Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
The EFFECT trial: evaluating exacerbations, biomarkers, and safety outcomes with two dose levels of fluticasone propionate/formoterol in COPD
Authors Papi A ![]() , Jones P
, Jones P ![]() , Dalvi P, McAulay K, McIver T, Dissanayake S
, Dalvi P, McAulay K, McIver T, Dissanayake S
Received 30 July 2015
Accepted for publication 3 October 2015
Published 9 November 2015 Volume 2015:10(1) Pages 2431—2438
DOI https://doi.org/10.2147/COPD.S93375
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Alberto Papi,1 Paul W Jones,2 Prashant S Dalvi,3 Kirsten McAulay,4 Tammy McIver,5 Sanjeeva Dissanayake3
1Department of Internal and CardioRespiratory Medicine, Research Centre on Asthma and COPD, University of Ferrara, Ferrara, Italy; 2Institute for Infection and Immunity, St George’s, University of London, London, UK; 3Medical Science – Respiratory, 4Medical Operations, 5Data Management and Statistics, Mundipharma Research Ltd, Cambridge, UK
Abstract: Inhaled corticosteroid/long-acting β2-agonist combination therapy is recommended in chronic obstructive pulmonary disease (COPD) patients at high risk of exacerbations. The EFFECT (Efficacy of Fluticasone propionate/FormotErol in COPD Treatment) trial is a Phase III, 52-week, randomized, double-blind study to evaluate the efficacy and safety of two doses of fluticasone propionate/formoterol compared to formoterol monotherapy in COPD patients with FEV1 ≤50% predicted and a history of exacerbations. The primary endpoint is the annualized rate of moderate and severe exacerbations. Secondary endpoints include pre-dose FEV1, EXACT-PRO (EXAcerbations of Chronic pulmonary disease Tool – Patient-Reported Outcome)-defined exacerbations, St George’s Respiratory Questionnaire for COPD, COPD Assessment Test, and EXACT-Respiratory Symptoms total score. Lung-specific biomarkers (surfactant protein D and CC chemokine ligand-18) will be measured in a subset of patients to explore their relationship to other clinical indices in COPD and their predictive utility. Pneumonia will be diagnosed per criteria defined by the British Thoracic Society community acquired pneumonia guideline, primarily by radiological confirmation and, additionally, using clinical criteria when a chest radiograph cannot be obtained. Serial measurements of serum potassium, vital signs and electrocardiograms, 24-hour Holter monitoring, and 24-hour urinary cortisol measurement will be performed in a subset of patients in addition to conventional safety assessments.
Keywords: chronic obstructive pulmonary disease, flutiform, inhaled corticosteroids, long-acting β2-agonist
Introduction
Exacerbations of chronic obstructive pulmonary disease (COPD) accelerate the rate of lung function decline, impair health-related quality of life, and are a common cause of health care utilization (HCU) and hospitalization in patients with COPD.1,2 Longitudinal observational studies have reported a mean of between 1.1 and 2.0 exacerbations per year in patients with severe to very severe COPD.3,4
The addition of an inhaled corticosteroid (ICS) to a long-acting β2-agonist (LABA) reduces exacerbation frequency and improves symptoms, quality of life, and lung function;5–7 hence, combination ICS/LABA therapy is recommended as first-line therapy in GOLD grade C and D disease.8
A fixed combination of fluticasone propionate and formoterol fumarate in a pressurized metered-dose inhaler (pMDI) (flutiform®; Napp Pharmaceuticals Limited, Cambridge, UK), which is licensed in Europe, Asia, and Australia for use in asthma, is now under development for the treatment of COPD. The present study evaluates the efficacy and safety of two dose levels of fluticasone propionate/formoterol pMDI over a twofold dose range for both ICS (250 and 500 μg) and LABA (10 and 20 μg) components. The annualized rate of moderate and severe exacerbations (defined per HCU criteria) is the primary endpoint, given that exacerbation risk reduction is the principal goal when initiating ICS/LABA treatment in COPD. As a very significant proportion of exacerbations go unreported,9 the study will use the EXACT (EXAcerbations of Chronic pulmonary disease Tool) electronic diary to facilitate enhanced reporting of exacerbations. In addition, the rate of EXACT-defined exacerbations will also be evaluated. Serum levels of the lung-specific biomarkers surfactant protein D (SP-D) and CC chemokine ligand-18 (CCL-18) will be assessed in a subgroup of patients. Changes in SP-D correlate with health status and lung function,10 whilst CCL-18 levels are associated with total and cardiovascular morbidity and mortality.11 Treatment effects upon these biomarkers will be examined to further explore their relationship to clinical outcomes and their predictive utility.
