Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
The genetic basis of amyotrophic lateral sclerosis: recent breakthroughs
Authors Eykens C, Robberecht W
Received 31 January 2015
Accepted for publication 30 March 2015
Published 5 October 2015 Volume 2015:5 Pages 327—345
DOI https://doi.org/10.2147/AGG.S57397
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr John Martignetti
Caroline Eykens,1,2 Wim Robberecht1–3
1Research Group Experimental Neurology, Department of Neurosciences, KU Leuven – University of Leuven, Leuven, Belgium; 2Laboratory of Neurobiology, Vesalius Research Center, VIB, Leuven, Belgium; 3Department of Neurology, University Hospitals Leuven, Leuven, Belgium
Abstract: Deciphering the genetic architecture of amyotrophic lateral sclerosis (ALS), an adult-onset neurodegenerative disorder of the motor neuron system, is important to understand the etiology of this fatal disease as well as to develop customized ALS therapies based on the patient's genetic fingerprint. In this review, we discuss the genetic basis of ALS, and attempt to link the causal genes to three highly interrelated pathogenic mechanisms: dysproteostasis, RNA dysregulation, and axon dysfunction. In addition, we address the clinical and biological implications of these genetic findings. Furthermore, we explore to what extent genetic knowledge can be converted into targeted and personalized treatments.
Keywords: amyotrophic lateral sclerosis, frontotemporal dementia, genetics, disease modifiers, personalized medicine
Introduction
Amyotrophic lateral sclerosis (ALS)
ALS is a severely disabling and lethal neurodegenerative disorder that affects ~2–3 per 100,000 individuals worldwide.1 It generally strikes people over the age of 50, although juvenile ALS occurs earlier.2 It is usually fatal within 3–5 years after symptom onset.3 Selective dysfunction and dying of motor neurons in the spinal cord, brainstem, and motor cortex are the key features of ALS.1 Consequently, the major symptoms consist of muscle weakness and paralysis, which emerge when motor axonal retraction occurs. Cognitive dysfunction is an additional finding in a subset of patients. The majority of patients (~90%) are classified as being sporadic ALS (SALS) because they are not aware of other affected family members. Only in 10% of ALS patients a family history of disease is evident; that form is referred to as familial ALS (FALS).4 The borders between FALS and SALS are gradually fading, as discussed in the “FALS and SALS” section. Despite the progress made in ALS research so far, current clinical practice fails to halt or even slow down the disease course significantly. Riluzole, the only FDA-approved drug for ALS, results in a survival benefit, which is however limited and not noticeable for patients.5
Disease variability
Although the clinical picture of ALS may seem remarkably stereotypic, phenotypic and genetic heterogeneity is a persistent feature of the disease.2,4,6,7 Age of onset, clinical manifestation, type of motor neuron involvement, disease duration, survival, and many other disease parameters vary significantly.2
Cognitive dysfunction is evident in some patients.8 Recent evidence indeed shows ALS to be closely related to frontotemporal dementia (FTD). FTD is a degenerative disease affecting large neurons in the frontal and temporal cortex.2,9,10 It usually manifests itself through behavioral or language abnormalities. Approximately 40% of FTD patients have a family history of the disorder,11 progranulin (PGRN )12–14 and MAPT15 mutations being the most frequently encountered causes for FTD until recently. Many ALS patients have neuropsychological (usually behavioral) abnormalities suggestive for FTD,10 a minority of them meeting the criteria for the additional diagnosis of FTD.16,17 The mirror finding also holds true: many FTD patients have motor abnormalities of variable severity (mostly amyotrophy).18,19 Therefore, ALS and FTD are thought to represent the extremes of one disease spectrum, called the ALS/FTD spectrum.2,6
In addition to the marked phenotypic variability, there also is marked genetic variability. The number of genes in which pathogenic mutations cause ALS is steadily increasing (Table 1). Hitherto, more than 25 ALS-related genes have been identified, explaining the cause of more than half of FALS.4 Thus, mutations in, at first glance, very different genes induce a very similar (or even identical) clinical picture, but the same mutation may induce quite variable phenotypes both in terms of onset and survival,20–22 or type of involvement.23,24 This generated an interesting but rather semantic debate on whether ALS is a true disease entity, or rather embodies a heterogeneous group of diseases with motor neuron degeneration as a common characteristic.4
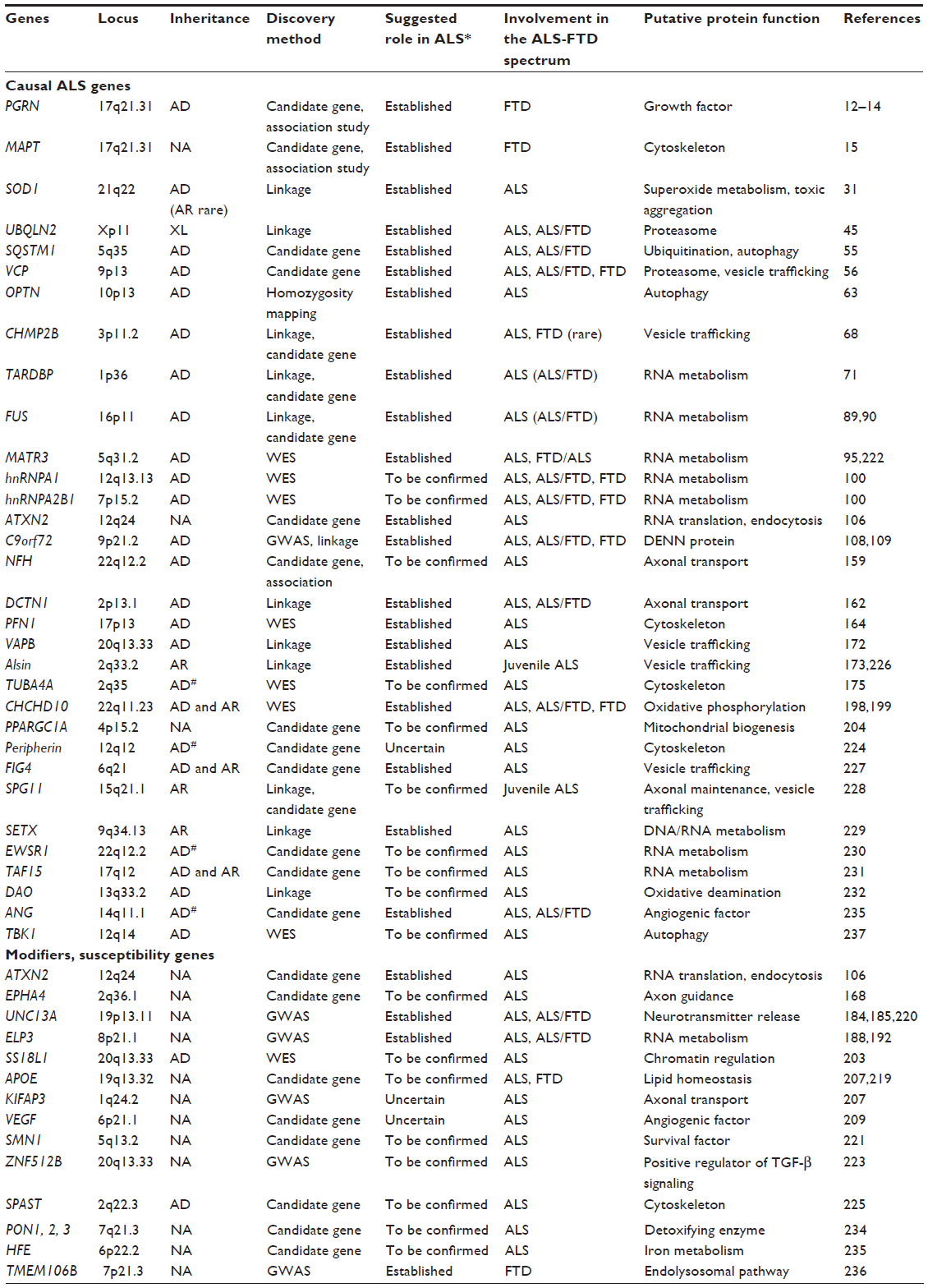 | Table 1 List of selected genes associated with ALS and FTD |
This progress in genetics has allowed the generation of disease models, which, hopefully, will serve as tools for the rational design and testing of therapeutic approaches. Furthermore, the boom in genetic discoveries has led to the notion that therapeutic interventions will need to be personalized, with ALS patients receiving the most suited therapy based on their genetic fingerprint.
