Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
The Exercise Training Modulatory Effects on the Obesity-Induced Immunometabolic Dysfunctions
Authors Soltani N ![]() , Marandi SM, Kazemi M, Esmaeil N
, Marandi SM, Kazemi M, Esmaeil N ![]()
Received 17 October 2019
Accepted for publication 18 February 2020
Published 19 March 2020 Volume 2020:13 Pages 785—810
DOI https://doi.org/10.2147/DMSO.S234992
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Nakisa Soltani,1 Sayed Mohammad Marandi,1 Mohammad Kazemi,2 Nafiseh Esmaeil3
1Department of Exercise Physiology, Faculty of Sport Sciences, University of Isfahan, Isfahan, Iran; 2Department of Genetics and Molecular Biology, Isfahan University of Medical Sciences, Isfahan, Iran; 3Department of Immunology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
Correspondence: Nafiseh Esmaeil
Department of Immunology, School of Medicine, Isfahan University of Medical Sciences, Isfahan 81744-176, Iran
Tel +98 31 37929097
Fax +98 3113 7929031
Email [email protected]
Sayed Mohammad Marandi
Department of Exercise Physiology, Faculty of Sport Sciences, University of Isfahan, Isfahan, Iran
Tel +983137932358
Fax +983136687572
Email [email protected]
Abstract: Reduced physical activity rate in people’s lifestyle is a global concern associated with the prevalence of health disorders such as obesity and metabolic disturbance. Ample evidence has indicated a critical role of the immune system in the aggravation of obesity. The type, duration, and production of adipose tissue-released mediators may change subsequent inactive lifestyle-induced obesity, leading to the chronic systematic inflammation and monocyte/macrophage (MON/MФ) phenotype polarization. Preliminary adipose tissue expansion can be inhibited by changing the lifestyle. In this context, exercise training is widely recommended due to a definite improvement of energy balance and the potential impacts on the inflammatory signaling cascades. How exercise training affects the immune system has not yet been fully elucidated, because its anti-inflammatory, pro-inflammatory, or even immunosuppressive impacts have been indicated in the literature. A thorough understanding of the mechanisms triggered by exercise can suggest a new approach to combat meta-inflammation-induced metabolic diseases. In this review, we summarized the obesity-induced inflammatory pathways, the roles of MON/MФ polarization in adipose tissue and systemic inflammation, and the underlying inflammatory mechanisms triggered by exercise during obesity.
Keywords: exercise training, immune system, adipose tissue, toll-like receptors, macrophage polarization, obesity, meta-inflammation
Introduction
A recent revival in two fields of metabolism and immunology, known as immunometabolism, links metabolic status with systemic inflammation.1 Discovering the tumor necrosis factor α (TNFα) production in adipose tissue (AT) was the first step towards developing the immunometabolism field.2 Afterwards, uncovering the infiltration of macrophages (MФ) into white adipose tissue (WAT) shed light on the immunometabolism field.3 AT acts as a sensor of the metabolic state and an activator of endocrine organs, due in part to the secretion of more than 50 bioactive factors named adipokines.1,4 Adipocytes, regarded as AT parenchymal cells, are the primary sources of AT energy storage and endocrine function. Maintaining AT homeostasis is managed by an interplay between immune, stromal (fibroblasts, adipocyte precursors, endothelial and endothelial smooth muscle cells), and parenchymal cells, named stromal vascular function (SVF).1,5,6 Accordingly, the immune system preserves the homeostasis through the inhibition of inflammation and control of AT remodeling.7
An imbalanced lifestyle leads to the storage of excess calories in adipocytes, adipocytes hypertrophy, and WAT expansion which induces the extracellular space limitation and micro hypoxia.8 Following these morphologic changes, the expression of stressful ligands is promoted by adipocytes and SVF.9 Eventually, these events induce the impairment of AT homeostasis causing the development of a chronic low-grade systemic inflammation (LGSI) and meta-inflammation, which leads to cardiometabolic diseases.2,10 Meta-inflammation, however, is considered to be different from a typical inflammation due to the type of activators, location of recruited immune cells, and duration and/or intensity of the immune system activities during an inflammatory state8 (Figure 1).
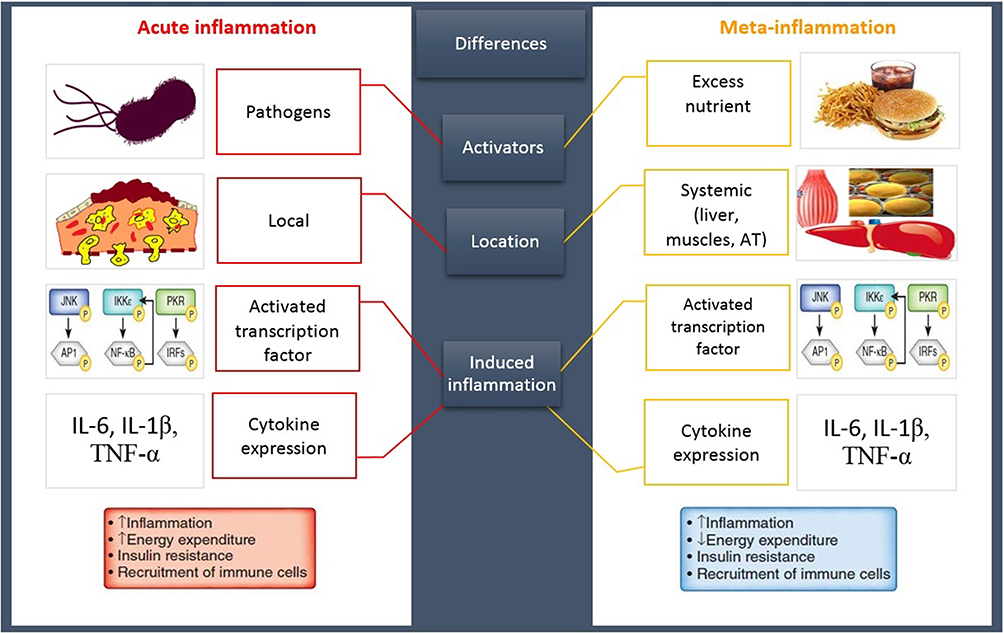 |
Figure 1 A classic inflammation VS meta-inflammation. Both pathways induce an inflammatory state following the upregulation of same inflammatory genes and cytokines. An acute inflammation has been promoted by invasive exogenous pathogens leading to the stimulation of immune system that induces a high-intensity short-term local inflammation. In contrast, a meta-inflammation has been developed by the excess nutrient, endogen stimulants, causing immune system provocation and promoting a chronic LGSI. Abbreviations: AT, adipose tissue; IL, interleukin; TNFα, tumor necrosis factor α; JNK, c-Jun N-terminal kinase; IKK, inhibitor of kappa light polypeptide gene enhancer in B-cells kinase; PKR, protein kinase R; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IRF, interferon regulatory factor. |
In recent years, due to uncovering the lower basal levels of inflammatory cells and cytokines among physically active individuals, exercise therapy has been frequently prescribed as a practical non-invasive approach for treating metabolic diseases.11,12 Physical activities promote several anti-inflammatory mechanisms leading to improvements in metabolic status and inflammatory profiles.13–15 Although doing an exhausting exercise like a marathon has been reported to have inflammatory features as a result of skeletal muscle damages, its valuable anti-inflammatory functions have been recognized even without an alteration of body weight.16,17 In this review, we meticulously reviewed recent immunometabolism concerns including adipose tissue macrophages (ATM) phenotypes and progenitors (monocytes) and their alteration during obesity and after exercise training. Then, we specifically described toll-like receptor (TLR) 4 signaling and its negative regulators as an important pathway in inflammation. Finally, we discussed whether inflammatory cells, cytokines, and TLR4 mediators have been affected by exercise training, suggesting the alteration of obesity-induced inflammatory status.
The Role of the Immune System in Adipose Tissue Homeostasis
A scientific revolution has been developed by revealing a strong association between the nutrient sensors and immune signals.18 Different types of immune cells accumulated in the lean adipose tissue (LAT) induce type 2 immune responses to inhibit the inflammatory pathways and maintain the tissue homeostasis. This is characterized by the anti-inflammatory cytokine accumulation, such as interleukin (IL)-5/13/4/10.19 The main source of IL-4 production is eosinophils (EOSs),20 and the visceral adipose tissue-resident EOSs survival is dependent on IL-5 produced by innate lymphoid cells-2 (ILC-2).21 Additionally, MФs are the prevalent cells in LAT, and M2 macrophages have anti-inflammatory features which mainly generate the anti-inflammatory mediators such as IL-10.22 Conversely, reduced EOSs in visceral adipose tissue23 have been accompanied by diminished frequencies of adipose tissue anti-inflammatory macrophages (M2ATM) in rats.20,23 Notably, exclusion of any part of the ILC2/EOS/M2ATM axis leads to the increase of the inflammatory cytokine levels, which develops insulin resistance (IR) in VAT. Several studies have shown that this axis is a critical regulatory mechanism in AT homeostasis.20,21,24 Additionally, VAT endothelial cells and fibroblasts predominately produce IL-33 belonging to the IL-1 superfamily. This cytokine preserves the AT homeostasis by maintaining the type 2 immune cells such as ILC2 and M2ATM.25 Interestingly, IL-33 receptor-blocking has reduced EOSs frequencies in VAT.26 Its exogenous consumption has been accompanied by an increase in ILCs and EOSs as well as IL-5 values.21 Besides, the invariant-chain natural killer T cells (iNKT), pronounced as the second group of immune cells, play an effective role in the maintenance of anti-inflammatory ATM levels. Surprisingly, iNKT cells omission has reduced IL-13 and IL-4 levels and increased inflammatory ATM in AT.27 On the other hand, the regulatory T cells (Treg) are a population of T cells directly involved in the inhibition of AT inflammation. These cells rely on the production of IL-33 in AT and produce anti-inflammatory cytokines like IL-10 to suppress the inflammatory immune cells activation.28,29 Also, IR improvement has been witnessed following a Treg cell transition to T cell-deficient animals with a diet-induced obesity (DIO).30 In addition to immune cells, adipocytes promote the LAT homeostasis by producing the anti-inflammatory adipokines such as adiponectin. The adiponectin expression reduces the inflammatory MФs (M1) rate, and the high levels of its receptors are expressed in M2. Adiponectin develops its immunomodulating impacts by IL-10 induction, interleukin-1 receptor antagonist (IL-1Ra) production, and nuclear factor-kappa B (NF-κB) pathway inhibition in M2ATM.31
In this sense, obesity-induced adipose tissue morphologic alterations affect the number and phenotype of immune cells. Preserving the M2ATM phenotype which predominantly depends on the anti-inflammatory cytokines and type 2 immune responses seems to have a critical role in modulating an inflammatory state in the AT.
Alteration of Macrophage Characters as a Result of Obesity-Induced Low-Grade Systemic Inflammation
A high caloric diet promotes many changes in AT which induce AT remodeling. Studies have shown that these changes lead to the formation of a significant number of crown-like structures (CLS).32,33 These structures have been demonstrated as a single dead adipocyte in human and rat obese adipose tissue (OAT) surrounded by MФs.34,35 It is noticed that CLSs are the prominent characters of AT homeostasis disturbance which lead to LGSI.8,34 In this context, the obesity classification based on the numbers of metabolic syndrome (MetS) disorders correlates with the percentage of CLSs.5 Furthermore, the association of CLSs with IR,36 cardiovascular diseases (CVD), and cancer prognosis32 has been reported. Importantly, the presence of dead adipocytes persists even up to 30 folds in OAT, while ATMs prominently gather around these cells.33
MФs, the heterogeneous group of innate immune cells, are the integral component of mammal tissues. More than 10–15% of cells in all tissues are MФs, which have a hematopoietic or an embryonic origin.32,37 Indeed, MФs are the strong arm of the innate immunity and perform a wide range of functions through phenotype distinction. Due to their phenotype polarization in a different environment and pathologic condition, they can adopt distinct functional phenotypes.38
