Back to Journals » OncoTargets and Therapy » Volume 9
The evolution of combined molecular targeted therapies to advance the therapeutic efficacy in melanoma: a highlight of vemurafenib and cobimetinib
Received 5 January 2016
Accepted for publication 1 May 2016
Published 22 June 2016 Volume 2016:9 Pages 3739—3752
DOI https://doi.org/10.2147/OTT.S86774
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Min Li
Theresa M Medina, Karl D Lewis
Cutaneous Oncology Program, Division of Medical Oncology, School of Medicine, University of Colorado – Anschutz Medical Campus, Aurora, CO, USA
Abstract: Metastatic melanoma is an aggressive, rapidly progressive disease which historically had very few effective treatment options. However, since 2011, the therapeutic landscape of melanoma has undergone a dramatic transformation with two distinct approaches and has catalyzed the successful advancement in the clinical field of immuno-oncology. In addition, the recognition of a key oncogenic driver mutation in melanoma, BRAF, stimulated the development of multiple potent kinase inhibitors which has also influenced the expansion and use of targeted agents in the practice of oncology. Vemurafenib, the initial BRAF inhibitor approved for the treatment of melanoma, was the first agent to demonstrate rapid clinical responses and significantly improved survival which was a clinical breakthrough in the treatment of melanoma. Although exciting and practice changing, the unparalleled responses with vemurafenib are usually not sustained. Further investigations delineated several mechanisms of acquired resistance which are most often mediated by the upregulation of the MAPK pathway. MEK inhibitors, another class of small-molecule inhibitors, were developed as an alternative agent to suppress the MAPK pathway downstream, independent from BRAF activation. Multiple studies have demonstrated the improvement in antitumor activity when MEK inhibitors are used in combination with BRAF inhibitors in the treatment of metastatic melanoma. This is a review of the investigations that led to the US Food and Drug Administration approval in 2015 of the combination of vemurafenib and cobimetinib, adding to the quickly growing armament for the treatment of advanced or metastatic melanoma with a BRAF V600 mutation.
Keywords: melanoma, vemurafenib, cobimetinib
Introduction
The incidence of melanoma has been increasing for several decades, particularly in the population over 50 years of age.1 It is estimated that in 2016, it will be the fifth most common cancer diagnosed in American men and the seventh most common cancer in American women.1 Most patients are diagnosed with localized disease and can be treated successfully early on with surgical resection; however, there are still individuals who present with or subsequently will develop disseminated disease. Historically, the prognosis of those with stage IV melanoma was poor with half of patients dying within 8–10 months of diagnosis.2,3 Now, however, the treatment of metastatic melanoma is rapidly evolving and translating to longer survival and improved outcomes for many patients. This is in part due to the increased understanding of the pathogenesis of melanoma, especially with the discovery that the MAPK pathway is the key signaling pathway in the development of melanoma. Better understanding of this mechanism of proliferation has led to the identification of multiple targeted therapies which has translated into a substantial improvement in the outcomes of patients with metastatic melanoma. It is worth noting that the advancements made in targeted therapy have coincided with the exciting evolution of immuno-oncology. This is a review of the use of vemurafenib and cobimetinib for the treatment of advanced or metastatic melanoma.
Origin of BRAF inhibitor therapy for metastatic melanoma
In the pathogenesis of melanoma, constituent activation of the MAPK pathway is often involved. This employs a complex signaling pathway through a small G protein (RAS) and three protein kinases (RAF, MEK, ERK). Oncogenic activation of this pathway can arise from mutations in any of the pathway components or promoters.4 Typically, the important downstream mediators in the MAPK cell signaling pathway are simulated by the activation of RAS which then signals downstream via the interaction between the serine–threonine kinases, BRAF and CRAF.5,6 In BRAF wild-type cells, the BRAF kinase is activated by homo- or heterodimer formation with other RAF isoforms such as ARAF or CRAF.5,6 In BRAF-mutated cells, the BRAF kinase remains constitutively activated in a monomeric state, with a >400-fold increase in kinase activity.5–7 Upon activation of RAF, there is an interaction with downstream MEK kinases (MEK1 and MEK2) which initiates MEK phosphorylation that in turn facilitates an activating phosphorylation of ERK which promotes oncogenesis.5,8,9 Ultimately, the activation of ERK results in cellular proliferation along with migration and angiogenesis, accompanied by a deterrence of apoptosis, thereby promoting malignant cell growth.10 In the pathogenesis of melanoma, there is staunch reliance upon this signaling cascade. The most common mechanism by which this occurs is through activating mutations in BRAF which is present in ~50% of all advanced cutaneous melanomas.11 The most common BRAF mutation is a single-nucleotide mutation, thymine to adenine, at position 1799 in the activation segment of the kinase domain which results in a substitution of glutamic acid for valine at codon 600 (V600E). This point mutation is identified in ~80% of BRAF mutations. The next most common mutation replaces two nucleotides, guanine and thymine, for adenine at positions 1798 and 1799, respectively, resulting in an alternate substitution of lysine for valine at codon 600 (V600K).2 The mutations at V600 result in increased kinase activity, and typically, these mutations are not associated with other oncogenic driver mutations, such as NRAS or KIT.2
After attempts to inhibit BRAF by using the multi-kinase inhibitor sorafenib were clinically unsuccessful, vemurafenib was developed as a potent kinase inhibitor with a particular specificity for the ATP-binding pocket of the activated, mutant BRAF kinase, particularly the V600E variant. Vemurafenib was the first selective, reversible, V600 BRAF kinase inhibitor developed. The Phase I trial (BRIM-1) that investigated escalating doses of vemurafenib recommended 960 mg twice daily as the maximum tolerated dose on the basis of safety, pharmacodynamic, pharmacokinetic, and efficacy data.12 From the Phase I trial of vemurafenib, there was an extension cohort of 32 patients with BRAF V600E mutations, which at the time demonstrated an unprecedented overall response rate of 56% (Table 1).12 The Phase II (BRIM-2) trial corroborated a response rate of 53% (including 6% with a complete response); the median overall survival (OS) was 15.9 months and the median progression-free survival (PFS) was 6.7 months.2 Of note, ~24% of patients in the Phase II trial went on to receive ipilimumab after progression, but when these patients were censored from analysis, it did not significantly change the median OS.2 Both trials reported similar toxicity profiles with the most common reported adverse events related to cutaneous toxicities such as photosensitivity, rash, and the unexpected development of cutaneous squamous cell carcinomas (SCCs). Arthralgia, fatigue, alopecia, and asymptomatic elevated transaminases were also reported.2,12 Of note, there was one patient who died due to rapid progression of melanoma and acute renal failure which may have been related to vemurafenib.2 The safety results reported in the Phase II, BRIM-2, trial showed that 45% of patients required a dose reduction and 64% of patients required dose interruptions which were most often made due to rash, arthralgia, elevated liver enzyme levels, and photosensitivity reactions.2 The median dose of vemurafenib was reported to be 1,740 mg/day which was 91% of the intended dose of 1,920 mg/day.2