FALS and SALS
Whether an individual patient has SALS or FALS is clinically decided based upon careful analysis of family history. However, mutations in typical ALS genes are found in a sizeable portion of patients labeled as sporadic. Incomplete penetrance,25 insufficient knowledge of family history, and diagnostic errors related to family members7 are, among others, reasons why the genetic predisposition is unnoticed. This has profound implications for clinicians in terms of correctly informing the family. Therefore, many ALS specialists consider genetic testing in all patients independent of a family history of disease. In addition, the finding that SALS patients actually have FALS, and the notion that some mutations have limited penetrance and thus may be considered as risk factors, have led to the hypothesis that all ALS is inheritable. A genetic basis for ALS without a familial context has indeed been addressed in twin studies26 and genome-wide association studies (GWAS),27 and is now being explored using recently developed sequencing techniques.27
In this review, we first discuss the genetic factors implicated in ALS and classify them in three possible disease mechanisms: protein homeostasis, RNA metabolism, and cytoskeletal integrity (Figure 1A–C). This classification is somewhat arbitrary and premature, as the exact mechanisms through which the mutations discussed induce motor neuron degeneration are still incompletely understood. Second, we assess the clinical and biological implications of neurogenetics and GWAS, and dig into genetic factors that act as disease modifiers. Third, we discuss what new genetic approaches may mean for the understanding of ALS. As the discovery of ALS-causing and modifying genes yields possible targets for intervention in cellular pathways, we critically look at the potential of translating this knowledge into target-based therapeutics. We conclude this review by looking into the possibility of customized ALS therapy based on genetic information.
 | Figure 1 Schematic overview of processes that contribute to ALS pathogenesis. |
ALS: collapse of the proteostasis machinery
A common hallmark of several neurodegenerative disorders is the accumulation of proteinaceous deposits.28 According to current thinking, accumulation of mutant, misfolding-prone proteins in vulnerable neurons and neighboring nonneuronal cells is induced by cellular stressors, of which one may be aging.29 By interfering with the proteostatic machinery, these protein aggregates fuel a self-reinforcing vicious circle of misfolded protein buildup and subsequent disturbance of various cellular processes.30 The convergence of the resulting effects within multiple cell types ultimately results in motor axonal retraction and motor neuronal death. There is ample experimental support for protein misfolding, protein accumulation, and proteostatic machinery dysfunction in ALS. However, the temporal relationship between these elements, their pathogenic contribution, the identity and role of cellular stresses, and the primary significance of these factors have been very difficult to firmly establish.
Superoxide dismutase 1 (SOD1): the old-timer among causal ALS genes
The groundbreaking discovery of the first ALS gene, SOD1, more than 20 years ago,31 turned the attention to hereditary ALS. Mutations in this gene account for ~12% of FALS and are found in ~1% of SALS.32,33 SOD1 is a ubiquitously expressed enzyme that catalyzes the dismutation of superoxide radicals and protects the cell against reactive oxygen species. So far, over 160 mutations scattered throughout all five exons have been identified (ALSoD Consortium; http://alsod.iop.kcl.ac.uk/), predominantly consisting of missense mutations; nonsense mutations or gene deletions/insertions appear to be rare.7 The majority of mutations are autosomal dominantly inherited, with the exception of the recessive D90A mutation in the Scandinavian population.34 For some mutations, genotype–phenotype correlations have been established. For example, carriers of the A4V mutation clinically mainly show lower motor neuron involvement and suffer from an aggressive ALS form with rapid disease progression and death on average occurring at 1.4 years after symptom onset.35 In contrast, D90A homozygous patients have a rather mild phenotype with mean disease duration of around 14 years.34 It remains to be seen whether all reported SOD1 variants are truly pathogenic.
The identification of SOD1 mutations has meant a momentous turnaround for ALS research. Rodent models overexpressing mutant SOD1 characterized by a fatal motor neuron degeneration reminiscent of ALS in humans were developed.36 In particular, the SOD1G93A mouse model has been widely used in order to get insight into the pathogenic mechanism of mutant SOD1.37 Several hypotheses regarding the mechanisms by which SOD1 aggregates contribute to the manifestation of motor neuron death have been proposed. These include perturbation of mitochondrial function, glutamate excitotoxicity, disturbance of axonal transport, SOD1 toxicity in nonneuronal cells surrounding the motor neurons, and impairment of protein homeostasis.38
One of the contributing mechanisms of mutant SOD1 may relate to protein homeostasis.6 Evidence suggests that the cell’s chaperone system is unable to refold misfolded mutant SOD1, resulting in abundant presence of toxic (likely oligomeric) SOD1 species. Consequently, the ubiquitin–proteasome system (UPS) and autophagy pathway appear to be unable to dispose these.6,30,39,40 In spinal cord motor neurons of ALS patients and rodent models, increased autophagosome expression was found, whereas proteasome activity was reduced.39 Moreover, the misfolded proteins impair the protein degradation machinery in the cell, which leads to a vicious circle of misfolded protein buildup through which a variety of important cellular processes are perturbed, resulting in axonal retraction and neuronal cell death.38
Still, even after 20 years of mutant SOD1 research, an unequivocal explanation for its toxicity has yet to be provided; a combination of different mechanisms is likely. Furthermore, most of data available in the literature relate to what happens in the mutant SOD1 mouse. Evidence for what happens in human ALS is less astounding.
Of notice, posttranslational modifications cause wild-type SOD1 to misfold and adopt an aggregation-prone and toxic conformation comparable to mutant SOD1.41–43 Using a conformation-specific antibody, aberrantly folded wild-type SOD1 has been described in the motor neurons in a subset of SALS cases (thus, without SOD1 mutations).43,44 These observations implicate that aberrantly modified forms of wild-type SOD1 may be at play in SALS pathogenesis.
Disturbance of protein degradation in ALS: a common theme
Strong evidence for a disturbance of protein degradation as a cellular disease mechanism in ALS comes from the exciting finding that ALS can be caused by mutations in proteins, which are directly or indirectly involved in regulated protein breakdown (Figure 2A).6
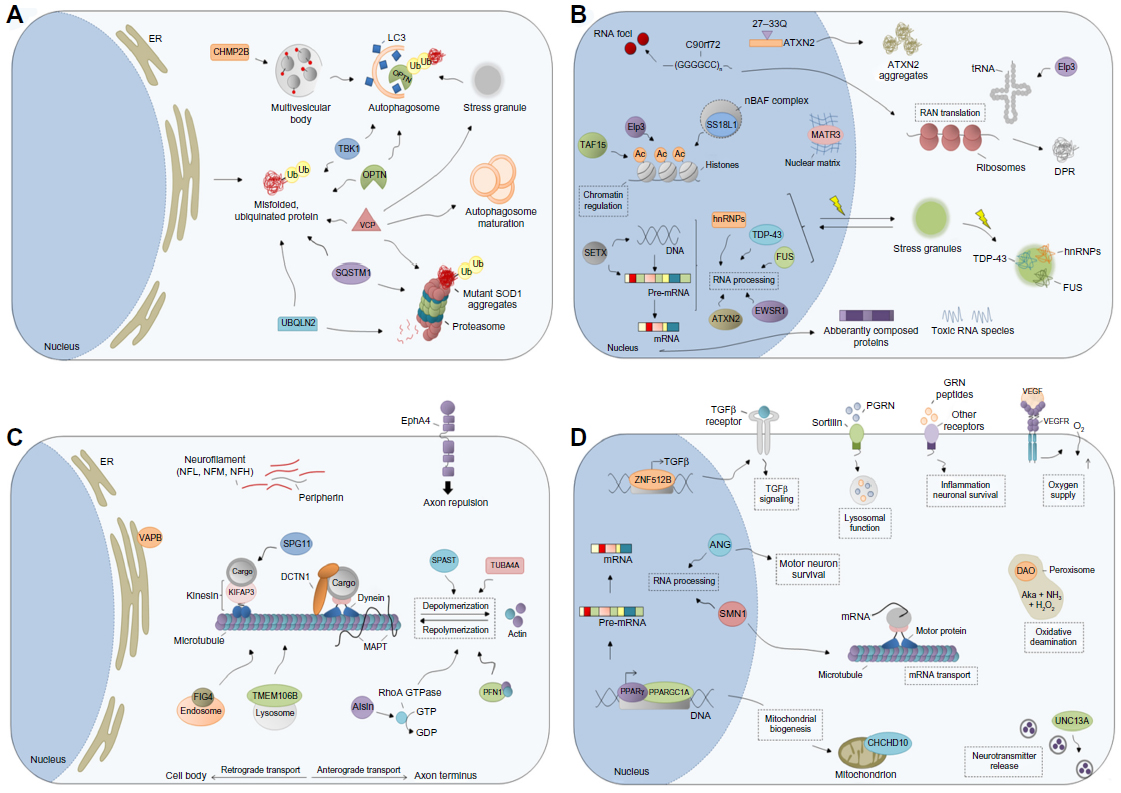 | Figure 2 Schematic overview of selected ALS genes according to their (neuronal) function. |
Ubiquilin 2 (UBQLN2)
In 2011, missense mutations in the UBQLN2 gene were found to underlie X-linked ALS and ALS/FTD.45 They appear to be rare. UBQLN2 is a member of the ubiquitin-like (UBL) protein family and consists of a C-terminal ubiquitin-associated (UBA) domain together with an N-terminal UBL domain.46 Through these domains, UBQLN2 binds poly-ubiquitin chains and associates with the proteasome and autophagosome.46 It thus mediates the delivery of ubiquitin-tagged proteins for degradation. Interestingly, the most prevalent UBQLN2 mutations affect the proline residue in a region containing 12 PXX tandem repeats (P497H, P497S, P506T, P509S, and P525S),45 but mutations outside this region have also been described.47,48 Of notice, UBQLN2 can also be found in skein-like inclusions in the spinal cord motor neurons and hippocampus of ALS and ALS/FTD patients not carrying mutations in this protein, suggesting that UBQLN2 may have a role in ALS that is not only attributable to mutations.45
Sequestosome 1 (SQSTM1)
SQSTM1, also known as p62, shares structural similarity with UBQLN2 and is found in inclusions in a variety of neurodegenerative disorders.49–51 The protein acts as a cargo receptor for the degradation of ubiquitinated proteins and regulates the activation of nuclear factor kappa-B (NF-κB) signaling.52 SQSTM1 mutations have been found in SALS, FALS, FTD, and FTD/ALS patients.53–55 Further screening and functional studies are needed to understand the pathogenic relevance of these SQSTM1 alterations for ALS.