In AT, MФs play multiple roles in host defense (via formation and elimination of an inflammatory response), tissue development, monitoring of tissue alternations (by sensing invading molecules with their various sensors) and particularly maintaining tissue homeostasis (via clearance of aged and apoptotic cell and/or tissue remodeling and repairing).32,37 Thus, it seems that ATMs act as a significant contributor in AT functions. The entire adipocyte counts are defined during childhood and remain unchanged during the lifespan.39 Therefore, a cycle of cell death and replacement of adipocytes regularly occurs in AT, where ATMs have pivotal roles in these processes.35 In other words, ATMs have considerable capacity for both eliminating the dead adipocytes and inhibiting the CLS formation in AT homeostasis.35 Although ATMs are the qualified phagocytic cells, they play a significant role during acute metabolic functions. A rapid ATM recruitment is revealed in AT during lipolysis. These cells acquire an inflammatory phenotype and start lipid buffering to supervise the gradient of the lipid released to circulation gradually by collecting and depositing excess released fatty acids.40 Both increased MФs accumulation in AT (from 10% in LAT to 40% in OAT) and/or lipid-enriched MФs in CLSs have been shown during obesity.3 Therefore, elevated serum levels of non-esterified fatty acid (NEFA), cholesterol, and lipopolysaccharide (LPS) are commonly found in obese individuals.41 It indicates a completely unsuccessful attempt of ATMs in buffering AT lipids.35,42 In this situation, the integrated interaction between the adipocytes and MФs of CLSs leads to generating and maintaining the chronic LGSI.33
As a result of the expression of chemoattractant factors, such as C-C motif ligand (CCL) 2/5, C-X-C motif ligand (CXCL) 1, and IL-6 in OAT, a cascade of immune cells moves toward the OAT. Interestingly, production of CCL2/monocyte chemoattractant protein-1 (MCP-1) has been frequently shown in adipocytes during obesity. Following the interplay between CCL2 and its receptor (CCR2) on monocytes (MONs), MONs chemotaxis and MФ accumulation are promoted constantly.35,43 IL-6 is known as a pro-inflammatory cytokine. The IL-6 receptor (IL-6R) trans-signaling develops through the proteolysis of IL-6R from the invading neutrophils (NEUs) to the AT. The soluble form of IL-6R (sIL-6R) provokes recruitment of other MONs chemoattractant factors including CCL2, CCL8, CXCL5, and CXCL6.44 Moreover, enhanced MON to MФ differentiation has been reported in IL-6 trans-signaling, leading to increased production of macrophage-colony stimulating factor (M-CSF) receptor (M-CSFR).45 Additionally, ATM recruitment has been decreased after high-fat diet (HFD) through blocking of IL-6 trans-signaling.46 The fractalkine, named C-X3-C motif ligand (CX3CL) 1, is also another adipokine which is associated with diabetes. Several investigators have demonstrated a direct relationship between fractalkine and glucose and insulin levels. Thus, the serum levels of CX3CL1 could be anticipated in MetS progression.47,48 Additionally, the fractalkine receptor (CX3CR1) has been regarded as an impressive mediator for recruiting the leukocyte/MONs into the AT during obesity.49 Interestingly, prescribing TNFα inhibitor to reduce inflammation is suggested for individuals with obesity and diabetes since it reduces the circulating plasma levels of other inflammatory factors like C-reactive protein (CRP) and IL-6.50 Furthermore, omission of the TNFα converting enzyme (TACE) in OB mice has revealed protecting functions of TACE against IR and T2D so that it can be considered a potential treatment approach in obesity-induced metabolic disorders.51
Although the control of MONs recruitment in AT is essential to inhibit LGSI, the expression of inflammatory cytokines (IL-6/1β and TNFα) from hypertrophied adipocytes and obesity-induced serum lipids at high levels such as free fatty acids (FFAs) can also promote the macrophage polarization. These new MФs have distinct features from resident ones.32,52 Indeed, MФs can perform their functions by an intricate phenotype distinction from an anti-inflammatory healing/proliferating phenotype (M2 MФs, alternative) to an inflammatory inhibiting/killing phenotype (M1 MФs, classical).53 Many studies have shown influential regulatory cascades connected to M1/M2 polarization. At the cell level, these pathways are intracellular signals promoted by regulatory molecules, including ligands, receptors, kinases, and cytokines.32,54,55 The M1/M2 pattern describes a various integrated program of MФ activation and function and demonstrates a better understanding of the MФ activities.37,56
The axis of M2 polarization refers to the type 2 cytokines, including IL-4/10/13 that are activated by recruiting anti-inflammatory transcription factors, while the M1 polarization axis is activated by both pathogen-associated molecular patterns (PAMPs) like LPS and damage-associated molecular patterns (DAMPs) including oxidized-low-density lipoprotein (ox-LDL) and FFAs32,42,56 (Table 1). Indeed, the M1 relates to an inflammatory profile because of stimulating the inflammatory cytokine expression, which is accompanied by a destructive function in tissues that either derive or maintain chronic LGSI and IR consequently.56 Remarkably, the systemic IR is generated by both changing insulin receptor signaling in metabolic tissues33 and activating inflammatory mediators such as c-Jun N-terminal kinases (JNK)-1/2,57 I kappa-B kinase (IκBK),58 extracellular signal-regulated kinase (ERK)-1/2,59 and mitogen-activated protein kinase (MAPK) in MФs.60 However, the M2 group, which has an extreme phagocytic capacity and immune cell suppression capability, preserves the extracellular matrix components and AT homeostasis.56 The ratio of M2: M1 (4:1) in LAT demonstrates the ability of the tissue to maintain the homeostasis.61
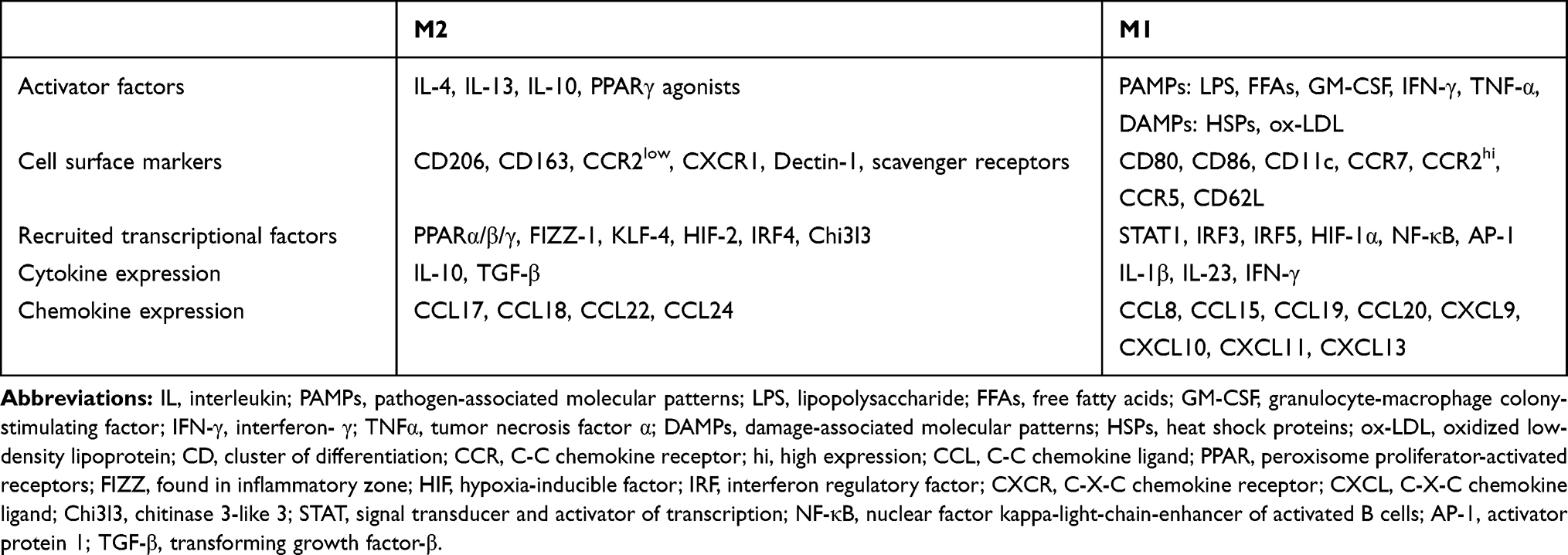 |
Table 1 M1/M2 Macrophage Differences |
Initiation of Adipose Tissue Inflammation
Considering the histological characteristics, AT inflammation has been determined by an increase in AT-infiltrating immune cells, mainly MФs. Moreover, cytotoxic T lymphocyte (CTL) accumulation and a decrease in NEU frequencies have been found during inflammation in AT.8 Interestingly, the early stage of HFD-induced obesity is specified due to higher lipid level of adipocytes and accumulation of immune cells in VAT.7 In obesity, the LGSI persists following the impairment of a feed-forward cycle in immune cell recruitment. In other words, an increase in inflammatory cytokines leads to the recruitment, infiltration, differentiation, and activation of a higher number of immune cells. Animal surveys have reported that accumulated cells in AT following the first week of HFD are NEUs, MФs, and natural killer cells (NK cells).59 Indeed, HFD-induced microenvironment changes in AT are accompanied by both the accumulations of chemoattractive inflammatory proteins and the alterations of AT oxygen consumption. These acute stresses are critical events in NEU recruitment during the first few days of HFD.62 However, the other immune cells (ATM and NK cells) accumulate with a delay following the HFD.7 Circulating NEUs are the main source of infiltrated NEUs in AT63 even though the NK cells may originate from the proliferation of tissue-resident cells.64 The MФ influx is more complicated than NEU and NK cells because of the proliferation of tissue-resident MФs65 and their accelerated infiltration in VAT during chronic inflammation.66
Although the immune cell infiltration always induces the inflammatory factors in OAT, the phenotype of recruited leukocytes can influence the MФ polarity. Accordingly, the leukocyte subclasses (considered the VAT inflammatory markers), systemic inflammation, and obesity-related cardiometabolic disease risk factors have recently attracted researchers’ attention.
From Monocyte to M1/M2 Macrophage Differentiation
Sub-Classes of Receptors-Determined Monocyte Features
Cell biology investigations have frequently published the pleiotropic functions of MONs and MФs. MONs constitute 2–8% of the entire circulating leukocytes in bone marrow, spleen, and blood.67 Typical features of these cells include irregular cellular shapes with a kidney or an oval-like nucleus and a high ratio of cytoplasm to the nucleus.37 MONs have 1–3 days of half-life during steady-state conditions,68 and if they migrate into other tissues, they can differentiate into tissue MФs where they engulf molecules or cells via phagocytosis. MФs, then, possess a larger periphery, longer lifetime (up to several months), and more phagocytic features than MONs.69 Nevertheless, MONs which do not migrate into the tissues (in order not to participate in an immunity response) will die after these few days.37 They initially act as environmental sensors and also participate in the replenishment of dendritic cells (DC) and MФs.68 The hematopoietic function of the MON/MФ progenitor is regulated by the PU.1 transcription factor and important cytokines such as IL-34 and M-CSF.70 These cytokine interacts with M-CSFR and activates their target cells.71 However, their activities rely on granulocyte-macrophage colony-stimulating factor (GM-CSF) during inflammation.72 In recent years, a challenging issue is the function of various MONs sub-populations in different inflammatory status. In other words, the features of circulating MONs can determine their fate in the tissues, suggesting alterations of the MФs polarization pathway toward M1 or conversely being transitioned into M2 type.
The human’s MON heterogeneity is categorized based on the expression of two types of cell surface markers, including CD14 and CD16 (lipopolysaccharide-binding and Fc-γ ΙΙΙ receptor, respectively).73 The CD14⁺CD16⁻ MONs are greatly phagocytic and have a higher capacity of ROS production and expression of CX3CR1 (increasing the accumulation of MONs during inflammation) and lower expression of CCR2 (increasing the chemotactic features of MONs during inflammation) in comparison with CD14+CD16+ MONs.74 In recent years, a more accurate classification has been made for MONs sub-population: CD14⁺⁺CD16⁻ classical MONs (CMs), CD14⁺CD16⁺ intermediate MONs (IM), and CD14⁺CD16⁺⁺ non-classical MONs (NCM)75 (Table 2). Seventy to eighty percent of the entire MONs population are CMs, which produce CD62L, CCR2, and CX3CR1 in high and low levels, respectively. They are incredibly phagocytic as a result of providing a high level of IL-10 and a low level of TNFα following LPS stimulation.74,76 Regarding the gene’s profile, the expression of angiogenesis, wound healing, and inflammation-related genes have been shown in CMs.77 In contrast, IMs which include 2–11% of whole MONs, induce inflammatory responses through producing high levels of TNFα and IL-1β following LPS stimulation.76 The evaluation of their gene profile has revealed two functions including the presentation of antigens and activation of T-cells.77 During inflammation, these two groups of MONs initiate an inflammatory response following a connection to the cell membrane through CCR2/CCL2 and/or CCR5/CCL5. Then, these cells invade the surrounding tissues that depend on vascular cell adhesion molecule-1 (VCAM-1) or CD106 and its ligand (very late antigen-1).68 The NCMs make up 2–8% of the total circulating MONs. They participate in blood vessel patrolling and tissue infiltrating by CX3CR1/CCL3 depending on intercellular adhesion molecule-1 (ICAM-1) or CD54 and its ligand (leukocyte function-associated antigen-1). Additionally, NCM displays a high production of IL-1β and TNFα in response to pathogenic fragments.76 The IMs and NCMs are regarded as inflammatory MONs.68,78
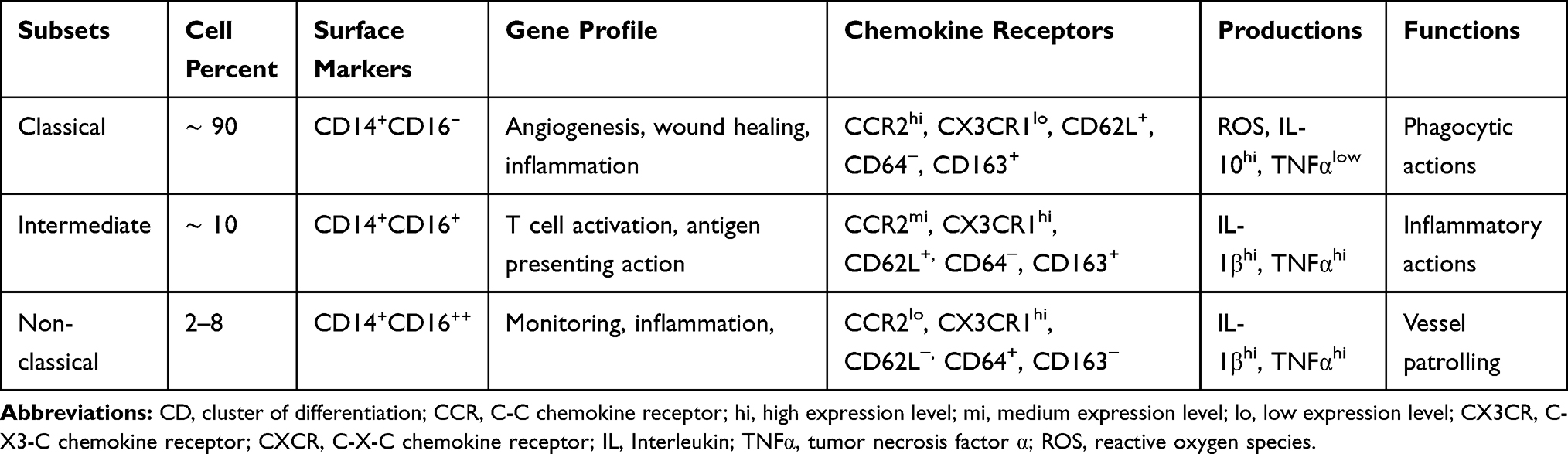 |
Table 2 Monocyte Subsets and Feature |
Although similar MONs sub-classes are thoroughly defined in humans and mice, their precise roles, markers, and the fate of each sub-class during an inflammatory status largely remains unclear. To shed light on the fate of MONs in humans, it is useful to evaluate the identical molecules between circulating MONs and tissue-resident MФs during inflammatory responses.