Methods
Study design
The study is of randomized, parallel-group, double-blind design (Figure 1). Patients will be randomized to one of three treatment arms for 52 weeks: fluticasone propionate/formoterol pMDI 250/10 μg bid (two puffs 125/5 μg) or 500/20 μg bid (2 puffs 250/10 μg) (flutiform®) or formoterol pMDI 12 μg bid (one puff 12 μg) (Atimos® Modulite; Chiesi Pharmaceutical GmbH, Vienna, Austria). All study treatments will be administered without a spacer.
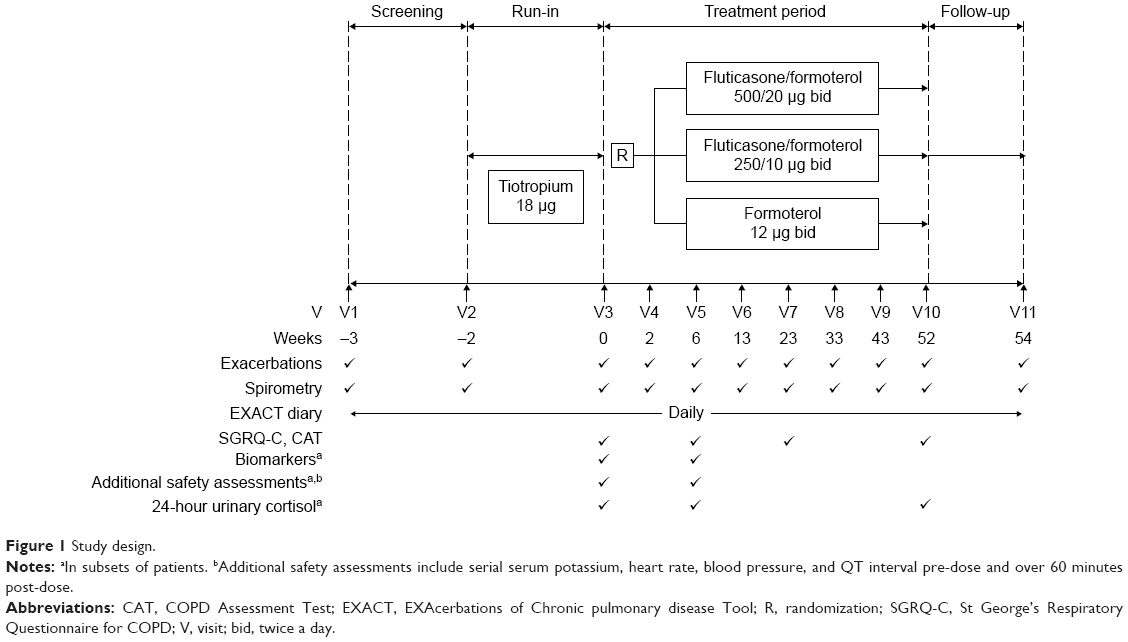 | Figure 1 Study design. |
Eligible patients will discontinue their existing COPD medications and receive tiotropium (Spiriva®; Boehringer Ingelheim, Ingelheim, Germany) 18 μg once daily during a 2-week run-in period in which a baseline EXACT score will be determined. The “baseline” EXACT score (used to identify subsequent periods of symptom worsening) will be reset throughout the 12-month study in accordance with EXACT user guidelines.12 Salbutamol pMDI (Ventolin® Evohaler®; Glaxo Wellcome, Uxbridge, UK) will be used as rescue medication.
Eligibility criteria
COPD patients ≥40 years with post-bronchodilator FEV1 ≤50% predicted and FEV1/FVC ratio <0.7 and a history of at least one moderate or severe COPD exacerbation in the last 12 months (requiring systemic corticosteroids and/or antibiotics and/or hospitalization) will be enrolled. A minimum 10-pack-year smoking history and the ability to correctly use a pMDI without a spacer are prerequisites for enrollment. Moderate or severe exacerbations at the time of screening (or during the run-in period) will render a patient ineligible as will the use of long-term oxygen therapy, uncontrolled cardiovascular disease, and clinically significant sleep apnea requiring the use of continuous positive airway pressure. During the treatment period, long-acting muscarinic antagonists, short-acting bronchodilators (other than study rescue medication), phosphodiesterase-4 inhibitors, non-cardioselective beta-blockers, xanthine derivatives, and systemic steroids (except those required for the short-term treatment of an exacerbation) are prohibited.