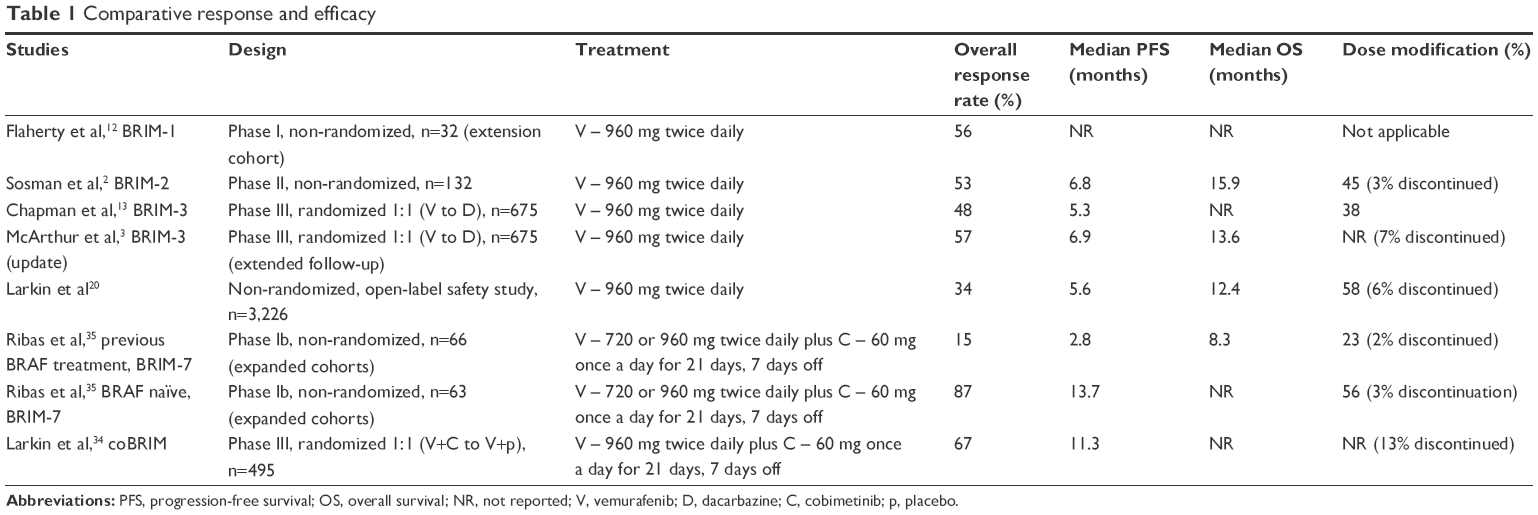 | Table 1 Comparative response and efficacy |
Vemurafenib monotherapy was further examined in BRIM-3, a Phase III multicenter, international trial which randomized 675 patients with melanoma who had V600 mutations (including 20 patients with atypical mutations, ie, V600K or V600D) to vemurafenib versus dacarbazine until disease progression.13 The response rates and toxicities associated with vemurafenib were similar to the results from the previous trials. Adverse events leading to dose modification or interruption were reported in 38% of patients treated with vemurafenib.13 This trial also demonstrated that patients with an alternative activating mutation in BRAF, that is, the V600K mutation, also benefited from vemurafenib with a reported response rate of 40%.13 After a planned interim analysis found a significant improvement in OS favoring vemurafenib, which showed a 63% relative reduction in the risk of death and a decrease of 74% in the risk of either death or disease progression (P<0.001), the protocol was amended such that patients in the dacarbazine arm were allowed to crossover to vemurafenib. About 25% of the patients did crossover but were then censored from further OS analysis.13 Extended follow-up results of the Phase III trial reported in 2014 after a follow-up of 12.5 months (censored at crossover) demonstrated the median OS for patients who received vemurafenib to be significantly better, 13.6 months as compared to 9.7 months with dacarbazine (hazard ratio [HR] 0.7, P=0.0008), and a PFS of 6.9 versus 1.6 months (HR 0.38, P<0.0001).14 Dabrafenib, another potent competitive inhibitor of the ATP-binding site of BRAF V600, has also been shown to demonstrate dramatic antitumor activity and was subsequently approved for the treatment of BRAF V600-mutated melanoma.15,16
Although the initial trials with the BRAF inhibitors given as monotherapy were practice changing, there were several recurring issues identified that warranted further investigation. An important toxicity recognized in all of the trials with single-agent BRAF inhibitors, as previously mentioned, was the development of cutaneous SCCs and keratoacanthomas. This was reported in 10%–14% of patients treated with dabrafenib and 19%–26% of patients treated with vemurafenib, and the onset typically occurred in the first 2–3 months of therapy.2,3,12–15 It was later recognized that this developed due to a paradoxical activation of the MAPK pathway in keratinocytes which did not harbor a BRAF mutation but had upstream oncogenic mutations, particularly preexisting HRAS mutations.13,16 The use of BRAF inhibitors thus hastens the progression of preexisting subclinical cancerous lesions but does not initiate tumorigenesis. Though cutaneous SCCs can be easily managed with local therapy, this corollary raised concern that other precancerous or occult cancer cells that are BRAF wild type but have altered MAPK signaling from activating RAS mutations could be stimulated to proliferate as a result of BRAF inhibitor therapy. Indeed, there were case reports of other secondary malignancies developing while on BRAF inhibitor monotherapy including an NRAS-mutated chronic myelomonocytic leukemia,17 progression of a KRAS-mutated pancreatic adenocarcinoma,18 and a KRAS-mutated colon adenocarcinoma.19
An extensive international, multicenter, open-label trial was designed to assess the safety of vemurafenib in a broader population, including patients with poor prognostic features such as an elevated lactate dehydrogenase (LDH) level and asymptomatic brain metastases, the elderly, and those with a poor performance status as rated by the Eastern Cooperative Oncology Group (ECOG) as ≥2.20 There were 3,226 patients enrolled in this single-arm study which confirmed the toxicity profile previously shown with vemurafenib therapy, and the incidence of grade 3 or 4 events was similar.20 This trial demonstrated that the presence of brain metastases or elevated LDH did not change the incidence of adverse events. Though, elderly patients (defined as ≥75 years) and those with an ECOG performance status ≥2 did have higher incidence of grade 3 and 4 toxicities and also were more likely to discontinue therapy due to adverse effects.20 The most frequent adverse effects listed as cause of dose modification or discontinuation were the development of cutaneous SCCs and keratoacanthomas, and QTc prolongation.20 In addition, there were 20 patients who died due to adverse events from vemurafenib.20 The most common grade 5 adverse events reported were general deterioration in health (five patients), cerebral hemorrhage (four out of five had existing central nervous system [CNS] metastases), cerebrovascular accident (four patients), and pulmonary embolism (four patients).20 Also of note, there were ten new primary malignancies reported that developed while on treatment including non-cutaneous SCCs, cutaneous T cell lymphomas, adenocarcinoma of the colon, Kaposi’s sarcoma, and urothelial carcinomas.20
Identified mechanisms of acquired resistance to BRAF inhibitor monotherapy
Another crucial issue that arose with selective BRAF inhibition was the limited durability of response. The activity of vemurafenib was characterized by a rapid response initially with several patients attaining substantial tumor reduction after just 14 days of therapy. Unfortunately however, the vast majority of patients treated with a BRAF inhibitor eventually progress due to the development of resistance. For instance, the Phase III trial comparing vemurafenib to dacarbazine greatly favored vemurafenib, though the median PFS was only ~7 months.3,15 In addition, there were 10%–15% of patients treated with vemurafenib without an initial response to therapy suggesting an intrinsic mechanism of resistance.21 The development of an acquired resistance was similarly reported in the Phase III investigation with dabrafenib versus dacarbazine with half of patients developing resistance ~5 months after starting treatment.15 Thus, further investigation into the underlying mechanisms of resistance was essential.