Valosin-containing protein (VCP)
Exome sequencing revealed mutations in the VCP gene in ~1%–2% of FALS and <1% of SALS patients.56 Such mutations had already been found in a syndrome characterized by inclusion body myopathy (IBM) with Paget’s disease of the bone (PDB) and FTD (IBMPFD).57 VCP (also known as p97) is a multifunctional AAA+-ATPase that orchestrates a number of processes, including autophagy and proteasomal degradation. More precisely, VCP directs ubiquitin-tagged proteins to the proteasome and is essential for the maturation of autophagosomes.58 Interestingly, nearly all mutations reside in the N-terminal domain, which is crucial to carry out the proper function of VCP.56,57 Moreover, in mammalian cells, the depletion of pathogenic mutations in VCP reduced the autophagic clearance of stress granules,59 which are presumed to create a proaggregation environment.60 Therefore, it is plausible that VCP mutations disrupt VCP’s protein and stress granule removal activity in ALS, giving rise to the accumulation of ubiquitinated protein deposits within the cell.
Optineurin (OPTN)
OPTN mutations are associated with a very diverse phenotype. Polymorphisms have been described in Paget’s disease;61 missense and insertion mutations were found in primary open-angle glaucoma (POAG);62 and OPTN mutations have now been reported as a (rare) cause of ALS.63 OPTN is a protein with many functions; it is involved in membrane and vesicle trafficking,64 NF-κB regulation,65 and autophagic clearance of protein aggregates.66 Homozygous deletions, nonsense mutations, and heterozygous missense mutations have been found in both FALS and SALS patients.63 OPTN depletion enhances protein aggregation in Hela cells,66 and OPTN inclusions are found in the spinal cord of SALS patients.64 The exact mechanism through which OPTN mutations cause ALS still remains to be established.
Charged multivesicular body protein 2b (CHMP2B)
Although rarely so, mutations in CHMP2B originally found to be associated with FTD67 may give rise to an ALS phenotype.68 CHMP2B is required for the sorting of integral membrane proteins in multivesicular bodies and their subsequent autophagic degradation.69
The finding of ALS-causing mutations in these proteins suggests that disturbances of protein degradation play a mechanistic role in ALS, as mentioned earlier. However, it should be noted that almost all mutations are missense mutations giving rise to proteins that, at most, have lost some of their function. This means that in the cell (apart from the UBQLN2 mutations), at least 50% of the normal function is preserved. Therefore, it remains to be convincingly shown that these mutations actually cause failure of the protein breakdown sufficient to induce cell dysfunction and death. It is fairly well possible that they are pathogenic not through such a loss-of-function mechanism, but rather through a gain-of-function mechanism such as described for SOD1 mutants. Obviously, both mechanisms may be at play.
ALS: dysregulation of RNA householding
The identification of disease-linked mutations in genes involved in RNA processing marked the beginning of a paradigm shift and challenged the proteocentric thinking of the ALS field. May aberrant RNA metabolism contribute to ALS pathogenesis (Figure 2B)?
TAR DNA-binding protein (TARDBP)
The TARDBP (TDP-43) is a major constituent of ubiquitinated inclusions in a variety of neurodegenerative disorders, including FTD and ALS.70 This observation prompted the mutational screening of the TARDBP gene (which encodes the TDP-43 protein) in a cohort of FALS and SALS cases.71 To this day, up to 47 missense mutations and one truncated variant have been identified.72 Nearly all of the missense mutations reside in exon 6, which encodes the C-terminal glycine-rich part.73 TARDBP mutations have been reported in 4% of FALS and 1% of SALS cases.4
TDP-43 is a multifunctional nuclear protein that is, among other things, involved in multiple levels of RNA processing including transcription, splicing, transport, and translation.73 Besides the C-terminal glycine-rich domain, TDP-43 also contains two RNA recognition motifs (RRMs). RRM1 is essential for binding to single-stranded RNA with a minimum of five GU repeats.74,75 GU-rich sequences are predominantly present in pre-mRNAs encoded by more than 6,000 genes, which are consequently targeted by TDP-43.75 Therefore, it is not surprising that depletion of TDP-43 with the aid of antisense oligonucleotides affected the expression of ~600 mRNAs and altered 965 splicing events in the mouse brain.75 Of note, long intronic sequences containing GU repeats are typically found in neuronally expressed genes, and thus many of these can be found on the list of genes affected by TDP-43 downregulation.76
Although TDP-43 is predominantly present in the nucleus, nuclear-cytoplasmic shuttling has been shown, a process controlled by its bipartite nuclear localization signal (NLS) and nuclear export signal (NES). Neurons and glial cells of the ALS nervous system display a redistribution of TDP-43 from the nucleus to the cytoplasm. In the cytoplasmic compartment, TDP-43 associates with stress granules that appear in times of cellular stress (Figure 2B).77 In these granules, mRNAs are sorted during a triage process that determines mRNA fate: translation, sequestration in the stress granule, or degradation.78 Stress granules in ALS presumably create a proaggregation environment and hereby contribute to the observed inclusion buildup and TDP-43 depletion from the nucleus.60
Based upon this, a multistep model has been suggested to explain TDP-43 toxicity.6,79 First, ALS-associated mutations augment cytoplasmic TDP-43 accumulation (loss-of-function in the nucleus) and enhance its aggregation propensity (gain-of-function).80 Second, stress granule formation further reinforces TDP-43 accumulation and aggregation.60 Their toxicity may further be enhanced by acting as a sink for other proteins and RNAs (loss-of-function). It is likely, based on what has been found in cellular and animal models, that both TDP-43 deficiency81–83 and overexpression84–88 are detrimental for the living organism. However, in at least one mouse model, TDP-43 overexpression induced a phenotype without any evidence for aggregate formation or TDP-43 mislocalization.88 The understanding of the interplay between TDP-43 aggregation, stress granules, and TDP-43 pathology needs further clarification.
Fused in sarcoma (FUS)
The interest in RNA processing as a mechanistic factor in motor neuron degeneration grew rapidly with the discovery of FUS mutations.89,90 FUS shares structural homology with TDP-43 and is also involved in RNA metabolism. The protein binds over 5,500 genes through a GUGGU-binding motif and FUS depletion alters the splicing of more than 950 mRNAs.91 Remarkably, most of them are distinct from the mRNAs targeted by TDP-43.91 Similar to what is seen in TDP-43, nearly all mutations are clustered in the C-terminal region encoded by exons 14 and 15.73 FUS variants account for ~4% of FALS and ~1% of SALS.4 Cognitive impairment is rare in ALS patients with FUS mutations and the overall survival is significantly shorter compared with other ALS forms.92 Of note, a possible association between FUS mutations (c. 1475delG and c. 1542G > T) and mental retardation was recently discovered.93
Under physiological conditions, FUS is predominantly present in the cell nucleus. Similar to what is seen for TDP-43, cytoplasmic mislocalization and FUS-positive inclusions can be found in ALS patients with FUS mutations.89,90 It is thought that this is explained by mutations in the NLS, which compromise transportin-mediated nuclear import.94 Interference with this transport results in cytoplasmic redistribution and subsequent recruitment of FUS protein into stress granules (Figure 2B).94 As stress granules are known to be stress-sensitive dynamic entities, it seems possible that they form insoluble aggregates under stress conditions. A similar multistep model as for TDP-43 toxicity has been proposed for FUSopathies.79 Again, it remains to be seen whether a loss-of-function or gain-of-function mechanism or both explain what happens in patients.
Matrin3 (MATR3)
In early 2014, Johnson et al identified missense mutations in MATR3 that segregated with disease in several families with multiple members affected by ALS.95 Interestingly, a MATR3 variant (Ser85Cys) had been found to underlie a distal, asymmetrical myopathy with vocal cord weakness in two large families of different origin,96,97 but it turns out that these patients actually suffered from a slowly progressive motor neuron disorder better classified as ALS.95
MATR3 is a highly conserved protein that is predominantly associated with the nuclear matrix.98 It has structural similarity with other ALS-linked genes, ie, TDP-43 and FUS. With its bipartite NLS, two zinc finger domains, and two RRMs, MATR3 is involved in RNA metabolism. It binds and stabilizes several mRNA species including that of TDP-43.99 Interestingly, one MATR3 variant (Ser85Cys) displayed a higher affinity for TDP-43 while its interaction with other partners remained unaltered.95 Cytoplasmic mislocalization of mutant MATR3 is found in motor neurons of some ALS patients, even without these patients carrying MATR3 mutations.95 It is tempting to speculate that MATR3 variants cause dysfunction of TDP-43. Despite being a rare cause of ALS, the discovery of MATR3 mutations underscores once again the importance of RNA metabolism in ALS.
Heterogeneous nuclear ribonucleoprotein complex proteins (hnRNPs): infectious proteins?
Recently, mutations in the prion-like domain of hnRNPA2/B1 and hnRNPA1 have been reported in patients suffering from ALS and IBMPFD.100 Notably, these mutations enhance hnRNPA2/B1 and hnRNPA1 incorporation into stress granules and exacerbate their intrinsic tendency to assemble into self-seeding fibrils.100 Multiple lines of evidence link hnRNPs to neurodegeneration. They directly interact with TDP-43 to regulate RNA metabolism101 and two of them (hnRNPA2/B1 and hnRNPA1) are suppressors of VCP-induced degeneration in a Drosophila model for multisystem proteinopathy.102 hnRNPA2/B1 and hnRNPA1 mutations are rare in ALS.100 Finding more families in which they segregate with the disease would be reassuring, but their identification has contributed to the generation of a hypothesis for a prion-like mechanism contributing to ALS.28 Pathogenic mutations strengthen a steric zipper motif in the prion-like domain of hnRNP proteins, which accelerates the formation of self-seeding fibrils that induce the polymerization of wild-type hnRNP.100 It is thought that such motifs may induce aggregation by acting as a template, forcing the conversion of natively folded proteins into an abnormal form. The resulting aggregates may be released and taken up by neighboring cells, thus explaining the spatially progressive nature of ALS.44,103 Interestingly, not only hnRNP proteins, but also SOD1, TDP-43, and FUS have been shown to contain such a prion-like domain.28,104 It should be noted that these findings strongly link RNA householding dysregulation and dysproteostasis as two pathogenic pathways for motor neuron degeneration.