Exacerbation assessments
As the primary outcome employs an HCU-based definition of exacerbations, the definition, management, and interpretation of these events has been standardized. Moderate exacerbations are those requiring treatment with systemic corticosteroids and/or antibiotics whilst severe exacerbations are events requiring hospitalization or resulting in death. Exacerbations requiring systemic steroids should be managed with a prednisolone regimen of 30–40 mg for 7–14 days;8 however, if clinically necessary, for example where concerns as to glycemic control arise, investigators may consider treatment for a shorter duration. Two events separated by an interval of at least 7 days will be identified as two distinct exacerbations.
Exacerbations will also be assessed using the EXACT. When validated exacerbation thresholds12 are met, in addition to the event being defined as an EXACT exacerbation, alerts will be sent to both the investigator (via an email) and the patient (via the electronic diary). The patient’s alert advises them to contact the site as soon as possible. If the patient fails to contact the site within 2 days, the site will contact the patient to ascertain whether an unscheduled site visit is necessary for clinical review. Although concordance between HCU-defined and EXACT-defined events is modest,13 this approach should increase physician–patient interaction during the study, thereby increasing the likelihood of prompt treatment institution for disease worsening.
Other efficacy assessments
Spirometry will be performed at all visits, whilst the St George’s Respiratory Questionnaire for COPD (SGRQ-C) and the COPD Assessment Test (CAT) will be administered at baseline and week 6, 23, and 52 (Figure 1). Throughout the study, patients will complete an electronic diary daily, which includes the EXACT-Patient-Reported Outcome (EXACT-PRO) questionnaire and additional questions on rescue medication use, sleep disturbance, and study medication compliance. Blood samples for the assessment of serum SP-D (sandwich ELISA; BioVendor, Brno, Czech Republic) and CCL-18 (sandwich ELISA; Myriad RBM, Austin, TX, USA) levels will be collected at baseline and week 6 in a subset of approximately 300 patients. Efficacy endpoints in the study are summarized in Table 1.
 | Table 1 Summary of study endpoints |
Safety assessments
In addition to routine safety monitoring, the occurrence of pneumonia will be carefully evaluated per two definitions both based upon British Thoracic Society (BTS) diagnostic criteria.14 The primary level of diagnosis will require radiological confirmation of pneumonia with sites encouraged to obtain chest radiographs whenever pneumonia is suspected. A secondary definition of pneumonia, based upon BTS-defined clinical criteria alone, will also be employed to capture events where a chest radiograph cannot be obtained.14 To evaluate potentially deleterious systemic effects associated with the higher doses of ICS (500 versus 250 μg bid) and LABA (20 versus 10 μg bid) employed in the study, additional tests will be performed in subsets of patients, including: serial serum potassium, heart rate, blood pressure, and QT interval measurements pre-dose and for 60 minutes post-dose (125 patients/arm); 24-hour Holter monitoring (100 patients/arm); and, in a subset of patients without ICS exposure at screening, 24-hour urinary cortisol collections (50 patients/arm).
Statistics
A sample size of 556 patients per treatment group provides 80% power to detect a 20% reduction in the rate of moderate and severe COPD exacerbations, assuming an exacerbation rate of 0.8 exacerbations per year in the formoterol group and a two-sided alpha of 0.05. Assuming 5% of patients will not be a part of the modified intent-to-treat population (the primary analysis population), a total of 1,758 patients will be randomized (586 per treatment group). The study protocol was amended to increase the original sample size of 486 patients per group, as a planned interim review of the pooled exacerbation rate when 50% of patients had been recruited revealed a lower than expected event rate.
The primary endpoint is the annualized rate of moderate and severe exacerbations (defined per HCU criteria), analyzed using a negative binomial regression model with fixed terms for treatment, FEV1 % predicted category, number of exacerbations in the previous year category, smoking status, prior ICS use, and country, and the logarithm of time on treatment as an offset variable. The secondary comparison (fluticasone propionate/formoterol 250/10 μg versus formoterol 12 μg) will be analyzed in a confirmatory manner only if the primary comparison (fluticasone propionate/formoterol 500/20 μg versus formoterol 12 μg) is significant at the 0.05 alpha level. Only if both these primary endpoint results are significant at the 0.05 level will the first key secondary endpoint (Table 1) be analyzed in a confirmatory manner using a Hochberg closed testing procedure. This stepwise approach, ie, primary comparison followed by secondary comparison and the requirement for both to be significant before proceeding to test the next endpoint in a confirmatory manner, will be employed hierarchically throughout the key secondary endpoints per the order listed in Table 1.