In general, the development of resistance to targeted kinase inhibitors in all tumor types is thought to occur by three different mechanisms: 1) a secondary reactivating mutation in the kinase, 2) the development of a mutation in an associated gene that bypasses the inhibited kinase, or 3) activation of another growth pathway. Most tumor types develop resistance to a targeted therapy via the first mechanism. For example, the T790M mutation in EGFR-mutant adenocarcinoma of the lung.21 Conversely, the mechanisms described for acquired BRAF inhibitor resistance are diverse but ultimately lead to reactivation of the MAPK pathway, ie, by the second mechanism. For instance, this was demonstrated from molecular analyses performed on biopsy samples from the BRIM-2 trial were performed at baseline, during treatment (cycle 1, day 15), and at progression to evaluate for potential biomarkers and mechanisms of resistance while on therapy.22 A clear observation was the reactivation of independent oncogenic signaling through the MAPK pathway at progression exhibited by increased levels of phosphorylated ERK.22
As mentioned, there are several diverse mechanisms described leading to MAPK pathway reactivation after treatment with a BRAF inhibitor which have been further categorized as MAPK-redundant or MAPK-related mechanisms.22 The MAPK-redundant mechanisms that have been described lead to upregulation or dysregulation of alternative bypass pathways such as the PI3K/AKT pathway (eg, by activating mutations of PI3K or AKT).21–23 Alternatively, loss of the inhibitor function of PTEN or mutational inactivation of PTEN can also lead to dysregulation of the PI3K/AKT pathway.24 The upregulation of cell surface receptor tyrosine kinases such as EGFR, insulin-like growth factor-1 receptor, or platelet-derived growth factor receptor-β can also lead to subsequent signaling through PI3K/AKT.21,22,24–27
However, it is much more common to find mechanisms that lead to MAPK-related resistance.21 The miscellany of mechanisms described includes 1) BRAF V600 gene amplification, 2) alternate splicing of BRAF V600E mRNA leading to enhanced dimerization, 3) development of NRAS (or less frequently K/H-RAS mutations), 4) MEK1/2 mutations, 5) COT1 overexpression, 6) CRAF overexpression, or 7) CDKN2A loss.21,22,25,27,28 The relative abundance and clinical significance of each of these mutations is not well understood. It is widely recognized that there is significant intra- and inter-tumor heterogeneity in the mechanisms of acquired BRAF resistance that develop,24,29 which demonstrates the tremendous genetic selective pressure to restore MAPK signaling for tumor proliferation. Although, despite this heterogeneity, for the majority of progressive tumors, each subclonal population of cells often develop only a single mechanism of resistance.29
A study evaluating the molecular profile of five BRAF-resistant metastases compared to a pretreatment sample obtained from a single patient treated with vemurafenib demonstrated the extent and variation of independently arising resistance mutations in a single patient.24 In addition, there was evidence of a small subpopulation of cells that harbored a resistance-conferring mutation prior to starting therapy, thus allowing a proliferative advantage once a BRAF inhibitor was started.24 This has previously been described for tumor samples that contain both NRAS and BRAF mutations. At baseline, mutations in NRAS and BRAF in a single cell are thought to be mutually exclusive, although coexisting mutations have been identified in rare circumstances, which again may allow for clonal expansion of resistant tumor cells once BRAF inhibitor therapy is initiated and may confer an intrinsic resistant phenotype.22
The clinical outcome associated with specific resistance mechanisms is another area that requires further exploration. A meta-analysis performed on a large cohort of tumor biopsies after progression, obtained from 100 patients enrolled in prior studies with vemurafenib and dabrafenib, was able to demonstrate a few clinically relevant associations with particular mutations.29 For instance, patients treated with vemurafenib were more likely to develop NRAS mutations (odds ratio [OR] 3.53, P=0.045).29 In addition, patients with NRAS mutations were more likely to have CNS metastases at baseline (OR 4.57, P=0.037) and also showed a marginal association with disease progression in the brain (OR 3.05, P=0.066).29 Patients with MEK1/2 mutations were more likely to demonstrate progressive disease in the liver (OR 7.61, P=0.011).29 Of note, there were no statistically significant differences in OS identified among patients with tumors harboring particular resistance mechanisms.29
The burgeoning role of MEK inhibitors
MEK inhibitors in preclinical studies were promising as an alternative targeted approach to therapy for melanoma. In vitro, MEK inhibitors demonstrated a stronger inhibition of both BRAF- and NRAS-mutated melanomas in comparison to vemurafenib.30 In the initial Phase III trial evaluating the use of the MEK inhibitor trametinib (the first US Food and Drug Administration [FDA]-approved small-molecule, selective, allosteric inhibitor of MEK1 and MEK2) in the first-line setting as a single agent, there was a significant improvement in the clinical response as compared to dacarbazine, despite allowing for crossover at progression.31 The median PFS was 4.8 versus 1.5 months (HR 0.45, P<0.001) and the median OS had not yet been reached at publication, but risk of death was lower in the trametinib arm (HR 0.54, P=0.01).31 However, indirect comparison showed that the response rate and PFS was lower than previously demonstrated for the BRAF inhibitors. In addition, the Phase II trial had a cohort of patients who were enrolled to the sequential use of trametinib after progression on a BRAF inhibitor, which showed minimal clinical activity with only 20% of patients demonstrating tumor reduction.32 The median PFS was only 1.8 months (95% confidence interval [CI], 1.8–2.0 months), and the median OS was 5.8 months (95% CI, 4.1–9.0 months).32 Though trametinib was approved as a single agent in the treatment of metastatic melanoma with a BRAF V600 mutation, it was rarely used as such due to the perceived inferior efficacy compared to the single-agent BRAF inhibitors. Cobimetinib is a potent, selective MEK1/2 inhibitor that maintains its inhibitory effect when MEK is phosphorylated and ultimately leads to decreased ERK1/2 phosphorylation.33 The most common toxicities reported with trametinib and cobimetinib include rash, diarrhea, fatigue, and peripheral edema.32,34
Improving treatment efficacy with a combined targeted therapy approach
As previously discussed, the upregulation of the MAPK pathway is the predominant way tumor cells overcome BRAF inhibitors. Transitioning to a MEK inhibitor upon resistance did not offer a significant therapeutic benefit. Thus, both the molecular mechanisms of acquired resistance and the toxicities associated with paradoxical MAPK activation with single agent BRAF inhibitor therapy led to the consideration of using a combined targeted therapy approach using both BRAF and MEK inhibitors. The aim was to further suppress tumor growth, delay or prevent acquired resistance by silencing tumor subclones and preventing the resurgence of MAPK upregulation, as well as inhibit the proliferation of other nonmelanoma subclinical tumor cells harboring RAS mutations.
Cobimetinib was first investigated in combination with vemurafenib in the Phase Ib trial (BRIM-7), an open-label, multicenter, dose escalation study, to assess the safety, tolerability, pharmacokinetics, and activity of cobimetinib in combination with vemurafenib. Initially, only patients who had progressed on vemurafenib were enrolled, but the protocol was later amended to include patients with a BRAF V600 mutation who were BRAF or MEK inhibitor treatment naïve.35 The results from these two cohorts were assessed separately.
