Back to Journals » Clinical Interventions in Aging » Volume 17
The Effect of Metabolites on Mitochondrial Functions in the Pathogenesis of Skeletal Muscle Aging
Authors Gu X ![]() , Wang W, Yang Y, Lei Y
, Wang W, Yang Y, Lei Y ![]() , Liu D
, Liu D ![]() , Wang X, Wu T
, Wang X, Wu T
Received 30 May 2022
Accepted for publication 11 August 2022
Published 22 August 2022 Volume 2022:17 Pages 1275—1295
DOI https://doi.org/10.2147/CIA.S376668
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Maddalena Illario
Xuchao Gu,1,2,* Wenhao Wang,1,2,* Yijing Yang,1,2,* Yiming Lei,1,2 Dehua Liu,1,2 Xiaojun Wang,1,2 Tao Wu1,2
1Department of Traditional Chinese Medicine, Huadong Hospital Affiliated to Fudan University, Shanghai, 200040, People’s Republic of China; 2Shanghai Key Laboratory of Clinical Geriatric Medicine, Huadong Hospital Affiliated to Fudan University, Shanghai, 200040, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Tao Wu; Xiaojun Wang, Department of Traditional Chinese Medicine, Huadong Hospital Affiliated to Fudan University, No. 221 West Yan-An Road, Shanghai, 200040, People’s Republic of China, Tel +86 13641673915 ; +86 13062796919, Email [email protected]; [email protected]
Abstract: Sarcopenia is an age-related systemic disease characterized by skeletal muscle aging that generally severely affects the quality of life of elderly patients. Metabolomics analysis is a powerful tool for qualitatively and quantitatively characterizing the small molecule metabolomics of various biological matrices in order to clarify all key scientific problems concerning cell metabolism. The discovery of optimal therapy requires a thorough understanding of the cellular metabolic mechanism of skeletal muscle aging. In this review, the relationship between skeletal muscle mitochondria, amino acid, vitamin, lipid, adipokines, intestinal microbiota and vascular microenvironment has been separately reviewed from the perspective of metabolomics, and a new therapeutic direction has been suggested.
Keywords: sarcopenia, metabolomics, mitochondria, intestinal microbiota, vascular endothelium
Introduction
Sarcopenia is a geriatric syndrome characterized by skeletal muscle aging.1 According to different diagnostic criteria, the global prevalence of sarcopenia in people aged over 65 years is between 10–20%, and that in those over 80 years is up to 50%.2 Sarcopenia often severely affects the quality of life in elderly people and is associated with serious health consequences in terms of falls, fractures, hospitalization, disability and mortality.3,4 Since the onset of sarcopenia is hidden and easily ignored, different diagnostic methods have been proposed successively for different regions and racial groups. The Asian Working Group for Sarcopenia has issued two consensus reports urging medical professionals to pay attention to it. Sarcopenia is one of the world’s major public health problems, posing a significant burden on health-care spending in various countries as the global elderly population grows. As a result, it is critical to focus on the pathogenesis, preventive measures, and treatment of sarcopenia.
Skeletal muscle is the largest organ in the body, accounting for 30–40% of body weight. It not only controls human movement, physical strength, and temperature, but it also has a significant impact on overall body metabolism via the anabolic/catabolic balance.5 In this way, it directly contributes to the progression of chronic diseases and aging. Previous studies6–8 have shown that mitochondrial dysfunction, oxidative stress, inflammation, abnormal autophagy, DNA methylation, and other factors can alter muscle cell metabolism. These can result in muscle cell damage and aging, and lead to skeletal muscle quality and function decline.
Mitochondria are metabolic organelles found in muscle cells that play an important role in the pathogenesis of sarcopenia. Mitochondria not only play an important role in oxidative phosphorylation, ATP synthesis, and energy metabolism, but they also have the ability to regulate the myonuclear domain of senescent muscle cells and skeletal myofiber plasticity under physiological and pathological conditions,9 indicating that these organelles are central to the maintenance of muscle cell activity. Given this situation, mitochondria are considered to participate in the regulation of aging-related muscle atrophy. In fact, with aging and loss of muscle function, the degeneration of mitochondria is accompanied by comprehensive pathophysiological modifications. These modifications include significant declines in mitochondrial content,10 resting and maximal oxygen (O2) consumption,11 activities of tricarboxylic acid cycle enzymes, and oxidative phosphorylation (OXPHOS) activity.12 The mitochondrial decay process is an established biomarker of aging and is also considered to be the main driving force in skeletal muscle aging.13
Metabolomics is a method to address all key scientific issues related to cell metabolism by qualitatively describing and quantitatively characterizing the small molecular metabolites of different biological substrates. Here, we review the literature on the metabolomics of sarcopenia, which are shown in Table 1, focusing on the interaction between the characteristic metabolites and mitochondria of skeletal muscles. Furthermore, we also discuss the relationship between characteristic metabolites and the vascular microenvironment of sarcopenia. We hope that this work can provide a fresh perspective for new diagnostic methods and treatment strategies.
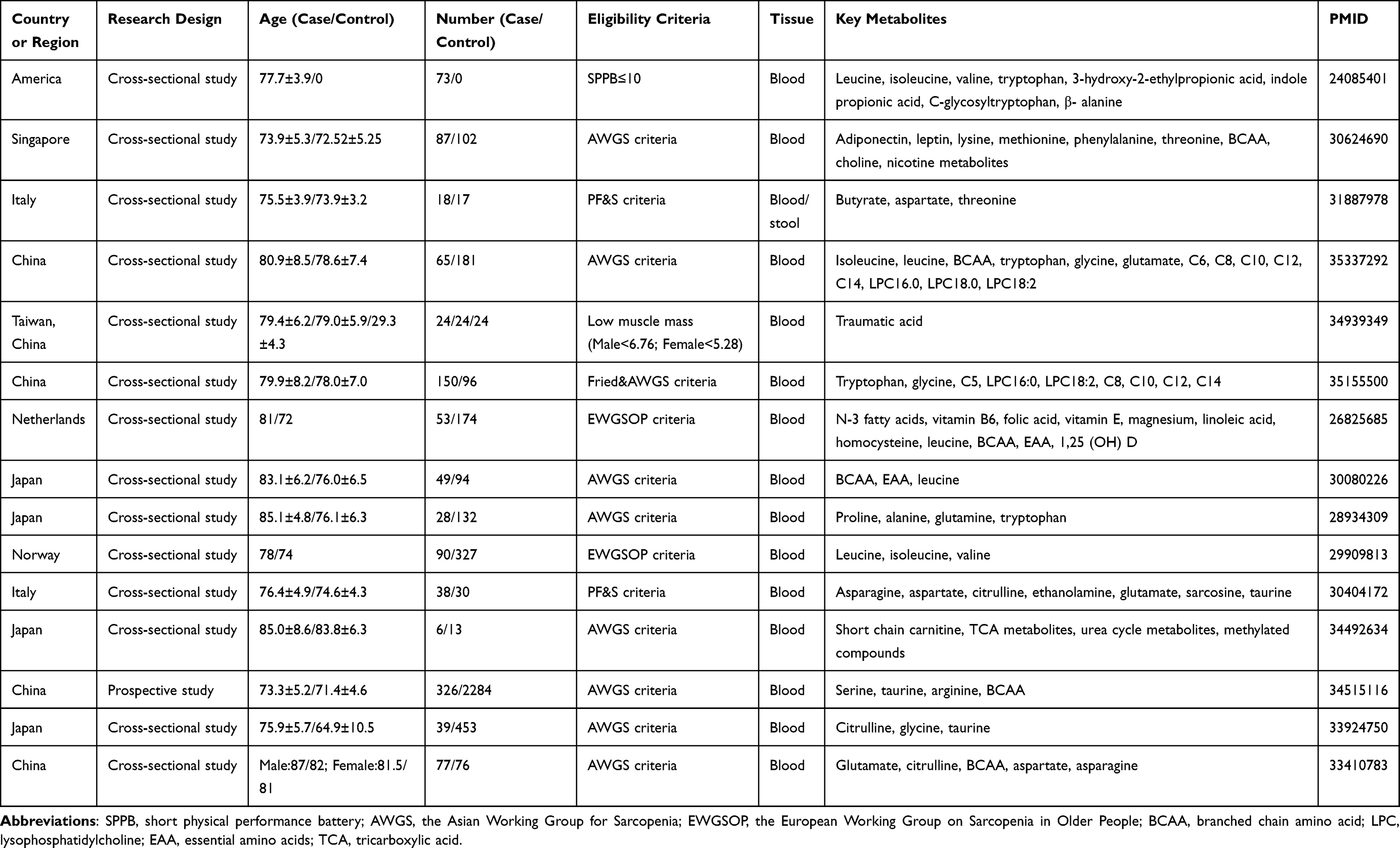 |
Table 1 Clinical Study on Metabolomics of Sarcopenia |
The Effect of Amino Acids and Vitamins on Mitochondria in Skeletal Muscles
The normal physiological function of mitochondria in energy metabolism is controlled by numerous complex interactions, which are also closely related to the pathogenesis of sarcopenia. With the innovation and development of metabolomics technology, multiple metabolites associated with sarcopenia have been identified, which include branched-chain amino acids (BCAAs), tryptophan, serine, methionine, sarcosine, aspartate, glutamate, taurine, and vitamin D. Their involvement in mitochondrial energy metabolism is briefly described as follows.
BCAAs
Multiple studies have demonstrated that the serum concentration of BCAAs decreases in patients with sarcopenia in comparison to elderly people without sarcopenia.14–17 BCAAs refer to leucine, valine, and isoleucine, which are essential amino acids that can only be obtained from dietary sources and cannot be synthesized indigenously. They are involved in muscle protein synthesis, satellite cell activation, and proteolysis inhibition.18 Although most amino acids are degraded in the liver, BCAAs are primarily metabolized in muscle tissue by mitochondrial dehydrogenase and branch-chain ketoacid dehydrogenase.19 BCAA degradation can be divided into two steps.
The first step involves the production of branched-chain keto acids (BCKAs), glutamine, and alanine from BCCAs under the action of more than 40 mitochondrial enzymes. Of which the two most important enzymes are branched-chain aminotransferases (BCATs) and branched-chain α-ketoacid dehydrogenase complex (BCKDH). These products are released into the blood circulation, among which glutamine and alanine are considered indicators of increased catabolism of BCAAs. The second step involves the production of gluconeogenic substrates and ketogenic substrates from BCKAs under the action of BCKDH. These products can enter the tricarboxylic acid (TCA) cycle or be stored in other forms to provide compensatory metabolism to organs during disease or trauma.19–21
In addition to the metabolic function, BCAAs also play a key role in signal transduction. BCAAs, especially leucine, are involved in regulating the mammalian target of rapamycin (mTOR) pathway, which has a key involvement in mitochondrial energy metabolism. Although the mechanism has not been thoroughly studied, leucine has been known to effectively activate the mTOR complex 1 (mTORC1) in at least two ways. Firstly by directly binding to sestrin 2 and releasing GTPase-activating protein activity for Rags 2 to promote the complete activation of mTORC1, and secondly, by increasing sirtuin 1 (SIRT1) and peroxisome proliferator-activated receptor gamma (PPARγ) coactivator-1α (PGC-1α) to activate the mTOR pathway. The activated mTOR pathway can increase mitochondrial biogenesis partially by increasing the nitric oxide generating system.22–24
In addition, some studies have shown that Leucyl-tRNA synthetase (LeuRS) can be used as a direct sensor of leucine and participate in the activation of mTORC1. However, in the absence of relevant evidence, further investigation is needed to validate this concept.25 BCAAs are also associated with other pathophysiological processes that are involved in the genesis of skeletal muscle aging, such as inflammation and insulin resistance.26–28 Therefore, the impairment of metabolism of BCAAs may be one of the basic factors contributing to sarcopenia.