Spirometry, SGRQ-C, CAT, and EXACT-Respiratory Symptoms (E-RS) will be analyzed using repeated measures analysis of covariance (ANCOVA) with fixed terms for treatment, FEV1 % predicted category, number of exacerbations in the previous year category, baseline value, smoking status, prior ICS use, country, time-point, and treatment by time-point interaction. The rate of EXACT-PRO exacerbations will be analyzed using the same model as the primary endpoint analysis. Changes in the biomarker levels (SP-D and CCL-18) from baseline will be analyzed using a Kruskal–Wallis test. The changes from baseline to week 6 will also be divided into quintiles to explore the relationship between the biomarkers and clinical indices. For SP-D, the change from baseline to week 6 across the quintile categories will be compared to changes in FEV1, SGRQ-C, and E-RS breathlessness score at week 6 and week 52: differences between quintiles will be evaluated using an ANCOVA. The relationship between SP-D quintiles and the number of HCU exacerbations over the 52-week treatment period will be analyzed using a negative binomial model. For CCL-18, the relationship between the change from baseline to week 6 and the number of patients with cardiovascular events over the 52-week treatment period will be compared between quintiles using a logistic regression model, whilst the time to first cardiovascular event will be compared using a Cox regression model. The incidence of all adverse events, including those of special interest, pneumonia, and cardiovascular events, will be presented. Summary descriptive statistics will be presented for laboratory and electrocardiograph indices.
Discussion
Very few COPD studies have previously compared two or more dose levels of an ICS/LABA combination.15–17 Thus, for example, fluticasone propionate doses of either 250 μg or 500 μg twice daily have been evaluated within ICS/LABA combinations5,18,19 but not in a head-to-head manner, leading to different doses of fluticasone propionate/salmeterol being licensed in the United States (250/50 μg bid) and Europe (500/50 μg bid). More recently, three dose levels of fluticasone furoate in combination with vilanterol have been evaluated within the same settings. Numerical differences in exacerbation rate (and pneumonia) were observed across fluticasone furoate doses of 50, 100 and 200 μg, albeit results were not entirely consistent across two studies.17
No COPD studies have previously compared an ICS/LABA combination over a dose range for both constituent components, the standard paradigm being the evaluation of a single bronchodilator dose. Interestingly, however, evidence from published studies suggests that modest benefits in non-spirometric parameters, including rescue medication use, symptom scores, premature study discontinuations, and exacerbations, may be obtained with formoterol 24 μg versus 12 μg twice daily without any apparent increase in safety risk.20,21 The safety of the higher dose of formoterol is further supported by the fact that daily formoterol doses of up to 48 μg (including use as rescue medication) have been licensed in Europe for COPD.
The above observations explain why two dose levels of fluticasone propionate/formoterol pMDI were evaluated in the present study, over a twofold dose range for both ICS (250 and 500 μg) and LABA (10 and 20 μg) components. Whilst an overt pairwise dose response is not anticipated (the primary comparison is between fluticasone propionate/formoterol and formoterol monotherapy), it will be of interest to see whether greater gains in efficacy versus LABA monotherapy can be achieved with the higher (500/20 μg) versus lower (250/10 μg) fluticasone propionate/formoterol dose.
Ideally, a factorial design (3×2) would have been preferable to ascertain the effect of ICS therapy and dose increments for each component of the combination. However, given the scale and associated cost of such a study, it was not feasible to implement this design. For similar reasons, the lowest available dose level of fluticasone propionate/formoterol was not evaluated in this study, albeit, on the basis of equipotency considerations,22 the recent fluticasone furoate/vilanterol data suggest that a fluticasone propionate dose of 100 μg bid may be suboptimal in COPD.17 Nonetheless, multiple ICS- and LABA-specific safety assessments in the present study will elucidate whether the higher doses of each active component are associated with any worsening in safety profile. Furthermore, the study will allow a comparison of both dose levels of fluticasone/formoterol versus the formoterol dose currently licensed for regular maintenance treatment of COPD in both Europe and the United States.
The study includes a 2-week tiotropium run-in period. Tiotropium was selected as run-in therapy rather than steroid-based therapies to avoid potential enrichment of the study population via retention of patients more likely to respond favorably to ICS treatment in the pre-study treatment period. A tiotropium run-in also avoids concerns as to an ICS withdrawal-induced increase in exacerbations in the formoterol arm23 – although Lapperre et al showed no evidence of an increase in proinflammatory markers or exaggerated clinical deterioration in COPD patients treated for 6 months with inhaled fluticasone followed by ICS withdrawal.24 The recent WISDOM study also suggests concerns as to rebound phenomena with ICS withdrawal may be unfounded.25 Finally, tiotropium is also recommended in this patient population and will provide a more effective run-in treatment than short-acting bronchodilators which have been employed in previous studies – limiting the risk of significant dropouts during this pre-randomization phase.