This study had two stages, an initial dose escalation stage followed by a cohort expansion. Vemurafenib was given in two different dosing schedules either at 720 mg twice daily or at the FDA-approved dose of 960 mg twice daily in a continuous 28-day cycle.35 This was given in combination with escalating doses of cobimetinib in three different 28-day cycle regimens: 14 consecutive days of cobimetinib followed by a 14-day drug holiday (14/14), 21 consecutive days of cobimetinib followed by a 7-day drug holiday (21/7), or as a continuous daily schedule (28/0).35 In a Phase I multicenter, open-label dose escalation study of single-agent cobimetinib, the maximum tolerated dose given on a 21-day, 7-day-off schedule was 60 mg once daily.34
Initially, most patients were enrolled in the cohorts with vemurafenib and cobimetinib given on the 14/14 schedule at escalating doses.35 Of note, the majority of the patients enrolled in the cohorts with the 14/14 schedule were those who had recently progressed on vemurafenib monotherapy. For some patients enrolled in the cohorts with the cobimetinib 14/14 schedule, it seemed that there was an initial tumor response while taking cobimetinib, but during the 14-day off period, there was evidence of tumor regrowth; thus, no further patients were accrued into the cohorts with a cobimetinib 14/14 schedule.35 There was no evidence of tumor regrowth among the patients enrolled in the cohorts with the 21/7-day schedule of cobimetinib.35 In the cohort assigned to vemurafenib 960 mg twice daily with cobimetinib 60 mg daily (28/0), there were two out of four patients who had a dose-limiting toxic effect (grade 3 stomatitis and fatigue, or grade 3 arthralgia and myalgia); thus, vemurafenib 960 mg twice daily and cobimetinib 60 mg daily (21/7) (cohort 1B) was defined as the maximum tolerated dose of the combination.35
Cohorts 1A (vemurafenib 720 mg twice daily with cobimetinib 60 mg daily, 21/7) and 1B (vemurafenib 960 mg twice daily with cobimetinib 60 mg daily, 21/7) were chosen for the expansion stage as they were both declared safe and tolerable. In addition, each of these cohorts provided optimal dosing of therapy. For instance, in cohort 1A this delivered the maximum tolerated dose and schedule of cobimetinib when investigated as a single agent and for cohort 1B, the previously approved dose and schedule of vemurafenib.35 There were no dose-limiting toxic effects recorded in the expansion cohort of either 1A or 1B.35 The expansion cohorts included both patients who had recently progressed on vemurafenib monotherapy and BRAF inhibitor-naïve patients.
In total, 129 patients were enrolled; 66 patients had recently progressed on vemurafenib monotherapy, and 63 patients had never received a BRAF inhibitor. There were more patients who were BRAF inhibitor naïve (62%) treated at the maximum tolerated dose of each drug compared to the 41% of patients previously treated with a BRAF inhibitor.35 When comparing the characteristics of each population, the patients who had previously progressed on vemurafenib monotherapy were more likely to have an ECOG performance status of 1, an elevated LDH, and stage IV M1c disease.35
Pharmacokinetic data showed no variation in the dose of vemurafenib when coadministered with cobimetinib. The patients who had recently progressed on vemurafenib were allowed to remain on vemurafenib beyond progression, up until cycle 1 day 1 of the combination. The day −1 maximum drug plasma concentration and area under the concentration–time curve at steady state for single-agent vemurafenib dosing were comparable to the cycle 1 day 14 concentrations of vemurafenib in combination with cobimetinib.35 Likewise, the maximum drug plasma concentration and area under the concentration–time curve at steady state were similar for cobimetinib single-agent dosing and cobimetinib in combination with vemurafenib. Vemurafenib is primarily metabolized by CYP3A and is a mild inducer of CYP3A.35 Cobimetinib is not an inducer or inhibitor of CYP3A and is metabolized by CYP3A4 and UGT2B7.35
Overall, there were more reported adverse events with greater severity for patients who had not received prior BRAF inhibitor therapy (Table 2). There were several postulated explanations for this observation. The patients who were BRAF inhibitor naïve were on therapy for a longer duration and thus with longer exposure had more opportunity to develop toxicities. Another reason may have been that for those previously treated with vemurafenib, the cohort that they were randomized to may have been appropriated to a lower dose of vemurafenib.35 In addition, patients previously exposed to vemurafenib may already have had ongoing management of vemurafenib-related toxicities or, for example, as in the case with photosensitivity, awareness of how to avoid development (ie, sunscreen or skin covering prior to sun exposure).35 When the known MEK inhibitor-related toxicities were evaluated, for example, diarrhea, cardiac events, and retinopathies, these were also found to be more common in the BRAF-naïve population. The most common MEK-associated adverse events were diarrhea, acneiform rash, and an increase in creatine phosphokinase activity (without rhabdomyolysis).35 There was a patient in the BRAF-naïve population who developed grade 3 symptomatic congestive cardiomyopathy which resulted in permanent discontinuation of cobimetinib, and the cardiomyopathy improved when transitioned to vemurafenib monotherapy.35 Of note, intermittent surveillance of cardiac function by echocardiography was not performed as part of this trial; thus, the detection of asymptomatic cardiac dysfunction may be underrepresented.
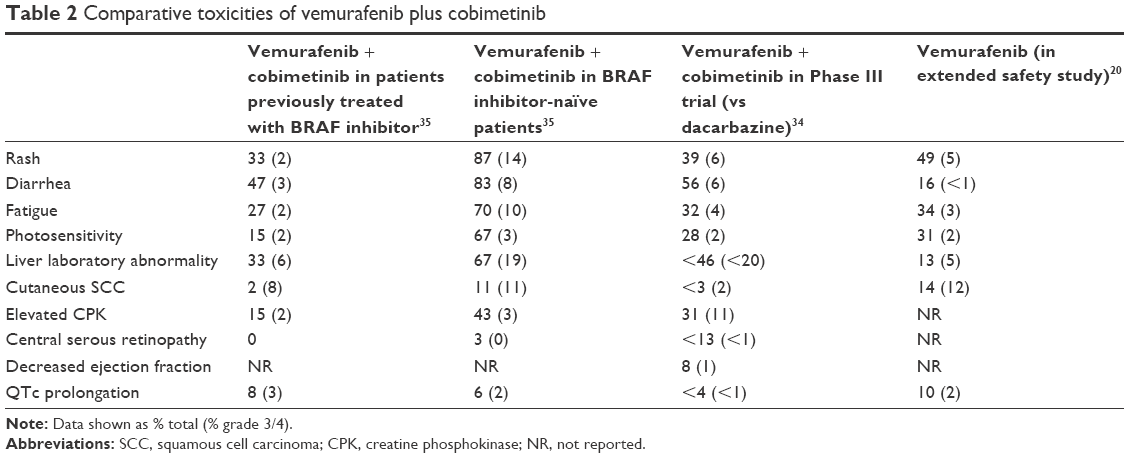 | Table 2 Comparative toxicities of vemurafenib plus cobimetinib |
As would be expected, given the trend for more adverse events in the vemurafenib-naïve group, there were more dose modifications with either interruptions or reductions made in the BRAF-naïve patients. For patients who had previous exposure to BRAF monotherapy, 27% required dose interruption or reduction of vemurafenib, 21% required interruption or reduction in cobimetinib, and 20% required dose interruption or reduction of both agents.35 In the BRAF-naïve group, 71% required dose interruption or reduction of vemurafenib, 57% required dose interruption or reduction of cobimetinib, and 54% required a dose interruption or reduction of both agents.35 In the BRAF-naïve population, there were two patients who had permanent withdrawal of both vemurafenib and cobimetinib, due to grade 3 rash in one and grade 3 QTc interval prolongation in the other. Vemurafenib was permanently discontinued in two patients, due to a grade 4 rise in γ-glutamyltransferase activity in one and a grade 2 rise in creatinine in the other.35 Despite the frequent dose modifications, only 5% of patients in cohort 1B (the recommended Phase III dose) required permanent discontinuation.35
A clinically relevant observation was that the BRAF-naïve population demonstrated a better response and clinical outcome in comparison to the patients who had recently progressed on a BRAF inhibitor. This is likely related to the tumor heterogeneity that arises in BRAF inhibitor therapy. Separate subclones with varying mechanisms of resistance may be working in parallel to upregulate the MAPK pathway, which then cannot be overcome with the addition of a MEK inhibitor after development, thereby allowing subpopulations unresponsive to MEK inhibition to have a survival advantage and be selected out among the heterogeneous population of tumor cells.35 Thus, the combined inhibition of BRAF and MEK is most effective when used at the onset of treatment to delay selective pressures that can induce varying genomic or potentially epigenetic changes.35