Tryptophan
Tryptophan is an essential amino acid for the human body. In a study of elderly patients in Massachusetts, it was observed that those with poor muscle mass are associated with low serum levels of tryptophan and related metabolites, such as indole propionic acid.29 Similar results were reported in the other two studies in Tokyo and Baltimore.14,30
Tryptophan is essential for protein synthesis, and normal protein turnover in skeletal muscles and lack of tryptophan has been observed to affect embryonic development, especially that of skeletal muscles.31 Ninomiya et al demonstrated that a diet deficient in tryptophan negatively regulated glycolysis and reduced muscle fiber diameter in mice, which recovered after switching to a standard diet, indicating that tryptophan plays a critical role in the regulation of skeletal muscle mass.32
Low levels of glycolysis inhibit mTOR signaling by enhancing the binding of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) to Ras homologous (Rheb).33 However, the supplementation of tryptophan was observed to increase muscle-derived insulin-like growth factor 1 (IGF-1) and the expression of genes involved in the eif4/p70s6 k/mTOR pathway to reverse muscle atrophy caused by low serum tryptophan levels.32,34 IGF-1 is an upstream activator of the mTORC1 and regulates the translational apparatus by activating P13K/AKT pathway through insulin receptors and associated adaptor molecules.35 In this process, Rheb is reduced to its inactive GDP-bound form.36 These results strengthen the correlation between IGF-1 and tryptophan, better explaining how IGF-1 activates the mTORC1 pathway.
In addition, tryptophan also plays a role in the biosynthesis of NAD+, a coenzyme involved in the redox reactions, which form the core of energy metabolism and are very important to maintain mitochondrial function.37 The NAD+ synthesized from tryptophan through the kynurenine pathway in cells is transported to mitochondria and is mainly consumed by sirtuin3, sirtuin4, and sirtuin5 to regulate mitochondrial acylation which is closely associated with lipid metabolism, ketone body biosynthesis, urea cycle and oxidative stress.38 Other metabolites produced upon tryptophan degradation through the kynurenine pathway can prevent excessive inflammation.39 However, it must be mentioned that the compounds produced in this pathway can regulate neuronal excitability and initiate immune tolerance, which is closely related to neurodegenerative disorders, pain syndromes and autoimmune diseases.40 It may adversely affect on the application of tryptophan in the treatment of sarcopenia. For instance, Connell et al demonstrated that the supplementation of tryptophan fails to effectively improve muscle strength in elderly patients with sarcopenia.38
Serine, Methionine, and Sarcosine
Serine, methionine, and sarcosine are also considered essential amino acids. Their levels in the serum of patients with sarcopenia have been observed to be decreased than in elderly people without sarcopenia.41,42 Calvani et al believed that the decrease in serum levels of these three amino acids is due to malnutrition caused by decreased in total energy and protein intake, as it is often thought to be the risk factor for sarcopenia.43
The connection between these three amino acids and mitochondria is connected by one-carbon metabolism. Methionine can be metabolized to form sarcosine. Serine, methionine, and sarcosine are directly or indirectly involved in one-carbon metabolism mediated by the folic acid cofactor, which includes biosynthesis of purines and thymidine, amino acid homeostasis, epigenetic maintenance, and redox defense.44 Previous studies have shown that mitochondria are also involved in one-carbon metabolism and play a role as the site of oxidation of these three amino acids,45 suggesting the involvement of these amino acids in synthesizing one-carbon units in mitochondria.
Formic acid is one of the basic products of one-carbon metabolism and plays an important role in the mitochondrial-cytosolic one-carbon cycle, mitochondrial tracing, and cytosolic serine catabolism.46 A complete set of enzymes linking serine to formic acid exists in both the cytosol and mitochondria. These enzymes allow parallelly running metabolic processes and a complete oxidative/reductive cycle, which catabolizes serine in the mitochondria and synthesizes serine in the cytosol. The physiological significance of the cycle that catabolizes serine in the mitochondria and synthesizes serine in the cytosol is to allow one-carbon metabolism and NAD+ metabolism to operate normally. The process is thermodynamically driven by the difference in electrochemical potential between mitochondrial nicotinamide adenine dinucleotide (NADH; and nicotinamide adenine dinucleotide phosphate [NADPH]) and cytosolic NADPH. A high cytosolic NADPH/NADP+ ratio favors flux from formic acid toward serine in the cytosol.47
Recently, studies have shown that mitochondrial dysfunction and mitochondrial DNA variation lead to changes in methionine metabolism, reducing s-adenosylmethionine (SAM) so that DNA methyltransferase (DNMT) fails to get enough SAM for the methylation of cysteine residues on DNA. Thus, nuclear DNA methylation is interrupted, and tissues show aging characteristics.48 Other studies have shown that the changes induced by mitochondrial respiratory chain deficiencies can activate genes related to serine synthesis (PHGDH, PSAT1, and PSPH) via activating transcription factor 4 (ATF4), resulting in increased serine levels but impairs formic acid release and remodels one-carbon metabolism. The disadvantages of this one-carbon remodeling may be extended to systemic metabolism.49,50 In summary, this evidence suggests that the metabolic changes of serine, methionine, and sarcosine may not be the cause but the result of sarcopenia.
Aspartate
Two studies enrolling elderly people living in communities in Italy with physical frailty and sarcopenia showed that high serum levels of aspartate and asparagine could characterize the aging and atrophy of skeletal muscle.41,51 Aspartate and asparagine are amino acids metabolized in resting muscles and are important substrates for nucleotide synthesis in the human body. Both of them provide amino and ammonia for the synthesis of glutamine and alanine, and their carbon skeleton can be used for the de novo synthesis of TCA cycle intermediates, a nitrogenous form of oxaloacetate, and α-ketoglutarate.52 Through analysis of comprehensive metabolomic data of human blood in relation to sarcopenia patients living in Japan, Masahiro et al showed that TCA metabolites are a panel of metabolic markers of sarcopenia.53 This suggests that the anabolism of aspartate and asparagine is related to the pathogenesis of sarcopenia from another perspective.
Several recent studies have implicated that cells provide necessary electron receptors and carbon for the biosynthesis of aspartate through the mitochondrial electron transport chain (ETC) and TCA cycle. The biosynthesis of aspartate is believed to be the essential function of mitochondrial respiration in mammalian cells.54–56 The biological processes involving aspartate are closely related to the mTORC1 signaling pathway. When the intracellular TCA cycle is normal, and aspartate delivery is sufficient, aspartate activates the mTORC1 pathway and promotes anabolism and cell growth.57,58 However, when the TCA cycle is inhibited, insufficient aspartate delivery leads to apoptosis.59
In addition to this, aspartate can cause an imbalance in mitochondrial homeostasis through key mechanisms of O2 sensing provided by the hypoxia-inducible factors (HIF) pathway. Generally speaking, mitochondrial complex III is believed to be the initiator of the HIF pathway.60 During hypoxia, mitochondrial complex III generates a mass of intracellular reactive oxygen species (ROS) in the cytoplasm to oxidize Fe2+ to Fe3+. Thereby inactivates Fe2+ as an essential cofactor of prolyl hydroxylase domain enzymes (PHDs) and the factor-inhibiting HIF (FIH). After the inhibition of PHDs and FIH (the major regulatory subunit of HIFα, β heterodimer), the HIFα subunit is stabilized instead of being continuously degraded by the proteasome upon hydroxylation at proline and asparagine residues.61 Stable HIFα forms a complex with HIFβ, which serves as a master transcription regulator of a hypoxia-induced gene to stimulate the expression of pyruvate dehydrogenase kinase-1 (PDK1), then inactivates pyruvate dehydrogenase, thereby suppressing the TCA cycle and mitochondrial respiration.62 Furthermore, HIF can stimulate the expression of lactate dehydrogenase-A, promote pyruvate to lactic acid conversion, and inhibit mitochondrial metabolism.63 In general, aspartate and asparagine can have a variety of effects on mitochondrial and muscle function. However, there is no evidence that aspartate and asparagine supplementation can improve the symptoms of sarcopenia patients; therefore, further research is needed to confirm this.
Glutamate
Serum levels of glutamate were found to differ between elderly patients with sarcopenia and those without sarcopenia.17,41,64 Li et al observed that the serum level of glutamate was positively correlated with gait speed in elderly male patients with sarcopenia living in Beijing communities.17 Gait speed is an important index to measure the life span and physical function of the elderly.64 Stefano et al reported that serum glutamate level was significantly associated with muscle mass and strength in Caucasian women.65 These observations suggest the potential role of glutamate in regulating skeletal muscle metabolism in elderly patients.
Glutamate is an important intermediate of the muscle energy metabolism under physiological and pathological conditions66 as it can fuel respiration and participate as an anaplerotic substrate to maintain optimum levels of TCA cycle intermediates through its conversion to α-ketoglutarate.67 Therefore, the changes in glutamate serum levels observed in patients with sarcopenia may suggest a disorder of muscle energy metabolism.
Although aspartate/glutamate carriers (AGCs) are widely believed to be the main transporters of glutamate entry into the mitochondrial matrix,68 some studies have shown that glutamate enters mitochondria through the sodium-calcium-exchanger (NCX) and sodium-dependent excitatory amino-acid transporters (EAATs) to stimulate calcium-dependent dehydrogenases activity and to drive ATP production in ischemic settings.67 It is possible that the discovery of this mechanism can help to improve the vascular microenvironments of aged skeletal muscles.
Glutamate is also closely related to mitochondrial NAD+ metabolism through glutamic oxaloacetic transaminase 1 (GOT1). The function of GOT1 is to catalyze the interconversion of aspartate and α-ketoglutarate to oxaloacetic acid and glutamate to transfer electrons to mitochondria. The downregulation of GOT1 indicates the low efficiency of mitochondrial NAD+ metabolism in muscles and apoptosis.54,69
Furthermore, glutamate is the most common neurotransmitter in the mammalian nervous system and is related to the regulation of synaptic homeostasis at the neuromuscular junction.70,71 Recent findings have implicated the neuromuscular junction to be an important locus in the development and progression of sarcopenia, while it is still unclear whether glutamate is involved in the adaptive remodeling of the neuromuscular junction in patients with sarcopenia.72 Altogether, the current evidence supports that the change in glutamate metabolism level is one of the reasons for the decrease in muscle energy metabolism. However, whether glutamate can become a potential therapeutic product to improve the symptoms of sarcopenia remains a question for further research.
Taurine
Taurine is a ubiquitous non-proteinogenic sulfur-containing amino acid, whose serum level has been observed to decrease significantly in patients with sarcopenia and seems to be associated with gait speed.41,42,73 Nutritional supplementation with taurine has been observed to hinder the development and progression of sarcopenia in human subjects.74
Taurine is widely expressed in human tissues, appearing to be the most abundant free amino acid in the heart, brain, and skeletal muscle, accounting for approximately 0.1% of total body weight.75 About 70% of taurine is present in skeletal muscles. This amino acid is involved in the regulation of ion channel functions, membrane stability, mitochondrial quality control, and calcium homeostasis.74 It is well known that the low activity of mitochondrial complex I and mitochondrial complex III is the main driver of ROS damage, and the generation of ROS can cause cell death. The consumption of cellular taurine leads to a decrease in the activity of mitochondrial complex I and mitochondrial complex III.76 Some studies have shown that taurine is released from cells after oxidative stress and chronic inflammatory stimulation,77 resulting in its high concentrations in tissues exposed to high levels of oxidants, suggesting its role in weakening oxidative stress. The supplementation of taurine can reduce the production of superoxide-free radicals in skeletal muscles to decrease lipid peroxidation and inflammation.78 Jong et al have proposed a new mechanism for the antioxidant effect of taurine.79 Taurine forms a complex with the mitochondrial tRNAs of leucine80 and lysine, which makes the efficient translation of mitochondrial-encoded proteins. Also, the assembly of the respiratory chain complex also depends on a sufficient supply of mitochondrial taurine. The reduction of mitochondrial translation and respiratory chain complex formation has been observed to increase the production of mitochondrial ROS.81 Therefore, the antioxidant effect of taurine appears to be mediated by taurine-driven mitochondrial protein translation. In addition, invitro studies have demonstrated that taurine can stimulate P13K/Akt/mTOR pathway and block nuclear factor kappa B (NF-κB) and Forkhead Box subfamily O transcription factors (FOXO) signaling to inhibit muscle-specific ubiquitin ligases Atrogin1 and MuRF1, which gets up-regulated during skeletal muscle atrophy, so as to prevent muscle atrophy.82 Overall, the presented evidence supports the potential benefit of taurine in sarcopenia.