With regards to the formoterol arm in the study, its inclusion satisfies regulatory requirements as to the comparison of a fixed dose combination versus its monocomponents (an ICS monotherapy arm being unethical in COPD patients).26 In addition, should fewer exacerbations be observed with fluticasone propionate/formoterol versus formoterol, it will confirm the study’s assay sensitivity.27 By contrast, if the study had compared fluticasone propionate/formoterol to tiotropium and evidenced similar exacerbation rates with each as in, for example, the INSPIRE study (which compared fluticasone/salmeterol and tiotropium),28 an inherent limitation would be that the sensitivity of the study to detect differences, should they exist, would remain unknown. Atimos® Modulite was selected as the specific formoterol comparator in order that patients are required to use a single device type only (pMDI) during the study, thus avoiding patient confusion likely to occur when patients are required to use devices which require distinctly different operational and inspiratory techniques.29 This is likely to be more prominent in the relatively elderly COPD population with a high prevalence of comorbidities.
Exacerbations will be defined using a conventional HCU definition. It is, however, recognized that as many as 50%–70% of exacerbations go unreported, leading to marked underestimation of exacerbation rates in clinical studies.9 Furthermore, HCU-defined exacerbation rates vary widely across different geographical territories.30 Such differences are likely related to differences in access to health care, different costs of access (which may prompt different rates of self-medication) and cultural differences.31 Given the inherent limitations of the HCU-anchored exacerbation definition, even though this remains the most conventional and accepted approach to date, it is hoped that the use of the EXACT tool to trigger patient–physician interaction after symptom worsening will increase the reporting of events. However, concordance between HCU and EXACT-defined exacerbations is modest: in a recent study, approximately two-thirds of the patients experiencing an EXACT-defined exacerbation did not have an HCU event and approximately half the number of patients experiencing an HCU exacerbation did not have an EXACT-defined exacerbation.13 The reason behind this lack of agreement is not clear, although the authors attributed a number of factors to it, such as missing EXACT scores during the HCU exacerbation in some cases. Despite this imperfect concordance, patients who experience EXACT-defined but untreated exacerbations exhibit similar short- to medium-term deterioration in health status as compared to those experiencing HCU exacerbations.13 Thus, although EXACT-defined exacerbations are not expected to be qualitatively distinct from HCU exacerbations, it is relevant to assess treatment effect on both types of exacerbation event and appropriate that EXACT-defined exacerbations will serve as a key secondary efficacy variable in this study.
Two serum biomarkers, SP-D and CCL-18, both primarily of pulmonary origin, were selected for inclusion in the study from amongst the many biomarkers evaluated in recent years on the basis of their consistent associations with important clinical indices or disease states, relative specificity, and potential for modification with ICS-based treatment. Both will be measured at baseline and week 6 in a subgroup of patients.
Associations between SP-D and health status, FEV1, symptoms,10,32 and exacerbations33,34 in COPD patients are well documented, with correlations between SP-D and clinical indices32 appearing stronger than for fibrinogen, another promising biomarker.35 Further, in their analysis of a panel of 34 potential COPD biomarkers, Dickens et al determined that SP-D levels were amongst the most reproducible over time.35 Serum SP-D levels (unlike fibrinogen)36 are reduced by ICSs.10 It is hypothesized that ICSs ameliorate lung inflammation, thereby reducing respiratory membrane permeability, hence the leakage of SP-D from the lung into the systemic compartment. Our study will shed further light on the correlation between SP-D levels and several clinical indices in COPD, and will evaluate the effect on SP-D of two dose levels of ICS. More importantly, the study will ascertain whether short-term changes in SP-D (after 6 weeks of treatment) predict long-term clinical benefits in outcomes such as lung function, health status, symptom severity, and exacerbation rate. If so, SP-D could potentially complement blood eosinophil counts in COPD management, as eosinophil counts predict clinical response to ICS treatment37 but are not appreciably altered by the ICS treatment. Hence, blood eosinophils appear more suited to initial screening than subsequent identification of response or disease monitoring.