C9orf72 mutations in ALS: secrets unlocked?
Since the discovery of repeat expansions as a mechanism of disease in the 1990s, at least 24 neurological disorders characterized by such mutations have been described.105 Repeat expansions in the Ataxin-2 (ATXN2) gene106 and the non-imprinted Prader–Willi/Angelman syndrome region protein 1 (NIPA1) gene107 had previously already been implicated in ALS and/or FTD. But recently, major progress in the field of ALS was made by the discovery of a noncoding hexanucleotide repeat expansion (G4C2) in the 5′ region of C9orf72 as a very prevalent cause of ALS and FTD.108,109 While in the normal population the number of repeats never exceeds 30, their number in C9orf72 mutation carriers increases to up to thousand repeats.108,109
The exact function of the cytoplasmic C9orf72 protein remains to be elucidated at present. The structural similarity to DENN-like proteins suggested that the protein may belong to this family of GDP–GTP exchange factors for Rab-GTPases, which are involved in the regulation of membrane trafficking.110 At least three different C9orf72 transcripts exist and are expressed in most tissues including brain.108
An impressive percentage of ALS and FTD can currently be explained by C9orf72 mutations: nearly 40% of FALS and almost 10% of SALS; more than 25% of familial FTD; and approximately 5% of sporadic FTD.111 The G4C2 expansion is associated with a variable phenotype as addressed in recent reviews.2,24,111 C9orf72-ALS has a high incidence of bulbar onset and cognitive dysfunction is frequent with C9orf72 mutations.2,24,111 Almost a third of C9orf72 patients have both ALS and FTD.111 Additional phenotypes include Parkinsonism, ataxia, and psychosis.112,113 In addition, a large variation in disease onset (27–83 years) and duration (3–264 months) is evident among C9orf72 subjects.111
Interestingly, some C9orf72 patients harbor variations in other ALS- and/or FTD-associated genes,111,114 a finding of possible pathogenic significance.111 It is thought, but far from proven, that the C9orf72 expansion mutation establishes a susceptibility for neurodegeneration and that additional factors (such as these mutations or other modifiers) shape the phenotype, acting as disease modifiers.111 Of note, in that regard, the motor component appears to be modified by genes different from those that modify the cognitive component.111
Nuclear RNA foci containing both sense108,115–117 and antisense116,118,119 repeats have been found in neurons of the frontal cortex, and the spinal cord of patients with C9orf72 mutations.108 Three plausible mechanisms have been proposed to explain the pathogenic effect of the C9orf72 expansions and they may exist simultaneously.
First, the expansion may interfere with C9orf72 expression. Several, but certainly not all studies have found a reduction of mRNA expression in patients with the C9orf72 expansion, possibly resulting in haploinsufficiency (loss-of-function).120,121 Motor axonal degeneration was indeed seen upon knockdown of C9orf72 in zebrafish.122 In addition, hypermethylation of the C9orf72 promoter in C9orf72 mutation carriers was found.123 This methylation-driven gene silencing also occurs in other repeat disorders, including Friedreich’s ataxia,124 fragile X mental retardation,125 and myotonic dystrophy (DM).126–128 However, promoter hypermethylation was associated with reduced accumulation of RNA foci and dipeptide repeat (DPR) protein aggregates in C9orf72 patient brains129 and a shorter disease duration,123 suggesting that hypermethylation actually represents a protective response.129 Furthermore, homozygosity is not associated with more severe disease.130,131 These findings, together with the fact that C9orf72 coding mutations have not been described yet, make a loss-of-function mechanism less likely to have a major contribution to the molecular pathogenesis of C9orf72 expansion mutations.132
In contrast, a body of evidence has emerged that favors a toxic RNA gain-of-function mechanism. The C9orf72 RNA repeat expansion may exert its toxic properties via the sequestration of multiple RRM-containing proteins and their subsequent depletion.117,133,134 Proteins involved in splicing, mRNA nuclear export, and/or translation were significantly enriched as binding partners of the G4C2 repeat expansion. hnRNPA3, hnRNPA2/B1, SFPQ, ILF3, NONO, hnRNP L, IL2BP1, ILF-2, FUS,133 and Puraα135 displayed strong and selective binding to the G4C2 repeat. Furthermore, immunohistochemical colocalization of RNA foci with SRSF2, hnRNPH1/F, and ALYREF in neuronal cells was observed.134 The avid and dynamic binding of these proteins to the expanded G4C2 sequence is presumably facilitated by the tendency of the repeat to form G-quadruplexes.136 Such deleterious gain-of-function of the G4C2 repeat expansion is similar to what is thought to occur in DM. This neuromuscular disorder is caused by a CTG repeat in the 3′UTR region of the myotonic dystrophy protein kinase (DMPK) gene (type one) or by a CCTG repeat in intron 1 of the zinc finger protein 9 (ZNF9) gene (type two).137–139 These repeats act as a sink for RNA binding proteins, resulting in the dysregulation of the (developmentally regulated) splicing of a variety of mRNAs such as the one of tau, the CIC-1 chloride channel, and troponin, explaining the clinical findings in DM patients.140–142 Sequestration of muscleblind-like 1 (MBNL1) appears to be crucial, as MBNL1-deficient mice display a phenotype reminiscent to that of DM.143,144
Major attention has recently been devoted to yet another possible disease mechanism, the contribution of repeat-associated non-ATG (RAN) translation products (Figure 2B).145 This ATG-independent translation from both sense and antisense C9orf72 repeat transcripts gives rise to five different DPR species: poly-(Gly-Ala), poly-(Gly-Arg), poly-(Gly-Pro), poly-(Pro-Arg), and poly-(Pro-Ala).146,147 These DPRs are found in (TDP-43 negative, p62/ubiquitin positive inclusions) the central nervous system of C9orf72 mutation carriers.147,148 When overexpressed in cells, yeast, or Drosophila, some of these DPRs evoke toxicity.149,150 Most but not all reports suggest poly-(Gly-Arg) and poly-(Pro-Arg) to be toxic, possibly through inducing nucleolar stress.148,151 It looks as if the G4C2 repeat toxicity in Drosophila eye is mediated by the generation of DPR species,150 but their pathological significance for the human condition still remains to be demonstrated. Still, even if their pathogenic contribution is limited, they may turn out to be very useful biomarkers.
Cytoskeletal defects in ALS: an emerging theme
With their axons extending more than one meter, motor neurons are highly dependent on axonal transport to shuttle organelles and vesicles between soma and synapses for their proper function and survival.152 Axonal swelling with cytoskeletal disarrangement is a hallmark of ALS pathology.153 Not surprisingly, a number of factors involved in axonal architecture and function have been documented to play a role in ALS (Figure 2C).154
Neurofilaments are essential for normal nerve cell function and are assembled from light (NFL), medium (NFM), and heavy (NFH) subunits. Decreased expression levels of NFL (encoded by the NEFL gene) were previously demonstrated in ALS patients.155 Interestingly, TDP-43 and FUS bind to NEFL mRNA,75,91,156 which is sequestered in stress granules in ALS motor neurons.157 Furthermore, NFH mutations were found in ALS patients.158,159 Overexpression of the intermediate filament peripherin, which is a component of inclusion bodies associated with degenerating motor neurons, induces motor neuron abnormalities in mice,160 and a frameshift deletion in the peripherin gene has been reported in ALS.161 The significance of both types of mutations requires more study.
Dynactin1 (DCTN1) mutations have been identified in a lower motor neuron disease consisting of vocal fold paralysis162 and were later reported in FALS and SALS patients.163 DCTN1 stabilizes the binding of cargos to the motor protein dynein and DCTN1 variants may therefore contribute to the axonal transport deficits seen in ALS.
Notwithstanding, it is the recent discovery of profilin1 (PFN1) mutations as a cause for ALS that strongly points out the possible involvement of the cytoskeleton in ALS pathogenesis.164,165 PFN1 is an important regulator of actin dynamics.166 Mutant PFN1 may contribute to ALS by its reduced actin binding, thus diminished actin polymerization, and subsequent cytoskeletal disturbance.164 In addition, PFN1 mutations may link the cytoskeletal defects and RNA aggregation seen in ALS. Mutant PFN1 colocalizes with and enhances the formation of stress granules.167 As the cytoskeletal machinery is crucial in the formation and disassemblage of RNA-containing stress granules, it is tempting to speculate that mutant PFN1 may play a pivotal role in the impairment of this process. Furthermore, PFN1 mutants form insoluble and ubiquitinated aggregates, which are enhanced by ubiquitin–proteasome impairment.164 These observations further emphasize that the artificially separated pathogenic mechanisms in ALS are highly interrelated.
There is additional evidence for a role of axonal factors in the mechanism of motor neuron degeneration.154 EphA4 expression has been found to inversely correlate with disease onset and survival.168 EphA4 is a receptor in the ephrin axonal repellent system169 and induces cytoskeleton remodeling through RhoA GTPase.170 Its expression in ALS patients was studied because its pharmacological and genetic inhibition rescued the ALS phenotype in zebrafish and increased the survival in ALS mice and rats.168 It was suggested that EphA4 is a determinant of the re-innervating capacity of motor neurons and contributes to the differential vulnerability of these neurons in ALS. Interestingly, ephrin signaling had already been found to be involved in the pathogenic mechanism of mutant vesicle-associated membrane protein-associated protein B and C (VAPB),171 which is a rare cause of ALS.172 Of notice, the alsin gene, in which loss-of-function mutations cause an unusual form of ALS,173 belongs to the RhoA GTPase family.174 These observations suggest that the dynamics of the cytoskeleton and axonal outgrowth may be involved in constituting ALS and its phenotype.