Monocyte-Derived Macrophages and Its Subsets During Obesity
During the last two decades, the role of ATMs in the development of metabolic disorders has been well documented. A significant rise of MФ counts has demonstrated in OAT in comparison with LAT.3,79 In this context, peripheral blood mononuclear cells (PBMCs), MONs and LYMs, which are affected by systemic environmental factors, might be pre-programmed due to the prolonged exposure to dyslipidemia, hyperglycemia, and inflamed metabolic organs-induced inflammatory molecules. These cells are recruited into the AT and generate the tissue-resident MФs.80 In other words, the tissue-resident M2ATM which may experience a phenotype distinction toward M1ATM during obesity42,54,81 can adopt the inflammatory features of the circulating MONs, causing alteration of MФs pathway polarization.33,37,67 This is in line with the development of tissue infiltration of MONs to different organs including liver, pancreas, and AT, which undergo a complete transformation toward tissue-resident MФs and develop a local inflammatory state.81 As a result of the high capability of infiltration and accumulation of MONs in inflamed sites, it is logical that these pre-programmed cells mainly participate in chronic LGSI.68 Notably, the gene expression of PBMCs participated in energy metabolism and IR are different in subjects with obesity, suggesting a relationship between these cells and obesity-induced immunometabolic disorders.82 Due to the higher number of MONs in individuals with overweight (OW) and OB compared to lean subjects, it is noticed that higher recruitment of MONs, in spite of macrophage proliferation, could be more critical in obesity-induced inflammation.83 The average number of MONs associates with body mass index (BMI)23 and a higher prevalence of MetS.84 However, these relations have not been reported in all surveys,78 and even a negative association has been demonstrated between the percentage of MONs and BMI.85 A sex-dependent association, furthermore, has been noticed between the average number of MONs and BMI.23 More investigators were interested in these immunometabolic-dependent responses when a higher expression of specific MONs markers (CD11b) was reported in the OB state compared with lean. This increase also correlated with a higher insulin resistance index (homeostasis model assessment of insulin resistance (HOMA-IR)).85 CD11b is a cell surface receptor of MONs that promotes their infiltration into the endothelium by binding to both ICAM-1 and VCAM-1.86 Remarkably, a significant rise in CD11b expression is accompanied by MetS incident-related factors. Moreover, its expression level correlates with glucose and triglyceride (TG) levels.87 Specifically, a significant development of CD16+ frequencies (IM and NCM) has been evidenced during OB state, which associated with morbidities ranging among patients with obesity.78 MONs of the patients with obesity with no comorbidities, in vitro, produce higher TNFα at basal level and also acquire increased sensitivity to LPS stimulation compared with lean subjects.88 Evaluation of the specific markers of M1 (TNFα and IL-6) and M2 (IL-4 and IL-10) in MONs of the non-diabetic individuals demonstrates their inflammatory features in comparison with lean individuals as a result of the higher level of M1 markers in their MONs.89 Interestingly, evaluation of the M1 marker messenger ribonucleic acid (mRNA) expression has clarified an association between IL-6 and low-density lipoprotein cholesterol (LDLc) levels. Also, IL-6 and TNF-α concentrations have correlated with BMI values.89
Beside the evaluation of M1 markers, the mRNA expression of M2 markers associates with some clinical parameters. The MONs of individuals with obesity and diabetes are less responsive to differentiation into M2 than lean individuals.80 Garton et al reported that the IL-10 mRNA expression levels associate with an increase in either the diastolic blood pressure or the glycated hemoglobin (HbA1c) values. Also, mRNA expression of CD163, which contributes to anti-inflammatory signals via IL-10 mediated-protein kinase B pathway,90 has decreased in patients with obesity and diabetes, which is associated with fasting insulin, HbA1c, and endothelial function index (pales wave velocity (PWV)).89 Increasing TNF-α production following LPS stimulation has also associated with TG and insulin concentrations in OB individuals without obesity-induced comorbidities.88
More specifically, CD14⁺⁺CD16⁺ cells are reported to have a positive association with the cardiometabolic disorder risk factor, including total cholesterol, LDL, high-density lipoprotein (HDL)/LDL, and TG/HDL ratio. On the contrary, a negative relation of CD14⁺CD16⁺⁺ cells with total cholesterol and TG has been demonstrated in individuals with OW and obesity.91 Additionally, a higher frequency of IM MONs has been shown in individuals who had three or more MetS criteria compared to metabolically healthy people.92 Adiponectin is an anti-inflammatory adipokine that contributes to maintaining the anti-inflammatory status through the expression of two types of the receptor (adipoR1/R2) on many kinds of cells including liver, MONs, skeletal muscle, and AT cells.93–95 The adiponectin plasma values have inversely correlated with CRP and TNF-α levels.5 Taking into consideration that MФs participate in both the lipolysis homeostasis35 and the foam cells formation in AT,41 adiponectin inhibits the transformation of MФs into foam cells and reduces lipid accumulation into MONs-derived MФs.96 Also, it has been reported that reduction of MФs infiltration, promotion of M2 polarization, and decline of chemokine expression are provoked by adiponectin.22,97,98
Considering the accumulation of MФs in AT during obesity, investigators have demonstrated other pathways resulting in the higher number of ATM. Additionally, the proliferation of tissue-resident MФs has been shown to be fully independent from AT MONs influx. In this regard, the populations of hematopoietic progenitor cells have been detected in WAT. These progenitors differentiate into cell lineages, including myeloid and lymphoid. This capacity of local precursors is remarkably similar to hematopoietic cells derived from bone marrow progenitors.99,100 In vitro isolated pre-adipocytes are also involved in ATM production.101 Interestingly, these cells carry out MФ functions such as phagocytosis and production of some specific antigens of MФs such as CD45, macrophage-1 antigen and F4/80.102 Furthermore, a local tissue-resident proliferation of MФs is revealed during obesity. Due to the reduction of ATM apoptosis during obesity, other recruitment mechanisms have suggested that they are independent of circulating MONs influx to AT, causing an increase in ATM count in OAT.103
On the whole, in recent years, discovering more ATM and distinction of MФ phenotypes in OAT have started a debate among researchers about the ATM origin in lean and obese AT. During obesity, recognizing the identical cell surface receptors among tissue-resident MФs and circulating immune cells, evaluation of inflammation-associated gene expression changes in ATMs and circulating immune cells, and origin tracing of tissue-resident MФs by cutting-edge technology may be helpful for a thorough understanding of ATMs origins during obesity. Both bone-marrow-derived pathways and local mechanisms are known as possible determinant ways which alter the tissue-resident MФ counts and phenotypes. It is interesting to determine the ways that are activated by obesity-induced inflammatory status. Thus, recognizing the primary regulatory factors in ATM counts and their phenotype distinction may provide a new therapeutic approach to inhibit and suppress the obesity-induced inflammation.34
Underlying Cellular Mechanisms of LGSI During Obesity
Pattern Recognition Receptors
As previously mentioned, there is a chronic low-grade systemic inflammation in the development of cardiometabolic disorders and other obesity-related diseases.18,104,105 Since this inflammation has inner-body sources, which is initiated by endogenous elements such as FFAs, cholesterol, heat shock proteins (HSP), and glucose, it is named sterile inflammation.106–108 At the cell level, these molecules provoke inflammation through two entirely distinct pathways, including TLRs and inflammasome receptor engagement.106,109 These are pattern recognition receptors (PRR) that originally belong to the innate immune system, modulating an appropriate immune response between both innate and adaptive immunities. They can recognize some parts of molecules, including PAMPs and DAMPs. Indeed, these molecules are produced by invaded pathogens or damaged cells, respectively.110,111 The recognition of both PAMPs and DAMPs provides massive protection against exogenous pathogens through eliminating them, which enhances homeostasis status via producing an appropriate response to endogenous components.112
Briefly, the inflammasome is a multiprotein complex with the oligomerization of two proteins, including nucleotide-binding domain-like receptor and apoptosis-related speck-like protein.113 The inflammasome complex is triggered by sensing some intracellular events such as cell death, metabolic changes, and inflammation.110 Eventually, activation of this multiprotein engages caspase-1 (an enzyme involved in IL-18 and IL-1β maturation process), promoting the initiation of inflammatory cascades.114
The critical role of TLRs is understood in both innate and adaptive immunity.111 Interestingly, TLRs activities have increased in obesity and MetS that probably contribute to chronic inflammatory status.104,115-117 TLRs family are regarded as one of the main cellular transmembrane PRRs which include 13 members 10 of which are only discovered in humans (type 1 to type 10).112,118 TLR family are expressed in different cells such as MФs, NK cells, MONs, LYMs, fibroblasts, endothelium, and epithelium (immune and non-immune tissues).111,117 Cells use different processes to recognize their specific ligands depending on their type.110 TLR1/2/4/5/6/9 are detected on the cell membrane. Nevertheless, TLR3/7/8/9 are expressed on internal components of the cells, such as endosomes.104,110 In recent years, TLRs are classified based on ligand features, for instance, TLR1/2/6/10 belong to the same category.112 Moreover, some types are reported to participate in autoimmune diseases, including TLR2/3/4/7/8/9.119 TLR1-9 types are expressed in hepatocytes, kupffer cells, hepatic DCs, and adipocytes during HFD, suggesting their possible involvement in liver fibrosis and meta-inflammation.120,121
TLRs Signaling Pathway
Following the PAMPs or DAMPs recognition by TLRs, various transcriptional genes upregulate depending on the cells and TLR types.110 In fact, TLRs are the type І integral transmembrane glycoprotein which consists of three parts: 1) an extracellular domain arranging the ligand distinction; 2) a single transmembrane domain localizing the receptor on cell; and 3) a critical cytoplasmic tail, named toll/IL-1 receptor (TIR) domain, conducting the intracellular cascades. Due to the ability of TIR domain to recruit the different adaptors, it is known as the major TLR domain, which determines downstream cascades of each TLR.122 There are five different protein adaptors recruited by TIR domain including myeloid differentiation factor 88 (MyD88), TIR-domain containing adaptor-inducing interferon-β (TRIF) also known as TIR-domain containing molecule 1 (TCAM1), TIR-domain containing adaptor protein (TIRAP) also known as MyD88 adaptor-like (Mal), TRIF-related adaptor molecule (TRAM), sterile α- and armadillo-motif containing protein (SARM).110,111,122 Different signaling pathways triggered by TLRs activation are generally dependent on MyD88 recruitment. They are recruited in two entirely distinct signaling cascades: TRIF-dependent pathway also named non-MyD88 or MyD88-independent pathway, and MyD88-dependent pathway inducing interferon (IFN) regulatory factors (IRF), activator protein 1 (AP-1) and NF-κB activation in downstream112,115 (Figure 2B). MyD88-dependent pathway is activated following the recruitment of IL-1R-associated kinase (IRAK) family,123 including IRAK-1/2/3/4. IRAK4 is reported to be the most important one in TLR response since individuals with IRAK4 deficiency showed a considerable reduction in TLR activities.124 Consequently, TNF-receptor-associated factor 6 (TRAF6) joins to this complex, which results in NF-κB activation via phosphorylation of its inhibitor kinases (inhibitor of kappa light polypeptide gene enhancer in B-cells kinases, IKK protein complex). However, in the non-MyD88 pathway, TIRF recruitment leads to IRF3 activation.122,125 All types of TLRs, except TLR3, only activate the MyD88-dependent pathway.
 |
Figure 2 LPS transferring pathway and modulation of TLR4 signaling cascade. (a) TLR4-MD2 complex and LPS recognizing: TLR4 signaling cascade is activated following LPS sensing mediated by three different protein activities, causing both ligand recognizing and transferring to receptor. The LBP-LPS complex is recognized by CD14, TLR4 co-receptor, leading to TLR4 cascade activation. (b) TLR4 protein adaptors and negative regulators: TLR4 signaling necessitates recruiting two main adaptors, including MyD88 and TRIF. Activation of other mediators causes the phosphorylation of some proteins which leads to the translocation of inflammatory transcription factors into nucleus, inducing effective inflammatory genes upregulation. Modulating of TLR4 is mediated by the negative regulator proteins in different stages (TLR4 sensing ligand, CD14, adaptor recruitment, transcriptional factor activation, transcriptional factor translocation, gene transcription) to either control the excessive response of TLR4 or suppress its activities at an effective time following an inflammatory response. Abbreviations: TLR4, toll-like receptor 4; LPS, lipopolysaccharide; LBP, LPS-binding protein; CD, cluster of differentiation; MD2, myeloid differentiation factor 2; MyD88, myeloid differentiation factor 88; TRIF, TIR domain-containing adaptor-inducing IFN-β; TIRAP, TIR domain-containing adaptor protein; TRAM, TRIF-related adaptor molecule; IRAK, IL-1R-associated kinase; TRAF6, TNF-receptor-associated factor 6; IKK, inhibitor of kappa light polypeptide gene enhancer in B-cells kinase; IL, interleukin; MAPK, mitogen-activated protein kinase; TNFα, tumor necrosis factor α; JNK, c-Jun N-terminal kinase; AP-1, activator protein 1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IRF, interferon regulatory factor; p, phosphorylation. |
TLR4, however, recruits four types of main adaptors and activates both downstream pathways.122,126 Hence, TLR4 is a crucial TLR and it has even been recognized as an M1 marker due to participating in M1/M2 ATM polarization during obesity.38,127 Additionally, gene expression of TLR4 has been associated with the concentration of cardiometabolic markers.128 TLR4 and TRIF gene expression, however, have not been changed among obese individuals with or without MetS and abdominal obesity.129 TLR4 endogenous ligands such as LPS, saturated fatty acid, and ox-LDL are abundantly produced during obesity.130–134 HSPs are another TLR ligand which restores tissue homeostasis. HSP72 levels that have a protective role in IR during obesity drop in individuals with diabetes.108 In this case, upregulating both TLR2/4 transcription factors and their adaptors have been demonstrated in either in-vitro or animal studies during obesity.131,132,135,136 Despite MyD88 down-regulation, adipocytes treatment in a high concentration of SFA (2mM, 48 hrs) has indicated an upregulation in NF-κB and TLR4 levels.132 It seems that TLR4 acts as an intermediate molecule among the excess nutrient, innate immunity, and inflammation since glucose metabolism and obesity-induced IR are ameliorated in TLR4 knock-out in mice with HFD.131 Adipocytes treatment with LPS in individuals with obesity and T2D has stimulated secretion of a high concentration of IL-6 and TNFα, while NF-κB blockage has led to reducing these levels.135 Furthermore, TLR2 expression has increased in MONs and adipocytes in HFD mice.136 Further human findings are summarized in Table 3.
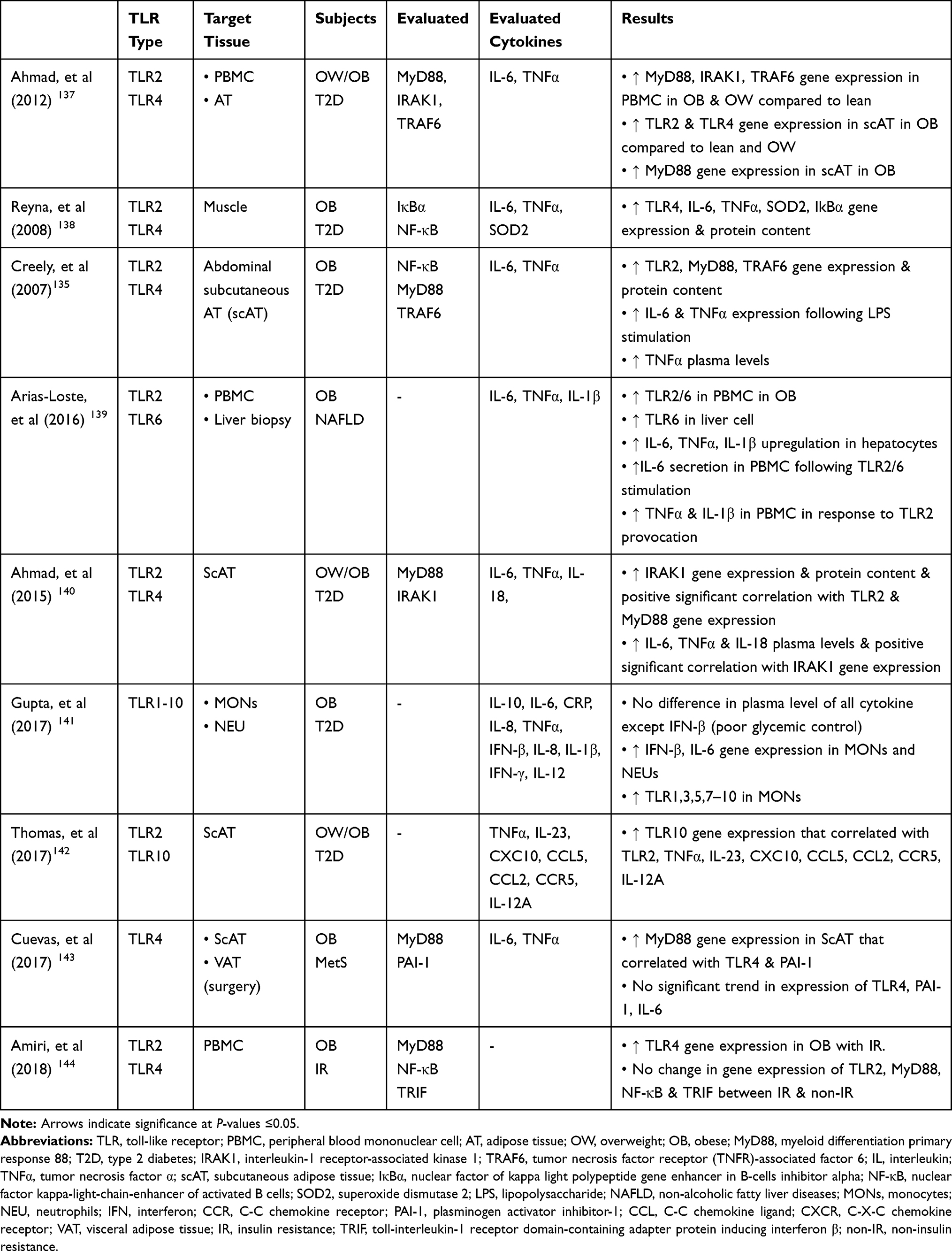 |
Table 3 Human Studies Outcome Related to TLRs |
LBP/TLR4/CD14 Axis
The activation of TLR4 signaling is associated with LPS representative proteins, including LPS-binding protein (LBP), and LPS chaperon protein (CD14 or LPS receptor) (Figure 2A). LBP is demonstrated to accelerate the connection of ligand to the receptor. In other words, initiation of TLR4 signaling cascades relies on the recognition of the LPB-LPS complex by the membrane-bound CD14 (mCD14).145 In this context, the higher levels of both LBP and CD14 can promote TLR4 activities. Interestingly, obesity-induced increased level of LPS serum has shown some difficulties in modulating inflammation expansion, suggesting a noticeable role of TLR4 in obesity and metabolic dysfunction.130 In this regard, the soluble form of CD14 (sCD14), mainly produced by MONs and hepatocyte,146 can represent the LPS to TLR4 in certain types of cells that do not express mCD14 abundantly such as non-myeloid lineage cells.147 Although LBP serum levels are not changed in OB individuals without MetS, TLR4, and TRIF mRNA expression levels have significantly correlated with LPB serum levels among them.129 Importantly, some evidence has shown the roles of CD14 and sCD14 in obesity-related diseases such as IR, steatohepatitis, and MetS.148–150 TLR4 signaling also depends on a co-receptor named myeloid differentiation factor-2 (MD2), so that MD2 deficiency leads to hepatocyte resistance in diet-induced steatohepatitis.151 Thus, interrupting the cooperation of the LBP/TLR4/CD14 axis by any molecules such as the soluble form of TLR4 could ameliorate the TLR4 signaling.152
Negative Regulators of TLR4
As far as TLR4-associated domain activities are concerned, modifying super strong inflammatory productions of TLR4 signaling, including IFN-γ, TNFα, IL-6/8/12/1β, and inducible nitric oxide (iNOS) is noticeably significant.104,120 Accordingly, the activities of the TLR4 signaling cascade must be tightly regulated to avoid its harmful effects. In this case, there are various intracellular integrated mechanisms, named negative regulators, which naturally modulate the excessive responses of TLR4 signaling111,120,125,152,153 (Figure 2B). Different features of these negative regulator proteins, which determine their functions, are dependent on the cell type and duration of secretion. Even though some of these proteins are expressed continuously in some cells, others are secreted during the activation of TLR4 pathway itself.153 These molecules are shown to promote distinct inhibitory mechanisms in different stages of TLR4 signaling cascade, including the connection of ligand and receptor, signaling transduction by adaptor recruitment, molecule translocation, transcriptional phase of proteins, and TLR4 co-receptor sensing.125,153 These various functional abilities of negative regulators give them a considerable power to control the cascade activities, the magnitude and peak of the response, duration of signaling, and sensitivity to initiate an immune response153 (Figure 2). For instance, the gene expression-associated IRF5 is suppressed by IRF4 connection to MyD88.154
Furthermore, a direct connection of SARM adaptor protein to TRIF suppresses the activities of TRIF.155 The decoy receptors of cell surface such as growth stimulation expressed gene 2 (ST2) inhibits TLR4 signaling in MONs through disconnecting MyD88 or Mal and the receptor.156 Also, NF-κB activation is inhibited by ubiquitin-specific protease 4 connection to TRAF6.157 A20 protein, also known as TNF alpha-induced protein 3 (TNFAIP3), has shown to inhibit both dependent and independent MyD88 pathways of TLR4 by the suppression of TRAF6 and NF-κB activation.158,159 Also, mice MФs with lack of TNFAIP3 produce a higher concentration of inflammatory cytokines.160
Therefore, the obvious understanding of TLRs domain function during meta-inflammation could offer a treatment for obesity-associated LGSI and disorders. Besides, there is scarce evidence about negative regulator expression and function during obesity. The interaction of different negative regulators during meta-inflammation has made an exciting research gap since the existence of such chronic inflammation is probably related to the inefficient functions of negative regulators.