Vitamin D
A study of patients with sarcopenia in Maastricht demonstrated that serum levels of 1,25-hydroxyvitamin D are significantly lower in such patients.83 1,25-hydroxyvitamin D, a bioactive metabolite activated by two hydroxylation reactions of vitamin D in the liver and kidney, is often used to assess vitamin D status.84 This indicates that the differential metabolism of vitamin D in patients may be one of the causes of sarcopenia.
In fact, vitamin D plays a crucial role in maintaining muscle health, mainly through the activation of the mTOR signaling pathway. Both in vitro and in vivo models have shown that vitamin D can reduce the production of ROS, enhance antioxidant capacity, and prevent muscle damage caused by oxidative stress when it is bound to vitamin D receptor (VDR) in skeletal muscle through 1,25-hydroxyvitamin D.85,86 Vitamin D deficiency can increase the activities of superoxide dismutase (SOD) and glutathione peroxidase (GPX), which may compensate for the decrease in antioxidant capacity caused by vitamin D deficiency.87,88 VDR also has a physiological function in promoting protein synthesis and muscle cell growth. Ashcroft et al have demonstrated that activated VDR can regulate mitochondrial respiration in muscle cells and promote energy metabolism, while the knockout of VDR reduced the ability of mitochondrial oxidative phosphorylation.89
Khalid et al demonstrated that the ultrastructure of mitochondria in mice is seriously degraded when fed a high-fat diet deficient in vitamin D. They used various animal models to study the effects of vitamin D treatment on changes in skeletal muscle under different diet conditions, indicating that vitamin D also plays a key role in maintaining mitochondrial morphological stability.90 Glerup et al discovered that vitamin D supplementation can improve muscle mass in people with vitamin D deficiency,91 and Bauer et al discovered that combining vitamin D and leucine-rich whey protein oral liquid can improve muscle quality in patients with sarcopenia.92 Altogether, this evidence suggests that vitamin D may play a role in the treatment of sarcopenia; however, it is not clear whether vitamin D supplementation alone can be beneficial. Currently, there is a lack of relevant, high-quality research evidence.
The Effect of Lipids, Adipokines, and Intestinal Microbiota on the Mitochondria in Skeletal Muscles
Lipid metabolism interacts with the intestinal microbiota and has a far-reaching impact on skeletal muscle mitochondrial energy metabolism of skeletal muscle, which is regulated primarily by fatty acid metabolism, phospholipid metabolism, and adipokines (adiponectin and leptin).
Fatty Acid Metabolism System
Fatty acids are produced by the microbiota, absorbed by the intestine, and enter the circulatory and muscular system through portal veins or lymphatic vessels. There are multiple mechanisms involved in the regulation of fatty acid metabolism in muscles. The coordinated control of fatty acid uptake, β-oxidation, and mitochondrial oxidative phosphorylation is very sensitive to the efficient generation of adenosine triphosphate. Fatty acids transported to muscles are not only used as substrates for energy production but also used in the synthesis of triglycerides and cell membrane phospholipids.
Fatty acid oxidation and muscle metabolic disorders are interrelated and play an important role in causing muscle loss,93 mainly due to the accumulation of intramyocellular lipid in skeletal muscles, which is a common feature of skeletal muscle aging. Studies have shown that the size and density of intramyocellular lipid increase significantly in aging muscles than in young muscles, although the number dose not increase much.10 Most of these lipids are biochemical fatty acids with toxic effects on cells and their respective L-carnitine derivatives. These toxic effects are partly due to the inhibition of respiratory chain complexes that causes an increase in ROS formation and imbalance of calcium homeostasis and finally result in swelling and rupturing of the mitochondria. In addition, these toxic effects activate signaling pathways that lead to apoptosis or necrosis and exacerbate the patient’s energy deficiency. The existing results from metabolomic research have shown that the disorder of fatty acid metabolism plays an important role in the pathogenesis of sarcopenia.
The carnitine system is an important part of the mitochondrial fatty acid metabolism system. All endogenous carnitines are categorized as L-carnitine if non-acylated; however, if acylated, then these are further categorized as short, medium, and long-chain acylcarnitine based on the length of the bound fatty acid chain, which provides indirect evidence of mitochondrial metabolic changes. L-carnitine is a naturally occurring compound found in the human body. Its most important biological function is to transport fatty acids from cytosol to mitochondria for follow-up β-oxidation and thereby regulates the ratio of free coenzyme A to acyl-coenzyme A in cells and mitochondria in the process of transporting short-chain and medium-chain acyl groups.94 Acylcarnitine can build up in cells with low mitochondrial activity and enter the bloodstream. As a result, elevated circulating acyl carnitine levels can be used as a marker of impaired mitochondrial energy metabolism.95 These processes are accelerated during aging, which impairs carnitine flux through the mitochondrial pathway and reflects mitochondrial dysfunction.96
Medium and Long-Chain Fatty Acids, and Medium and Long-Chain Acylcarnitine
Studies have shown that serum levels of medium and long-chain fatty acid (LCFA), as well as medium and long-chain acylcarnitine (derivatives of fatty acid metabolism), vary significantly in the geriatric population with sarcopenia and those without sarcopenia. The higher the serum concentration of these metabolites, the worse the physical function of patients with sarcopenia.17,97
The toxic effects of fatty acids and their acylcarnitine derivatives mainly depend on the saturation and chain length of fatty acids, and thus, saturated long-chain fatty acids are the most lipotoxic. LCFA comprise all fatty acids with more than 12 carbon atoms in the chain, of which palmitic acid and stearic acid and their acylcarnitine derivatives are the most harmful to muscle function. One of the reasons behind their harmful effects is their influence on mitochondrial energy metabolism. Through animal studies, Tonsgard et al demonstrated that the increase of palmitic acid and stearic acid in serum is associated with a decrease in mitochondrial ATP production.98 Additionally, Hirabara et al demonstrated that increased plasma lipid levels, mainly those of palmitic acid and stearic acid, can alter insulin-stimulated glucose metabolism (including a reduction in glycogen synthesis, glucose oxidation, and lactate production) in skeletal muscle cells leading to mitochondrial dysfunction.99
However, recent studies have shown that the LCFA is necessary for the human body. LCFA plays an important role in promoting fatty acid oxidation and mitochondrial dynamics. Gao et al have shown that the lack of serine affects the level of acylcarnitine in cells and thereby disrupts mitochondrial energy metabolism, changes mitochondrial dynamics and causes an increase in mitochondrial fragmentation, whereas supplementation of palmitate could partially restore the mitochondrial fragmentation caused by serine deprivation.100 Through an observational study, Denzi et al demonstrated that dietary supplementation of stearic acid can quickly and effectively promote mitochondrial fusion in the human body, enhance fatty acid oxidation in vivo and reduce the level of acylcarnitine in blood circulation suggesting its benefits to the human body.101 In addition, in vitro studies have confirmed that stearic acid and human transferrin receptor 1 (TRF1) play an important role in the regulation of mitochondrial dynamics. Stearic acid inhibits the activation of c-Jun N-terminal kinase signaling through the stearoylation of TRF1 and reduces mitofusin ubiquitination through HECT domain-containing ubiquitin E3 ligase, which finally promotes mitochondrial fusion and function.102
Lysophosphatidylcholine
Observational studies have shown a significant downward trend in the serum levels of lysophosphatidylcholine (LPC) in patients with sarcopenia, indicating that LPC is one of the characteristic metabolites distinguishing elderly people with and without sarcopenia.17,97,103 The serum levels of LPCs contributing significantly to this discrimination between elderly people with and without sarcopenia are LPC 16:0, LPC 18:0, and LPC 18:2, which are also the most abundant LPC present in human plasma.
The LPC is the main glycerophospholipid found in human blood and has both positive and negative effects on mitochondria under specific conditions. The role of LPC in oxidative stress and inflammation is quite complex. On the one hand, LPC can induce NADPH oxidase 5 (NOX5)-dependent ROS production leading to inflammation and cell injury.104 On the other hand, LPC can reduce ROS production by either inhibiting the formation of 5-lipoxygenation products intended to downregulate pro-inflammatory lipid mediators or by activating peroxisome proliferator receptors δ (PPARδ) mediated regulation of mitochondrial metabolism and reduction of human skeletal muscle inflammation.105,106
LPC also plays a role in the synthesis of cardiolipin, which is a unique phosphatidylglycerol lipid exclusively found in the mitochondrial membrane and is essential for mitochondrial homeostasis. The products of LPC acylation, such as lysophosphatidic acid and phosphatidic acid, act as the precursors for the synthesis of cardiolipin.107 In terms of mitochondrial energy metabolism, cardiolipin plays an important role in oxidative phosphorylation by stabilizing the structure of the mitochondrial complex I–V108 and shaping the curvature of the mitochondrial ridge, thus ensuring the assembly and correct functioning of the electron transport chain complex. These processes affect ATP and ROS production by mitochondria.109 Cardiolipin can also regulate mitochondrial dynamics bidirectionally to meet the needs of cell metabolism. Cardiolipin not only stimulates dynamin-related protein 1 (DRP1) activity to promote membrane remodeling and mitochondrial division but also interacts with optic atrophy 1 (OPA1) to promote mitochondrial fusion.110,111 Additionally, cardiolipin can also regulate mitochondrial autophagy. Cardiolipin mediates autophagosome formation and fusion with lysosomes to eliminate dysfunctional mitochondria by recruiting microtubule-associated protein 1A/1B light chain 3 and other proteins associated with mitochondrial autophagy.112,113
Adiponectin and Leptin
Adiponectin and leptin are adipokines that regulate glucose and lipid metabolism, inflammation, muscle metabolism, and insulin sensitivity. Relevant metabolomic studies have shown that these adipokines are a group of metabolites that characterize decreased muscle strength and, therefore, can potentially be used in the diagnosis of sarcopenia.15 Several studies have reported a correlation between adiponectin, leptin, and muscle strength in the elderly. Bucci et al reported that circulating adiponectin is negatively correlated with quadriceps femoris torque and grip strength in the elderly, and the leptin/adiponectin ratio is positively correlated with quadriceps femoris torque and grip strength in the elderly.114 Lu et al reported that adiponectin is associated with decreased muscle function, whereas leptin has a protective effect on muscle mass.15
Although adiponectin appears to promote muscle aging and atrophy, adiponectin and leptin coordinate with each other to promote muscle function physiologically. Adiponectin and leptin have been observed to activate the AMP-activated protein kinase (AMPK) signaling pathway and promote mitochondrial biosynthesis. In animal models of obesity and type 2 diabetes mellitus (T2DM), increasing adiponectin activated the branched chain by stimulating the activity of mitochondrially localized 2C-type Ser/Thr protein phosphatase (PP2Cm) and by increasing the expression of branched-chain α-keto acid dehydrogenase (a key regulatory point of branched chain amino acid catabolism)115 to activate the AMPK-mediated catabolism of BCAAs, so as to decrease insulin resistance in vivo.116 Other studies have shown that adiponectin induces extracellular Ca2+ influx and activates Ca2+/calmodulin-dependent protein kinase (CaMKKβ), AMPK and SIRT1 through adiponectin receptor 1 (AdipoR1), which is widely expressed in muscle and myotube β. This process increases PGC-1α expression, decreases acetylation, and increases the mitochondria in muscle cells.117
In the same way, adiponectin can also inhibit NF-κB expression and thereby reducing inflammatory factors (TNF-α, IFN-Y) and increasing anti-inflammatory factors (IL-10, IL-1Ra), leading to reduced oxidative stress and inflammation in skeletal muscles.118 Adiponectin also regulates several transcription factors promoting muscle regeneration, such as Myf5 and MyoD. Leptin exerts its function through leptin receptors (LePR) comprising six subtypes, of which LepRb is the most important because it regulates most of the functions of leptin.119 The binding of Leptin to this receptor activates tyrosine kinase Janus kinase 2 (JAK2), which in turn participates in AMPK and ERK pathways to mediate energy homeostasis.120 Leptin is also involved in the induction of adiponectin protein expression, so its effect on the AMPK pathway may be partly mediated by adiponectin.121
In summary, from the perspective of the mechanism of action of adiponectin and leptin, high plasma levels of adiponectin and leptin are related to a better metabolic phenotype. However, in the actual analysis of blood metabolites in patients with sarcopenia, it was found that the level of adiponectin increases paradoxically. This indicates that the pathogenesis of sarcopenia triggers the compensatory increase of adiponectin; however, the increased adiponectin cannot successfully reduce the pathogenesis of sarcopenia. The increase of leptin caused by the stacking of fat in the process of aging may lead to leptin resistance. The reduction in the expression of leptin-sensitive receptors in muscles and the reduction of fatty acid oxidation in muscles lead to fat deposition in other organs, such as the liver, heart, and muscle, further reducing muscle quality in obese patients.119 Similarly, an adiponectin resistance mechanism may also exist in the human body, which possibly affects the normal function of adiponectin.