The overall objective response rate for those previously treated with a BRAF inhibitor was 15% (with no complete responses observed) compared to 87% (and 10% with complete response) for BRAF-naïve patients.35 Extended follow-up reported at the 2015 American Society of Clinical Oncology (ASCO) meeting noted an additional four patients in the BRAF-naïve cohort who later also had a complete response (16% complete response).36 The durability of response was better among the BRAF-naïve patients, and the median PFS did not change with extended follow-up (13.7 months, 95% CI, 10.1–17.5).35,36 This is a notable improvement in comparison to the patients who had previously progressed on a BRAF inhibitor alone with a PFS of 2.8 months (95% CI, 2.6–3.4) and to the historically reported PFS with vemurafenib monotherapy of 6.9 months.35 This again demonstrates the improved efficacy and outcome with delaying the emergence of resistant tumor clones and that the addition of a MEK inhibitor after developing resistance to a BRAF inhibitor was not able to overcome these mechanisms to lengthen the duration of response to the same meaningful degree. The median OS for the BRAF inhibitor pretreated group was 8.3 months (95% CI, 5.95–10.87) with an estimated 1-year survival of 32% (95% CI, 19–45). In comparison, the median OS for the BRAF-naïve group had not yet been reached at the time of publication, but the estimated 1-year survival was 83% (95% CI, 73–93),35 and the extended follow-up results showed that the median OS was 28.5 months.36
The combination of vemurafenib and cobimetinib was further investigated in the Phase III trial (coBRIM), an international, multicenter randomized trial that again made evident the improvement in response rate and PFS when cobimetinib was used in combination with vemurafenib. In the coBRIM trial, 495 previously untreated patients with unresectable, locally advanced stage IIIC or stage IV melanoma with a V600 mutation were randomized to the combination of vemurafenib 960 mg twice daily plus cobimetinib 60 mg daily (21/7) compared to vemurafenib 960 mg twice daily plus placebo.34 The groups were evenly matched in regard to stage, performance status, visceral metastatic disease, history of brain metastasis, and elevated LDH (43% in the control group, 46% in the combination group).34
The objective response rate was much higher in the combination group as compared to the control group (68% versus 45%, P<0.001), and there was a higher rate of complete responses seen in the combination group as well (10% versus 4%).34 The median duration of response was not reached in the combination group, but in the control group, it was 7.3 months (similar to previously reported vemurafenib monotherapy). The PFS again favored the combination group, with half of patients in the combination arm demonstrating a PFS of 9.9 months compared to 6.2 months in the control group (HR 0.51, P<0.001).34 The interim analysis of the OS was not yet mature at publication, but the rate of OS reported at 9 months for the combination was 81% versus 73% (HR 0.65, P=0.046).34
There was an update from extended follow-up presented at the 2015 ASCO meeting after a median follow-up of 14.2 months that showed the median PFS for the combination group was 12.3 versus 7.2 months for the control group (HR 0.58, 95% CI, 0.46–0.72).37 The overall response rate was as similarly reported (70% versus 50% with a complete response rate of 16% versus 11%).37 The final update on OS was presented at the 2015 Society for Melanoma Research (SMR) conference after a median follow-up of 18.5 months. The patients treated with the combination of cobimetinib and vemurafenib had a median OS of 22.3 months (95% CI, 20.3–not reached), in comparison to 17.4 months of the vemurafenib plus placebo arm (HR 0.70, P=0.005).38
In terms of safety, most of the toxicities observed with the combination of vemurafenib and cobimetinib were mild to moderate (grade 1–2) with the most common adverse events reported being photosensitivity and rash, elevated liver transaminases, elevated creatine phosphokinase, and diarrhea (Table 2).39 The toxicities appeared to improve with time on therapy with many toxicities resolving after 4 months of therapy and the most severe toxicities were typically reported in the first 28 days of treatment.39 There were equivalent rates of grade 3 adverse events (49%) between the two study groups. There were fewer grade 4 events reported (9% in the control group versus 13% in the combination group), and half of these were due to asymptomatic laboratory abnormalities including elevated liver transaminases and creatine phosphokinase levels.34 The most common grade 4 event in the combination arm was an asymptomatic elevation of the creatine phosphokinase level, which was reported in 4% of patients. There were more reported gastrointestinal events in the combination group, diarrhea (all grades, 56% versus 28%), nausea (all grades, 40% versus 24%), and vomiting (all grades, 21% versus 13%), compared to the control group. Despite the differences between the incidence and types of adverse events between the two groups, there was not a significant difference between the two groups in regard to the number of patients who required study drug discontinuation (13% in the combination versus 12% in the control group).34
When looking at MEK inhibitor class effects, the combination had a higher frequency of central serous retinopathy; though the majority of events were grade 1 or 2 and were reversible in most cases without treatment.34 A small fraction did require a dose reduction or discontinuation of cobimetinib.34 It is worth noting that a potential benefit of the 21/7-day schedule of cobimetinib was that this allowed time for recovery of the ocular toxicity during the drug holiday when present; thus, patients did not require additional intervention and could continue on therapy without dose interruption or reduction. Subjects did have intermittent surveillance of cardiac function by echocardiogram or multigated acquisition scans (MUGA) during the study, and the rate of clinically significant cardiac events was low and relatively similar between the two arms.34 There were some adverse events that were more common in the control group such as keratoacanthomas, alopecia, and arthralgias. Of particular interest again was the confirmation of decreased incidence of cutaneous SCC in the combination group (<3% versus 11% in the control group).34
There were six grade 5 events that were reported as being related to therapy in the combination group and three grade 5 events reported in the control group.34 The five deaths in the combination group were identified as being due to cardiac arrest (one patient), pneumonia (one patient), fatigue and asthenia (one patient), cerebral hemorrhage (one patient, although progressive disease was listed as cause of death), hemiparesis (one patient, progressive disease listed as cause of death), and one death was due to an unspecified event.34
This trial added further support to the benefit seen when BRAF and MEK inhibitors are used in combination upfront for the treatment of advanced melanoma with a BRAF V600 mutation, as a targeted therapeutic approach. The improvement in median PFS, response rate, and decreased incidence of secondary cutaneous SCCs seen with the combination of vemurafenib and cobimetinib are similar to the findings observed with the combination of dabrafenib and trametinib in the COMBI-D and COMBI-V trials.40,41 In addition, all three trials showed that those with an atypical V600K mutation also derived clinical benefit from the combination therapy, though this was less robust than the benefit derived by those with a V600E mutation.39–41 In the coBRIM trial, the V600K mutation subgroup analysis showed a reduction in the risk of progression with an HR or 0.27 favoring the combination of vemurafenib and cobimetinib.39 In the COMBI-V trial, the V600K mutation subgroup demonstrated an HR for progression or death of 0.71.41 In the COMBI-D trial, the V600K subgroup showed an HR for progression or death of 0.68.40
Another significant point that was demonstrated with all three of the combination BRAF/MEK inhibitor Phase III trials was the identification of patient factors that might predict worse outcomes. One high-risk group is the subset with an elevated LDH which has long been recognized as a poor prognostic indicator in patients with metastatic melanoma. From the update presented of the coBRIM trial at the 2015 SMR conference, after extended follow-up, the median OS in patients with an elevated LDH treated with combination cobimetinib and vemurafenib was 14.8 months as compared to 11.2 months for the vemurafenib plus placebo arm (HR 0.77).38 In contrast, for those patients with a normal LDH in the combination arm, the median OS was not yet reached, and in the vemurafenib plus placebo arm, the median OS was 23.3 months (HR 0.59).38