Exercise Training as a Therapeutic Intervention in Obesity-Induced Immunometabolic Disturbance
Exercise Training and Chronic Inflammation
Worldwide change in lifestyles, a combination of being sedentary and consuming high caloric nutrients, has accelerated obesity. An inactive lifestyle explains obesity and metabolic disorders.161 As obesity is accompanied with various morbidities, it has put a burden on different societies in recent years. The excess nutrients play the role of metabolic ligands, which promote chronical activation of the inflammatory pathways leading to immunometabolic disturbance.104,115 An intriguing relation has been demonstrated between CRP values and a lower percentage of body fat among men with obesity.162 In this sense, the inverse association of aerobic fitness and physical activities with inflammation levels and lower share of inflammatory MONs (CD14+CD16+) has been reported, respectively.11,163 Besides, a lower TNFα, CRP production and CD14+CD16+ percentage have been demonstrated in physically active individuals compared to inactive people.11,12 It seems that exercise training induces calorie usage accompanies by metabolic health and inflammatory status improvement.164 However, its potential immunomodulatory molecular mechanisms are not thoroughly evaluated. The produced myokines through exercise training probably interfere with the inflammatory pathways.165,166 For instance, irisin, an adipomyokine and exercise mediator, correlates with metabolic condition14 and interferes in TLR4/MyD88 activities.15 These positive effects of exercise could be independent of adiposity alterations.17 Also, inflammatory condition improvement has been reported in elder subjects through MyD88/non-MyD88 TLR2/4 cascades following resistance training.13 Several animal studies have demonstrated exercise benefits to inhibit inflammation in OB mice through the reduction of MФs infiltration, an increase of M1 to M2 polarization, and amelioration of insulin pathway.167–169
Furthermore, recent investigations on humans have revealed the provocation of mechanisms associated with M2 markers following exercise.170,171 Additionally, the improvement of chemotactic status, leukocyte functions,171,172 MONs differentiation,92,173 insulin sensitivity,174 the reduction of inflammatory markers, metabolic health-related risk factors,174–176 and circulating inflammatory MONs11,12 emerge following different exercise training protocols. In fact, the beneficial impacts of exercise training are induced by both immune and metabolic-related cells, such as adipocytes, hepatocytes, and myocytes.166 Exercise training prescription for improving the immunometabolic disorders is explained by AT differences between lean and obese conditions. In other words, it should be noted that not only the increase of adiposity but also the imbalance of immune cell counts have been demonstrated in OAT compared with LAT. Obesity is accompanied by a higher MФ percentage (40% in obese and 10% in lean), immune cell infiltration, remodeling, and death of adipocytes.34,35 In this sense, the positive impacts of a volunteered exercise training protocol are shown to be related to the reduction of adipocyte size, CLS formation, and immune cell recruitment in OAT in mice.177
In contrast, an acute swimming protocol does not affect CLS,168 suggesting exercise intensity-dependent immunomodulatory responses.178 CLS reduction is followed by a decrease in the gene expression of inflammatory markers, and consequently, suppression of inflammation. In this way, inflammatory status and insulin function have been reported to be ameliorated in SVF even after an exercise session because of TNFα, CCL2, and IL-1β downregulation.168 Reduction of TNFα and IL-6 gene expression and also amelioration of insulin signaling have induced in AT, skeletal muscles, and liver following a session of swimming.179 Additionally, a 6-week running has led to TNFα and CCL2 downregulation in HFD-induced obesity180 that is equal to 15 weeks of walking, which reduce TNFα in abdominal scAT of individuals with obesity.174 Men with OW and obesity who participated in a 2-week short-term high-intensity interval training (HIIT) (6 sessions) have also shown a reduction of IL-6 and adiponectin in AT. In this study, not only is ICAM-1 mRNA level constant but also mRNA of TNFα, IL-10, and CCL2 are not detected.176 Participating in a 3-month moderate aerobic exercise training has been reported to upregulate HSP60, IL-6, and TNFα among people with obesity and T2D.181 However, reduction of HSp60 and HSP70, higher IL-10, and unchanged values of TNFα mRNA expression have been reported following 8-week resistance training in elder healthy subjects.13 In this state, a significant reduction of TNFα protein levels is found following a whole-body vibration in dynamic or static exercise among elder subjects with OW. The increase of IL-10/TNFα ratio has also been noticed due to the rise of IL-10 gene expression and protein contents.182
Exercise Training and M1/M2 Polarization Markers
In addition to exercise impacts on AT inflammatory status, exercise training greatly influences MФs infiltration and M1/M2 polarization markers in AT. The chemotactic state of leukocytes is essential in recruiting the leukocytes into inflammatory sites.35,43 For instance, CCL2, which contributes to obesity-associated MФs infiltration into the AT,66 is downregulated following a moderate-intensity protocol. Infiltration of NEUs, MONs, and T cells, in contrast, rises by high-intensity training among subjects with obesity and sedentary lifestyle due to higher CCR5 expression on CM and IM. Unchanged body weight demonstrated that AT alterations have not affected the plasma concentration of these chemoattractant proteins, indicating a direct impact of exercise on the pathways-induced expression of chemotactic chemokine receptors.172 In this way, the lower expression of inflammatory markers such as CD14 and CD68 has been found in subjects with obesity following exercise.174 On the contrary, a 12-week endurance exercise has increased the CD68+ inflammatory MФs in scAT in men with OW.183 Evaluating of exercise-promoted M1/M2 gene expression has shown anti-inflammatory impacts of moderate-intensity endurance training. M2 markers, including IL-10 and dectin-1, are upregulated following 8-week moderate treadmill running in MONs of females with OW and obesity. Despite increasing TNFα gene expression, CCL2 expression, known as the M1 marker, is reduced after the exercise. M2 marker upregulation could be partially associated with the peroxisome proliferator-activated receptors (PPAR)-γ pathway. The PPAR-γ mRNA levels of obese MONs, however, have not changed significantly following exercise. Interestingly, the upregulation of either PPAR-γ target genes (CD63 and liver X receptor α) or its intracellular ligand generation has increased considerably following an 8-week running in MONs.170
Exercise Training and mRNA Expression Changes
Exercise-induced AT and ATM mRNA expression changes are reflected in circulating plasma markers. The baseline concentration values of IFN-γ, IL-6/17a, and CRP correlate with BMI and IL-10 among adults with OW and obesity.11,171 Also, an acute HIIT treadmill running has led to a fall in IL-8 (CXCL8) and a simultaneous increase of IL-10 plasma concentrations in OW/OB compared to the lean group. IL-8, CCL2, and IL-6 baseline values, however, have been reported to be unequal among the subjects.178 Individuals with OW and obesity have initially shown higher values compared to the lean. Patients with obesity and T2D have the higher circulating concentrations of high-sensitive CRP (hsCRP), endothelin-1, and soluble ICAM-1 (sICAM-1) compared with lean and obese. Also, the endothelial function did not improve after a 2-week aerobic cycling protocol in these subjects and no change has been found in the circulating levels of endothelin-1, and sICAM-1 following exercise training.184 A single session of HIIT cycling training has induced a 20% reduction of produced TNFα following LPS stimulation among individuals with obesity and T2D. In contrast, a 12-week resistance training (with/without diet control) has affected neither TNFα nor IL-6 levels following LPS stimulation among post-menopausal subjects with OW and obesity.12 Also, the plasma levels of these inflammatory cytokines have decreased immediately after exercise in both subjects with obesity and T2D (14%) and healthy normal weight (44%).185 Dorneles et al186 revealed that IL-10, IL-6, and transforming growth factor (TGF)-β serum levels are increased following an acute HIIT protocol while the circulating values of TNFα and IL-17a have shown no significant change after exercise. Despite rising plasma level of IL-6 in both acute high-intensity and moderate-intensity training, circulating levels of leptin and IFN-γ decreased immediately after HIIT protocol in men with obesity.
Meanwhile, the concentration level of IL-4 has increased immediately after training. Thus, HIIT-induced reduction in the ratio of IFN-γ/IL-4 shows a positive immunomodulatory response of exercise training.187 In contrast, based on a study on the exercise training-promoted M2 markers (IL-4, IL-13, etc.), no changes were observed in serum concentrations of either IL-4 or IL-13 following an 8-week moderate aerobic training protocol in female adults with OW and obesity.170 Barry et al188 have reported no alteration in TNFα, IL-6, and IL-10 serum concentrations after both short-term HIIT and moderate-intensity interval training (MICT). Also, two short protocols with different intensities do not affect the IL-10/6/1β and TNFα serum concentrations in prediabetic OW/OB females.189
Additionally, the inhibitory function of IL-10 on TNFα expression has decreased after two different protocols.188 However, exercise-induced positive effects on immunity have been shown by other studies where the exercise has reduced circulating MCP-1 and CRP and increased adiponectin in subjects with obesity.174 These results are in line with the significant reduction of sIL-6R and MCP-1 reported after 2 weeks of HIIT in men with OW and obesity. However, lower circulating adiponectin concentration and unchanged values of TNFα and IL-10 are also reported in this study.176 Although the plasma values of IL-17a/6 and IFN-γ have shown no change following a 12-week concurrent training, the concentration of hsCRP and IL-10 increased significantly.171 Liu et al175 have demonstrated higher serum values of IL-18, a marker associated with T2D development, in individuals with T2D than in the healthy ones. Following a combined aerobic and resistance training (IT group), IL-18 levels have decreased compared with T2D subjects who only received a diet control intervention and continued their conventional drugs (CT group). They have also shown lower circulating levels of IL-33, a regulatory marker of NF-κB, in both groups in comparison with the healthy control group, and interestingly, these values have increased in IT compared to CT.175 However, the levels of cytokines related to diabetes initiation and development (Pentraxin 3 and chitinase-3-like-protein 1)190,191 have introduced no significant changes among the three groups before and after exercise intervention.175 Although the serum levels of HSP60 have increased following a 3-month aerobic exercise, a significant reduction is shown in individuals with obesity and diabetes.181 The potential impacts of exercise training on the immune system are also found by increasing the serum level of adiponectin and IL-10 in OW/OB women after water running.192 In addition to increased adiponectin serum concentration, the IL-15 levels have increased following a 24-week combined exercise in adult men with obesity.162 Obesity-induced reduction of IL-15 myokine levels has correlated with the percentage of body fat. This noticable result has presented an axis from muscle to AT (muscle-to-fat axis) which suugests the regulatory roles of this myokine in body fat function.193 However, aerobic training has increased the leptin and IL-6 circulating levels in individuals with OW.183 In this way, the increasing level of hsCRP and TNFα emerge after a 12-week combined exercise,171 a finding which is not consistent with Park et al. They have reported a significant decrease in TNFα serum concentration following a 12-week combined exercise in postmenopausal women with obesity.194 Also, a 24-week combined exercise has reduced CRP serum levels by about 118% in adult men with obesity that led to an improvement in their CVD risk classification to the low-risk category.162 Neither resistance training nor diet restriction with resistance training has changed the CRP and adiponectin serum levels.12 Additionally, neither HIIT nor MICT has significantly influenced the chemokine-induced AT leukocyte infiltration because of unchanged serum levels of CCL2, CCL3, and CXCL8 in adults with obesity.172 A significant decrease in IL-10 levels has been reported following a 20-week combined training, although the positive effects of this protocol are demonstrated by greater IL-10/TNFα ratio.195 Due to the preventive effect of resistance training with higher volume and/or frequency on increase of IL-6 and CRP levels,196,197 an exercise training method-induced higher energy expenditure has been proposed to be more effective in reducing inflammatory mediators.198,199 Serum analysis by the nuclear magnetic resonance technique has provided some signals used in evaluating the clinical conditions.200 GlycA is one of the novel biomarkers which attributes in systemic inflammation. It is associated with circulating levels of fibrinogen, TNFα, CRP, and IL-6.201 Also, its reduction has been demonstrated among sedentary prediabetic adults following a six-month exercise training. Interestingly, this reduction is associated with visceral adiposity reduction, suggesting a mechanism related to VAT modulation for reducing GlycA.202
Exercise Training and Circulating Leukocyte Features