Physical activity is the golden intervention to improve the metabolism of adiponectin and leptin. The reason is that physical activity increases the sensitivity of muscle tissues to these metabolites. Kim et al showed that the combined intervention of physical activity and diet could improve the gait speed and leptin level of patients with sarcopenia.122 Christina et al reported that 16 weeks of aerobic exercise and resistance exercise could significantly improve adiponectin and leptin levels in patients with sarcopenia compared with routine care.123 These studies suggest in part the biological mechanism of exercise in the treatment of sarcopenia.
Intestinal Microbiota
Intestinal microbiota and the metabolites produced by these play a key role in maintaining the physiological and metabolic homeostasis of the host.124 Intestinal microbiota has been considered as a hidden metabolic organ of human beings.125 With aging, the microbiome composition and function of intestinal microbiota undergo significant changes called intestinal microbiota imbalance, which is mainly characterized by high inter-individual variability, reduced biodiversity, and pathogen colonization.126
Although the pathological relationship between intestinal flora and human diseases has not been thoroughly investigated, an increasing body of evidence suggests a link between changes in intestinal flora, age-related inflammatory response, and energy anabolic resistance in muscle atrophy. The interaction between intestinal microbiota and skeletal muscle is called the gut-muscle axis. Metabolomic studies have revealed great differences in the intestinal microbiota among elderly people with and without sarcopenia.51
The imbalance in intestinal flora composition promotes the reduction of Bacteroides and excessive growth of phylum Firmicutes, enhances the permeability of intestinal mucosa, promotes the metabolic disorder of systemic inflammation, subclinical immune activation, and insulin resistance, and finally leads to the injury of mitochondria and muscle.127
Conversely, the gut-muscle axis is not only a pathway that connects pathogenic factors and skeletal muscle degeneration but also provides a channel for the treatment of skeletal muscle aging. Studies have demonstrated that the intake of probiotics or fecal microbial transplantation can regulate mitochondria energy metabolism and skeletal muscle functions.128–130 Munukka et al reported that probiotic supplementation in mice fed on a high-fat diet can regulate intestinal microbial composition, reduce systemic inflammation and oxidative stress, and increase muscle mass.131 Chen et al showed that the Lactobacillus casei supplementation in elderly mice could produce anti-ROS and anti-inflammatory effects as well as maintain muscle and mitochondrial functions.132 The mechanism of intestinal microbiota on mitochondria is shown in Figure 1. Although the underlying mechanism needs to be studied further, the existing research evidence shows that the metabolites of intestinal microbiota, especially short-chain fatty acids (SCFA), are key intermediates affecting the gut-muscle axis.133
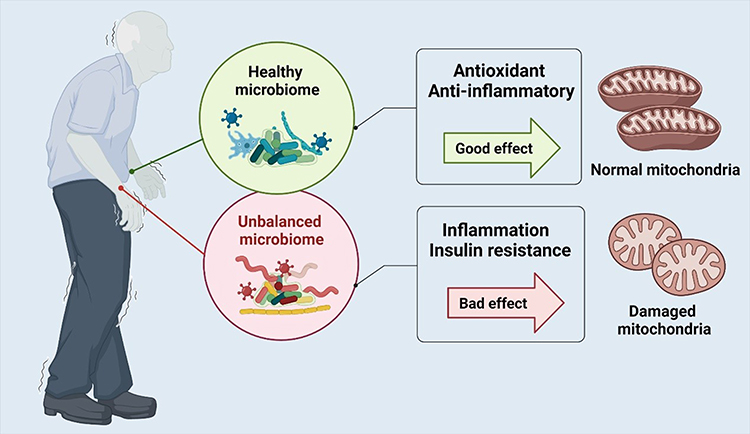 |
Figure 1 Under normal circumstances, the intestinal microbiota, such as Faecalibacterium prausnitzii and Lactobacillus casei, promotes the physiology of mitochondria to protect skeletal muscle by maintaining antioxidant and anti-inflammatory capacity. However, dysregulated intestinal flora, the reduction of Bacteroides and excessive growth of phylum Firmicutes, leads to the release of inflammatory factors. These inflammatory factors spread from the intestine and damage mitochondria and skeletal muscle. |
SCFA
In the upper gastrointestinal tract, carbohydrates escape host digestion and are fermented to produce functional oligosaccharides. Microorganisms in the cecum and proximal colon centrally metabolize these functional oligosaccharides, producing SCFA as the end product.134 SCFA comprises organic fatty acids with 1–6 carbon atoms in the chain. Under the synergistic effect of the intestinal microbiota, the derived SCFA mainly includes acetate, propionate, and butyrate (the proportion is approximately 60:20:20, respectively) that account for more than 95% of total SCFA.135,136
SCFA plays a crucial role in the regulation of systemic inflammation. Recent studies have shown that the main receptors of SCFA are free fatty acid receptors 2/3 (FFAR2/3) and G protein-coupled receptors 41/43 (GPR41/43).130,137 FFAR2 and FFAR3 are differentially expressed in cells and are involved in the regulation of a variety of cell functions. FFAR2 plays a major role in inflammation and immune response. It is mainly expressed in immune cells, such as neutrophils, eosinophils, dendritic cells, and monocytes.138 FFAR3 is mainly expressed in adipose tissue and is related to obesity and other metabolic diseases.132 GPR41 and GPR43, the main receptors of acetate, are expressed in almost all metabolically active tissues of the human body. Therefore, acetate may be one of the most important metabolites of the interaction between intestinal microbiota and host metabolites. This was confirmed by a study, which reported that systemic inflammation tends to decrease after the delivery of acetate to the distal colon, while butyrate is mainly absorbed by colonic cells, with no significant change in systemic inflammation observed.139 SCFA regulates mitochondrial metabolism and oxidative stress in the skeletal muscles through the AMPK signaling pathway. Studies have shown that SCFA can induce AMPK phosphorylation in myotubes and skeletal muscles, which is manifested by an increase in the AMP/ATP ratio in tissues.140,141 This leads to increased energy consumption, increased glucose uptake, and sugar production, and the transformation of lipid synthesis into fat oxidation. This process also activates p38 mitogen-activated protein kinases (p38 MAPKs) and PGC1-α, which improves mitochondrial content and function to promote energy metabolism and antioxidant capacity in skeletal muscles.142
The Effect of Vascular Microenvironment on Muscle Function
As mentioned above, a stable vascular microenvironment can result in sufficient muscle fiber perfusion, which is vital for maintaining skeletal muscle survival and function. The skeletal muscle is a highly vascularized tissue, and the normal physiological activity of the skeletal muscle is dependent on the material exchange between muscle cells and capillaries.
Vascular endothelial cells (EC), located in the inner walls of vessels, are involved in the regulation of the exchange of substances in the skeletal muscles, which largely determines the homeostasis of the vascular microenvironment. Therefore, the changes in EC biological function are related to muscle quality. Wang et al reported that apoptosis of EC accounts for more than 75% of muscle cell apoptosis in elderly mice.143 Studies have shown that the decrease in microvascular oxygenation and the linear density of perfusion capillaries caused by aging aggravates the dyshomeostasis of the vascular microenvironment.9 This leads to an increase in hypoxic areas in the muscle, inhibition of skeletal muscle regeneration, and obstruction of skeletal muscle anabolism.144
The destruction process of the vascular microenvironment is mediated mainly by vascular endothelial growth factor (VEGF), which is responsible for angiogenesis in skeletal muscles. Studies have shown that the delivery of VEGF can determine the size of the damaged area of the skeletal muscle and the time required for its complete regeneration.145 Hypoxia-inducible factor-1α (HIF-1α) is significantly up-regulated in aged or damaged blood vessels, inhibiting VEGF expression. An imbalance in the NAD+ metabolome can decrease EC sensitivity to VEGF and angiogenesis.146,147 Furthermore, VEGF can regulate changes in SC and their microenvironment, as well as promote muscle growth. There is no doubt about the importance of SC in muscle hypertrophy and regeneration. It has been confirmed that the spatial association between EC and SC affects the activity of SCs.148 Some studies have reported that ECs are closer to activated and differentiated SCs than static SCs.149
The harmful effect of oxidative stress on the vascular microenvironment has been widely studied. Oxidative stress can lead to EC apoptosis, imbalance in monocyte dynamics, and nitric oxide (NO) inactivation, leading to inflammation and endothelial-dependent vasodilation.150 Oxidative stress also results hyperphosphatemia through p65 nuclear translocation, leading to vascular calcification.151 Vascular calcification can significantly affect muscle fiber perfusion, accelerate muscle loss, and cause functional decline. The activation of mitochondrial autophagy in EC responds to skeletal muscle regeneration by reducing the effect of inhibition of oxidative stress on angiogenesis. Wu et al demonstrated that the phosphatase and tensin homolog-induced putative kinase 1 (PINK1) along with Parkin signaling is the keyway for EC to protect mitochondrial structure and function and promote the regulation of mitochondrial autophagy under metabolic stress. Activation of this pathway can prevent mitochondrial dysfunction, ROS production, and apoptosis.152 Other studies suggest that TFEB, as a key regulator of lysosomal biogenesis and autophagy, is up-regulated in ischemic skeletal muscles, activates AMPK signaling pathway, and promotes autophagy, which may be a key factor mediating autophagy of ECs.153
Insulin resistance is also an important factor that destroys the vascular microenvironment.154 Under normal circumstances, EC can increase NO bioavailability by phosphorylating NO synthase, while insulin resistance can reduce NO bioavailability, leading to oxidative stress, inflammation, glycotoxicity, and EC dysfunction.155
Vitamin D is essential for the EC function as its deficiency can lead to impaired endothelium-dependent vasodilation, and vascular calcification, while its supplementation can reverse these by regulating matrix Gla protein (MGP).156
The disorder of lipid metabolism inhibited the function of EC to a great extent. For example, high levels of palmitic acid can inhibit the expression and activity of cathepsin and induce EC apoptosis.157 It can also inhibit angiogenesis by inducing mitochondrial damage and dysfunction of the Hippo-Yap pathway.158 LPC can induce EC calcium influx, drive NOX5-dependent oxidative stress, and then lead to EC dysfunction.104 LPC can also induce apoptosis and inflammatory injury through GPR4-activated NLRP3 inflammasome.159 Adiponectin and leptin can also promote angiogenesis. Adiponectin promotes angiogenesis by inducing cross-talk between AMPK and Akt signaling.160 Leptin can up-regulate mitochondrial autophagy by increasing the expression of VEGF and activating the SIRT1-PINK1 pathway to induce angiogenesis in tissues.161 SCFA can prevent vascular dysfunction and vascular endothelial inflammation, and lead to vasodilation by activating GPR41/43 and inhibiting histone deacetylase.162
Discussion
Upon reviewing the metabolomic research related to sarcopenia in recent years, many characteristic metabolites related to the pathogenesis of sarcopenia have been identified in this paper, including amino acids, vitamins, fatty acids, carnitine, choline, adipokines, and so on. We described how they participate in the pathological process of sarcopenia and focused on how they cause the aging and atrophy of skeletal muscles and contribute to sarcopenia by affecting mitochondria. We also discussed the role of the homeostasis of the vascular microenvironment in sarcopenia. To the best of our knowledge, this is the first review to systematically discuss the relationship between metabolomic characteristics and mitochondrial function in sarcopenia.
