au/Information/Genetics-Fact-Sheets/DNAGeneticTestingTestingforGeneticConditionsandGeneticSusceptibilityFS21. Accessed September 30, 2013. | |
Reznik-Wolf H, Treves TA, Shabtai H, et al. Germline mutational analysis of presenilin 1 and APP genes in Jewish-Israeli individuals with familial or early-onset Alzheimer disease using denaturing gradient gel electrophoresis (DGGE). Eur J Hum Genet. 1998;6(2):176–180. | |
Reznik-Wolf H, Treves TA, Davidson MA, et al. A novel mutation of presenilin 1 in familial Alzheimer’s disease in Israel detected by denaturing gradient gel electrophoresis. Hum Genet. 1996;98(6):700–702. | |
Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci U S A. 1989;86(8):2766–2770. | |
Hayashi K. PCR-SSCP: a simple and sensitive method for detection of mutations in the genomic DNA. PCR Methods Appl. 1991;1(1):34–38. | |
Qiu P, Shandilya H, D’Alessio JM, O’Connor K, Durocher J, Gerard GF. Mutation detection using Surveyor nuclease. Biotechniques. 2004;36(4):702–707. | |
Bannwarth S, Procaccio V, Paquis-Flucklinger V. Surveyor Nuclease: a new strategy for a rapid identification of heteroplasmic mitochondrial DNA mutations in patients with respiratory chain defects. Hum Mutat. 2005;25(6):575–582. | |
Mitani N, Tanaka S, Okamoto Y. Surveyor nuclease-based genotyping of SNPs. Clin Lab. 2006;52(7–8):385–386. | |
Yang YG, Kim JY, Park SJ, Kim SW, Jeon OH, Kim DS. Apolipoprotein E genotyping by multiplex tetra-primer amplification refractory mutation system PCR in single reaction tube. J Biotechnol. 2007;131(2):106–110. | |
Nauck M, Hoffmann MM, Wieland H, März W. Evaluation of the apo E genotyping kit on the LightCycler. Clin Chem. 2000;46(5):722–724. | |
Wu YY, Delgado R, Costello R, Sunderland T, Dukoff R, Csako G. Quantitative assessment of apolipoprotein E genotypes by image analysis of PCR-RFLP fragments. Clin Chim Acta. 2000;293(1–2):213–221. | |
Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006296. | |
Lin B, Wang J, Cheng Y. Recent Patents and Advances in the Next-Generation Sequencing Technologies. Recent Pat Biomed Eng. 2008;2008(1):60–67. | |
Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92(5):255–264. | |
Jin SC, Pastor P, Cooper B, et al; Ibero-American Alzheimer Disease Genetics Group Researchers, Cruchaga C. Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimers Res Ther. 2012;4(4):34. | |
Vallania FL, Druley TE, Ramos E, et al. High-throughput discovery of rare insertions and deletions in large cohorts. Genome Res. 2010;20(12):1711–1718. | |
Twine NA, Janitz K, Wilkins MR, Janitz M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLoS One. 2011;6(1):e16266. | |
Sutherland GT, Janitz M, Kril JJ. Understanding the pathogenesis of Alzheimer’s disease: will RNA-Seq realize the promise of transcriptomics? J Neurochem. 2011;116(6):937–946. | |
Neumann PJ, Hammitt JK, Mueller C, et al. Public attitudes about genetic testing for Alzheimer’s disease. Health Aff (Millwood). 2001;20(5):252–264. | |
Aisen PS, Vellas B, Hampel H. Moving towards early clinical trials for amyloid-targeted therapy in Alzheimer’s disease. Nat Rev Drug Discov. 2013;12(4):324. | |
Marguerat S, Wilhelm BT, Bähler J. Next-generation sequencing: applications beyond genomes. Biochem Soc Trans. 2008;36(Pt 5):1091–1096. | |
Lill CM, Bertram L. Towards unveiling the genetics of neurodegenerative diseases. Semin Neurol. 2011;31(5):531–541. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.