In the COMBI-D trial, the median PFS in the subgroup with an elevated LDH in the dabrafenib plus trametinib combination group was also lower at 7.1 months (as compared to 9.3 months in the entire combination group).40 This was in comparison to the dabrafenib monotherapy subgroup with an elevated LDH which had a median PFS of 3.8 months (as compared to 8.8 months in the entire monotherapy group), with an HR for death or progression of 0.64 (95% CI, 0.42–0.95).40 In the COMBI-V trial, for the combination group treated with dabrafenib plus trametinib in comparison to vemurafenib monotherapy, the HR for progression or death was 0.56 (95% CI, 0.46–0.69).41 This was in comparison to the subgroup with an elevated LDH which had an HR of progression or death of 0.72.41
Though these trials cannot be directly compared, it is worth noting that there are some baseline differences between the two groups in these trials. In both the COMBI-D and COMBI-V trials, there were about a quarter of patients enrolled who had received immunotherapy prior to enrollment.40,41 Though these patients had progressed prior to enrollment, it is unclear whether there may be any influence on the durability of response for those patients previously treated with immunotherapy prior to enrollment. In the coBRIM trial, all patients enrolled were treatment naïve. In addition, the proportion of patients with an elevated LDH (known to be associated with decreased response and durability to combination therapy as described) ranged between 32% and 37% in the COMBI-V and COMBI-D trials.40,41 In comparison, for the coBRIM trial, the vemurafenib and cobimetinib arm enrolled 46% of patients with an elevated LDH, while the vemurafenib control arm had 43% of patients with an elevated LDH.39
Additional strategies to overcome acquired resistance mechanisms
Though the combination of vemurafenib and cobimetinib improved the response, PFS, and OS, unfortunately, the majority of patients eventually developed resistance to the combination as well. The reported mechanisms underlying acquired resistance to the combination BRAF/MEK inhibitor therapy are similar to the mechanisms for BRAF inhibitor monotherapy with restoration of MAPK signaling. A study using whole-exome sequencing and whole-transcriptome sequencing (RNA-seq) on tumor samples from five patients with acquired resistance to combined dabrafenib and trametinib therapy demonstrated that some of the MAPK pathway mutations that arise in the setting of monotherapy such as MEK1/2 mutations, BRAF amplification, and BRAF splice isoforms were also identified after exposure to combination therapy and thus may confer cross-resistance to combination therapy.42 This may help explain the intrinsic resistance displayed in those patients who received combination BRAF/MEK inhibitor after progression on BRAF inhibitor monotherapy. In addition, there continued to be several MAPK pathway alterations demonstrating that BRAF-mutant melanoma maintains a continued reliance on MEK/ERK signaling despite combined pathway inhibition.42 Thus, the development of small-molecule ERK inhibitors may provide a way to overcome multiple varying resistance mechanisms that arise in the MAPK pathway and these agents have already begun to enter clinical trials.42 Alternative pathway upregulation may also play a role; there is evidence that in vitro dual-BRAF/MEK-resistant cell lines demonstrate hyperactivation of the PI3K/AKT pathway as well.43
Recalling the pathogenesis of cutaneous SCCs that can arise on BRAF inhibitor monotherapy and the paradoxical activation of MAPK signaling that drives cellular proliferation demonstrates the concept of drug dependency for cellular proliferation. It has been demonstrated that intermittent dosing of BRAF inhibitors in vitro may delay the onset of acquired resistance by selecting out the subclones of resistant tumor cells that may remain dependent on BRAF inhibitor therapy for cellular proliferation. Thus, it has been postulated whether intermittent dosing of BRAF inhibitors may be another potential strategy to delay the onset of acquired resistance.44 In fact, a case report of a patient with melanoma with a BRAF V600E mutation who developed an NRAS-mutated chronic myelomonocytic leukemia on vemurafenib was treated with intermittent dosing of vemurafenib monotherapy and later with intermittent vemurafenib and cobimetinib due to cytopenias. This patient had a sustained complete clinical response for 85 weeks that was ongoing without evidence of developing resistance.45
Although most patients develop acquired resistance to targeted therapy, there are a select number of patients with melanoma treated with targeted therapy who maintain a long-term durable clinical response with a significant survival benefit much like what is seen with immunotherapy. Extended follow-up from the Phase I trial with vemurafenib monotherapy demonstrated that eight patients (out of 32 in the expansion cohort) treated with vemurafenib 960 mg twice daily were still alive >3 years from treatment initiation and six of these patients had still only received vemurafenib therapy.46 There were five patients who survived >4 years and continued to receive only vemurafenib monotherapy.46 Further subgroup analyses were performed on the patients in the extension cohort who survived >3 years, and the characteristics delineated that were associated with a median PFS of >12 months were an ECOG performance status of 0, low volume burden of disease (average sum of baseline target lesions was 65 mm, standard deviation ±37 mm), and non-CNS metastatic disease.46
The extended follow-up results from the Phase I vemurafenib trial also found that 20 patients (45.5%) continued to receive treatment with vemurafenib for >30 days after initial disease progression. Most of the progressive sites were considered to be controlled (or limited) by the investigator and thus amenable to local therapy with either surgery or radiation.46 The most common site of disease progression was skin and soft tissue (53%) followed by CNS metastasis (32%).46 The median PFS was similar in patients who received vemurafenib for <30 or >30 days after disease progression (6.4 versus 6.7 months).46 However, recall that the OS for all 48 patients enrolled in the Phase I trial was 14 months. The median OS for all patients with progressive disease after initial progression was 6.1 months. For those patients who discontinued vemurafenib <30 days after progression, the OS was 3.4 months (range 0–26.9) as compared to 10.0 months for those who received vemurafenib for >30 days after progression (range 3.6–41.0).46 Half of the patients who continued treatment after initial progression continued to take vemurafenib for ~3.8 months (range 1.1–26.6 months).46 These findings further support the concept of inter-tumor heterogeneity and divergent clonal populations at separate metastatic sites. Of note, similar long-term findings for a small subset of those enrolled in the initial trials with dabrafenib monotherapy (BREAK-3) have also been described.47
In addition, a retrospective, single-institution analysis of 95 patients treated with vemurafenib or dabrafenib monotherapy also showed a benefit of continuation of BRAF inhibitors beyond progression with improvement in median OS.48 It was reported that 31% of patients developed progressive disease at a single site in that analysis,48 which may be amenable to local therapy. However, it should be noted that there have been several case reports indicating that both BRAF inhibitors may act as radiation sensitizers and thus concomitant use with radiation therapy should be considered with caution.49–51 Further investigations regarding the optimal management of patients with progressive disease on the combination BRAF/MEK inhibitor therapy are ongoing.