CCL-18 is of interest given the prominent association between cardiovascular disease and COPD, albeit mechanisms underlying this association are not well understood. Comorbid cardiovascular disease independently increases exacerbation,38 hospitalization,38 and mortality risk39 in COPD. Half of COPD deaths are attributed to cardiovascular disease,40 whilst COPD increases the risk of sudden cardiac death.41 Serum CCL-18 levels are elevated in COPD patients, with higher levels linked to an increased risk of cardiovascular morbidity and mortality and all-cause mortality.11 Prednisolone reduces CCL-18 levels,11 but the effect of ICS on CCL-18 has not been reported. Given these observations, the present study will evaluate changes in CCL-18 level with treatment and will also evaluate the relationship between levels of this biomarker and the occurrence of a constellation of arteriosclerotic events. It is of note that in the INSPIRE study,28 treatment with fluticasone/salmeterol was associated with significantly lesser mortality as compared to tiotropium, the difference mainly driven by deaths due to cardiovascular causes. Furthermore, the TORCH study42 also showed the lowest number of cardiovascular events in the fluticasone/salmeterol group compared to the monotherapy and placebo groups. Therefore, it is of interest to elucidate the effect of ICS-based treatment on CCL-18 and relate the change in this measure to cardiovascular events observed during the treatment period.
Finally, the occurrence of pneumonia will be quantified based on criteria specified in the BTS community acquired pneumonia guideline,14 so avoiding the limitations of earlier randomized controlled trials which did not employ a prospective standardized definition of pneumonia.43,44 The use of accepted clinical and particularly radiological criteria to confirm the diagnosis of pneumonia in our study will limit misdiagnosis. This trial will thus further assess the association between the use of ICSs and risk of pneumonia in COPD,45,46 although, importantly, a recent review concluded that respiratory mortality and all-cause mortality are either unchanged or reduced by ICS administration.47 It will also be of interest to examine whether there is any difference in the rate of pneumonia events in our study of a fluticasone propionate/formoterol suspension pMDI formulation compared to those previously seen with fluticasone propionate/salmeterol dry powder. The head-to-head comparison of both fluticasone propionate/formoterol doses in the present study will additionally allow assessment of any dose-related signal for pneumonia, which is not evident in between-trial comparisons of fluticasone propionate/salmeterol,5,18 in contrast to fluticasone furoate/vilanterol over the dose range 50/25 to 200/25 μg once daily.17
In summary, this study will compare the efficacy and safety of two dose levels of fluticasone propionate/formoterol (over a twofold dose range for both components) with formoterol in COPD patients. Use of the EXACT should enhance the reporting of exacerbations over the 52-week treatment period and further experience of this instrument. Two lung-derived biomarkers will be measured and their clinical associations examined, and the occurrence of pneumonia will be rigorously evaluated.
Trade marks
®FLUTIFORM is a registered trade mark of Jagotec AG and is used under licence. ®ATIMOS is a registered trade mark of Chiesi Farmaceutici S.p.A. ®SPIRIVA and HandiHaler are registered trade marks of Boehringer Ingelheim Pharma GmbH & Co. KG. ®VENTOLIN and EVOHALER are registered trade marks of Glaxo Group Limited.
Disclosure
This study is sponsored by Mundipharma Research Ltd. Professor Papi reports personal fees from Mundipharma, during the conduct of the study; grants, personal fees, non-financial support and other from Chiesi, Astrazeneca, GlaxoSmithKline, Boehringer Ingelheim, Merck Sharp & Dohme, Pfizer, Takeda, and Mundipharma (outside the submitted work); personal fees and non-financial support from Menarini, Novartis, and Zambon. Professor Jones reports personal fees from Mundipharma during the conduct of the study; personal fees from AstraZeneca, Chiesi, GSK, Novartis, Sandoz, outside the submitted work. Dr Dalvi, Ms McAulay and Dr Dissanayake are employees of the study sponsor, Mundipharma Research Limited. Mrs McIver was an employee of Mundipharma Research Limited at the time the study was designed.