More direct evidence, however, comes from the finding that tubulin alpha 4A (TUBA4A) mutations are associated with ALS.175 TUBA4A mutants disrupt microtubule dynamics and stability, strengthening the hypothesis that alterations of cytoskeletal integrity may have a major role in ALS.175 Interestingly, at least one mutant (W407X) displayed aggregation propensities reminiscent of other ALS-associated mutant proteins.175 Its pathogenic effect may thus be explained in terms of overburdening of the UPS and trapping of tubulin-binding proteins.175 However, the relevance of these aggregates in vivo still remains to be seen.
A genetic basis for SALS and phenotypic variability: a complex puzzle with missing pieces
Although unraveling of the genetic basis of some forms of ALS has provided insights in the mechanisms underlying motor neuron degeneration, many questions remain. One major issue relates to the genetic basis of ALS/FTD that occurs in patients who have no affected family members. Some of this may be Mendelian hereditary as explained earlier. Some of it may be polygenic. Genetic variants may indeed convey susceptibility, while other factors (genetic or environmental) may be needed to establish phenotypic expression. The C9orf72 expansion mutation already points in that direction, as mentioned earlier. Twin studies, GWAS, and the recent exome and whole-genome sequencing efforts address these issues.
Twin studies in ALS
Twin studies are valuable tools to study the genetic and environmental contribution in the etiology of complex disorders.176 Hereby, the frequency of disease occurring in both members of the twin pair is calculated (concordance) whereafter the concordance rates of identical and dizygotic twins are compared.177 High concordance rates in monozygotic twins and much lower concordance rates in dizygotic twins imply a strong genetic influence. Equal concordance in monozygotic and dizygotic twins suggests that environmental factors are more important.177
Twin studies have been employed to test the assumption that there is a genetic component to all ALS. One study included 171 twin pairs by combining the British MND data with data from twins identified via an ALS registry in the United Kingdom and from the National Swedish Twin Registry.26 The heritability of SALS was estimated as 0.61, strongly suggesting that the majority of SALS cases also have a genetic basis. This is however an overestimation compared to two independent studies that applied genome-wide complex trait analysis (GCTA) to unrelated ALS individuals.178,179 GCTA differs from traditional GWAS studies in such a way that the overall effect of multiple single-nucleotide polymorphisms (SNPs), which may be nonsignificant on their own but additive together, can be assessed.180 The heritability estimates of SALS ranged from 20%–25%, implying that about one-fifth instead of more than half of disease risk comes from mutant genes in SALS.
It appears that the methodological differences make a precise estimate of ALS heritability difficult.181 The low number of possible study subjects remains a main limitation for the power of a classical twin study. In addition, recent evidence reveals the environmental, genetic, and epigenetic complexity of twin studies.181,182
GWAS conveying candidate ALS genes: food for thought
GWAS have been successful in identifying the genetic basis for a long list of human diseases. Essentially, this approach screens the genome for SNPs that occur more frequently in patients with a particular disease than in control individuals. Thousands of SNPs across the genome can be examined, making GWAS an interesting tool to identify the variations contributing to a person’s risk and modifying the disease. The most consistent result obtained in GWAS in ALS is the linkage to the chromosome 9p21 locus that later was found to contain the expanded C9orf72 gene.183 Only few of the many other GWAS results in ALS reported could be replicated in different populations.
UNC13A was identified as a risk factor for ALS184,185 and a modifier of ALS survival in general186,187 and in C9orf72 patients.188 UNC13A is a presynaptic protein that regulates the release of neurotransmitters, such as glutamate, at neuromuscular synapses.186 Upon exocytosis, presynaptic vesicles are recruited to the membrane and primed for membrane fusion.189 Disturbance of this priming process as a result of altered UNC13A function could lead to impairment in neurotransmitter release and ultimately to the death of motor neurons.190 As UNC13A regulates glutamate release, this mechanism may support the glutamate excitotoxicity hypothesis in ALS.191
Polymorphisms in the elongator acetyltransferase complex subunit 3 (Elp3) gene have been found to be associated with ALS,192 and with survival after onset in C9orf72 expansion carriers.188 Elp3 is a member of a complex involved in RNA processing through mRNA elongation, histone acetylation, and modification of transfer RNA wobble nucleosides.193 Knockdown of Elp3 resulted in shortening and abnormal branching of motor neuron axons in zebrafish and in synaptic abnormalities in Drosophila.192 The effect of the polymorphisms on Elp3 function is unknown, and how Elp3 affects neuronal function is uncertain. In Drosophila, Elp3 acetylates Bruchpilot, a synaptic active zone protein involved in synaptic vesicle release.194 This may link its mechanism to that of UNC13A, discussed earlier. However, it is unknown whether Elp3 also affects the function of active zone proteins in mammalian cells. It is fairly well possible, given the significance of RNA metabolism as a factor in the mechanism of ALS, that it is the effect of Elp3 on transfer RNA function that explains its effect in ALS.195
Disappointingly, many of the GWAS hits in ALS did not stand the test of replication.196 Several factors may be invoked to explain this. GWAS requires thousands of patient and control samples in order to obtain sufficient statistical power. Recently generated large DNA banks and the public availability of results obtained in individual studies, allowing meta-analyses, are necessary for this.197 Furthermore, in view of the disease heterogeneity explained earlier, it may be necessary to study phenotypically or genetically more homogenous patient populations. Finally, it should be mentioned that the commonly studied SNPs only cover the most common genetic variation4 and that we may have to step away from this common disease/common variant hypothesis in ALS. To that end, new genotyping platforms are currently used to detect rare variants (minor allele frequency <5% instead of >5% for common variants).4
Whole-genome sequencing projects and exome sequencing efforts
It is unlikely that one single locus drives the genetic risk for so-called SALS; a number of different genes may add to the risk, each with a relatively modest contribution.4 Exome and whole-genome sequencing covers most of all variations possible.
The use of exome sequencing, which aims to identify coding variants, led to the discovery of a missense mutation in, for example, the VCP gene that segregated with ALS56 and in the CHCHD10 gene that causes ALS and FTD.198,199 Exome sequencing in parent-case offspring trios has already been employed for the investigation of de novo mutations in SALS.200–202 To this end, de novo mutations have been found in genes encoding chromatin regulators, including the neuronal chromatin remodeling complex (nBAF) component SS18L1 (also known as CREST).203 Furthermore, the usage of exome-wide rare variant burden analysis recently resulted in the discovery of TUBA4A mutations in ALS.175
Whole-genome sequencing represents a promising approach for the detection of rare variants that establish ALS risk. To this end, the “whole-genome sequencing project” aims to sequence over 1,500 genomes, making use of samples available in the UK MND DNA bank. This study collaborates with Project MinE, an international initiative that aims to sequence 15,000 motor neuron disease genomes in several participating countries (including the Netherlands, Belgium, France, the US, and Australia). The detailed clinical information linked to these samples will allow the search for genetic factors that are associated with certain phenotypes such as slow disease progression or extended survival, cognitive involvement, etc.
Disease heterogeneity and disease modifiers: hope or hype?
The finding that the same mutation can be associated with quite variable phenotypes, as mentioned earlier, suggests the existence of (genetic and/or environmental) factors that modify the phenotype. Such factors are important to identify, as they may represent therapeutic targets, which would enable us to tackle disease severity, even without knowing the exact cause of ALS. Genetic factors such as UNC13A, Elp3, and EphA4 have been mentioned earlier. In addition, effects of PGRN,14 PPARGC1A,204 APOE,205 MAO-B,206 KIFAP3,207 ZNF512B,208 VEGF,209 and ATXN2210,211 on the age of onset or survival have been reported.
The intermediate-length CAG expansion in ATXN2 (≥34 repeats, encoding polyglutamine [polyQ]) was initially identified as a cause of spinocerebellar ataxia type 2 (SCA2).212 Afterward, the gene was put forward as a modifier of mutant TDP-43 toxicity in yeast.106 Based on this observation, a genetic study was performed in humans, which showed that a polyQ intermediate expansion (27–33 repeats) on one allele in ATXN2 is a risk factor for ALS.106,213–216 This raises the possibility that SCA2 and ALS represent opposite ends of a clinical spectrum, further underlining that ALS most likely consists of a group of diseases that has motor neuron degeneration in common. In ALS, the expanded repeat alleles are interrupted with 1–3 CAA codons that reside within the CAG repeat.210 The number of interruptions is a determinant of disease onset.210 In addition, patients bearing ≥31 polyQ ATNX2 repeats had a shorter survival than those with <31 repeats.211 Disease-modifying therapies targeting ATXN2 may therefore represent a promising therapeutic approach for ALS.
Finally, a possible modifying effect of VEGF in ALS has been reported. VEGF overexpression217 or intracerebrovascular administration218 in SOD1-based rodent models attenuated onset of paralysis, improved motor performance, and prolonged survival. A clinical trial is ongoing in which VEGFa is delivered intracerebroventricularly using an infusion pump. The clinical significance of all these modifying factors identified in genetic studies or in animal models needs further study. An overview of genes for which there is more than anecdotal evidence is represented in Table 1 and Figure 2.219–237
Therapeutic targets and potential for personalized treatment?