Although a thorough evaluation of adipocyte functions is needed in the homeostasis of the immune system, the VAT biopsy is not easily accessible in human studies.67 As ATM polarization is initially followed by a distinction of various subsets of circulating immune cells, it seems that the potential immunomodulatory abilities of exercise could be evaluated by investigating the circulating leukocyte features. ATMs are mostly generated from bone-marrow-derived circulating MONs during inflammation,37 and their pre-programming could occur by being exposed to obesity-caused hyperglycemia and dyslipidemia.80 In this sense, a wide range of responses is found among leukocytes after different exercise protocols. A half-marathon running by trained individuals has led to higher number of NEUs and lower percentage of MONs and LYMs, 30 mins after exercise remaining even after 3 hrs.203 Besides, an increased total number of leukocytes have been shown immediately and 2 hrs after a treadmill running bout with either low-intensity or high-intensity VO2peak among healthy young subjects. In this condition, only the increase in LYMs, MONs, and NEUs has been significant.204 However, exercise-caused leukocyte mobilizing is shown to be independent of metabolic conditions (one or more MetS criteria existence). A very short bout of cycling training has induced similar effects on circulating leukocyte among individuals with obesity and with/without MetS.92 Additionally, a 2-week combined endurance and resistance training protocol have not affected the MONs subset proportion.205 Leukocyte, LYMs, and MONs numbers of elderly subjects are also affected by a 12-week concurrent training.206 Also, neither MICT nor HIIT induced significant impacts on the number of leukocytes, granulocytes, and MONs in OB adults in a ten-week period.172 Meanwhile, in a study that compared two bouts of HIIT and MIIT protocols, the leukocyte, LYMs, and MONs numbers significantly increased after two protocols. But these results are shown only after HIIT in the lean group, where a significant increase is reported in the total number of leukocytes and MONs.178 Matos et al173 have evaluated the CD16+ inflammatory MONs among people with both obesity and different insulin sensitivity. They have notified a higher CD16+ MONs percentage in subjects with insulin resistance that had a positive correlation with fasting insulin and BMI. However, this higher percentage is not found in obese individuals with T2D.185 Exercise training has been shown to modify CD16+ MONs in the obese with insulin resistance. The percentage of this MONs subset fell 1 hr after a moderate cycling bout in the lean and obese subjects with/without IR.173 These results were partially in line with another study that evaluated the acute response of leukocyte numbers by a HIIT protocol among individuals with obesity and T2D. The total leukocyte and NEU counts of people with T2D obese have been significantly higher than healthy subjects with normal weight. The exercise-promoted immunity positive impacts have also been notified when the counts of total leukocytes (CD16+ MONs, CMs, and NEUs) that significantly increased immediately after exercise reduced 1 hr after training in comparison with before-exercise levels.185 In this regard, Dorneles et al186 have found similar results. Following an HIIT session, the total number of leukocytes has shown a 27% increase as a result of the 41% and 59% increase in LYMs and MONs, respectively.186 However, a comparison of two short-term protocols (HIIT and MIIT) has demonstrated no changes in CD15+ NEUs, CD14+ MONs, and LYMs counts in prediabetic females with OW and obesity.189 Also, two weeks of HIIT could not influence EOS, lymphocyte, NEU, basophils, and MON counts as well as CM, IM, NCM percentage among young adults with sedentary lifestyle.205 It has been suggested that both human body defense system and disease susceptibility could improve by moderate-intensity exercise. In fact, the improvement of leukocyte function has been revealed by a moderate concurrent exercise that induced a reduction in CD4+ LYMs.171 Especially, the percentage of inflammatory MONs has reduced about 64% following a long-term combined exercise protocol in elderly sedentary individuals with obesity11 while it has not been reported after a resistance one. They found that exercise intervention with diet restriction could not decrease the percentage of CD14+CD16+ MONs in subjects with OW and obesity. Only individuals with OW (not OB) showed a reduction of CD14+CD16+ MONs frequency, where they participated in the exercise training group without a restricted diet.12 These exciting results verify the potential of exercise training compared with diet intervention. Additionally, obesity-caused CD4+ dysfunction is reported to limit the tumor suppression and promoting tumor growth activities.105 Even though the counts of T cell subsets (CD4+, CD3+, and CD8+) are not affected by exercise, T-cells-related immunity is improved by higher CD28 (a critical receptor for T cell-associated immune response) and CD80 expression levels on CD8+ and CD14+ cells, respectively.206
Exercise Training Affects TLR4 Pathway
Phenotype alterations of immune cells are accompanied by the molecular changes in immune cells influencing on systemic inflammatory status even without alterations of circulating cytokine concentrations.172 Investigations of PBMC-related transcriptome have shown various pathways that play notable roles in energy metabolism, inflammation, IR, and cancer. Associations of PBMCs with development and exacerbation of obesity have been shown by diversity in PBMC gene expression profiles (TLR, MAPK, and NF-κB pathways) among individuals with normal weight and OW. The increased gene expression involved in inflammatory pathways such as CD19 is demonstrated in PBMCs in OW subjects. The metabolic condition variations of these subjects are associated with the different status of their obesity.82 Immunomodulating roles of the both acute and chronic exercise training protocols are displayed following the suppression of these inflammatory pathways, including TLR2/4 activities.13,185 However, there is conflicting evidence about the inhibitory effects of exercise in animal and human studies.117 TLR4 gene expression has been decreased following a 16-week treadmill running and an 8-week swimming in AT of OB mice.167,179 This reduction is also demonstrated in the liver and skeletal muscles that all express the higher levels of TLR4 at the baseline following HFD. Besides, TLR4 downstream activities evaluating by MyD88 and IKKβ expression fell after exercise in these tissues.179 Due to the release of anti-inflammatory myokines such as irisin, IL-10, and adiponectin after exercise, it seems that exercise modulates immune responses through suppressing the TLR4 activities.15,207 However, there is no clear evidence in this area because increased IRAK-1 mRNA levels in DIO mice are not affected by exercise.179 Based on the effects of physical activity on the human body, investigators proposed different mechanisms to evaluate the TLR4 cascade in response to exercise. In this context, exercise-training-induced reduction of FFA circulating levels has been shown following a 12-week cycling,208 suggesting an explanation for reducing the TLR4 activities. Also, concerning obesity-increased LPS concentration,130 the reduction of both LPS and TLR4 co-receptor (CD14) is revealed following a 12-week combined exercise training among postmenopausal women with obesity, suggesting exercise-induced immune function amelioration.194 Table 4 illustrates the results of human studies evaluating the TLR2/4 responses to the exercise training.
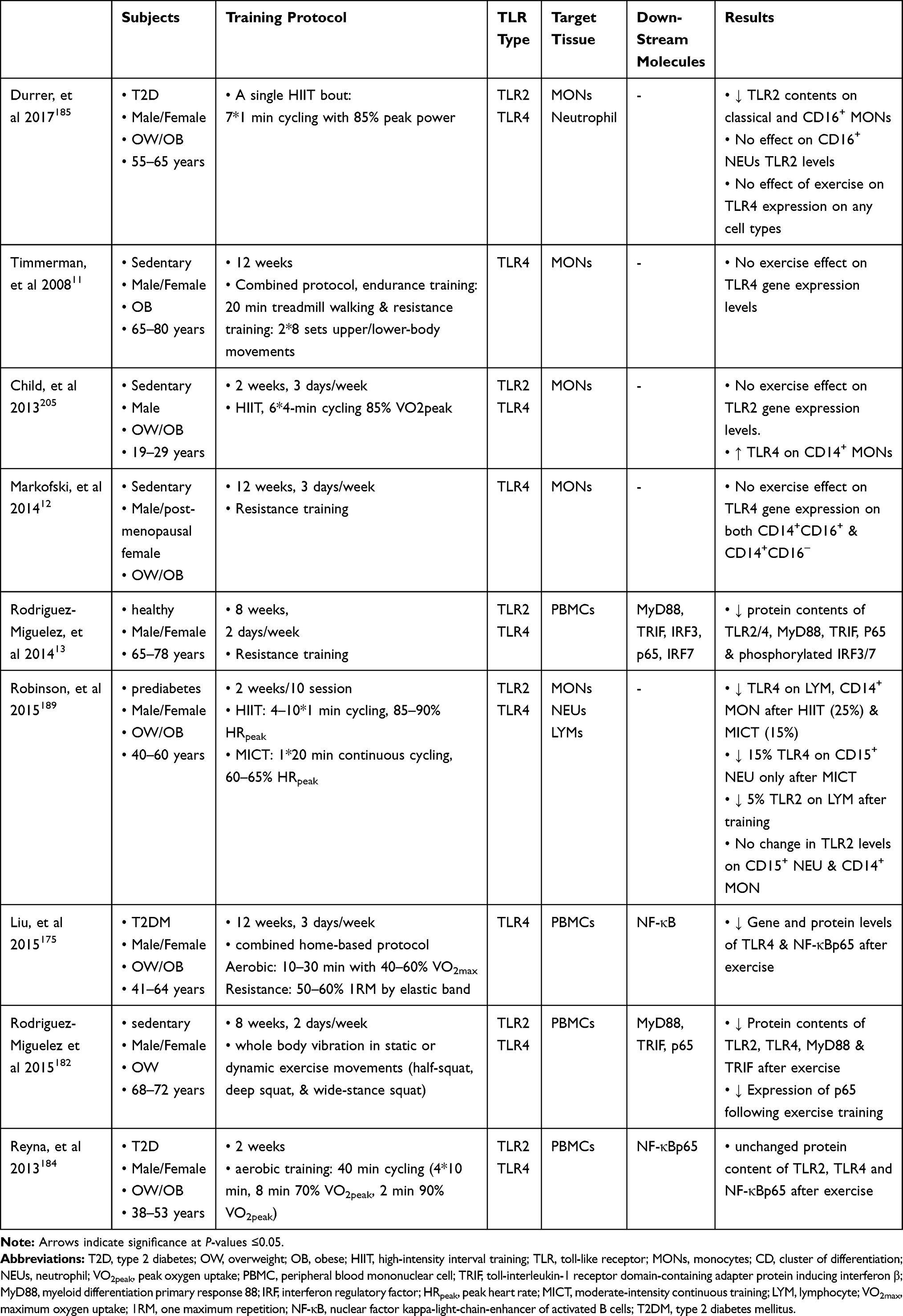 |
Table 4 Outcomes of Human Studies on TLR2/4 Following the Exercise Interventions |
Exercise-induced immunomodulatory impacts are reported to be associated with TLR2/4 activities, which could be independent of adipose tissue weight loss. Exercise-associated TLR4 downregulation can develop as a result of the suppression of M1 MON infiltration, reduction of TLR4 ligand, blunting of the activators of TLR4 cascades, and promotion of M1 to M2 distinction in obese AT. The mechanisms require further detailed studies.
Conclusion
It is frequently proved that obesity is accompanied by the various disorders including systemic hypertension, T2D, dyslipidemia, and fatty liver due to the development of chronic inflammation. In other words, a systematic low-intensity inflammation is promoted during obesity, which is remarkably dissimilar to the classic one. It is recognized that this type of inflammation develops by the inner stimulants causing systemic inflammation. In this context, the storage of excessive nutrients in adipocytes of persons who are physically inactive leads to adipose tissue morphologic changes such as adipocyte hypertrophy. As a result, these hypertrophied cells provide hypoxic microenvironment and a constant increase of lipolysis leading to accelerated production of the inner motivators, including fatty acids and chemotaxis-related ligands.
On the one hand, as it is depicted in detail in Figure 3, circulating immune cells are probably stimulated by the enormous amounts of FFAs. Consequently, these stimulated receptors initiate the inflammatory signaling cascades and provoke down-stream gene transcription of inflammatory cytokines. Additionally, the hypertrophied adipocytes-induced hypoxia leads to the gradual cell death, creation of CLS, and activation of hypoxia-associated cytokines, providing a chemotactic environment. Therefore, the MON/MФ origin and their distinction pathways, which play pivotal roles in immunity induction and modulation, are affected by these AT changes, causing considerable M1ATM distinction. The increased expression of influential inflammatory transcription factors occurs due to the complicated relationship between the dead adipocytes and surrounding M1ATM in the OAT. Furthermore, the balance of inflammatory/anti-inflammatory immune cells has been reported to be disturbed following these events. The inflammatory cytokines produced by FFAs stimulates circulating MONs accumulation around the inflamed AT. Not only do these cytokines recruit the abundance of immune cells, but they are also infiltrated into the AT, and consequently, induce the development of the FFAs production, inflammatory cytokine expression, and M2 to M1 distinction. Hence, this vicious circle leads to a severe immunometabolic disturbance.
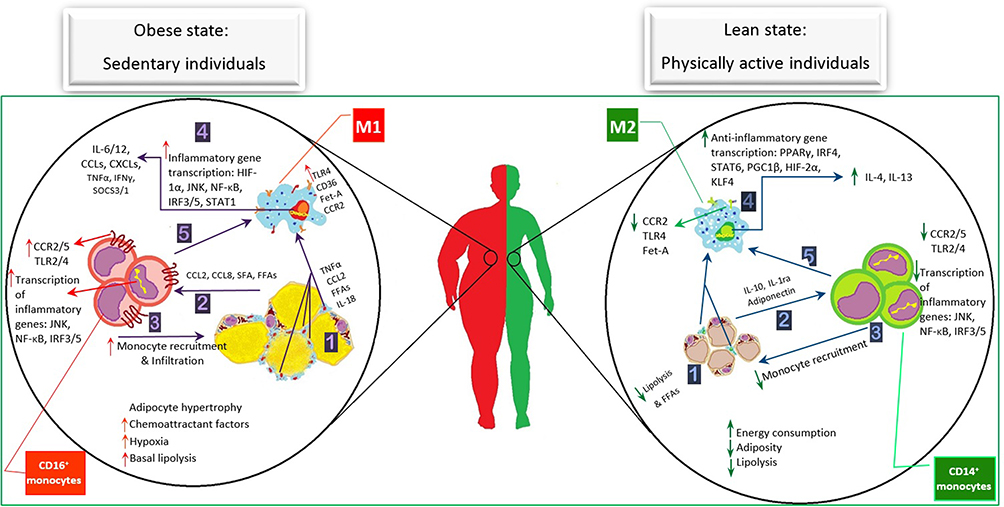 |
Figure 3 Physical activity alters circulating monocyte features and obese adipose tissue macrophage distinction. Obese state: The excess nutrients-caused adipocyte hypertrophy and cell death induce the micro hypoxia in AT. ① Although MФs accumulation is increased, constantly elevated levels of circulating non-esterified fatty acid reveal an inefficient function of MФs to maintain the AT homeostasis. These complex interactions of the immune system and adipocyte cause the release of inflammatory cytokines into the blood. Both circulating cytokines and the fatty-acids provide a chemoattractive environment. ② The increased CCL2 release and hypoxia result ③ the MON recruitment and more intensive infiltration into the AT. In AT, this inflammatory condition promotes M1 polarization leading to ④ higher number of surface receptors such as TLR4 and CCR2 which activate the influential inflammatory transcription factors, resulting in the upregulation of several inflammatory cytokine genes such as CCL and CXCL family, IFNγ, IL-6/12, and TNFα. ⑤ Circulating MONs are affected by releasing the abundant inflammatory cytokines and FFAs into the bloodstream. They might be altered toward the inflammatory types due to the increase of inflammatory receptors and, consequently, activation of inflammatory signaling cascades, causing the upregulation of more inflammatory genes. This impaired feed-forward cycle aggravates the inflammatory status in individuals with obesity. Lean state: Exercise training improves the inflammatory state through the inhibition of AT expansion (increasing energy consumption and modulation of inflammatory cascade activities (anti-inflammatory cytokine expression). It seems that the improvement of the inflammatory condition in either AT or bloodstream is induced by the reduction of the chemoattractant factors like CCL2 and FFAs. This has influenced the MФ polarization pathway toward M2. Consequently, it leads to a decrease in both the adipocyte death and the expression of inflammatory cytokine receptors on the surface of cell membrane, which resulted in the downregulation of inflammatory pathways. The circulating MONs which experience the anti-inflammatory condition obtain the features of anti-inflammatory MON subsets. Moreover, producing the strong anti-inflammatory myokines could interrupt these pathways following the exercise training, resulting in the improvement of obesity induced-inflammatory cycle. Abbreviations: TLR4, toll-like receptor 4; CD, cluster of differentiation; fet-A, fetuin-A; CCR2, C-C chemokine receptor type 2; HIF1α, hypoxia-inducible factor 1 α; JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IRF, interferon regulatory factor; STAT, signal transducer and activator of transcription; IL, interleukin; CCL, C-C motif chemokine ligand; CXCL, C-X-C motif chemokine ligand, TNFα, tumor necrosis factor α; IFNγ, interferon regulatory factor γ; SOCS, suppressor of cytokine signaling; CCR, C-C motif chemokine receptor; FFA, free fatty-acid; SFA, saturated fatty-acid; PGC1β, peroxisome proliferator-activated receptor co-activator 1 β; PPARγ, peroxisome proliferator-activated receptor γ; KLF4, kruppel like factor 4. |
Since obesity-induced adipose tissue MФ polarization is suggested as a new target of obesity and related disorder treatment, investigating practical therapeutic strategies-related MФ polarization is on the rise. Exercise training as a non-invasive low-cost strategy has abundantly been recommended for interrupting the obesity-induced inflammatory mechanisms because of its potential to trigger these mechanisms at the early stage of inflammation in circulating immune cells. Initially, the adipocyte hypertrophy is inhibited by exercise training due to higher consumption of excessive energy. In physically active people, a regular cycle of adipocytes death/regeneration causes the inhibition of CLS forming, inducing a balance in the ratio of anti-inflammatory/inflammatory immune cells. Additionally, transcription of the anti-inflammatory genes is demonstrated during exercise training, causing an anti-inflammatory influx of cytokines/adipokines. Consequently, the lower concentration of chemoattractant factors in adipose tissue leads to the reduction of MON recruitment and infiltration. This anti-inflammatory environment either maintains the polarization mechanisms of M2ATM or suppresses the distinction of M1ATM. Moreover, expression of inflammatory myokines during exercise activities is shown to be valuable for maintaining the lean state. A complicated network of the anti-inflammatory activities is provoked by exercise-induced myokines, including downregulation of inflammatory factors, alterations of immune cell counts, increase of anti-inflammatory genes, and functional changes in immune cells. The precise role of these events has not been clarified in the prevention, susceptibility, suppression, and treatment of immunometabolic disorders at molecular levels. It is further practical that future studies be conducted on the early stages of obesity-induced inflammation, such as the systemic changes of circulating immune cells. The prolonged exposure to both dyslipidemia and the inflammatory environment could promote a new distinction among circulating immune cells leading to provoking the inflammatory MФ polarization. A systematic anti-inflammatory environment demonstrated following the exercise training can trigger obesity-induced immunometabolic changes in these circulating cells, indicating the inhibition of the aggravation of this inflammatory state. These changes may also be developed by exercise-induced modulatory effects on the negative regulators of inflammatory signaling pathways. Additionally, concentrating on the duration (acute, short term, and long term), calorie usage, type (resistance, endurance, and combined training), training method (cycling, bandage, water training, etc.), volume (sessions per week), and intensity (low, moderate, and high) of the exercise protocols could be useful to improve the exercise effects on immunity-related metabolic dysfunction.