It is worth highlighting that an increasing number of studies have reported that the cross-talk between characteristic metabolites and mitochondria plays a key role in the pathogenesis of sarcopenia. This is mainly determined on the basis of the following aspects. Firstly, amino acids (mainly BCAA, tryptophan, taurine, and aspartate) and LCFA (mainly palmitic acid and stearic acid) regulate the biogenesis, function, and dynamics of mitochondria through the mTOR signaling pathway. Amino acids are the positive regulatory drivers of the mTORC1. mTORC1 activates mitochondrial respiration and ATP production by stimulating the translation of nuclear-encoded mitochondrial related mRNA to meet the high-energy demands of muscle cells. mTORC1 is also involved in selective mitochondrial autophagy, which is a quality control mechanism that can ensure the recovery and removal of damaged mitochondria.163 LCFA has a negative regulatory effect on mTORC1. They can cause insulin resistance, prevent the binding of insulin to its surface receptor, and inhibit the activity of mTORC1 through IGF-1 signaling. Secondly, amino acids (mainly tryptophan, glutamate, serine, and sarcosine) are also necessary for NAD+ metabolism. Tryptophan is synthesized outside the mitochondria as the raw material for NAD+ synthesis and then transported into the mitochondria to participate in metabolism. Glutamate, serine, and sarcosine keep NAD + metabolism running normally via their respective metabolic pathways. NAD+ metabolism serves as a link between various metabolic pathways such as fatty acid oxidation, the TCA cycle, and mitochondrial respiration. The disorder of NAD+ will seriously affect mitochondrial energy metabolism. In addition, SCFA, adiponectin, and leptin can comprehensively regulate mitochondrial homeostasis by activating the AMPK signaling pathway. AMPK has many downstream effectors, and no single substrate has been found to mediate most of its effects. But in simpler terms, AMPK can participate in mitochondrial biosynthesis and increase energy output through PGC-1α and PPARγ. It can also regulate mitochondrial fission through DRP1 and control mitochondrial autophagy by activating autophagy kinase 1 to maintain mitochondrial function. Interestingly, AMPK and mTOR can regulate mitochondrial functions in not only parallel but also in cross-talk with each other.163 Additionally, there is a need to mention the regulatory effect of characteristic metabolites on ROS. Abnormal levels of ROS destroy the redox environment in muscles, interrupt cell signal transduction, alter mitochondrial biogenesis and remodeling, increase apoptosis, and lead to skeletal muscle aging and atrophy. Aspartate, taurine, and vitamin D all help to stabilize the level of ROS in the mitochondria, reduce oxidative stress and inflammation, and promote mitochondrial metabolism. LCFA and its carnitine derivatives can inhibit the activity of mitochondrial complexes and generate ROS. LPC can regulate the production of ROS bidirectionally. LPC can up-regulate the production of ROS through NOX5 or downregulate it through PPARδ. It is worth emphasizing that a moderate amount of ROS actually plays a beneficial role in maintaining mitochondrial homeostasis. For example, lower ROS levels can activate multiple pathways to protect mitochondria from stress, delay aging, and inhibit cell death.164 It can also maintain the balance between cell quiescent differentiation and self-renewal.165 The mechanism of characteristic metabolites on mitochondria of skeletal muscle is shown in Figure 2.
 |
Figure 2 The effect of amino acid and lipid metabolism on mitochondrial function of skeletal muscle. (A) The process of BCAA and taurine degraded to Acetyl-CoA in mitochondrial energy metabolism is explained. Tryptophan and aspartate act as the core components of NAD+ metabolism. BCAA, taurine, tryptophan and aspartate can activate mTORC1 signaling and promote mitochondrial function. Glutamate participates in NAD+ metabolism through GOT1. Serine, methionine and sarcosine are involved in mitochondria related single carbon metabolism. (B) LCFA, MCFA and SCFA can promote β-oxidation to increase ATP production, but LCFA and MCFA can also inhibit mTORC1 and reduce mitochondrial function. LPC leads to the production of ROS in mitochondria through NOX5. Cardiolipin produced by LCFA, MCFA and LPC can promote mitochondrial division and fusion. SCFA, LPC, adiponectin and leptin can improve mitochondrial energy metabolism through AMPK. |
Further, the importance of intestinal microbiota and vascular microenvironment in the pathology of sarcopenia should be reiterated. Several pieces of research evidence showed that the imbalance of the intestinal microbiota is highly correlated with the degree of human muscle aging, suggesting the role of the intestinal microbiota in reducing the risk of sarcopenia. The intestinal microbiota can enhance the development of sarcopenia by changing the immune response, inducing low-grade inflammation, and disrupting host mitochondrial energy metabolism. Supplementation with specific probiotics can enhance amino acid availability and reduce inflammation which is largely achieved by increasing the production of SCFA. This interaction between intestinal microbiota and skeletal muscle is called the gut-muscle axis. Understanding the pathophysiology of the gut-muscle axis is the key to carrying out transformation research. The vascular microenvironment strongly affects the nutritional supply to the skeletal muscles. Vascular endothelial dysfunction limits the muscle fiber perfusion, weakens substrate transport, interferes with mitochondrial autophagy, and leads to the atrophy and loss of function of skeletal muscles. Active maintenance of vascular microenvironment homeostasis and the development of interventions to weaken vascular functions are of great significance in improving skeletal muscle status and treat sarcopenia.
There are also some limitations to this review. The metabolomic studies reviewed in this paper often have the following defects. Firstly, most of these are cross-sectional studies. Due to the lack of complete baseline and long-term follow-up data, it is difficult to accurately analyze the relationship between sarcopenia and these characteristic metabolites. Due to the particularity of elderly patients, an individual often suffers from a variety of diseases, such as diabetes, hypertension, and osteoporosis, which are common diseases, and sarcopenia is closely related to insulin resistance, inflammatory diseases, obesity, and weakness. These factors can disturb the results of the metabolomic analysis. Secondly, although the age gap of the research population in the referenced studies is small, there are great differences in the number, gender, nationality, and country. Due to the differences in social culture and living customs, the research results may not apply to all populations. Thirdly, most of the studies do not deal with the batch effect. Although it must be recognized that minimizing the batch effect is a very challenging task from the perspective of experiment and data processing, neglecting or ignoring the batch effect can mislead the research results and even make it impossible to reproduce. Finally, it should be noted that the metabolomics research on sarcopenia is still in its infancy. Repeated small sample size and low-quality research is a waste of public resources. To understand the metabolic mechanism of sarcopenia, prospective and large-sample metabolomics research is highly warranted.
The treatment for sarcopenia is also a topic worthy of discussion. Due to the complex pathology and treatment of sarcopenia, many experts recommend and emphasize the role of physical activity in preventing sarcopenia.166 Physical activity is considered the preferred strategy for the treatment of sarcopenia, which improves the physical performance and muscle strength in patients with sarcopenia and reduces the risks of falls and related injuries.167 It is well-known that mitochondria are the powerhouse to drive human movement and physical activity regulates mitochondrial function. For example, physical activity can improve mitochondrial electron transport in skeletal muscle to improve oxidative phosphorylation, reversing the reduction in autophagy level, and improving muscle antioxidant capacity.168 Contrary to the grand occasion of physical activity therapy, there is no drug approved by FDA for the treatment of sarcopenia. Previous studies tried to treat sarcopenia through DHEA, HGH, testosterone, and clenbuterol, but all failed because either the effect was not ideal or the side effects were obvious.169 Currently, most of the clinical trials on the treatment of sarcopenia registered in the US National Library of Medicine are interventional therapies and only a few studies have tried using drugs for its treatment, such as Rooks et al using bimalumab and Regeneron Pharmaceuticals using troglozumab. The rise of metabolomics research is an opportunity to identify the operating mechanism of metabolites of sarcopenia in the human body. In the future, these characteristic metabolites may be used as unique targets to help us in developing drugs for the treatment of sarcopenia. According to the current situation, the regulation of the mitochondrial function using metabolites is a field with considerable potential.
In conclusion, there has been a lot of research on sarcopenia and metabolomics, but the results of this research are only at a preliminary stage. It is hoped that more research can combine clinical and animal experiments, giving full play to the unique advantages of metabolomics and helping us better understand the correlation between characteristic metabolites and skeletal muscle aging, which will be helpful in developing new diagnostic methods and treatment strategies.
Abbreviations
O2, oxygen; OXPHOS, oxidative phosphorylation; BCAAs, branched-chain amino acids; BCKAs, branched chain keto acids; BCATs, branched chain aminotransferases; BCKDH, branched chain α-ketoacid dehydrogenase complex; TCA, tricarboxylic acid; mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; SIRT1, sirtuin 1; PGC-1α, PPARγ coactivator-1α; LeuRS, Leucyl-tRNA synthetase; Rheb, Ras homologous; IGF-1, insulin-like growth factor 1; SAM, s-adenosylmethionine; DNMT, DNA methyltransferase; ATF4, activating transcription factor 4; ETC, electron transport chain; HIF, hypoxia-inducible factors; ROS, reactive oxygen species; PHDs, prolyl hydroxylase domain enzymes; FIH, factor-inhibiting HIF; PDK1, pyruvate dehydrogenase kinase-1; AGCs, aspartate/glutamate carriers; NCX, sodium-calcium-exchanger; EAATs, excitatory amino-acid transporters; GOT1, glutamic oxaloacetic transaminase 1; VDR, vitamin D receptor; GPX, glutathione peroxidase; LCFA, long-chain fatty acid; SCFA, short-chain fatty acid; TRF1, transferrin receptor 1; LPC, lysophosphatidylcholine; PPARδ, peroxisome proliferator receptors δ; DRP1, dynamin-related protein 1; OPA1, optic atrophy 1; AMPK, AMP-activated protein kinase; T2DM, type 2 diabetes mellitus; PP2Cm, mitochondrial localized 2C-type Ser/Thr protein phosphatase; CaMKKβ, Ca2+/calmodulin-dependent protein kinase; AdipoR1, adiponectin receptor 1; LePR, leptin receptor; JAK2, tyrosine kinase Janus kinase 2; GPR41, G protein-coupled receptor 41; FFAR2/3, free fatty acid receptors 2/3; GPR43, G protein coupled receptors 43; p38 MAPKs, p38 mitogen-activated protein kinases; GLUT4, glucose transporter 4; EC, endothelial cell; VEGF, vascular endothelial growth factor; HIF-1α, hypoxia inducible factor-1α; SC, satellite cell; NO, nitric oxide; PINK1, putative kinase 1; NOX5, NADPH oxidase 5; SPPB, short physical performance battery; AWGS, the Asian Working Group for Sarcopenia; EWGSOP, the European Working Group on Sarcopenia in Older People; EAA, essential amino acids.