References
Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57:847–852. | ||
Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1418–1422. | ||
Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363:1128–1138. | ||
Jenkins CR, Jones PW, Calverley PM, et al. Efficacy of salmeterol/fluticasone propionate by GOLD stage of chronic obstructive pulmonary disease: analysis from the randomised, placebo-controlled TORCH study. Respir Res. 2009;10:59. | ||
Calverley PM, Anderson JA, Celli B, et al; TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356:775–789. | ||
Calverley P, Pauwels R, Vestbo J; TRial of Inhaled STeroids ANd long-acting beta2 agonists study group. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2003;361(9356):449–456. | ||
Szafranski W, Cukier A, Ramirez A, et al. Efficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary disease. Eur Respir J. 2003;21(1):74–81. | ||
Global Strategy for the Diagnosis, Management and Prevention of Chronic Pulmonary Obstructive Disease. Global Initiative for Chronic Obstructive Lung Disease; updated 2014. Available from: http://www.goldcopd.org/uploads/users/files/GOLD_Report_2014_Jan23.pdf. Accessed January 24, 2014. | ||
Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161(5):1608–1613. | ||
Sin DD, Man SF, Marciniuk DD, et al; ABC (Advair, Biomarkers in COPD) Investigators. The effects of fluticasone with or without salmeterol on systemic biomarkers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(11):1207–1214. | ||
Sin DD, Miller BE, Duvoix A, et al; ECLIPSE Investigators. Serum PARC/CCL-18 concentrations and health outcomes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(9):1187–1192. | ||
EXACT-PRO Initiative. The Exacerbations of Chronic Pulmonary Disease Tool (EXACT) User Manual Version 5. Bethesda: Evidera; 2013. | ||
Jones PW, Lamarca R, Chuecos F, et al. Characterisation and impact of reported and unreported exacerbations: results from ATTAIN. Eur Respir J. 2014;44(5):1156–1165. | ||
Lim WS, Baudouin SV, George RC; Pneumonia Guidelines Committee of the BTS Standards of Care Committee. BTS guidelines for the management of community acquired pneumonia in adults: update 2009. Thorax. 2009;64 Suppl 3:iii1–iii55. | ||
Tashkin DP, Rennard SI, Martin P, et al. Efficacy and safety of budesonide and formoterol in one pressurized metered-dose inhaler in patients with moderate to very severe chronic obstructive pulmonary disease: results of a 6-month randomized clinical trial. Drugs. 2008;68(14):1975–2000. | ||
Rennard SI, Tashkin DP, McElhattan J, et al. Efficacy and tolerability of budesonide/formoterol in one hydrofluoroalkane pressurized metered-dose inhaler in patients with chronic obstructive pulmonary disease: results from a 1-year randomized controlled clinical trial. Drugs. 2009;69(5):549–565. | ||
Dransfield MT, Bourbeau J, Jones PW, et al. Once-daily inhaled fluticasone furoate and vilanterol versus vilanterol only for prevention of exacerbations of COPD: two replicate double-blind, parallel-group, randomised controlled trials. Lancet Respir Med. 2013;1(3):210–223. | ||
Anzueto A, Ferguson GT, Feldman G, et al. Effect of fluticasone propionate/salmeterol (250/50) on COPD exacerbations and impact on patient outcomes. COPD. 2009;6(5):320–329. | ||
Ferguson GT, Anzueto A, Fei R, Emmett A, Knobil K, Kalberg C. Effect of fluticasone propionate/salmeterol (250/50 microg) or salmeterol (50 microg) on COPD exacerbations. Respir Med. 2008;102(8):1099–1108. | ||
Rossi A, Kristufek P, Levine BE, et al; Formoterol in Chronic Obstructive Pulmonary Disease (FICOPD) II Study Group. Comparison of the efficacy, tolerability, and safety of formoterol dry powder and oral, slow-release theophylline in the treatment of COPD. Chest. 2002;121(4):1058–1069. | ||
Dahl R, Greefhorst LA, Nowak D, et al; Formoterol in Chronic Obstructive Pulmonary Disease I Study Group. Inhaled formoterol dry powder versus ipratropium bromide in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164(5):778–784. | ||
Bateman ED, Bleecker ER, Lötvall J, et al. Dose effect of once-daily fluticasone furoate in persistent asthma: a randomized trial. Respir Med. 2012;106(5):642–650. | ||
Jarad NA, Wedzicha JA, Burge PS, Calverley PM. An observational study of inhaled corticosteroid withdrawal in stable chronic obstructive pulmonary disease. ISOLDE Study Group. Respir Med. 1999;93(3):161–166. | ||
Lapperre TS, Snoeck-Stroband JB, Gosman MM, et al; Groningen Leiden Universities Corticosteroids in Obstructive Lung Disease Study Group. Effect of fluticasone with and without salmeterol on pulmonary outcomes in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med. 2009;151(8):517–527. | ||