Abbreviations
AT, adipose tissue; ATM, adipose tissue macrophages; adipoR, adiponectin receptor; AP-1, activator protein 1; BMI, body mass index; CLSs, crown-like structures; CRP, c-reactive protein; CVD, cardiovascular diseases; CCL, C-C motif ligand; CXCL, C-X-C motif ligand; CCR, C-C motif receptor; CX3CL, C-X3-C motif ligand; CMs, classical monocyte; CD, cluster of differentiation; DIO, diet induced-obesity; DAMPs, damaged-associated molecular patterns; DC, dendritic cell; EOSs, eosinophils; ERK, extracellular signal-regulated kinase; FFAs, free fatty acids; GM-CSF, macrophage colony-stimulating factor; HFD, high-fat diet; HOMA-IR, homeostasis model assessment of insulin resistance; HbA1c, glycated hemoglobin; HDL, high-density lipoprotein; HSP, heat shock proteins; hsCRP, high-sensitive CRP; HIIT, high-intensity interval training; IL, interleukin; ILC-2, innate lymphoid cells-2; IR, insulin resistance; iNKT, invariant-chain natural killer T cells; IL-1Ra, interleukin-1 receptor antagonist; IL-6R, IL-6 receptor; IκBK, I kappa-B kinase; IM, intermediate monocyte; ICAM-1, intercellular adhesion molecule-1; IFN, interferon regulatory factor; IRAK, IL-1R-associated kinase; IKK, inhibitor of kappa light polypeptide gene enhancer in B-cells kinase; iNOS, inducible nitric oxide; JNK, c-Jun N-terminal kinases; LGSI, low-grade systemic inflammation; LAT, lean adipose tissue; LPS, lipopolysaccharides; LBP, LPS-binding protein; MФs, macrophages; MetS, metabolic syndrome; MONs, monocytes; M-CSF, macrophage-colony stimulating factor; M-CSFR, receptor; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemoattractant protein-1; mRNA, messenger ribonucleic acid; MyD88, myeloid differentiation factor 88; Mal, MyD88 adaptor-like; mCD14, membrane-bound CD14; MD2, myeloid differentiation factor 2; MICT, moderate-intensity interval training; NF-κB, nuclear factor-kappa B; NEFA, non-esterified fatty acid; NK cells, natural killer cells; NCM, non-classical monocyte; OW, overweight; OB, obese/obesity; PAMPs, pathogen-associated molecular patterns; PBMC, peripheral blood mononuclear cells; PRR, pattern recognition receptors; VAT, visceral adipose tissue; WAT, white adipose tissue; ox-LDL, oxidized low-density lipoprotein; PWV, pales wave velocity; OAT, obese adipose tissue.
Acknowledgments
Appreciations are extended to Dr. Hossein Khanahmad (Department of Genetics and Molecular Biology, Isfahan University of Medical Science, Isfahan, Iran) for his advice.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors have no conflicts of interest to declare.
References
1. Man K, Kutyavin VI, Chawla A. Tissue immunometabolism: development, physiology, and pathobiology. Cell Metab. 2017;25(1):11–26. doi:10.1016/j.cmet.2016.08.016
2. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-: direct role in obesity-linked insulin resistance. Sci NY Washinton. 1993;259:87. doi:10.1126/science.7678183
3. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi:10.1172/JCI200319246
4. Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflammation. 2013;2013:1–12. doi:10.1155/2013/139239
5. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97. doi:10.1038/nri2921
6. Boa B, Yudkin J, Hinsbergh V, Bouskela E, Eringa E. Exercise effects on perivascular adipose tissue: endocrine and paracrine determinants of vascular function. Br J Pharmacol. 2017;174(20):3466–3481. doi:10.1111/bph.v174.20
7. Wensveen FM, Valentić S, Šestan M, Turk Wensveen T, Polić B. The “Big Bang” in obese fat: events initiating obesity‐induced adipose tissue inflammation. Eur J Immunol. 2015;45(9):2446–2456. doi:10.1002/eji.201545502
8. León-Pedroza JI, González-Tapia LA, del Olmo-Gil E, Castellanos-Rodríguez D, Escobedo G, González-Chávez A. Low-grade systemic inflammation and the development of metabolic diseases: from the molecular evidence to the clinical practice. Cirugía y Cirujanos. 2015;83(6):543–551. doi:10.1016/j.circen.2015.11.008
9. Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab. 2007;293(4):E1118–E1128. doi:10.1152/ajpendo.00435.2007
10. Kitamura H, Kimura S, Shimamoto Y, et al. Ubiquitin-specific protease 2–69 in macrophages potentially modulates metainflammation. FASEB J. 2013;27(12):4940–4953. doi:10.1096/fsb2.v27.12
11. Timmerman KL, Flynn MG, Coen PM, Markofski MM, Pence BD. Exercise training-induced lowering of inflammatory (CD14+ CD16+) monocytes: a role in the anti-inflammatory influence of exercise? J Leukoc Biol. 2008;84(5):1271–1278. doi:10.1189/jlb.0408244
12. Markofski MM, Flynn MG, Carrillo AE, Armstrong CL, Campbell WW, Sedlock DA. Resistance exercise training-induced decrease in circulating inflammatory CD14+ CD16+ monocyte percentage without weight loss in older adults. Eur J Appl Physiol. 2014;114(8):1737–1748. doi:10.1007/s00421-014-2902-1
13. Rodriguez-Miguelez P, Fernandez-Gonzalo R, Almar M, et al. Role of toll-like receptor 2 and 4 signaling pathways on the inflammatory response to resistance training in elderly subjects. Age. 2014;36(6):9734. doi:10.1007/s11357-014-9734-0
14. Bonfante ILP, Chacon-Mikahil MPT, Brunelli DT, et al. Combined training, FNDC5/irisin levels and metabolic markers in obese men: a randomised controlled trial. Eur J Sport Sci. 2017;17(5):629–637. doi:10.1080/17461391.2017.1296025
15. Mazur-Bialy AI, Pocheć E, Zarawski M. Anti-inflammatory properties of irisin, mediator of physical activity, are connected with TLR4/MyD88 signaling pathway activation. Int J Mol Sci. 2017;18(4):701. doi:10.3390/ijms18040701
16. Ostrowski K, Rohde T, Asp S, Schjerling P, Pedersen BK. Pro‐and anti‐inflammatory cytokine balance in strenuous exercise in humans. J Physiol. 1999;515(1):287–291. doi:10.1111/j.1469-7793.1999.287ad.x
17. Balducci S, Zanuso S, Nicolucci A, et al. Anti-inflammatory effect of exercise training in subjects with type 2 diabetes and the metabolic syndrome is dependent on exercise modalities and independent of weight loss. Nutr Metab Cardiovasc Dis. 2010;20(8):608–617. doi:10.1016/j.numecd.2009.04.015
18. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177–185. doi:10.1038/nature21363
19. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2009;28:445–489. doi:10.1146/annurev-immunol-030409-101212
20. Wu D, Molofsky AB, Liang H-E, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243–247. doi:10.1126/science.1201475
21. Molofsky AB, Nussbaum JC, Liang H-E, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210(3):535–549. doi:10.1084/jem.20121964
22. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. doi:10.1172/JCI29881
23. Ilavská S, Horváthová M, Szabová M, et al. Association between the human immune response and body mass index. Hum Immunol. 2012;73(5):480–485. doi:10.1016/j.humimm.2012.02.023
24. Patsouris D, Li -P-P, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8(4):301–309. doi:10.1016/j.cmet.2008.08.015
25. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity. 2015;42(6):1005–1019. doi:10.1016/j.immuni.2015.06.006
26. Hashiguchi M, Kashiwakura Y, Kojima H, Kobayashi A, Kanno Y, Kobata T. IL‐33 activates eosinophils of visceral adipose tissue both directly and via innate lymphoid cells. Eur J Immunol. 2015;45(3):876–885. doi:10.1002/eji.201444969
27. Mathis D. Immunological goings-on in visceral adipose tissue. Cell Metab. 2013;17(6):851–859. doi:10.1016/j.cmet.2013.05.008
28. Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–939. doi:10.1038/nm.2002
29. Vasanthakumar A, Moro K, Xin A, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. 2015;16(3):276–285. doi:10.1038/ni.3085
30. Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15(8):921–929. doi:10.1038/nm.2001
31. Barnes MA, Carson MJ, Nair MG. Non-traditional cytokines: how catecholamines and adipokines influence macrophages in immunity, metabolism and the central nervous system. Cytokine. 2015;72(2):210–219. doi:10.1016/j.cyto.2015.01.008
32. Qiu Y, Shan B, Yang L, Liu Y. Adipose tissue macrophage in immune regulation of metabolism. Sci China Life Sci. 2016;59(12)1–9.
33. Bai Y, Sun Q. Macrophage recruitment in obese adipose tissue. Obesity Rev. 2015;16(2):127–136. doi:10.1111/obr.2015.16.issue-2
34. Thomas D, Apovian C. Macrophage functions in lean and obese adipose tissue. Metabolism. 2017;72:120–143. doi:10.1016/j.metabol.2017.04.005
35. Boutens L, Stienstra R. Adipose tissue macrophages: going off track during obesity. Diabetologia. 2016;59(5):879–894. doi:10.1007/s00125-016-3904-9
36. Bigornia S, Farb M, Mott M, et al. Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr Diabetes. 2012;2(3):e30. doi:10.1038/nutd.2012.3
37. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Fron immunol. 2014;5. doi:10.3389/fimmu.2014.00514
38. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1–m2 polarization balance. Front Immunol. 2014;5. doi:10.3389/fimmu.2014.00614
39. Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–787. doi:10.1038/nature06902
40. Kosteli A, Sugaru E, Haemmerle G, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest. 2010;120(10):3466–3479. doi:10.1172/JCI42845
41. Shapiro H, Pecht T, Shaco-Levy R, et al. Adipose tissue foam cells are present in human obesity. J Clin Endocrinol Metab. 2013;98(3):1173–1181. doi:10.1210/jc.2012-2745
42. Castoldi A, de Souza CN, Câmara NOS, Moraes-Vieira PM. The macrophage switch in obesity development. Front Immunol. 2016;6:637.
43. Kamei N, Tobe K, Suzuki R, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem. 2006;281(36):26602–26614. doi:10.1074/jbc.M601284200
44. Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175(6):3463–3468. doi:10.4049/jimmunol.175.6.3463
45. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro-and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta Mol Cell Res. 2011;1813(5):878–888. doi:10.1016/j.bbamcr.2011.01.034
46. Kraakman MJ, Kammoun HL, Allen TL, et al. Blocking IL-6 trans-signaling prevents high-fat diet-induced adipose tissue macrophage recruitment but does not improve insulin resistance. Cell Metab. 2015;21(3):403–416. doi:10.1016/j.cmet.2015.02.006
47. Xueyao Y, Saifei Z, Dan Y, et al. Circulating fractalkine levels predict the development of the metabolic syndrome. Int J Endocrinol. 2014;2014.