Acknowledgment
Xiaojun Wang and Tao Wu are co-corresponding authors.
Funding
This work was supported by the National Natural Science Foundation of China (82104790) and Three-Year Action Plan of Shanghai for Further Accelerating the Inheritance, Innovation and Development of Traditional Chinese Medicine (ZY (2021-2023)-0302).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Cruz-Jentoft AJ, Baeyens JP, Bauer JM, et al. Sarcopenia: European consensus on definition and diagnosis: report of the European Working Group on sarcopenia in older people. Age Ageing. 2010;39(4):412–423. doi:10.1093/ageing/afq034
2. Shafiee G, Keshtkar A, Soltani A, et al. Prevalence of sarcopenia in the world: a systematic review and meta- analysis of general population studies. J Diabetes Metab Disord. 2017;16:21. doi:10.1186/s40200-017-0302-x
3. Goodpaster BH, Park SW, Harris TB, et al. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci. 2006;61(10):1059–1064. doi:10.1093/gerona/61.10.1059
4. Beaudart C, Zaaria M, Pasleau F, et al. Health outcomes of sarcopenia: a systematic review and meta-analysis. PLoS One. 2017;12(1):e0169548. doi:10.1371/journal.pone.0169548
5. Dardevet D, Mosoni L, Savary-Auzeloux I, et al. Important determinants to take into account to optimize protein nutrition in the elderly: solutions to a complex equation. Proc Nutr Soc. 2021;80(2):207–220. doi:10.1017/S0029665120007934
6. Cho MR, Lee S, Song SK. A review of sarcopenia pathophysiology, diagnosis, treatment and future direction. J Korean Med Sci. 2022;37(18):e146. doi:10.3346/jkms.2022.37.e146
7. Fulop T, Larbi A, Dupuis G, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. 2017;8:1960. doi:10.3389/fimmu.2017.01960
8. Ferri E, Marzetti E, Calvani R, et al. Role of age-related mitochondrial dysfunction in sarcopenia. Int J Mol Sci. 2020;21(15):5236. doi:10.3390/ijms21155236
9. Marzetti E, Calvani R, Cesari M, et al. Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol. 2013;45(10):2288–2301. doi:10.1016/j.biocel.2013.06.024
10. Crane JD, Devries MC, Safdar A, et al. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J Gerontol A Biol Sci Med Sci. 2010;65(2):119–128. doi:10.1093/gerona/glp179
11. Short KR, Vittone JL, Bigelow ML, et al. Age and aerobic exercise training effects on whole body and muscle protein metabolism. Am J Physiol Endocrinol Metab. 2004;286(1):E92–101. doi:10.1152/ajpendo.00366.2003
12. Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300(5622):1140–1142. doi:10.1126/science.1082889
13. Kim Y, Triolo M, Hood DA. Impact of aging and exercise on mitochondrial quality control in skeletal muscle. Oxid Med Cell Longev. 2017;2017:3165396. doi:10.1155/2017/3165396
14. Lustgarten MS, Price LL, Chale A, et al. Branched chain amino acids are associated with muscle mass in functionally limited older adults. J Gerontol a Biol Sci Med Sci. 2014;69(6):717–724. doi:10.1093/gerona/glt152
15. Lu Y, Karagounis LG, Ng TP, et al. Systemic and metabolic signature of sarcopenia in community-dwelling older adults. J Gerontol A Biol Sci Med Sci. 2020;75(2):309–317. doi:10.1093/gerona/glz001
16. Yamada M, Kimura Y, Ishiyama D, et al. Plasma amino acid concentrations are associated with muscle function in older Japanese women. J Nutr Health Aging. 2018;22(7):819–823. doi:10.1007/s12603-018-1014-8
17. Meng L, Yang R, Wang D, et al. Specific lysophosphatidylcholine and acylcarnitine related to sarcopenia and its components in older men. BMC Geriatr. 2022;22(1):249. doi:10.1186/s12877-022-02953-4
18. Le Couteur DG, Solon-Biet SM, Cogger VC, et al. Branched chain amino acids, aging and age-related health. Ageing Res Rev. 2020;64:101198. doi:10.1016/j.arr.2020.101198
19. Holecek M. Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr Metab. 2018;15:33. doi:10.1186/s12986-018-0271-1
20. Biswas D, Duffley L, Pulinilkunnil T. Role of branched-chain amino acid-catabolizing enzymes in intertissue signaling, metabolic remodeling, and energy homeostasis. FASEB J. 2019;33(8):8711–8731. doi:10.1096/fj.201802842RR
21. Neinast M, Murashige D, Arany Z. Branched chain amino acids. Annu Rev Physiol. 2019;81:139–164. doi:10.1146/annurev-physiol-020518-114455
22. D’Antona G, Ragni M, Cardile A, et al. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab. 2010;12(4):362–372. doi:10.1016/j.cmet.2010.08.016
23. Saxton RA, Knockenhauer KE, Wolfson RL, et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science. 2016;351(6268):53–58. doi:10.1126/science.aad2087
24. Zhang L, Li F, Guo Q, et al. Leucine supplementation: a novel strategy for modulating lipid metabolism and energy homeostasis. Nutrients. 2020;12(5):1299. doi:10.3390/nu12051299
25. Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14(3):133–139. doi:10.1038/nrm3522
26. Zhenyukh O, Civantos E, Ruiz-Ortega M, et al. High concentration of branched-chain amino acids promotes oxidative stress, inflammation and migration of human peripheral blood mononuclear cells via mTORC1 activation. Free Radic Biol Med. 2017;104:165–177. doi:10.1016/j.freeradbiomed.2017.01.009
27. Yoon MS. The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients. 2016;8(7):405. doi:10.3390/nu8070405
28. Li CW, Yu K, Shyh‐Chang N, et al. Pathogenesis of sarcopenia and the relationship with fat mass: descriptive review. J Cachexia Sarcopenia Muscle. 2022;13(2):781–794. doi:10.1002/jcsm.12901
29. Moaddel R, Fabbri E, Khadeer MA, et al. Plasma biomarkers of poor muscle quality in older men and women from the baltimore longitudinal study of aging. J Gerontol A Biol Sci Med Sci. 2016;71(10):1266–1272. doi:10.1093/gerona/glw046
30. Toyoshima K, Nakamura M, Adachi Y, et al. Increased plasma proline concentrations are associated with sarcopenia in the elderly. PLoS One. 2017;12(9):e0185206. doi:10.1371/journal.pone.0185206
31. Dideriksen K, Reitelseder S, Holm L. Influence of amino acids, dietary protein, and physical activity on muscle mass development in humans. Nutrients. 2013;5(3):852–876. doi:10.3390/nu5030852
32. Ninomiya S, Nakamura N, Nakamura H, et al. Low levels of serum tryptophan underlie skeletal muscle atrophy. Nutrients. 2020;12(4):978. doi:10.3390/nu12040978
33. Lee MN, Ha SH, Kim J, et al. Glycolytic flux signals to mTOR through glyceraldehyde-3-phosphate dehydrogenase-mediated regulation of Rheb. Mol Cell Biol. 2009;29(14):3991–4001. doi:10.1128/MCB.00165-09
34. Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev. 2009;89(2):381–410. doi:10.1152/physrev.00016.2008
35. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. doi:10.1038/nrg1879
36. Roux PP, Topisirovic I. Regulation of mRNA translation by signaling pathways. Cold Spring Harb Perspect Biol. 2012;4(11):a012252–a012252. doi:10.1101/cshperspect.a012252
37. Nacarelli T, Azar A, Sell C. Aberrant mTOR activation in senescence and aging: a mitochondrial stress response? Exp Gerontol. 2015;68:66–70. doi:10.1016/j.exger.2014.11.004
38. Connell NJ, Grevendonk L, Fealy CE, et al. NAD+-precursor supplementation with L-tryptophan, nicotinic acid, and nicotinamide does not affect mitochondrial function or skeletal muscle function in physically compromised older adults. J Nutr. 2021;151(10):2917–2931. doi:10.1093/jn/nxab193
39. Sorgdrager FJH, Naudé PJW, Kema IP, et al. Tryptophan metabolism in inflammaging: from biomarker to therapeutic target. Front Immunol. 2019;10:2565. doi:10.3389/fimmu.2019.02565
40. Vécsei L, Szalárdy L, Fülöp F, Toldi J. Kynurenines in the CNS: recent advances and new questions. Nat Rev Drug Discov. 2013;12(1):64–82. doi:10.1038/nrd3793
41. Calvani R, Picca A, Marini F, et al. A distinct pattern of circulating amino acids characterizes older persons with physical frailty and sarcopenia: results from the BIOSPHERE study. Nutrients. 2018;10(11):1691. doi:10.3390/nu10111691
42. Yeung SSY, Zhu ZL, Kwok T, et al. Serum amino acids patterns and 4-year sarcopenia risk in community-dwelling chinese older adults. Gerontology. 2021;68:1–10.
43. Calvani R, Miccheli A, Landi F, et al. Current nutritional recommendations and novel dietary strategies to manage sarcopenia. J Frailty Aging. 2013;2(1):38–53.
44. Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017;25(1):27–42. doi:10.1016/j.cmet.2016.08.009
45. Tibbetts AS, Appling DR. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr. 2010;30:57–81. doi:10.1146/annurev.nutr.012809.104810
46. Herbig K, Chiang E-P, Lee L-R, et al. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J Biol Chem. 2002;277(41):38381–38389. doi:10.1074/jbc.M205000200
47. Yang XM, MacKenzie RE. NAD-dependent methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase is the mammalian homolog of the mitochondrial enzyme encoded by the yeast MIS1 gene. Biochemistry. 1993;32(41):11118–11123. doi:10.1021/bi00092a022
48. Lopes A. Mitochondrial metabolism and DNA methylation: a review of the interaction between two genomes. Clin Epigenetics. 2020;12(1):182. doi:10.1186/s13148-020-00976-5
49. Bao XR, Ong S-E, Goldberger O, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife. 2016;5. doi:10.7554/eLife.10575
50. Nikkanen J, Forsström S, Euro L, et al. Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 2016;23(4):635–648. doi:10.1016/j.cmet.2016.01.019
51. Picca A, Ponziani FR, Calvani R, et al. Gut microbial, inflammatory and metabolic signatures in older people with physical frailty and sarcopenia: results from the BIOSPHERE study. Nutrients. 2019;12(1):65. doi:10.3390/nu12010065
52. Wagenmakers AJ. Muscle amino acid metabolism at rest and during exercise: role in human physiology and metabolism. Exerc Sport Sci Rev. 1998;26:287–314. doi:10.1249/00003677-199800260-00013
53. Kameda M, Teruya T, Yanagida M, et al. Reduced uremic metabolites are prominent feature of sarcopenia, distinct from antioxidative markers for frailty. Aging. 2021;13(17):20915–20934. doi:10.18632/aging.203498
54. Birsoy K, Wang T, Chen W, et al. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162(3):540–551. doi:10.1016/j.cell.2015.07.016
55. Sullivan LB, Gui D, Hosios A, et al. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. 2015;162(3):552–563. doi:10.1016/j.cell.2015.07.017
56. Cardaci S, Zheng L, MacKay G, et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat Cell Biol. 2015;17(10):1317–1326. doi:10.1038/ncb3233
57. Krall AS, Xu S, Graeber TG, et al. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun. 2016;7:11457. doi:10.1038/ncomms11457
58. Krall AS, Mullen PJ, Surjono F, et al. Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab. 2021;33(5):1013–1026 e6. doi:10.1016/j.cmet.2021.02.001
59. Alkan HF, Walter KE, Luengo A, et al. Cytosolic aspartate availability determines cell survival when glutamine is limiting. Cell Metab. 2018;28(5):706–720 e6. doi:10.1016/j.cmet.2018.07.021
60. Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008;15(4):660–666. doi:10.1038/sj.cdd.4402307
61. Jezek P, Plecitá-Hlavatá L, Smolková K, et al. Distinctions and similarities of cell bioenergetics and the role of mitochondria in hypoxia, cancer, and embryonic development. Int J Biochem Cell Biol. 2010;42(5):604–622. doi:10.1016/j.biocel.2009.11.008
62. Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. doi:10.1016/j.cmet.2006.02.002
63. Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405(1):1–9. doi:10.1042/BJ20070389
64. Ng TKS, Kovalik J-P, Ching J, et al. Novel metabolomics markers are associated with pre-clinical decline in hand grip strength in community-dwelling older adults. Mech Ageing Dev. 2021;193:111405. doi:10.1016/j.mad.2020.111405
65. Sensi SL. Alzheimer’s disease, time to turn the tide. Aging. 2018;10(10):2537–2538. doi:10.18632/aging.101581
66. Gancheva S, Jelenik T, Álvarez-Hernández E, et al. Interorgan metabolic crosstalk in human insulin resistance. Physiol Rev. 2018;98(3):1371–1415. doi:10.1152/physrev.00015.2017
67. Magi S, Piccirillo S, Amoroso S. The dual face of glutamate: from a neurotoxin to a potential survival factor-metabolic implications in health and disease. Cell Mol Life Sci. 2019;76(8):1473–1488. doi:10.1007/s00018-018-3002-x
68. Fiermonte G, Palmieri L, Todisco S, et al. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem. 2002;277(22):19289–19294. doi:10.1074/jbc.M201572200
69. Schantz PG, Henriksson J. Enzyme levels of the NADH shuttle systems: measurements in isolated muscle fibres from humans of differing physical activity. Acta Physiol Scand. 1987;129(4):505–515. doi:10.1111/j.1748-1716.1987.tb08090.x
70. Verma M, Lizama BN, Chu CT. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl Neurodegener. 2022;11(1):3. doi:10.1186/s40035-021-00278-7
71. Zhao K, Hong H, Zhao L, et al. Postsynaptic cAMP signalling regulates the antagonistic balance of Drosophila glutamate receptor subtypes. Development. 2020;147(24). doi:10.1242/dev.191874
72. Deschenes MR, Flannery R, Hawbaker A, et al. Adaptive remodeling of the neuromuscular junction with aging. Cells. 2022;11(7):1150. doi:10.3390/cells11071150
73. Miyamoto K, Hirayama A, Sato Y, et al. A metabolomic profile predictive of new osteoporosis or sarcopenia development. Metabolites. 2021;11(5):278. doi:10.3390/metabo11050278
74. Scicchitano BM, Sica G. The beneficial effects of taurine to counteract sarcopenia. Curr Protein Pept Sci. 2018;19(7):673–680. doi:10.2174/1389203718666161122113609
75. Huxtable RJ. Physiological actions of taurine. Physiol Rev. 1992;72(1):101–163. doi:10.1152/physrev.1992.72.1.101
76. Ramos-Mandujano G, Hernandez-Benitez R, Pasantes-Morales H. Multiple mechanisms mediate the taurine-induced proliferation of neural stem/progenitor cells from the subventricular zone of the adult mouse. Stem Cell Res. 2014;12(3):690–702. doi:10.1016/j.scr.2014.02.009
77. Lambert IH, Kristensen DM, Holm JB, et al. Physiological role of taurine–from organism to organelle. Acta Physiol. 2015;213(1):191–212. doi:10.1111/apha.12365
78. Silva LA, Silveira PCL, Ronsani MM, et al. Taurine supplementation decreases oxidative stress in skeletal muscle after eccentric exercise. Cell Biochem Funct. 2011;29(1):43–49. doi:10.1002/cbf.1716
79. Jong CJ, Azuma J, Schaffer S. Mechanism underlying the antioxidant activity of taurine: prevention of mitochondrial oxidant production. Amino Acids. 2012;42(6):2223–2232. doi:10.1007/s00726-011-0962-7
80. Suzuki T, Suzuki T, Wada T, et al. Taurine as a constituent of mitochondrial tRNAs: new insights into the functions of taurine and human mitochondrial diseases. EMBO J. 2002;21(23):6581–6589. doi:10.1093/emboj/cdf656
81. Ricci C, Pastukh V, Leonard J, et al. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am J Physiol Cell Physiol. 2008;294(2):C413–22. doi:10.1152/ajpcell.00362.2007
82. De Luca A, Pierno S, Camerino DC. Taurine: the appeal of a safe amino acid for skeletal muscle disorders. J Transl Med. 2015;13:243. doi:10.1186/s12967-015-0610-1
83. Ter Borg S, de Groot LC, Mijnarends DM, et al. Differences in nutrient intake and biochemical nutrient status between sarcopenic and nonsarcopenic older adults-results from the maastricht sarcopenia study. J Am Med Dir Assoc. 2016;17(5):393–401. doi:10.1016/j.jamda.2015.12.015
84. Holick MF. Vitamin D status: measurement, interpretation, and clinical application. Ann Epidemiol. 2009;19(2):73–78. doi:10.1016/j.annepidem.2007.12.001
85. Latham CM, Brightwell CR, Keeble AR, et al. Vitamin D promotes skeletal muscle regeneration and mitochondrial health. Front Physiol. 2021;12:660498. doi:10.3389/fphys.2021.660498
86. Ricca C, Aillon A, Bergandi L, Alotto D, Castagnoli C, Silvagno F. Vitamin D receptor is necessary for mitochondrial function and cell health. Int J Mol Sci. 2018;19:1672.
87. Dzik K, Skrobot W, Flis DJ, et al. Vitamin D supplementation attenuates oxidative stress in paraspinal skeletal muscles in patients with low back pain. Eur J Appl Physiol. 2018;118(1):143–151. doi:10.1007/s00421-017-3755-1
88. Bhat M, Ismail A. Vitamin D treatment protects against and reverses oxidative stress induced muscle proteolysis. J Steroid Biochem Mol Biol. 2015;152:171–179. doi:10.1016/j.jsbmb.2015.05.012
89. Ashcroft SP, Bass JJ, Kazi AA, et al. The vitamin D receptor regulates mitochondrial function in C2C12 myoblasts. Am J Physiol Cell Physiol. 2020;318(3):C536–C541. doi:10.1152/ajpcell.00568.2019
90. Alkharfy KM, Al-Daghri NM, Ahmed M, et al. Effects of vitamin D treatment on skeletal muscle histology and ultrastructural changes in a rodent model. Molecules. 2012;17(8):9081–9089. doi:10.3390/molecules17089081
91. Glerup H, Mikkelsen K, Poulsen L, et al. Hypovitaminosis D myopathy without biochemical signs of osteomalacic bone involvement. Calcif Tissue Int. 2000;66(6):419–424. doi:10.1007/s002230010085
92. Bauer JM, Verlaan S, Bautmans I, et al. Effects of a vitamin D and leucine-enriched whey protein nutritional supplement on measures of sarcopenia in older adults, the PROVIDE study: a randomized, double-blind, placebo-controlled trial. J Am Med Dir Assoc. 2015;16(9):740–747. doi:10.1016/j.jamda.2015.05.021
93. Fearon KC. Cancer cachexia and fat-muscle physiology. N Engl J Med. 2011;365(6):565–567. doi:10.1056/NEJMcibr1106880
94. Reuter SE, Evans AM. Carnitine and acylcarnitines: pharmacokinetic, pharmacological and clinical aspects. Clin Pharmacokinet. 2012;51(9):553–572. doi:10.1007/BF03261931
95. McGill MR, Li F, Sharpe MR, et al. Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after Acetaminophen overdose in mice and humans. Arch Toxicol. 2014;88(2):391–401. doi:10.1007/s00204-013-1118-1
96. Rizza S, Copetti M, Rossi C, et al. Metabolomics signature improves the prediction of cardiovascular events in elderly subjects. Atherosclerosis. 2014;232(2):260–264. doi:10.1016/j.atherosclerosis.2013.10.029
97. Meng L, Shi H, Wang D-G, et al. Specific metabolites involved in antioxidation and mitochondrial function are correlated with frailty in elderly men. Front Med. 2022;9:816045. doi:10.3389/fmed.2022.816045
98. Tonsgard JH, Getz GS. Effect of Reye’s syndrome serum on isolated chinchilla liver mitochondria. J Clin Invest. 1985;76(2):816–825. doi:10.1172/JCI112039
99. Hirabara SM, Curi R, Maechler P. Saturated fatty acid-induced insulin resistance is associated with mitochondrial dysfunction in skeletal muscle cells. J Cell Physiol. 2010;222(1):187–194. doi:10.1002/jcp.21936
100. Gao X, Lee K, Reid MA, et al. Serine availability influences mitochondrial dynamics and function through lipid metabolism. Cell Rep. 2018;22(13):3507–3520. doi:10.1016/j.celrep.2018.03.017
101. Senyilmaz-Tiebe D, Pfaff DH, Virtue S, et al. Dietary stearic acid regulates mitochondria in vivo in humans. Nat Commun. 2018;9(1):3129. doi:10.1038/s41467-018-05614-6
102. Senyilmaz D, Virtue S, Xu X, et al. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature. 2015;525(7567):124–128. doi:10.1038/nature14601
103. Tian Q, Mitchell BA, Zampino M, et al. Longitudinal associations between blood lysophosphatidylcholines and skeletal muscle mitochondrial function. Geroscience. 2022. doi:10.1007/s11357-022-00548-w
104. da Silva JF, Alves J, Silva-Neto J, et al. Lysophosphatidylcholine induces oxidative stress in human endothelial cells via NOX5 activation - implications in atherosclerosis. Clin Sci. 2021;135(15):1845–1858. doi:10.1042/CS20210468
105. Hung ND, Sok DE, Kim MR. Prevention of 1-palmitoyl lysophosphatidylcholine-induced inflammation by polyunsaturated acyl lysophosphatidylcholine. Inflamm Res. 2012;61(5):473–483. doi:10.1007/s00011-012-0434-x
106. Klingler C, Zhao X, Adhikary T, et al. Lysophosphatidylcholines activate PPARdelta and protect human skeletal muscle cells from lipotoxicity. Biochim Biophys Acta. 2016;1861(12 Pt A):1980–1992. doi:10.1016/j.bbalip.2016.09.020
107. Schlame M, Greenberg ML. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(1):3–7. doi:10.1016/j.bbalip.2016.08.010