The use of targeted therapy in CNS metastatic disease
The development of CNS metastatic disease is a frequent complication that arises in patients with metastatic melanoma and remains a major therapeutic challenge. The prognosis of patients with CNS disease remains poor. While the systemic response rates of vemurafenib and dabrafenib for those with BRAF V600-mutated melanoma can be dramatic, the response and outcome for those with CNS metastatic disease is still under investigation. In the initial trials evaluating the use of BRAF inhibitor monotherapy, patients with CNS disease were excluded from study. There was a Phase II trial (BREAK-MB) investigating the use of dabrafenib in patients with CNS metastatic disease which demonstrated an intracranial response rate of 30.8% for those who had progressed after prior therapy for brain metastasis.52 There are a few retrospective case series that have reported clinical activity of vemurafenib in patients with CNS metastases which prompted further prospective studies.53,54 There is a Phase II trial evaluating the outcome for those with a BRAF V600 mutation and CNS metastatic disease treated with vemurafenib that is ongoing, but preliminary results presented at the 2013 SMR congress demonstrated an intracranial response rate of 26%.55 A small, two-center study using vemurafenib as a neoadjuvant-like therapy in patients with poor prognosis and symptomatic, unresectable lesions in the brain again demonstrated potential activity of vemurafenib in the treatment of CNS metastatic melanoma.56 In this study, over half of the patients had had prior CNS metastatic disease that had been previously treated with whole-brain radiation prior to enrollment with new metastases. The median treatment duration was 3.8 months (range 0.1–11.3 months), the median PFS was 3.9 months (95% CI, 3–5.5 months), and half of patients had an OS of 5.3 months (95% CI, 3.9–6.6 months).56 Partial responses at both intracranial and extracranial sites were observed in 42% of patients, and stable disease observed in 38% of patients.56 In addition, the use of corticosteroids decreased (defined as reduction of at least 33% of the dose) in 67% of the patients.56
In the Phase III trials investigating the combination of vemurafenib and cobimetinib as well as the Phase III trials evaluating the combination of dabrafenib and trametinib, patients were eligible to enroll with a history of CNS metastatic disease that had been treated, and were stable for a defined period of time (3 weeks in coBRIM and 12 weeks in COMBI-D and COMBI-V).40,41,44 As of yet, there have not been subgroup analyses reported on the outcomes of these patients in regard to control of CNS disease. There was a phase II clinical trial using the combination of vemurafenib and cobimetinib in patients with active CNS disease (coBRIM-B; NCT02230306), which was closed due to slow accrual, and results have not yet been published.
The initial interplay of combined immunotherapy and targeted therapy
There is a growing recognition and understanding of the role kinase inhibitors may have in modulating the tumor-associated immune response. Specifically, in regard to BRAF inhibitor therapy with vemurafenib, it has been shown that there is a significant increase in CD8+ tumor infiltrating T lymphocytes, with increased clonality which can occur within 10–14 days after starting treatment.57–61 There is also a corresponding increase of cytotoxic activity by CD8+ T cells and NK cells as demonstrated by increased Granzyme B and perforin staining.59 However, there is also evidence that there may be increased markers of T cell exhaustion in preclinical studies of the lymphocytic infiltrate after treatment with BRAF inhibitors.59 It should also be noted that there have been preclinical studies that suggest that MEK inhibitors may promote a T cell-suppressive environment, but whether this has clinical relevance is unknown.58,64
From studies looking at both patient tumor samples and melanoma cell lines after treatment with vemurafenib (and some studies after treatment with dabrafenib and trametinib), it has been shown that there is an increase in the expression of melanoma-associated antigens and major histocompatibility complex class I and II receptors on melanoma cells.57–59,62 However, when BRAF inhibitor resistance develops, there is loss of melanocyte differentiation antigen expression.58 From several preclinical studies, there is also immune modulation of tumor microenvironment upon treatment with BRAF inhibitors with decreased levels of immunosuppressive cytokines or mediators, such as IL-10, IL-6, and vascular endothelial growth factor, early after treatment.57,59,63,64 Furthermore, there have been several preclinical studies demonstrating an upregulation of PD-L1 expression in response to BRAF inhibitor therapy (both with vemurafenib and dabrafenib).59,65,66 Thus, it may be that immune evasion is another mechanism of acquired BRAF inhibitor resistance.
It appears that there may be a potential synergistic relationship that could improve the durability of response with a treatment approach that uses combined targeted therapy in combination with a checkpoint inhibitor either in sequence or concurrently. The first reported combination using a BRAF inhibitor and immunotherapy was a Phase I trial using the combination of vemurafenib and ipilimumab. Unfortunately, due to significant hepatic toxicity, the trial was terminated early.67 Preliminary findings from another trial that combined dabrafenib and trametinib versus dabrafenib with ipilimumab found that several patients treated with the combination of dabrafenib and trametinib with ipilimumab had an increased incidence of colonic perforation.68 However, there are several ongoing trials looking at safety and efficacy of various combinations of targeted therapy including vemurafenib alone or with cobimetinib and an anti-PD-L1 antibody. There are other studies using dabrafenib alone or in combination with trametinib and nivolumab.
Conclusion and future directions
The combination of vemurafenib and cobimetinib clearly has demonstrated a significant improvement in the response and OS of patients with BRAF V600-mutated melanoma and confirms that BRAF or MEK inhibitor monotherapy should no longer be the standard first-line treatment approach. It is unclear whether there are significant differences in outcomes between the two different BRAF/MEK inhibitor combinations. When deciding which combination regimen to use, the difference in toxicities may help guide the treatment decision. For instance, there is a higher incidence of pyrexia which can be debilitating for some patients treated with the dabrafenib and trametinib combination requiring dose interruptions and modifications, whereas cutaneous toxicities are more prevalent with the combination of vemurafenib and cobimetinib.
While combined BRAF/MEK inhibitor therapy can induce rapid responses, which can be especially advantageous in those with significantly symptomatic disease, the durability of response is often limited due to the development of acquired resistance. This is in contrast to those who respond to immunotherapy which has demonstrated that the durability of response makes a significant impact on OS. There are a number of ongoing trials using other small-molecule, targeted agents in combination with BRAF and MEK inhibitors as a strategy to prevent or overcome the development of resistance. Additional therapeutic strategies are also needed for those patients with intrinsic resistance mechanisms to targeted therapy which diminish initial response and durability. Furthermore, it is warranted to consider strategies using both targeted therapy and immunotherapy in combination for the treatment of BRAF-mutated melanoma. It remains unclear what the optimal approach should be with the available therapeutic strategies for those with advanced BRAF mutated melanoma. For example, should combination BRAF and MEK inhibitors be used first, which combination should be used, when should immunotherapy be considered? Can the combination of BRAF and MEK inhibitors be safely combined with immunotherapy, or is there a role for using the combination of BRAF and MEK inhibitors in the adjuvant setting? There are multiple clinical trials underway to help address some of these uncertainties and with the hope of continuing to improve long-term survival and therapeutic options for patients with advanced melanoma.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. | ||
McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF V600E and BRAF V600K mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomized, open-label study. Lancet Oncol. 2014;15(3):323–332. | ||
Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9(17):6483–6488. | ||
Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol. 2005;223(27):6771–6790. | ||