48. Shah R, Hinkle CC, Ferguson JF, et al. Fractalkine is a novel human adipochemokine associated with type 2 diabetes. Diabetes. 2011;60(5):1512–1518. doi:10.2337/db10-0956
49. Polyák Á, Ferenczi S, Dénes Á, et al. The fractalkine/Cx3CR1 system is implicated in the development of metabolic visceral adipose tissue inflammation in obesity. Brain Behav Immun. 2014;38:25–35. doi:10.1016/j.bbi.2014.01.010
50. Dominguez H, Storgaard H, Rask-Madsen C, et al. Metabolic and vascular effects of tumor necrosis factor-α blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res. 2005;42(6):517–525. doi:10.1159/000088261
51. Serino M, Menghini R, Fiorentino L, et al. Mice heterozygous for tumor necrosis factor-α converting enzyme are protected from obesity-induced insulin resistance and diabetes. Diabetes. 2007;56(10):2541–2546. doi:10.2337/db07-0360
52. Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18(6):816–830. doi:10.1016/j.cmet.2013.11.001
53. Mills C. M1 and M2 macrophages: oracles of health and disease. Crit Reviews™ in Immunol. 2012;32(6).
54. Hill DA, Lim H-W, Kim YH, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc National Acad Sci. 2018;115(22):E5096–E5105. doi:10.1073/pnas.1802611115
55. Peterson KR, Cottam MA, Kennedy AJ, Hasty AH. Macrophage-Targeted therapeutics for metabolic disease. Trends Pharmacol Sci. 2018;39(6):536–546. doi:10.1016/j.tips.2018.03.001
56. Porta C, Riboldi E, Ippolito A, Sica A Molecular and epigenetic basis of macrophage polarized activation.
57. Han MS, Jung DY, Morel C, et al. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. 2013;339(6116):218–222. doi:10.1126/science.1227568
58. Gao Z, Hwang D, Bataille F, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor κB kinase complex. J Biol Chem. 2002;277(50):48115–48121. doi:10.1074/jbc.M209459200
59. Banks AS, McAllister FE, Camporez JPG, et al. An ERK/Cdk5 axis controls the diabetogenic actions of PPAR [ggr]. Nature. 2015;517(7534):391–395. doi:10.1038/nature13887
60. Carlson CJ, Koterski S, Sciotti RJ, Poccard GB, Rondinone CM. Enhanced basal activation of mitogen-activated protein kinases in adipocytes from type 2 diabetes: potential Role of p38 in the downregulation of GLUT4 expression. Diabetes. 2003;52(3):634–641. doi:10.2337/diabetes.52.3.634
61. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008;57(12):3239–3246. doi:10.2337/db08-0872
62. Jeffery E, Church CD, Holtrup B, Colman L, Rodeheffer MS. Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol. 2015;17(4):376–385. doi:10.1038/ncb3122
63. Talukdar S, Bandyopadhyay G, Li D, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18(9):1407–1412. doi:10.1038/nm.2885
64. Wensveen FM, Jelenčić V, Valentić S, et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol. 2015;16(4):376–385. doi:10.1038/ni.3120
65. Amano SU, Cohen JL, Vangala P, et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014;19(1):162–171. doi:10.1016/j.cmet.2013.11.017
66. Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116(6):1494–1505. doi:10.1172/JCI26498
67. Pecht T, Gutman‐Tirosh A, Bashan N, Rudich A. Peripheral blood leucocyte subclasses as potential biomarkers of adipose tissue inflammation and obesity subphenotypes in humans. Obesity Rev. 2014;15(4):322–337. doi:10.1111/obr.12133
68. Yang J, Zhang L, Yu C, Yang X-F, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomarker Res. 2014;2(1):1. doi:10.1186/2050-7771-2-1
69. Hedl M, Abraham C. Negative regulation of human mononuclear phagocyte function. Mucosal Immunol. 2013;6(2):205–223.
70. Celada A, Borràs FE, Soler C, et al. The transcription factor PU. 1 is involved in macrophage proliferation. J Exp Med. 1996;184(1):61–69. doi:10.1084/jem.184.1.61
71. Ma X, Lin WY, Chen Y, et al. Structural basis for the dual recognition of helical cytokines IL-34 and CSF-1 by CSF-1R. Structure. 2012;20(4):676–687. doi:10.1016/j.str.2012.02.010
72. Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood. 1980;56(6):947–958. doi:10.1182/blood.V56.6.947.947
73. Ziegler-Heitbrock H. Heterogeneity of human blood monocytes: the CD14+ CD16+ subpopulation. Immunol Today. 1996;17(9):424–428. doi:10.1016/0167-5699(96)10029-3
74. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi:10.1016/S1074-7613(03)00174-2
75. Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116(16):e74–e80. doi:10.1182/blood-2010-02-258558
76. Cros J, Cagnard N, Woollard K, et al. Human CD14 dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33(3):375–386. doi:10.1016/j.immuni.2010.08.012
77. Wong KL, Tai JJ-Y, Wong W-C, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118(5):e16–e31. doi:10.1182/blood-2010-12-326355
78. Rogacev KS, Ulrich C, Blömer L, et al. Monocyte heterogeneity in obesity and subclinical atherosclerosis. Eur Heart J. 2010;31(3):369–376. doi:10.1093/eurheartj/ehp308
79. Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. doi:10.1172/JCI200319451
80. Bories G, Caiazzo R, Derudas B, et al. Impaired alternative macrophage differentiation of peripheral blood mononuclear cells from obese subjects. Diabetes Vasc Dis Res. 2012;9(3):189–195. doi:10.1177/1479164111430242
81. Kraakman MJ, Murphy AJ, Jandeleit-Dahm K, Kammoun HL. Macrophage polarization in obesity and type 2 diabetes: weighing down our understanding of macrophage function? Front Immunol. 2014;5:470. doi:10.3389/fimmu.2014.00470
82. Jung UJ, Seo YR, Ryu R, Choi M-S. Differences in metabolic biomarkers in the blood and gene expression profiles of peripheral blood mononuclear cells among normal weight, mildly obese and moderately obese subjects. Br J Nutr. 2016;116(6):1022–1032. doi:10.1017/S0007114516002993
83. Kullo IJ, Hensrud DD, Allison TG. Comparison of numbers of circulating blood monocytes in men grouped by body mass index (<25, 25 to <30, ≥30). Am J Cardiol. 2002;89(12):1441–1443. doi:10.1016/s0002-9149(02)02366-4
84. Babio N, Ibarrola-Jurado N, Bulló M, et al. White blood cell counts as risk markers of developing metabolic syndrome and its components in the PREDIMED study. PLoS One. 2013;8(3):e58354. doi:10.1371/journal.pone.0058354
85. Viardot A, Heilbronn L, Samocha‐Bonet D, Mackay F, Campbell L, Samaras K. Obesity is associated with activated and insulin resistant immune cells. Diabetes Metab Res Rev. 2012;28(5):447–454. doi:10.1002/dmrr.2302
86. Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;18(6):228–232. doi:10.1016/j.tcm.2008.11.004
87. Jialal I, Adams-Huet B, Devaraj S. Monocyte cell adhesion molecule receptors in nascent metabolic syndrome. Clin Biochem. 2016;49(6):505–507. doi:10.1016/j.clinbiochem.2015.12.009
88. Tanaka SI, Isoda F, Ishihara Y, Kimura M, Yamakawa T. T lymphopaenia in relation to body mass index and TNF‐α in human obesity: adequate weight reduction can be corrective. Clin Endocrinol (Oxf). 2001;54(3):347–354.
89. Satoh N, Shimatsu A, Himeno A, et al. Unbalanced M1/M2 phenotype of peripheral blood monocytes in obese diabetic patients. Diabetes Care. 2010;33(1):e7–e7. doi:10.2337/dc09-1315
90. Garton T, Keep RF, Hua Y, Xi G. CD163, a hemoglobin/haptoglobin scavenger receptor, after intracerebral hemorrhage: functions in microglia/macrophages versus neurons. Transl Stroke Res. 2017;8(6):612–616.
91. Kim JE, Lin G, Zhou J, Mund JA, Case J, Campbell WW. Weight loss achieved using an energy restriction diet with normal or higher dietary protein decreased the number of CD14++ CD16+ proinflammatory monocytes and plasma lipids and lipoproteins in middle-aged, overweight, and obese adults. Nutr Res. 2017;40:75–84. doi:10.1016/j.nutres.2017.02.007
92. Wonner R, Wallner S, Orsó E, Schmitz G. Effects of acute exercise on monocyte subpopulations in metabolic syndrome patients. Cytometry Part B. 2018;94(4):596–605. doi:10.1002/cyto.b.v94.4
93. Yamauchi T, Kamon J, Ito Y, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423(6941):762. doi:10.1038/nature01705
94. Ghadge AA, Khaire AA, Kuvalekar AA. Adiponectin: a potential therapeutic target for metabolic syndrome. Cytokine Growth Factor Rev. 2018;39:151–158. doi:10.1016/j.cytogfr.2018.01.004
95. Savage DB, Petersen KF, Shulman GI. Mechanisms of insulin resistance in humans and possible links with inflammation. Hypertension. 2005;45(5):828–833. doi:10.1161/01.HYP.0000163475.04421.e4
96. Ouchi N, Kihara S, Arita Y, et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation. 2001;103(8):1057–1063. doi:10.1161/01.CIR.103.8.1057
97. Yokota T, Oritani K, Takahashi I, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96(5):1723–1732. doi:10.1182/blood.V96.5.1723
98. Okamoto Y, Folco EJ, Minami M, et al. Adiponectin inhibits the production of CXC receptor 3 chemokine ligands in macrophages and reduces T-lymphocyte recruitment in atherogenesis. Circ Res. 2008;102(2):218–225. doi:10.1161/CIRCRESAHA.107.164988
99. Poglio S, De Toni F, Lewandowski D, et al. In situ production of innate immune cells in murine white adipose tissue. Blood. 2012;120(25):4952–4962
100. Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157(4):832–844. doi:10.1016/j.cell.2014.04.016
101. Cousin B, Munoz O, André M, et al. A role for preadipocytes as macrophage-like cells. FASEB J. 1999;13(2):305–312. doi:10.1096/fsb2.v13.2
102. Charrière G, Cousin B, Arnaud E, et al. Preadipocyte conversion to macrophages: evidence of plasticity. J Biol Chem. 2003;278(11):9850–9855. doi:10.1074/jbc.M210811200
103. Hill AA, Anderson-Baucum EK, Kennedy AJ, Webb CD, Yull FE, Hasty AH. Activation of NF-κB drives the enhanced survival of adipose tissue macrophages in an obesogenic environment. Mol Metab. 2015;4(10):665–677. doi:10.1016/j.molmet.2015.07.005
104. Rogero MM, Calder PC. Obesity, inflammation, toll-like receptor 4 and fatty acids. Nutrients. 2018;10(4):432. doi:10.3390/nu10040432
105. De Angulo A, Travis P, Jolly C. Obesity-Induced T Cell Senescence Contributes to Prostate Cancer Progression. AACR; 2018.
106. Filgueiras LR, Serezani CH, Jancar S. Leukotriene B4 as a potential therapeutic target for the treatment of metabolic disorders. Front Immunol. 2015;6. doi:10.3389/fimmu.2015.00515
107. Chung J, Nguyen A-K, Henstridge DC, et al. HSP72 protects against obesity-induced insulin resistance. Proc National Acad Sci. 2008;105(5):1739–1744. doi:10.1073/pnas.0705799105
108. Kurucz I, Morva A, Vaag A, et al. Decreased expression of heat shock protein 72 in skeletal muscle of patients with type 2 diabetes correlates with insulin resistance. Diabetes. 2002;51(4):1102–1109. doi:10.2337/diabetes.51.4.1102
109. Ringseis R, Eder K, Mooren F, Krüger K. Metabolic signals and innate immune activation in obesity and exercise. inflammation. 2015;10(14):157–167.
110. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.022
111. Vidya MK, Kumar VG, Sejian V, Bagath M, Krishnan G, Bhatta R. Toll-like receptors: significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol. 2018;37(1):20–36. doi:10.1080/08830185.2017.1380200
112. Goulopoulou S, McCarthy CG, Webb RC. Toll-like receptors in the vascular system: sensing the dangers within. Pharmacol Rev. 2016;68(1):142–167. doi:10.1124/pr.114.010090
113. Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327(5963):296–300. doi:10.1126/science.1184003
114. Jo E-K, Kim JK, Shin D-M, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13(2):148–1459.
115. Jialal I, Kaur H, Devaraj S. Toll-like receptor status in obesity and metabolic syndrome: a translational perspective. J Clin Endocrinol Metab. 2013;99(1):39–48. doi:10.1210/jc.2013-3092
116. Salehian B, Mahabadi V. The crossroad of inflammation and diabetes: role of toll-like receptor. Int J Diabetes Clin Res. 2015;2(3). doi:10.23937/2377-3634/1410032
117. Rada I, Deldicque L, Francaux M, Zbinden-Foncea H. Toll like receptor expression induced by exercise in obesity and metabolic syndrome: a systematic review. Exerc Immunol Rev. 2018;24.
118. Zhang X-J, Zhang P, Li H. Interferon regulatory factor signalings in cardiometabolic diseases. Hypertension. 2015;66(2):222–247. doi:10.1161/HYPERTENSIONAHA.115.04898
119. Farrugia M, Baron B. The role of toll-like receptors in autoimmune diseases through failure of the self-recognition mechanism. Int J Inflam. 2017;2017.
120. Yang L, Seki E. Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front Physiol. 2012;3:138. doi:10.3389/fphys.2012.00138
121. Kim S-J, Choi Y, Choi Y-H, Park T. Obesity activates toll-like receptor-mediated proinflammatory signaling cascades in the adipose tissue of mice. J Nutr Biochem. 2012;23(2):113–122. doi:10.1016/j.jnutbio.2010.10.012
122. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi:10.1038/nri1391
123. Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7(6):837–847. doi:10.1016/S1074-7613(00)80402-1
124. Medvedev AE, Lentschat A, Kuhns DB, et al. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J Exp Med. 2003;198(4):521–531. doi:10.1084/jem.20030701
125. Kondo T, Kawai T, Akira S. Dissecting negative regulation of toll-like receptor signaling. Trends Immunol. 2012;33(9):449–458. doi:10.1016/j.it.2012.05.002
126. O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–364. doi:10.1038/nri2079
127. Orr JS, Puglisi MJ, Ellacott KL, Lumeng CN, Wasserman DH, Hasty AH. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes. 2012;61(11):2718–2727.
128. Lancaster GI, Febbraio MA. The immunomodulating role of exercise in metabolic disease. Trends Immunol. 2014;35(6):262–269. doi:10.1016/j.it.2014.02.008
129. Naghizadeh M, Baradaran B, Saghafi-Asl M, et al. Toll-like receptor signaling and serum levels of interferon β and lipopolysaccharide binding protein are related to abdominal obesity: a case-control study between metabolically healthy and metabolically unhealthy obese individuals. Nutr Res. 2018;55:11–20. doi:10.1016/j.nutres.2018.03.014
130. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi:10.2337/db06-1491
131. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi:10.1172/JCI28898
132. Youssef-Elabd EM, McGee KC, Tripathi G, et al. Acute and chronic saturated fatty acid treatment as a key instigator of the TLR-mediated inflammatory response in human adipose tissue, in vitro. J Nutr Biochem. 2012;23(1):39–50. doi:10.1016/j.jnutbio.2010.11.003
133. Jialal I, Huet BA, Kaur H, Chien A, Devaraj S. Increased toll-like receptor activity in patients with metabolic syndrome. Diabetes Care. 2012;35(4):900–904. doi:10.2337/dc11-2375
134. Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4–dependent and–independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25(6):1213–1219. doi:10.1161/01.ATV.0000159891.73193.31
135. Creely SJ, McTernan PG, Kusminski CM, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292(3):E740–E747. doi:10.1152/ajpendo.00302.2006
136. Caricilli AM, Nascimento PH, Pauli JR, et al. Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J Endocrinol. 2008;199(3):399–406. doi:10.1677/JOE-08-0354
137. Ahmad R, Al-Mass A, Atizado V, et al. Elevated expression of the toll like receptors 2 and 4 in obese individuals: its significance for obesity-induced inflammation. Journal of inflammation. 2012;9(1):48.
138. Reyna SM, Ghosh S, Tantiwong P, et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes. 2008;57(10):2595-2602.
139. Arias-Loste MT, Iruzubieta P, Puente Á, et al. Increased expression profile and functionality of TLR6 in peripheral blood mononuclear cells and hepatocytes of morbidly obese patients with non-alcoholic fatty liver disease. International journal of molecular sciences. 2016;17(11):1878.
140. Ahmad R, Shihab PK, Thomas R, et al. Increased expression of the interleukin-1 receptor-associated kinase (IRAK)-1 is associated with adipose tissue inflammatory state in obesity. Diabetology & metabolic syndrome. 2015;7(1):71.
141. Gupta S, Maratha A, Siednienko J, et al. Analysis of inflammatory cytokine and TLR expression levels in Type 2 Diabetes with complications. Scientific reports. 2017;7(1):7633.
142. Thomas RS, Kochumon S, Sindhu S, Ahmad R. Elevated expression of TLR10 in the adipose tissue in obesity: Implication in metabolic inflammation. In: Am Assoc Immnol; 2017.
143. Cuevas AM, Lazo M, Zuñiga I, et al. Expression of MYD88 in Adipose Tissue of Obese People: Is There Some Role in the Development of Metabolic Syndrome? Metabolic syndrome and related disorders. 2017;15(2):80-85.
144. Amiri P, Naghizadeh M, Baradaran B, Saghafi-Asl M, Shanehbandi D, Mirmajidi S. Insulin resistance in relation to inflammatory gene expression and metabolic features in apparently healthy obese individuals. International Journal of Diabetes in Developing Countries. 2018:1-8.
145. da Silva Correia J, Ulevitch RJ. MD-2 and TLR4 N-linked glycosylations are important for a functional lipopolysaccharide receptor. J Biol Chem. 2002;277(3):1845–1854.