108. Falabella M, Vernon HJ, Hanna MG, et al. Cardiolipin, mitochondria, and neurological disease. Trends Endocrinol Metab. 2021;32(4):224–237. doi:10.1016/j.tem.2021.01.006
109. Paradies G, Paradies V, De Benedictis V, et al. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim Biophys Acta. 2014;1837(4):408–417. doi:10.1016/j.bbabio.2013.10.006
110. Stepanyants N, Macdonald PJ, Francy CA, et al. Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol Biol Cell. 2015;26(17):3104–3116. doi:10.1091/mbc.E15-06-0330
111. Ban T, Ishihara T, Kohno H, et al. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol. 2017;19(7):856–863. doi:10.1038/ncb3560
112. Chu CT, Bayir H, Kagan VE. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy. 2014;10(2):376–378. doi:10.4161/auto.27191
113. Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15(10):1197–1205. doi:10.1038/ncb2837
114. Bucci L, Yani SL, Fabbri C, et al. Circulating levels of adipokines and IGF-1 are associated with skeletal muscle strength of young and old healthy subjects. Biogerontology. 2013;14(3):261–272. doi:10.1007/s10522-013-9428-5
115. Lu G, Sun H, She P, et al. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J Clin Invest. 2009;119(6):1678–1687. doi:10.1172/JCI38151
116. Bifari F, Nisoli E. Branched-chain amino acids differently modulate catabolic and anabolic states in mammals: a pharmacological point of view. Br J Pharmacol. 2017;174(11):1366–1377. doi:10.1111/bph.13624
117. Iwabu M, Yamauchi T, Okada-Iwabu M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464(7293):1313–1319. doi:10.1038/nature08991
118. Kalinkovich A, Livshits G. Sarcopenic obesity or obese sarcopenia: a cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res Rev. 2017;35:200–221. doi:10.1016/j.arr.2016.09.008
119. Alizadeh Pahlavani H. Exercise therapy for people with sarcopenic obesity: myokines and adipokines as effective actors. Front Endocrinol (Lausanne). 2022;13:811751. doi:10.3389/fendo.2022.811751
120. Peng J, Yin L, Wang X. Central and peripheral leptin resistance in obesity and improvements of exercise. Horm Behav. 2021;133:105006. doi:10.1016/j.yhbeh.2021.105006
121. Li L, Pan R, Li R, et al. Mitochondrial biogenesis and peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation by physical activity: intact adipocytokine signaling is required. Diabetes. 2011;60(1):157–167. doi:10.2337/db10-0331
122. Kim H, Kim M, Kojima N, et al. Exercise and nutritional supplementation on community-dwelling elderly Japanese women with sarcopenic obesity: a randomized controlled trial. J Am Med Dir Assoc. 2016;17(11):1011–1019. doi:10.1016/j.jamda.2016.06.016
123. Dieli-Conwright CM, Courneya KS, Demark-Wahnefried W, et al. Effects of aerobic and resistance exercise on metabolic syndrome, sarcopenic obesity, and circulating biomarkers in overweight or obese survivors of breast cancer: a randomized controlled trial. J Clin Oncol. 2018;36(9):875–883. doi:10.1200/JCO.2017.75.7526
124. The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi:10.1038/nature11234
125. Guinane CM, Cotter PD. Role of the gut microbiota in health and chronic gastrointestinal disease: understanding a hidden metabolic organ. Therap Adv Gastroenterol. 2013;6(4):295–308. doi:10.1177/1756283X13482996
126. Picca A, Calvani R, Cesari M, et al. Biomarkers of physical frailty and sarcopenia: coming up to the place? Int J Mol Sci. 2020;21(16):5635. doi:10.3390/ijms21165635
127. Murphy EA, Velazquez KT, Herbert KM. Influence of high-fat diet on gut microbiota: a driving force for chronic disease risk. Curr Opin Clin Nutr Metab Care. 2015;18(5):515–520. doi:10.1097/MCO.0000000000000209
128. van Krimpen SJ, Jansen FAC, Ottenheim VL, et al. The effects of pro-, pre-, and synbiotics on muscle wasting, a systematic review-gut permeability as potential treatment target. Nutrients. 2021;13(4):1115. doi:10.3390/nu13041115
129. Ticinesi A, Nouvenne A, Cerundolo N, et al. Gut microbiota, muscle mass and function in aging: a focus on physical frailty and sarcopenia. Nutrients. 2019;11(7):1633. doi:10.3390/nu11071633
130. Vrieze A, Van Nood E, Holleman F, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143(4):913–6 e7. doi:10.1053/j.gastro.2012.06.031
131. Munukka E, Rintala A, Toivonen R, et al. Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. ISME J. 2017;11(7):1667–1679. doi:10.1038/ismej.2017.24
132. Chen LH, Chang -S-S, Chang H-Y, et al. Probiotic supplementation attenuates age-related sarcopenia via the gut-muscle axis in SAMP8 mice. J Cachexia Sarcopenia Muscle. 2022;13(1):515–531. doi:10.1002/jcsm.12849
133. Sridharan GV, Choi K, Klemashevich C, et al. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat Commun. 2014;5:5492. doi:10.1038/ncomms6492
134. Zhang N, Jin M, Wang K, et al. Functional oligosaccharide fermentation in the gut: improving intestinal health and its determinant factors-a review. Carbohydr Polym. 2022;284:119043. doi:10.1016/j.carbpol.2021.119043
135. Verbeke KA, Boobis AR, Chiodini A, et al. Towards microbial fermentation metabolites as markers for health benefits of prebiotics. Nutr Res Rev. 2015;28(1):42–66. doi:10.1017/S0954422415000037
136. Cook SI, Sellin JH. Review article: short chain fatty acids in health and disease. Aliment Pharmacol Ther. 1998;12(6):499–507. doi:10.1046/j.1365-2036.1998.00337.x
137. Li M, van Esch BC, Wagenaar GTM, et al. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur J Pharmacol. 2018;831:52–59. doi:10.1016/j.ejphar.2018.05.003
138. Kim S, Kim J-H, Park BO, et al. Perspectives on the therapeutic potential of short-chain fatty acid receptors. BMB Rep. 2014;47(3):173–178. doi:10.5483/BMBRep.2014.47.3.272
139. McLoughlin RF, Berthon BS, Jensen ME, et al. Short-chain fatty acids, prebiotics, synbiotics, and systemic inflammation: a systematic review and meta-analysis. Am J Clin Nutr. 2017;106(3):930–945. doi:10.3945/ajcn.117.156265
140. den Besten G, Bleeker A, Gerding A, et al. Short-chain fatty acids protect against high-fat diet-induced obesity via a PPARgamma-dependent switch from lipogenesis to fat oxidation. Diabetes. 2015;64(7):2398–2408. doi:10.2337/db14-1213
141. Gao Z, Yin J, Zhang J, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58(7):1509–1517. doi:10.2337/db08-1637
142. Maruta H, Yoshimura Y, Araki A, et al. Activation of AMP-activated protein kinase and stimulation of energy metabolism by acetic acid in L6 myotube cells. PLoS One. 2016;11(6):e0158055. doi:10.1371/journal.pone.0158055
143. Wang H, Listrat A, Meunier B, et al. Apoptosis in capillary endothelial cells in ageing skeletal muscle. Aging Cell. 2014;13(2):254–262. doi:10.1111/acel.12169
144. Chen W, Chen Y, Liu Y, et al. Autophagy in muscle regeneration: potential therapies for myopathies. J Cachexia Sarcopenia Muscle. 2022;13:1673–1685. doi:10.1002/jcsm.13000
145. Arsic N, Zacchigna S, Zentilin L, et al. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol Ther. 2004;10(5):844–854. doi:10.1016/j.ymthe.2004.08.007
146. Das A, Huang GX, Bonkowski MS, et al. Impairment of an endothelial NAD(+)-H2S signaling network is a reversible cause of vascular aging. Cell. 2018;173(1):74–89 e20. doi:10.1016/j.cell.2018.02.008
147. Jeong IH, Bae W-Y, Choi J-S, et al. Ischemia induces autophagy of endothelial cells and stimulates angiogenic effects in a hindlimb ischemia mouse model. Cell Death Dis. 2020;11(8):624. doi:10.1038/s41419-020-02849-4
148. Nederveen JP, Joanisse S, Snijders T, et al. Skeletal muscle satellite cells are located at a closer proximity to capillaries in healthy young compared with older men. J Cachexia Sarcopenia Muscle. 2016;7(5):547–554. doi:10.1002/jcsm.12105
149. Christov C, Chrétien F, Abou-Khalil R, et al. Muscle satellite cells and endothelial cells: close neighbors and privileged partners. Mol Biol Cell. 2007;18(4):1397–1409. doi:10.1091/mbc.e06-08-0693
150. Silva BSA, Uzeloto JS, Lira FS, et al. Exercise as a peripheral circadian clock resynchronizer in vascular and skeletal muscle aging. Int J Environ Res Public Health. 2021;18(24):12949. doi:10.3390/ijerph182412949
151. Zhao MM, Xu M-J, Cai Y, et al. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011;79(10):1071–1079. doi:10.1038/ki.2011.18
152. Wu W, Xu H, Wang Z, et al. PINK1-parkin-mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS One. 2015;10(7):e0132499. doi:10.1371/journal.pone.0132499
153. Settembre C, Di malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi:10.1126/science.1204592
154. Duncan ER, Crossey PA, Walker S, et al. Effect of endothelium-specific insulin resistance on endothelial function in vivo. Diabetes. 2008;57(12):3307–3314. doi:10.2337/db07-1111
155. Kubota T, Kubota N, Kumagai H, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13(3):294–307. doi:10.1016/j.cmet.2011.01.018
156. Hou YC, Lu C-L, Zheng C-M, et al. The role of vitamin D in modulating mesenchymal stem cells and endothelial progenitor cells for vascular calcification. Int J Mol Sci. 2020;21(7):2466. doi:10.3390/ijms21072466
157. Zhang J, Shan Y, Li Y, et al. Palmitate impairs angiogenesis via suppression of cathepsin activity. Mol Med Rep. 2017;15(6):3644–3650. doi:10.3892/mmr.2017.6463
158. Yuan L, Mao Y, Luo W, et al. Palmitic acid dysregulates the Hippo-YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS-STING-IRF3 signaling mechanism. J Biol Chem. 2017;292(36):15002–15015. doi:10.1074/jbc.M117.804005
159. Liu T, Wang X, Guo F, et al. Lysophosphatidylcholine induces apoptosis and inflammatory damage in brain microvascular endothelial cells via GPR4-mediated NLRP3 inflammasome activation. Toxicol In Vitro. 2021;77:105227. doi:10.1016/j.tiv.2021.105227
160. Ouchi N, Kobayashi H, Kihara S, et al. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. 2004;279(2):1304–1309. doi:10.1074/jbc.M310389200
161. Jiang T, Liu T, Deng X, et al. Adiponectin ameliorates lung ischemia-reperfusion injury through SIRT1-PINK1 signaling-mediated mitophagy in type 2 diabetic rats. Respir Res. 2021;22(1):258. doi:10.1186/s12931-021-01855-0
162. Li M, van Esch BC, Henricks PAJ, et al. The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor alpha-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front Pharmacol. 2018;9:533. doi:10.3389/fphar.2018.00533
163. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121–135. doi:10.1038/nrm.2017.95
164. Zhang B, Pan C, Feng C, et al. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Rep. 2022;27(1):45–52. doi:10.1080/13510002.2022.2046423
165. Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9(4):298–310. doi:10.1016/j.stem.2011.09.010
166. Smith C, Woessner MN, Sim M, et al. Sarcopenia definition: does it really matter? Implications for resistance training. Ageing Res Rev. 2022;78:101617. doi:10.1016/j.arr.2022.101617
167. Bao W, Sun Y, Zhang T, et al. Exercise programs for muscle mass, muscle strength and physical performance in older adults with sarcopenia: a systematic review and meta-analysis. Aging Dis. 2020;11(4):863–873. doi:10.14336/AD.2019.1012
168. Harper C, Gopalan V, Goh J, et al. Exercise rescues mitochondrial coupling in aged skeletal muscle: a comparison of different modalities in preventing sarcopenia. J Transl Med. 2021;19(1):71. doi:10.1186/s12967-021-02737-1
169. Denison HJ, Cooper C, Sayer AA, et al. Prevention and optimal management of sarcopenia: a review of combined exercise and nutrition interventions to improve muscle outcomes in older people. Clin Interv Aging. 2015;10:859–869. doi:10.2147/CIA.S55842
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.