Magnussen H, Disse B, Rodriguez-Roisin R, et al; WISDOM Investigators. Withdrawal of inhaled glucocorticoids and exacerbations of COPD. N Engl J Med. 2014;371:1285–1294. | ||
European Medicines Agency. Guideline on Clinical Development of Fixed Dose Combination Medicinal Products. EMA/CHMP/281825/2015. London: European Medicines Agency; 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/05/WC500186840.pdf. Accessed July 16, 2015. | ||
ICH Harmonised Tripartite Guideline: Choice of Control Group and Related Issues in Clinical Trials. E10. ICH; 2000. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E10/Step4/E10_Guideline.pdf. Accessed July 16, 2015. | ||
Wedzicha JA, Calverley PM, Seemungal TA, Hagan G, Ansari Z, Stockley RA; INSPIRE Investigators. The prevention of chronic obstructive pulmonary disease exacerbations by salmeterol/fluticasone propionate or tiotropium bromide. Am J Respir Crit Care Med. 2008;177(1):19–26. | ||
van der Palen J, Klein JJ, van Herwaarden CL, Zielhuis GA, Seydel ER. Multiple inhalers confuse asthma patients. Eur Respir J. 1999;14(5):1034–1037. | ||
Vogelmeier C, Hederer B, Glaab T, et al; POET-COPD Investigators. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med. 2011;364:1093–1103. | ||
Langsetmo L, Platt RW, Ernst P, Bourbeau J. Underreporting exacerbation of chronic obstructive pulmonary disease in a longitudinal cohort. Am J Respir Crit Care Med. 2008;177(4):396–401. | ||
Sin DD, Leung R, Gan WQ, Man SP. Circulating surfactant protein D as a potential lung-specific biomarker of health outcomes in COPD: a pilot study. BMC Pulm Med. 2007;7:13. | ||
Lomas DA, Silverman EK, Edwards LD, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints study investigators. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur Respir J. 2009;34(1):95–102. | ||
Shakoori TA, Sin DD, Ghafoor F, Bashir S, Bokhari SN. Serum surfactant protein D during acute exacerbations of chronic obstructive pulmonary disease. Dis Markers. 2009;27(6):287–294. | ||
Dickens JA, Miller BE, Edwards LD, Silverman EK, Lomas DA, Tal-Singer R; Evaluation of COPD Longitudinally to Identify Surrogate Endpoints (ECLIPSE) study investigators. COPD association and repeatability of blood biomarkers in the ECLIPSE cohort. Respir Res. 2011;12:146. | ||
Duvoix A, Dickens J, Haq I, et al. Blood fibrinogen as a biomarker of chronic obstructive pulmonary disease. Thorax. 2013;68(7):670–676. | ||
Pascoe S, Locantore N, Dransfield MT, Barnes NC, Pavord ID. Blood eosinophil counts, exacerbations, and response to the addition of inhaled fluticasone furoate to vilanterol in patients with chronic obstructive pulmonary disease: a secondary analysis of data from two parallel randomised controlled trials. Lancet Respir Med. 2015;3(6):435–442. | ||
Papaioannou AI, Bartziokas K, Loukides S, et al. Cardiovascular comorbidities in hospitalised COPD patients: a determinant of future risk? Eur Respir J. 2015;46(3):846–849. | ||
Divo M, Cote C, de Torres JP, et al; BODE Collaborative Group. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(2):155–161. | ||
Huiart L, Ernst P, Suissa S. Cardiovascular morbidity and mortality in COPD. Chest. 2005;128(4):2640–2646. | ||
Lahousse L, Niemeijer MN, van den Berg ME, et al. Chronic obstructive pulmonary disease and sudden cardiac death: the Rotterdam study. Eur Heart J. 2015;36(27):1754–1761. | ||
Calverley PM, Anderson JA, Celli B, et al; TORCH Investigators. Cardiovascular events in patients with COPD: TORCH study results. Thorax. 2010;65(8):719–725. | ||
Crim C, Calverley PM, Anderson JA, et al. Pneumonia risk in COPD patients receiving inhaled corticosteroids alone or in combination: TORCH study results. Eur Respir J. 2009;34(3):641–647. | ||
Calverley PM, Stockley RA, Seemungal TA, et al; Investigating New Standards for Prophylaxis in Reduction of Exacerbations (INSPIRE) Investigators. Reported pneumonia in patients with COPD: findings from the INSPIRE study. Chest. 2011;139(3):505–512. | ||
Wedzicha JA, Singh D, Vestbo J, et al; FORWARD Investigators. Extrafine beclomethasone/formoterol in severe COPD patients with history of exacerbations. Respir Med. 2014;108(8):1153–1162. | ||
Kew KM, Seniukovich A. Inhaled steroids and risk of pneumonia for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2014;3:CD010115. | ||
Festic E, Scanlon PD. Incident pneumonia and mortality in patients with chronic obstructive pulmonary disease. A double effect of inhaled corticosteroids? Am J Respir Crit Care Med. 2015;191(2):141–148. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.