146. Pan Z, Zhou L, Hetherington CJ, Zhang D-E. Hepatocytes contribute to soluble CD14 production, and CD14 expression is differentially regulated in hepatocytes and monocytes. J Biol Chem. 2000;275(46):36430–36435. doi:10.1074/jbc.M003192200
147. Pugin JM, Schürer-Maly C, Leturcq D, Moriarty A, Ulevitch RJ, Tobias PS. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc National Acad Sci. 1993;90(7):2744–2748. doi:10.1073/pnas.90.7.2744
148. Fernández-Real JM. Pérez del Pulgar S, Luche E, et al,. CD14 modulates inflammation-driven insulin resistance. Diabetes. 2011;60(8):2179–2186. doi:10.2337/db10-1210
149. Ogawa Y, Imajo K, Yoneda M, et al. Soluble CD14 levels reflect liver inflammation in patients with nonalcoholic steatohepatitis. PLoS One. 2013;8(6):e65211. doi:10.1371/journal.pone.0065211
150. Jialal I, Rajamani U, Adams-Huet B, Kaur H. Circulating pathogen associated molecular pattern–binding proteins and high mobility group box protein 1 in nascent metabolic syndrome: implications for cellular toll-like receptor activity. Atherosclerosis. 2014;236(1):182–187. doi:10.1016/j.atherosclerosis.2014.06.022
151. Csak T, Velayudham A, Hritz I, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointestinal Liver Physiol. 2011;300(3):G433–G441. doi:10.1152/ajpgi.00163.2009
152. Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5(6):446–458. doi:10.1038/nri1630
153. Hamerman JA, Pottle J, Ni M, He Y, Zhang ZY, Buckner JH. Negative regulation of TLR signaling in myeloid cells—implications for autoimmune diseases. Immunol Rev. 2016;269(1):212–227. doi:10.1111/imr.12381
154. Negishi H, Ohba Y, Yanai H, et al. Negative regulation of toll-like-receptor signaling by IRF-4. Proc National Acad Sci. 2005;102(44):15989–15994. doi:10.1073/pnas.0508327102
155. Carty M, Goodbody R, Schröder M, Stack J, Moynagh PN, Bowie AG. The human adaptor SARM negatively regulates adaptor protein TRIF–dependent toll-like receptor signaling. Nat Immunol. 2006;7(10):1074. doi:10.1038/ni1382
156. Takezako N, Hayakawa M, Hayakawa H, et al. ST2 suppresses IL-6 production via the inhibition of IκB degradation induced by the LPS signal in THP-1 cells. Biochem Biophys Res Commun. 2006;341(2):425–432. doi:10.1016/j.bbrc.2005.12.206
157. Zhou F, Zhang X, van Dam H, Ten Dijke P, Huang H, Zhang L. Ubiquitin-specific protease 4 mitigates toll-like/interleukin-1 receptor signaling and regulates innate immune activation. J Biol Chem. 2012;287(14):11002–11010. doi:10.1074/jbc.M111.328187
158. Enesa K, Moll HP, Luong L, Ferran C, Evans PC. A20 suppresses vascular inflammation by recruiting proinflammatory signaling molecules to intracellular aggresomes. FASEB J. 2015;29(5):1869–1878. doi:10.1096/fsb2.v29.5
159. Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell. 2011;44(4):559–571. doi:10.1016/j.molcel.2011.09.015
160. Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of toll-like receptor responses. Nat Immunol. 2004;5(10):1052–1060. doi:10.1038/ni1110
161. Booth FW, Roberts CK, Laye MJ. Lack of exercise is a major cause of chronic diseases. Compr Physiol. 2012;2(2):1143.
162. Brunelli DT, Chacon-Mikahil M, Gáspari AF, et al. Combined training reduces subclinical inflammation in obese middle-age men. Med Sci Sports Exerc. 2015;47(10):2207–2215. doi:10.1249/MSS.0000000000000658
163. Lavie CJ, Church TS, Milani RV, Earnest CP. Impact of physical activity, cardiorespiratory fitness, and exercise training on markers of inflammation. J Cardiopulm Rehabil Prev. 2011;31(3):137–145. doi:10.1097/HCR.0b013e3182122827
164. You T, Arsenis NC, Disanzo BL, LaMonte MJ. Effects of exercise training on chronic inflammation in obesity. Sports Med. 2013;43(4):243–256. doi:10.1007/s40279-013-0023-3
165. Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol. 2011;11(9):607. doi:10.1038/nri3041
166. Goh J, Goh KP, Abbasi A. Exercise and adipose tissue macrophages: new frontiers in obesity research? Front Endocrinol (Lausanne). 2016;7. doi:10.3389/fendo.2016.00065
167. Kawanishi N, Yano H, Yokogawa Y, Suzuki K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc Immunol Rev. 2010;16.
168. Oliveira AG, Araujo TG, Carvalho BM, et al. Acute exercise induces a phenotypic switch in adipose tissue macrophage polarization in diet‐induced obese rats. Obesity. 2013;21(12):2545–2556. doi:10.1002/oby.20402
169. Xiong X-Q, Geng Z, Zhou B, et al. FNDC5 attenuates adipose tissue inflammation and insulin resistance via AMPK-mediated macrophage polarization in obesity. Metabolism. 2018;83:31–41. doi:10.1016/j.metabol.2018.01.013
170. Ruffino J, Davies N, Morris K, et al. Moderate-intensity exercise alters markers of alternative activation in circulating monocytes in females: a putative role for PPARγ. Eur J Appl Physiol. 2016;116(9):1671–1682. doi:10.1007/s00421-016-3414-y
171. Colato A, Abreu F, Medeiros N, et al. Effects of concurrent training on inflammatory markers and expression of CD4, CD8, and HLA-DR in overweight and obese adults. J Exercise Sci Fitness. 2014;12(2):55–61. doi:10.1016/j.jesf.2014.06.002
172. Barry JC, Simtchouk S, Durrer C, Jung ME, Little JP. Short-term exercise training alters leukocyte chemokine receptors in obese adults. Med Sci Sports Exerc. 2017;49(8):1631–1640. doi:10.1249/MSS.0000000000001261
173. Matos MA, Duarte TC, Ottone V, et al. The effect of insulin resistance and exercise on the percentage of CD16+ monocyte subset in obese individuals. Cell Biochem Funct. 2016;34(4):209–216. doi:10.1002/cbf.3178
174. Bruun JM, Helge JW, Richelsen B, Stallknecht B. Diet and exercise reduce low-grade inflammation and macrophage infiltration in adipose tissue but not in skeletal muscle in severely obese subjects. Am J Physiol Endocrinol Metab. 2006;290(5):E961–E967. doi:10.1152/ajpendo.00506.2005
175. Liu Y, Liu S-X, Cai Y, Xie K-L, Zhang W-L, Zheng F. Effects of combined aerobic and resistance training on the glycolipid metabolism and inflammation levels in type 2 diabetes mellitus. J Phys Ther Sci. 2015;27(7):2365–2371. doi:10.1589/jpts.27.2365
176. Leggate M, Carter WG, Evans MJ, Vennard RA, Sribala-Sundaram S, Nimmo MA. Determination of inflammatory and prominent proteomic changes in plasma and adipose tissue after high-intensity intermittent training in overweight and obese males. J Appl Physiol. 2012;112(8):1353–1360. doi:10.1152/japplphysiol.01080.2011
177. Haczeyni F, Barn V, Mridha AR, et al. Exercise improves adipose function and inflammation and ameliorates fatty liver disease in obese diabetic mice. Obesity. 2015;23(9):1845–1855. doi:10.1002/oby.21170
178. Dorneles GP, Haddad DO, Fagundes VO, et al. High intensity interval exercise decreases IL-8 and enhances the immunomodulatory cytokine interleukin-10 in lean and overweight–obese individuals. Cytokine. 2016;77:1–9. doi:10.1016/j.cyto.2015.10.003
179. Oliveira AG, Carvalho BM, Tobar N, et al. Physical exercise reduces circulating lipopolysaccharide and toll–like receptor 4 activation and improves insulin signaling in tissues of diet-induced obesity rats. Diabetes. 2011;60(3):784–796.
180. Vieira VJ, Valentine RJ, Wilund KR, Antao N, Baynard T, Woods JA. Effects of exercise and low-fat diet on adipose tissue inflammation and metabolic complications in obese mice. Am J Physiol Endocrinol Metab. 2009;296(5):E1164–E1171. doi:10.1152/ajpendo.00054.2009
181. Khadir A, Kavalakatt S, Cherian P, et al. Physical exercise enhanced heat shock protein 60 expression and attenuated inflammation in the adipose tissue of human diabetic obese. Front Endocrinol (Lausanne). 2018;9:16. doi:10.3389/fendo.2018.00016
182. Rodriguez-Miguelez P, Fernandez-Gonzalo R, Collado PS, et al. Whole-body vibration improves the anti-inflammatory status in elderly subjects through toll-like receptor 2 and 4 signaling pathways. Mech Ageing Dev. 2015;150:12–19. doi:10.1016/j.mad.2015.08.002
183. Auerbach P, Nordby P, Bendtsen LQ, et al. Differential effects of endurance training and weight loss on plasma adiponectin multimers and adipose tissue macrophages in younger, moderately overweight men. Am J Physiol Regul Integr Comp Physiol. 2013;305(5):R490–R498. doi:10.1152/ajpregu.00575.2012
184. Reyna SM, Tantiwong P, Cersosimo E, DeFronzo RA, Sriwijitkamol A, Musi N. Short-term exercise training improves insulin sensitivity but does not inhibit inflammatory pathways in immune cells from insulin-resistant subjects. J Diabetes Res. 2013;2013.
185. Durrer C, Francois M, Neudorf H, Little JP. Acute high-intensity interval exercise reduces human monocyte Toll-like receptor 2 expression in type 2 diabetes. Am J Physiol Regul Integr Comp Physiol. 2017;312(4):R529–R538. doi:10.1152/ajpregu.00348.2016
186. Dorneles GP, da Silva IRV, Korb A, et al. High intensity interval exercise enhances the global HDAC activity in PBMC and anti-inflammatory cytokines of overweight-obese subjects. Obesity Med. 2016;2:25–30. doi:10.1016/j.obmed.2016.05.004
187. de Souza DC, Matos VA. dos Santos VO, et al,. Effects of high-intensity interval and moderate-intensity continuous exercise on inflammatory, leptin, IgA, and lipid peroxidation responses in obese males. Front Physiol. 2018;9. doi:10.3389/fphys.2018.00567
188. Barry JC, Simtchouk S, Durrer C, Jung ME, Mui AL, Little JP. Short-term exercise training reduces anti-inflammatory action of interleukin-10 in adults with obesity. Cytokine. 2018;111:460–469. doi:10.1016/j.cyto.2018.05.035
189. Robinson E, Durrer C, Simtchouk S, et al. Short-term high-intensity interval and moderate-intensity continuous training reduce leukocyte TLR4 in inactive adults at elevated risk of type 2 diabetes. J Appl Physiol. 2015;119(5):508–516. doi:10.1152/japplphysiol.00334.2015
190. Yang HS, Woo JE, Lee SJ, Park SH, Woo JM. Elevated plasma pentraxin 3 levels are associated with development and progression of diabetic retinopathy in Korean patients with type 2 diabetes mellitus. Invest Ophthalmol Vis Sci. 2014;55(9):5989–5997. doi:10.1167/iovs.14-14864
191. Rathcke C, Johansen J, Vestergaard H. YKL-40, a biomarker of inflammation, is elevated in patients with type 2 diabetes and is related to insulin resistance. Inflammation Res. 2006;55(2):53–59. doi:10.1007/s00011-005-0010-8
192. Colato A, Fraga L, Dorneles G, Vianna P, Chies J, Peres A. Impact of aerobic water running training on peripheral immune-endocrine markers of overweight-obese women. Sci Sports. 2017;32(1):46–53. doi:10.1016/j.scispo.2016.04.003
193. Nielsen AR, Hojman P, Erikstrup C, et al. Association between interleukin-15 and obesity: interleukin-15 as a potential regulator of fat mass. J Clin Endocrinol Metab. 2008;93(11):4486–4493. doi:10.1210/jc.2007-2561
194. Park S-M, Kwak Y-S, Ji J-G. The effects of combined exercise on health-related fitness, endotoxin, and immune function of postmenopausal women with abdominal obesity. J Immunol Res. 2015;2015.
195. Chagas EFB, Bonfim MR, Turi BC, Brondino NCM, Monteiro HL. Effect of moderate intensity exercise on inflammatory markers among postmenopausal women. J Phys Act Health. 2017;14(6):479–485.
196. Nunes PRP, Barcelos LC, Oliveira AA, et al. Effect of resistance training on muscular strength and indicators of abdominal adiposity, metabolic risk, and inflammation in postmenopausal women: controlled and randomized clinical trial of efficacy of training volume. Age. 2016;38(2):40. doi:10.1007/s11357-016-9901-6
197. Orsatti FL, Nahas E, Maesta N, et al. Effects of resistance training frequency on body composition and metabolics and inflammatory markers in overweight postmenopausal women. J Sports Med Phys Fitness. 2014;54(3):317–325.
198. Geffken DF, Cushman M, Burke GL, Polak JF, Sakkinen PA, Tracy RP. Association between physical activity and markers of inflammation in a healthy elderly population. Am J Epidemiol. 2001;153(3):242–250. doi:10.1093/aje/153.3.242
199. Pischon T, Hankinson SE, Hotamisligil GS, Rifai N, Rimm EB. Leisure‐time physical activity and reduced plasma levels of obesity‐related inflammatory markers. Obes Res. 2003;11(9):1055–1064. doi:10.1038/oby.2003.145
200. Otvos JD, Shalaurova I, Wolak-Dinsmore J, et al. GlycA: a composite nuclear magnetic resonance biomarker of systemic inflammation. Clin Chem. 2015;61(5):714–723. doi:10.1373/clinchem.2014.232918
201. Dungan K, Binkley P, Osei K. GlycA is a novel marker of inflammation among non-critically ill hospitalized patients with type 2 diabetes. Inflammation. 2015;38(3):1357–1363. doi:10.1007/s10753-014-0107-8
202. Bartlett DB, Shepherd SO, Wilson OJ, et al. Neutrophil and monocyte bactericidal responses to 10 weeks of low-volume high-intensity interval or moderate-intensity continuous training in sedentary adults. Oxid Med Cell Longev. 2017;2017:1–12. doi:10.1155/2017/8148742
203. Abbasi A, de Paula Vieira R, Bischof F, et al. Sex-specific variation in signaling pathways and gene expression patterns in human leukocytes in response to endotoxin and exercise. J Neuroinflammation. 2016;13(1):289. doi:10.1186/s12974-016-0758-5
204. Neves PRDS, Tenório TRDS, Lins TA, et al. Acute effects of high-and low-intensity exercise bouts on leukocyte counts. J Exercise Sci Fitness. 2015;13(1):24–28. doi:10.1016/j.jesf.2014.11.003
205. Child M, Leggate M, Gleeson M. Effects of two weeks of high-intensity interval training (HIIT) on monocyte TLR2 and TLR4 expression in high BMI sedentary men. Int J Exerc Sci. 2013;6(1):10.
206. Shimizu K, Suzuki N, Imai T, et al. Monocyte and T-cell responses to exercise training in elderly subjects. J Strength Conditioning Res. 2011;25(9):2565–2572. doi:10.1519/JSC.0b013e3181fc5e67
207. Folco EJ, Rocha VZ, López-Ilasaca M, Libby P. Adiponectin inhibits pro-inflammatory signaling in human macrophages independent of interleukin-10. J Biol Chem. 2009;284(38):25569–25575. doi:10.1074/jbc.M109.019786
208. Kim D-Y, Jung S-Y, Seo B-D. Effect of exercise intervention on changes in free fatty acid levels and metabolic risk factors in stroke patients. J Phys Ther Sci. 2014;26(2):275–279. doi:10.1589/jpts.26.275
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.