It is hoped for that the discovery of disease-causing and modifying genes in ALS allows the development of tailored therapies based upon the patient’s genetic fingerprint. Approaches for this are emerging. Causal treatments directly target the factor considered to be the cause of ALS and aim to lower the expression of disease-causing proteins or RNA species. An immunological approach is being explored for mutant SOD1-associated ALS. Both passive and active immunization protocols have been shown to have a positive impact on disease progression and to delay the mortality in ALS mice by decreasing the abundance of misfolded SOD1.238–240 Genetic approaches use lentiviral vector-delivered siRNA or use intrathecal delivery of antisense oligonucleotides. They have been successfully studied in rodent models for mutant SOD1-associated ALS.241–243 Based on these findings, a Phase 1 trial was initiated and the intrathecal administration of antisense oligonucleotide against SOD1 was found to be safe and well tolerated.244 This study may pave the way for applying this approach to other forms of genetically determined motor neuron disorders and in fact several neurodegenerative diseases such as Huntington’s disease.245
The generation of iPSC-derived neurons from ALS patients now allows to generate and test novel therapeutic approaches. Some studies report these cells to show a phenotype that may be relevant to what happens in vivo,115,246–252 but more work is needed. One such highly interesting example is the use of neurons from patients with a C9orf72 expansion mutation.252 These were found to develop RNA foci reminiscent of what is seen in vivo. Antisense oligonucleotides targeting the C9orf72 transcript reduced the formation of such RNA foci suggesting that this is an approach to be considered in C9orf72 patients.115,116,247
Conclusion
ALS genetics has made remarkable progress and is essential to gain insight into the complex pathogenic mechanisms leading to motor neuron degeneration and the clinical entity we call ALS. Still, despite the boom of newly found ALS genes, the cause of approximately a third of FALS and most of SALS remains unknown.4 The identification of the remaining genes represents a substantial challenge, for which novel approaches are being explored. The genetic heterogeneity of ALS has led to the notion that therapeutic interventions will need to be personalized. Treatment options based on the genetic cause of ALS are on the way. In the long run, further efforts will presumably enable clinicians to treat ALS patients with the most suited therapy, based on their genetic fingerprint.
Acknowledgments
The authors receive funding from the Fund for Scientific Research Flanders (FWO), the University of Leuven (GOA/11/014), the Interuniversity Attraction Poles Programme of the Belgian Federal Science Policy Office (P7/16), the European Community’s Health Seventh Framework Programme, the Association Belge contre les Maladies neuro-Musculaires (ABMM), the ALS Therapy Alliance, the ALS Association (ALSA; 039CUK and 14-LGCA-181) and the European Research Council, and the European’s Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement number 340429. WR is supported through the E von Behring Chair for Neuromuscular and Neurodegenerative Disorders, the Laevers Fund for ALS Research, and the Een Hart voor ALS and Opening the Future funds of the University of Leuven.
Disclosure
The authors report no conflicts of interest in this work.
References
Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001; 2(11):806–819. | |
Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10(11):661–670. | |
Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344(22):1688–1700. | |
Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17–23. | |
Hugon J. Riluzole and ALS therapy. Wien Med Wochenschr. 1996; 146(9–10):185–187. | |
Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14(4):248–264. | |
Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7(11):603–615. | |
Kew JJ, Goldstein LH, Leigh PN, et al. The relationship between abnormalities of cognitive function and cerebral activation in amyotrophic lateral sclerosis. A neuropsychological and positron emission tomography study. Brain. 1993;116(pt 6):1399–1423. | |
Strong MJ, Yang W. The frontotemporal syndromes of ALS. Clinicopathological correlates. J Mol Neurosci. 2011;45(3):648–655. | |
Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6(11):994–1003. | |
Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73(18):1451–1456. | |
Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. | |
Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. | |
Sleegers K, Brouwers N, Maurer-Stroh S, et al. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology. 2008;71(4):253–259. | |
Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. | |
Rippon GA, Scarmeas N, Gordon PH, et al. An observational study of cognitive impairment in amyotrophic lateral sclerosis. Arch Neurol. 2006;63(3):345–352. | |
Mioshi E, Caga J, Lillo P, et al. Neuropsychiatric changes precede classic motor symptoms in ALS and do not affect survival. Neurology. 2014;82(2):149–155. | |
Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59(7):1077–1079. | |
Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134(pt 9):2582–2594. | |
Régal L, Vanopdenbosch L, Tilkin P, et al. The G93C mutation in superoxide dismutase 1: clinicopathologic phenotype and prognosis. Arch Neurol. 2006;63(2):262–267. | |
Penco S, Lunetta C, Mosca L, et al. Phenotypic heterogeneity in a SOD1 G93D Italian ALS family: an example of human model to study a complex disease. J Mol Neurosci. 2011;44(1):25–30. | |
Kim W, Kim JS, Lee KS, Gwoun YJ, Kim JM, Lee KH. Anticipation and phenotypic heterogeneity in korean familial amyotrophic lateral sclerosis with superoxide dismutase 1 gene mutation. J Clin Neurol. 2007;3(1):38–44. | |
Millecamps S, Boillée S, Le Ber I, et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet. 2012;49(4):258–263. | |
Cooper-Knock J, Shaw PJ, Kirby J. The widening spectrum of C9ORF72-related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathol. 2014;127(3):333–345. | |
Al-Chalabi A, Lewis CM. Modelling the effects of penetrance and family size on rates of sporadic and familial disease. Hum Hered. 2011; 71(4):281–288. | |
Al-Chalabi A, Fang F, Hanby MF, et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry. 2010;81(12):1324–1326. | |
Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: new genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2015;1607:75–93. | |
Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9(3):237–248. | |
Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71(1):35–48. | |
Bendotti C, Marino M, Cheroni C, et al. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog Neurobiol. 2012;97(2):101–126. | |
Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993; 364(6435):362. | |
Chiò A, Traynor BJ, Lombardo F, et al. Prevalence of SOD1 mutations in the Italian ALS population. Neurology. 2008;70(7):533–537. | |
Andersen PM, Nilsson P, Ala-Hurula V, et al. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet. 1995;10(1):61–66. | |
Andersen PM, Forsgren L, Binzer M, et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain. 1996;119(pt 4):1153–1172. | |
Cudkowicz ME, McKenna-Yasek D, Sapp PE, et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol. 1997;41(2):210–221. | |
Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85(1):94–134. | |
Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. | |
Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187(6):761–772. | |
Chen S, Zhang X, Song L, Le W. Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 2012;22(1):110–116. | |
Cheroni C, Marino M, Tortarolo M, et al. Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18(1):82–96. | |
Rakhit R, Cunningham P, Furtos-Matei A, et al. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277(49):47551–47556. | |
Ezzi SA, Urushitani M, Julien JP. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem. 2007;102(1):170–178. | |
Bosco DA, Morfini G, Karabacak NM, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396–1403. | |
Grad LI, Guest WC, Yanai A, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci U S A. 2011;108(39):16398–16403. | |
Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. | |
Ko HS, Uehara T, Tsuruma K, Nomura Y. Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 2004;566(1–3):110–114. | |
Williams KL, Warraich ST, Yang S, et al. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(10):2527. e3–e10. | |
Synofzik M, Maetzler W, Grehl T, et al. Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol Aging. 2012;33(12):2949. e13–e17. | |
Zatloukal K, Stumptner C, Fuchsbichler A, et al. p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160(1):255–263. | |
Nakaso K, Yoshimoto Y, Nakano T, et al. Transcriptional activation of p62/A170/ZIP during the formation of the aggregates: possible mechanisms and the role in Lewy body formation in Parkinson’s disease. Brain Res. 2004;1012(1–2):42–51. | |
Kuusisto E, Salminen A, Alafuzoff I. Early accumulation of p62 in neurofibrillary tangles in Alzheimer’s disease: possible role in tangle formation. Neuropathol Appl Neurobiol. 2002;28(3):228–237. | |
Bjorkoy G, Lamark T, Johansen T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy. 2006;2(2):138–139. | |
Rubino E, Rainero I, Chiò A, et al; TODEM Study Group. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79(15):1556–1562. | |
Le Ber I, Camuzat A, Guerreiro R, et al; French Clinical and Genetic Research Network on FTD/FTD-ALS. SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 2013;70(11):1403–1410. | |
Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–1446. | |
Johnson JO, Mandrioli J, Benatar M, et al; ITALSGEN Consortium. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–864. | |
Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36(4):377–381. | |
Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14(2):117–123. | |
Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153(7):1461–1474. | |
Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201(3):361–372. | |
Albagha OM, Visconti MR, Alonso N, et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet. 2010;42(6):520–524. | |
Rezaie T, Child A, Hitchings R, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295(5557):1077–1079. | |
Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–226. | |
Deng HX, Bigio EH, Zhai H, et al. Differential involvement of optineurin in amyotrophic lateral sclerosis with or without SOD1 mutations. Arch Neurol. 2011;68(8):1057–1061. | |
Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha-induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol. 2007;17(16):1438–1443. | |
Korac J, Schaeffer V, Kovacevic I, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126(pt 2):580–592. | |
Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–808. | |