Chong H, Guan KL. Regulation of Raf through phosphorylation and N terminus-C terminus interaction. J Biol Chem. 2003;278(38):36269–36276. | ||
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. | ||
Garnett MJ, Rana S, Paterson H, Marais R. Wild type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20(6):963–969. | ||
Terai K, Mastuda M. The amino-terminal B-Raf specific region mediates calcium-dependent homo- and hetero-dimerization of Raf. EMBO J. 2006;25(15):3556–3564. | ||
Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65(6):2412–2421. | ||
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. | ||
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. | ||
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. | ||
Ascierto PA, Minor D, Ribas A, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol. 2013;31(26):3205–3211. | ||
Haushchild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomized controlled trial. Lancet. 2012;380(9839):358–365. | ||
Oberholzer PA, Kee D, Dziunycz P, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012;30(3):316–321. | ||
Callahan MK, Rampal R, Harding JJ, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367:2316–2321. | ||
Grey A, Cooper A, McNeil C, et al. Progression of KRAS mutant pancreatic adenocarcinoma during vemurafenib treatment in a patient with metastatic melanoma. Intern Med J. 2014;44(6):597–600. | ||
Andrews MC, Behren A, Chionh F, et al. BRAF inhibitor-driven tumor proliferation in a KRAS-mutated colon carcinoma is not overcome by MEK1/2 inhibition. J Clin Oncol. 2013;31(35):e448–e451. | ||
Larkin J, Del Vecchio M, Ascierto PA, et al. Vemurafenib in patients with BRAFV600 mutated metastatic melanoma: an open-label, multicenter, safety study. Lancet Oncol. 2014;15(4):436–444. | ||
Luke JL, Hodi FS. Ipilimumab, vemurafenib, dabrafenib, and trametinib: synergistic competitors in the clinical management of BRAF mutant malignant melanoma. Oncologist. 2013;18:717–725. | ||
Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31(14):1767–1774. | ||
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. | ||
Kemper K, Krijgsman O, Cornelissen-Steijger P, et al. Intra- and inter-tumor heterogeneity in a vemurafenib-resistant melanoma patient and derived xenografts. EMBO Mol Med. 2015;7(9):1104–1118. | ||
Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. | ||
Girotti MR, Pedersen M, Sanchez-Laorden B, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3:158–167. | ||
Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. | ||
Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to BRAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. | ||
Johnson DB, Menzies AM, Zimmer L, et al. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behavior, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51(18):2792–2799. | ||
Joseph EW, Pratilas CA, Poulikakos PI, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF selective manner. Proc Natl Acad Sci U S A. 2010;107(33):14903–14908. | ||
Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. | ||
Kim KB, Kefford R, Pavlick AC, et al. Phase II study of the MEK1/MEK2 inhibitor trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(4):482–489. | ||
Rosen L, LoRusso P, Wee Ma W, et al. Abstract 4716: a first-in-human phase 1 study to evaluate the MEK1/2 inhibitor GDC-0973 administered daily in patients with advanced solid tumors. Presented at: AACR 102nd Annual Meeting; April 2–6, 2011; Orlando, FL. Cancer Res. 2011;71:4716. | ||
Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–1876. | ||
Ribas A, Gonzalez R, Pavlick A, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAF V600-mutated melanoma: a phase 1b study. Lancet Oncol. 2014;15:954–965. | ||
Pavlick AC, Ribas A, Gonzalez R, et al. Extended follow-up results of phase 1b study (BRIM7) of vemurafenib (VEM) with cobimetinib (COBI) in BRAF-mutant melanoma. J Clin Oncol. 2015;33(15):abstr 9020. | ||
Larkin JM, Yan Y, McArthur GA, et al. Update of progression-free survival (PFS) and correlative biomarker analysis from coBRIM: phase III study of cobimetinib (cobi) plus vemurafenib (vem) in advanced BRAF-mutated melanoma. J Clin Oncol. 2015;33 Suppl:abstr 9006. | ||
Atkinson V, Larkin J, McArthur GA, et al. Improved overall survival (OS) with cobimetinib (COBI) + vemurafenib (V) in advanced BRAF-mutated melanoma and biomarker correlates of efficacy. Presented at: The Society for Melanoma Research Congress; November 18–21, 2015; San Francisco, CA. | ||
Acierto PA, McArthur GA, Dreno B, et al. coBRIM: a phase 3, double blind, placebo-controlled study of vemurafenib versus vemurafenib + cobimetinib in previously untreated BRAF V600 mutation-positive patients with unresectable locally advanced or metastatic melanoma. J Transl Med. 2015;13(Suppl 1):O4. | ||
Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–1888. | ||
Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. | ||
Wagle N, Van Allen EM, Treacy DJ, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4(1):61–68. | ||
Wang VE, McCormick F, Settleman J. Hyperactive AKT pathway and reactivation of the MAPK/ERK pathway in melanoma cells resistant to dual BRAF and MEK inhibition. In: Proceedings of the 105th Annual Meeting of the American Association for Cancer Research; April 5–9, 2014; San Diego, CA. Cancer Res. 2014;74(19 Suppl):abstr 3698. | ||
Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. | ||
Abdel-Wahab O, Kilmek VM, Gaskell AA, et al. Efficacy of intermittent combined RAF and MEK inhibition in a patient with concurrent BRAF- and NRAS-mutant malignancies. Cancer Discov. 2014;4:538–545. | ||
Puzanov I, Amaravadi RK, McArthur GA, et al. Long-term outcome in BRAF V600E melanoma patients treated with vemurafenib: patterns of disease progression and clinical management of limited progression. Eur J Cancer. 2015;15:1435–1443. | ||
Hauschild A, Grobb J, Demidov L, et al. An update on overall survival (OS) and follow-on therapies in BREAK-3, a phase III, randomized trial: dabrafenib (D) vs dacarbazine (DTIC) in patients with BRAF V600E mutation-positive metastatic melanoma. Ann Oncol. 2014;25(Suppl 4):iv378. | ||
Chan MM, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:1323–1341. | ||
Anker CJ, Ribas A, Grossman AH, et al. Severe liver and skin toxicity after radiation and vemurafenib in metastatic melanoma. J Clin Oncol. 2013;31(17):e283–e287. | ||
Boussemart L, Boivin C, Claveau J, et al. Vemurafenib and radiosensitization. JAMA Dermatol. 2013;149:855–857. | ||
Satzger I, Degen A, Asper H, Kapp A. Serious skin toxicity with the combination of BRAF inhibitors and radiotherapy. J Clin Oncol. 2013;31:e220–e222. | ||
Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087–1095. | ||
Dzienis MR, Atkinson VG. Response rate to vemurafenib in patients with B-RAF-positive melanoma brain metastases: a retrospective review. Melanoma Res. 2014;24:349–353. | ||
Harding JJ, Catalanotti F, Munhoz RR, et al. A retrospective evaluation of vemurafenib as treatment for BRAF-mutant melanoma brain metastases. Oncologist. 2015;20:789–797. | ||
Kefford RF, Maio M, Arance A, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single arm, phase 2, multicenter study. Presented at: Society for Melanoma Research Congress Tenth Anniversary International Congress; November 17–20, 2013; Philadelphia, PA. | ||
Dummer R, Goldinger SM, Turtschi CP, et al. Vemurafenib in patients with BRAFV600 mutation-positive melanoma with symptomatic brain metastases: final results of an open-label pilot study. Eur J Cancer. 2014;50:611–621. | ||
Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18(5):1386–1394. | ||
Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–5219. | ||
Frederick DT, Piris A, Cogdil AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–1231. | ||
Liu C, Peng W, Xu C, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19:393–403. | ||
Cooper ZA, Frederick DT, Juneja VR, et al. BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology. 2013;2:e26615. | ||
Sapkota B, Hill CE, Pollack BP, et al. Vemurafenib enhances MHC induction in BRAF homozygous melanoma cells. Oncoimmunology. 2013;2(1):e22890. | ||
Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–1656. | ||
Ott PA, Henry T, Baranda SJ, et al. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother. 2013;62:811–822. | ||
Jiang X, Zhou J, Giobbie-Hurder A, et al. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19:598–609. | ||
Liu L, Mayes PA, Eastman S, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21:1639–1651. | ||
Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–1366. | ||
Puzanov I, Callahan MK, Linette GP, et al. Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation-positive unresectable or metastatic melanoma (MM). J Clin Oncol. 2014;32:abstr 2511. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.