Parkinson N, Ince PG, Smith MO, et al; MRC Proteomics in ALS Study; FReJA Consortium. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 2006; 67(6):1074–1077. | |
Filimonenko M, Stuffers S, Raiborg C, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179(3):485–500. | |
Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. | |
Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. | |
Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34(6):812–826. | |
Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19(R1):R46–R64. | |
Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4):452–458. | |
Polymenidou M, Lagier-Tourenne C, Hutt KR, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. | |
Buratti E, Romano M, Baralle FE. TDP-43 high throughput screening analyses in neurodegeneration: advantages and pitfalls. Mol Cell Neurosci. 2013;56:465–474. | |
Colombrita C, Zennaro E, Fallini C, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111(4):1051–1061. | |
Kedersha N, Anderson P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans. 2002;30(pt 6):963–969. | |
Dormann D, Haass C. TDP-43 and FUS: a nuclear affair. Trends Neurosci. 2011;34(7):339–348. | |
Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284(30):20329–20339. | |
Laird AS, Van Hoecke A, De Muynck L, et al. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS One. 2010;5(10):e13368. | |
Kraemer BC, Schuck T, Wheeler JM, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119(4):409–419. | |
Feiguin F, Godena VK, Romano G, D’Ambrogio A, Klima R, Baralle FE. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009;583(10):1586–1592. | |
Zhou H, Huang C, Chen H, et al. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010;6(3):e1000887. | |
Wils H, Kleinberger G, Janssens J, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107(8):3858–3863. | |
Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106(44):18809–18814. | |
Stallings NR, Puttaparthi K, Luther CM, Burns DK, Elliott JL. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis. 2010;40(2):404–414. | |
Arnold ES, Ling SC, Huelga SC, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A. 2013;110(8):E736–E745. | |
Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. | |
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. | |
Lagier-Tourenne C, Polymenidou M, Hutt KR, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15(11):1488–1497. | |
Sabatelli M, Conte A, Zollino M. Clinical and genetic heterogeneity of amyotrophic lateral sclerosis. Clin Genet. 2013;83(5):408–416. | |
Yamashita S, Mori A, Sakaguchi H, et al. Sporadic juvenile amyotrophic lateral sclerosis caused by mutant FUS/TLS: possible association of mental retardation with this mutation. J Neurol. 2012;259(6):1039–1044. | |
Dormann D, Rodde R, Edbauer D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 2010;29(16):2841–2857. | |
Johnson JO, Pioro EP, Boehringer A, et al; ITALSGEN Consortium. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17(5):664–666. | |
Feit H, Silbergleit A, Schneider LB, et al. Vocal cord and pharyngeal weakness with autosomal dominant distal myopathy: clinical description and gene localization to 5q31. Am J Hum Genet. 1998;63(6):1732–1742. | |
Senderek J, Garvey SM, Krieger M, et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet. 2009;84(4):511–518. | |
Belgrader P, Dey R, Berezney R. Molecular cloning of matrin 3. A 125-kilodalton protein of the nuclear matrix contains an extensive acidic domain. J Biol Chem. 1991;266(15):9893–9899. | |
Salton M, Elkon R, Borodina T, et al. Matrin 3 binds and stabilizes mRNA. PLoS One. 2011;6(8):e23882. | |
Kim HJ, Kim NC, Wang YD, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495(7442):467–473. | |
Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280(45):37572–37584. | |
Ritson GP, Custer SK, Freibaum BD, et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci. 2010;30(22):7729–7739. | |
Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011;147(3):498–508. | |
Munch C, O’Brien J, Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A. 2011;108(9):3548–3553. | |
La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11(4):247–258. | |
Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–1075. | |
Blauw HM, van Rheenen W, Koppers M, et al. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21(11):2497–2502. | |
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. | |
Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. | |
Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29(4):499–503. | |
van Blitterswijk M, DeJesus-Hernandez M, Rademakers R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol. 2012;25(6):689–700. | |
Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135(pt 3):693–708. | |
Byrne S, Elamin M, Bede P, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 2012;11(3):232–240. | |
van Blitterswijk M, van Es MA, Hennekam EA, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21(17):3776–3784. | |
Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–428. | |
Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530–E4539. | |
Lee YB, Chen HJ, Peres JN, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5(5):1178–1186. | |
Mizielinska S, Lashley T, Norona FE, et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126(6):845–857. | |
Zu T, Liu Y, Bañez-Coronel M, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110(51):E4968–E4977. | |
Waite AJ, Bäumer D, East S, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35(7):1779. e5–1779. e13. | |
Gijselinck I, Van Langenhove T, van der Zee J, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11(1):54–65. | |
Ciura S, Lattante S, Le Ber I, et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013;74(2):180–187. | |
Xi Z, Zinman L, Moreno D, et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet. 2013;92(6):981–989. | |
Evans-Galea MV, Carrodus N, Rowley SM, et al. FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann Neurol. 2012;71(4):487–497. | |
Sutcliffe JS, Nelson DL, Zhang F, et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992; 1(6):397–400. | |
Thornton CA, Wymer JP, Simmons Z, McClain C, Moxley RT 3rd. Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat Genet. 1997;16(4):407–409. | |
López Castel A, Nakamori M, Tomé S, et al. Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum Mol Genet. 2011;20(1):1–15. | |
Klesert TR, Otten AD, Bird TD, Tapscott SJ. Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat Genet. 1997;16(4):402–406. | |
Liu EY, Russ J, Wu K, et al. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 2014;128(4):525–541. | |
Fratta P, Poulter M, Lashley T, et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013;126(3):401–409. | |
Cooper-Knock J, Higginbottom A, Connor-Robson N, et al. C9ORF72 transcription in a frontotemporal dementia case with two expanded alleles. Neurology. 2013;81(19):1719–1721. | |
Harms MB, Cady J, Zaidman C, et al. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging. 2013;34(9):2234. e13–e19. | |
Mori K, Lammich S, Mackenzie IR, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125(3):413–423. | |
Cooper-Knock J, Walsh MJ, Higginbottom A, et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 2014;137(pt 7):2040–2051. | |
Xu Z, Poidevin M, Li X, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110(19):7778–7783. | |
Fratta P, Mizielinska S, Nicoll AJ, et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. 2012;2:1016. | |
Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255(5049):1253–1255. | |
Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293(5531):864–867. | |
Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;69(2):385. | |
Sergeant N, Sablonnière B, Schraen-Maschke S, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet. 2001;10(19):2143–2155. | |
Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280(5364):737–741. | |
Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10(1):35–44. | |
Suenaga K, Lee KY, Nakamori M, et al. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PLoS One. 2012;7(3):e33218. | |
Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302(5652):1978–1980. | |
Pearson CE. Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet. 2011;7(3):e1002018. | |
Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–646. | |
Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. | |
May S, Hornburg D, Schludi MH, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128(4):485–503. | |
Wen X, Tan W, Westergard T, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84(6):1213–1225. | |
Mizielinska S, Grönke S, Niccoli T, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345(6201):1192–1194. | |
Zhang YJ, Jansen-West K, Xu YF, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128(4):505–524. | |
Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68(4):610–638. | |
Hirano A. Cytopathology of amyotrophic lateral sclerosis. Adv Neurol. 1991;56:91–101. | |
Schmidt ER, Pasterkamp RJ, van den Berg LH. Axon guidance proteins: novel therapeutic targets for ALS? Prog Neurobiol. 2009; 88(4):286–301. | |
Bergeron C, Beric-Maskarel K, Muntasser S, Weyer L, Somerville MJ, Percy ME. Neurofilament light and polyadenylated mRNA levels are decreased in amyotrophic lateral sclerosis motor neurons. J Neuropathol Exp Neurol. 1994;53(3):221–230. | |
Strong MJ, Volkening K, Hammond R, et al. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35(2):320–327. | |
Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009;1305:168–182. | |
Tomkins J, Usher P, Slade JY, et al. Novel insertion in the KSP region of the neurofilament heavy gene in amyotrophic lateral sclerosis (ALS). Neuroreport. 1998;9(17):3967–3970. | |
Figlewicz DA, Krizus A, Martinoli MG, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3(10):1757–1761. | |
Beaulieu JM, Nguyen MD, Julien JP. Late onset of motor neurons in mice overexpressing wild-type peripherin. J Cell Biol. 1999;147(3):531–544. | |
Gros-Louis F, Larivière R, Gowing G, et al. A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J Biol Chem. 2004;279(44):45951–45956. | |
Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33(4):455–456. | |
Münch C, Sedlmeier R, Meyer T, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63(4):724–726. | |
Wu CH, Fallini C, Ticozzi N, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488(7412):499–503. | |
Smith BN, Vance C, Scotter EL, et al. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol Aging. 2015;36(3):1602. e17–e27. | |
Mockrin SC, Korn ED. Acanthamoeba profilin interacts with G-actin to increase the rate of exchange of actin-bound adenosine 5′-triphosphate. Biochemistry. 1980;19(23):5359–5362. | |
Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci. 2014;34(24):8083–8097. | |
Van Hoecke A, Schoonaert L, Lemmens R, et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med. 2012;18(9):1418–1422. | |
Klein R. Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat Neurosci. 2009;12(1):15–20. | |
Winning RS, Ward EK, Scales JB, Walker GK. EphA4 catalytic activity causes inhibition of RhoA GTPase in Xenopus laevis embryos. Differentiation. 2002;70(1):46–55. | |
Tsuda H, Han SM, Yang Y, et al. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell. 2008;133(6):963–977. | |
Nishimura AL, Mitne-Neto M, Silva HC, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004; 75(5):822–831. | |
Yang Y, Hentati A, Deng HX, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29(2):160–165. | |
Linseman DA, Loucks FA. Diverse roles of Rho family GTPases in neuronal development, survival, and death. Front Biosci. 2008;13: 657–676. | |
Smith BN, Ticozzi N, Fallini C, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84(2):324–331. | |
Hrubec Z, Robinette CD. The study of human twins in medical research. N Engl J Med. 1984;310(7):435–441. | |
Graham AJ, Macdonald AM, Hawkes CH. British motor neuron disease twin study. J Neurol Neurosurg Psychiatry. 1997;62(6):562–569. | |
Keller MF, Ferrucci L, Singleton AB, et al. Genome-wide analysis of the heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2014;71(9):1123–1134. | |
Fogh I, Ratti A, Gellera C, et al; SLAGEN Consortium and Collaborators. A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum Mol Genet. 2014;23(8):2220–2231. | |
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. | |
Talbot K. Do twin studies still have anything to teach us about the genetics of amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry. 2010;81(12):1299–1300. | |
Kaminsky ZA, Tang T, Wang SC, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41(2):240–245. | |
Laaksovirta H, Peuralinna T, Schymick JC, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9(10):978–985. | |
Diekstra FP, van Vught PW, van Rheenen W, et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(3):630. e3–e8. | |
Chiò A, Mora G, Restagno G, et al. UNC13A influences survival in Italian amyotrophic lateral sclerosis patients: a population-based study. Neurobiol Aging. 2013;34(1):e1–e5. | |
van Es A, Veldink JH, Saris CG, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41(10):1083–1087. | |
Shatunov A, Mok K, Newhouse S, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9(10):986–994. | |
van Blitterswijk M, Mullen B, Wojtas A, et al. Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol Neurodegener. 2014;9:38. | |
Zikich D, Mezer A, Varoqueaux F, et al. Vesicle priming and recruitment by ubMunc13-2 are differentially regulated by calcium and calmodulin. J Neurosci. 2008;28(8):1949–1960. | |
Varoqueaux F, Sons MS, Plomp JJ, Brose N. Aberrant morphology and residual transmitter release at the Munc13-deficient mouse neuromuscular synapse. Mol Cell Biol. 2005;25(14):5973–5984. | |
Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762(11–12):1068–1082. | |
Simpson CL, Lemmens R, Miskiewicz K, et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet. 2009;18(3):472–481. | |
Creppe C, Malinouskaya L, Volvert ML, et al. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell. 2009;136(3):551–564. | |
Miskiewicz K, Jose LE, Bento-Abreu A, et al. ELP3 controls active zone morphology by acetylating the ELKS family member Bruchpilot. Neuron. 2011;72(5):776–788. | |
Selvadurai K, Wang P, Seimetz J, Huang RH. Archaeal Elp3 catalyzes tRNA wobble uridine modification at C5 via a radical mechanism. Nat Chem Biol. 2014;10(10):810–812. | |
Schymick JC, Talbot K, Traynor BJ. Genetics of sporadic amyotrophic lateral sclerosis. Hum Mol Genet. 2007;16(Spec No 2):R233–R242. | |
Gwinn K, Corriveau RA, Mitsumoto H, et al; ALS Research Group. Amyotrophic lateral sclerosis: an emerging era of collaborative gene discovery. PLoS One. 2007;2(12):e1254. | |
Johnson JO, Glynn SM, Gibbs JR, et al. Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain. 2014;137(pt 12):e311. | |
Bannwarth S, Ait-El-Mkadem S, Chaussenot A, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014;137(pt 8):2329–2345. | |
DeJesus-Hernandez M, Kocerha J, Finch N, et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat. 2010;31(5):E1377–E1389. | |
Chiò A, Calvo A, Moglia C, et al. A de novo missense mutation of the FUS gene in a “true” sporadic ALS case. Neurobiol Aging. 2011;32(3):553. e23–e26. | |
Alexander MD, Traynor BJ, Miller N, et al. “True” sporadic ALS associated with a novel SOD-1 mutation. Ann Neurol. 2002;52(5):680–683. | |
Chesi A, Staahl BT, Jovicic A, et al. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci. 2013;16(7):851–855. | |
Eschbach J, Schwalenstöcker B, Soyal SM, et al. PGC-1alpha is a male-specific disease modifier of human and experimental amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22(17):3477–3484. | |
Zetterberg H, Jacobsson J, Rosengren L, Blennow K, Andersen PM. Association of APOE with age at onset of sporadic amyotrophic lateral sclerosis. J Neurol Sci. 2008;273(1–2):67–69. | |
Orrù S, Mascia V, Casula M, et al. Association of monoamine oxidase B alleles with age at onset in amyotrophic lateral sclerosis. Neuromuscul Disord. 1999;9(8):593–597. | |
Landers JE, Melki J, Meininger V, et al. Reduced expression of the kinesin-associated protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2009;106(22):9004–9009. | |
Tetsuka S, Morita M, Iida A, Uehara R, Ikegawa S, Nakano I. ZNF512B gene is a prognostic factor in patients with amyotrophic lateral sclerosis. J Neurol Sci. 2013;324(1–2):163–166. | |
Lambrechts D, Storkebaum E, Morimoto M, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34(4):383–394. | |
Yu Z, Zhu Y, Chen-Plotkin AS, et al. PolyQ repeat expansions in ATXN2 associated with ALS are CAA interrupted repeats. PLoS One. 2011;6(3):e17951. | |
Chiò A, Calvo A, Moglia C, et al. ATXN2 polyQ intermediate repeats are a modifier of ALS survival. Neurology. 2015;84(3):251–258. | |
Sanpei K, Takano H, Igarashi S, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14(3):277–284. | |
Van Damme P, Veldink JH, van Blitterswijk M, et al. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology. 2011;76(24):2066–2072. | |
Ross OA, Rutherford NJ, Baker M, et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum Mol Genet. 2011;20(16):3207–3212. | |
Lee T, Li YR, Ingre C, et al. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum Mol Genet. 2011;20(9):1697–1700. | |
Daoud H, Belzil V, Martins S, et al. Association of long ATXN2 CAG repeat sizes with increased risk of amyotrophic lateral sclerosis. Arch Neurol. 2011;68(6):739–742. | |
Wang Y, Mao XO, Xie L, et al. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J Neurosci. 2007;27(2):304–307. | |
Storkebaum E, Lambrechts D, Dewerchin M, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8(1):85–92. | |
Seripa D, Bizzarro A, Panza F, et al. The APOE gene locus in frontotemporal dementia and primary progressive aphasia. Arch Neurol. 2011;68(5):622–628. | |
Diekstra FP, Van Deerlin VM, van Swieten JC, et al. C9orf72 and UNC13A are shared risk loci for amyotrophic lateral sclerosis and frontotemporal dementia: a genome-wide meta-analysis. Ann Neurol. 2014;76(1):120–133. | |
Corcia P, Camu W, Halimi JM, et al. SMN1 gene, but not SMN2, is a risk factor for sporadic ALS. Neurology. 2006;67(7):1147–1150. | |
Millecamps S, De Septenville A, Teyssou E, et al; French Research Network on FTD and FTD-ALS. Genetic analysis of matrin 3 gene in French amyotrophic lateral sclerosis patients and frontotemporal lobar degeneration with amyotrophic lateral sclerosis patients. Neurobiol Aging. 2014;35(12):2882. e13–e15. | |
Iida A, Takahashi A, Kubo M, et al. A functional variant in ZNF512B is associated with susceptibility to amyotrophic lateral sclerosis in Japanese. Hum Mol Genet. 2011;20(18):3684–3692. | |
Leung CL, He CZ, Kaufmann P, et al. A pathogenic peripherin gene mutation in a patient with amyotrophic lateral sclerosis. Brain Pathol. 2004;14(3):290–296. | |
Meyer T, Schwan A, Dullinger JS, et al. Early-onset ALS with long-term survival associated with spastin gene mutation. Neurology. 2005;65(1):141–143. | |
Hadano S, Hand CK, Osuga H, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29(2):166–173. | |
Chow CY, Landers JE, Bergren SK, et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84(1):85–88. | |
Orlacchio A, Babalini C, Borreca A, et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain. 2010;133(pt 2):591–598. | |
Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004;74(6):1128–1135. | |
Couthouis J, Hart MP, Erion R, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21(13):2899–2911. | |
Couthouis J, Hart MP, Shorter J, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A. 2011;108(52):20881–20890. | |
Mitchell J, Paul P, Chen HJ, et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc Natl Acad Sci U S A. 2010;107(16):7556–7561. | |
Greenway MJ, Andersen PM, Russ C, et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38(4):411–413. | |
Wills AM, Cronin S, Slowik A, et al. A large-scale international meta-analysis of paraoxonase gene polymorphisms in sporadic ALS. Neurology. 2009;73(1):16–24. | |
Goodall EF, Greenway MJ, van Marion I, Carroll CB, Hardiman O, Morrison KE. Association of the H63D polymorphism in the hemochromatosis gene with sporadic ALS. Neurology. 2005;65(6):934–937. | |
Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42(3):234–239. | |
Cirulli ET, Lasseigne BN, Petrovski S, et al; FALS Sequencing Consortium. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436–1441. | |
Liu HN, Tjostheim S, Dasilva K, et al. Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J Neurosci. 2012;32(26):8791–8799. | |
Gros-Louis F, Soucy G, Lariviere R, Julien JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem. 2010;113(5):1188–1199. | |
Urushitani M, Ezzi SA, Julien JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104(7):2495–2500. | |
Smith RA, Miller TM, Yamanaka K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116(8):2290–2296. | |
Raoul C, Abbas-Terki T, Bensadoun JC, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11(4):423–428. | |
Ralph GS, Radcliffe PA, Day DM, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11(4):429–433. | |
Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–442. | |
Lu XH, Yang XW. “Huntingtin holiday”: progress toward an antisense therapy for Huntington’s disease. Neuron. 2012;74(6):964–966. | |
Zhang Z, Almeida S, Lu Y, et al. Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS One. 2013;8(10):e76055. | |
Sareen D, O’Rourke JG, Meera P, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. | |
Egawa N, Kitaoka S, Tsukita K, et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med. 2012;4(145):145ra104. | |
Dimos JT, Rodolfa KT, Niakan KK, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–1221. | |
Burkhardt MF, Martinez FJ, Wright S, et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. 2013;56:355–364. | |
Bilican B, Serio A, Barmada SJ, et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A. 2012;109(15):5803–5808. | |
Almeida S, Gascon E, Tran H, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126(3):385–399. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.