Back to Journals » OncoTargets and Therapy » Volume 11
The effect of additional chemotherapy on high-risk prostate cancer: a systematic review and meta-analysis
Authors Chen J, Zhang X, Sun G, Zhao J ![]() , Liu J, Zhao P, Dai J, Shen P, Zeng H
, Liu J, Zhao P, Dai J, Shen P, Zeng H ![]()
Received 12 September 2018
Accepted for publication 16 November 2018
Published 13 December 2018 Volume 2018:11 Pages 9061—9070
DOI https://doi.org/10.2147/OTT.S187239
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Junru Chen,* Xingming Zhang,* Guangxi Sun, Jinge Zhao, Jiandong Liu, Peng Zhao, Jindong Dai, Pengfei Shen, Hao Zeng
Department of Urology, Institute of Urology, West China Hospital, Sichuan University, Chengdu 610041, China
*These authors contributed equally to this work
Objective: The role of additional chemotherapy in the treatment of high-risk prostate cancer (PCa) remains a controversy. This meta-analysis aimed to investigate the effect of additional chemotherapy on high-risk PCa.
Methods: Randomized controlled trials (RCTs) about additional chemotherapy for high-risk PCa were searched in PubMed, MEDLINE, EMBASE, and the Cochrane Central Register of Controlled Trials. We extracted HRs of overall survival (OS) and progression-free survival (PFS) for each trial and performed the meta-analysis using Review Manager 5.3.
Results: Eight RCTs involving 4,007 patients were included. Data from four trials, which could collect OS, showed that additional chemotherapy could not significantly improve the OS in patients with high-risk PCa (HR: 0.93; 95% CI: 0.79–1.09; P=0.37). However, the pooled analysis suggested significantly longer PFS in high-risk PCa patients treated with additional chemotherapy (HR: 0.81; 95% CI: 0.74–0.90; P<0.0001). The meta-analysis showed additional chemotherapy to androgen-deprivation therapy improved PFS (HR: 0.82; 95% CI: 0.74–0.91; P=0.0002). Greater improvement in PFS was found in high-risk PCa patients treated with additional docetaxel-based chemotherapy (HR: 0.73; 95% CI: 0.64–0.83; P<0.00001). No prolonged PFS was observed in high-risk PCa patients with non-docetaxel-based chemotherapy (HR: 0.97; 95% CI: 0.83–1.14; P=0.74).
Conclusion: Additional chemotherapy, especially docetaxel-based chemotherapy, could significantly improve the PFS in high-risk PCa patients. More evidence about the effect of additional chemotherapy on OS is needed. Further investigations in PCa should also focus on the suitable population for chemotherapy as well as optimal use of chemotherapy.
Keywords: chemotherapy, high-risk, prostate cancer, systematic review, meta-analysis
Introduction
Prostate cancer (PCa) is the most common malignancy in men and the second leading cause of cancer death in male patients in the USA.1 Although most patients diagnosed with prostate cancer have favorable disease characteristics and can be cured by radical surgery or radiation, those with high-risk features have a high biochemical relapse rate and an increased mortality up to about 30%.2,3 Boorjian et al evaluated the records of 7,591 patients in Mayo Clinic and found that compared with patients with low-risk PCa, those with high-risk PCa had significantly higher risk of recurrence and cancer-specific mortality.4
Although high-risk PCa has been variously defined, the D’Amico classification is widely used at present.5 Current treatments for high-risk PCa include radical prostatectomy (RP), radiation therapy (RT), external-beam RT, brachytherapy, androgen-deprivation therapy (ADT), and chemotherapy, alone or in combination.6 Several studies indicated that RT plus adjuvant ADT could decrease the risk of progression and improve survival,7–9 but adjuvant ADT alone seemed underpowered to prolong survival in patients with high-risk PCa who received RP.10,11 In the end, high-risk patients still progressed after receiving radical therapies and adjuvant ADT. So, it is reasonable to apply more aggressive therapies to target the systemic components of the disease. Chemotherapy of docetaxel has been proved to improve clinical outcomes of patients with metastatic castrate-resistant prostate cancer (mCRPC).12,13 Furthermore, a recent systematic review and meta-analysis demonstrated that addition of docetaxel to standard of care improved survival in patients with metastatic hormone-sensitive PCa.14 Therefore, the question arose whether patients with high-risk PCa could benefit from additional chemotherapy. Results from the GETUG 12 trial showed that ADT plus docetaxel and estramustine could significantly improve relapse-free survival in patients with high-risk localized PCa.15 However, the RTOG 9902 trial demonstrated no significant differences in overall survival (OS) or progression-free survival (PFS) with additional chemotherapy to long-term androgen suppression plus RT.16 Therefore, more evidence on the validity of additional chemotherapy in high-risk PCa is needed.
The aim of this study was to conduct a systematic review and meta-analysis of relevant randomized controlled trials (RCTs) to evaluate the effects of additional chemotherapy in high-risk PCa and guide clinical practice.
Methods
Protocol and registration
We developed a protocol defining search strategy and data synthesis of the review, in accordance with the PRISMA statement. The review was registered on the International Prospective Register of Systematic Reviews (PROSPERO, registration number 42017064354). The protocol is available on https://www.crd.york.ac.uk/PROSPERO/display_record.asp?ID=CRD42017064354.
Search strategy
The search strategy is available in the protocol. EMBASE (1974 to 2018.5) and MEDLINE (1966 to 2018.5) were searched through OvidSP. Further searches were undertaken on the Cochrane Central Register of Controlled Trials (1948 to 2018.5) and PubMed (1950 to 2018.5). The last search was conducted on May 10, 2018. The references of included trials not identified by our electronic searches were also searched for extra possible studies. There were no limitations to language and publication status.
Inclusion and exclusion criteria
Inclusion criteria: 1) RCTs investigating the effects of additional chemotherapy in high-risk PCa (according to D’Amico criteria) patients;5 2) no restrictions in published languages.
Exclusion criteria: 1) patients with evidence of metastases on bone scan, CT scan, or MRI; 2) patients who received chemotherapy previously; 3) RCTs reporting only laboratory findings or without survival outcomes.
Data extraction
Two researchers (JRC and XMZ) assessed the titles and abstracts of the searched studies, respectively. Any disagreements were reconciled by a third researcher (PFS). Full texts of potential eligible studies were screened to identify the final included studies. For each included study, the information collected was as follows: study design, study duration, selection criteria, sample size, characteristics of participants (median age, TNM stage, Gleason score, and median prostate-specific antigen [PSA] concentration), follow-up time, controlled group treatments, chemotherapy (components, dosage, scheduling), outcomes, etc.
Outcomes
The primary outcome, OS, was defined as the time from randomization until death from any cause. The secondary outcomes were PFS and adverse events (AEs). PFS was defined as the time from randomization to biochemical failure, onset of metastases on imaging, proven local relapse, or death from any cause.
Quality assessment
Two reviewers (JRC and XMZ) used RevMan 5.3 software to evaluate the risk of bias of the included studies according to the Cochrane Handbook version 5.1.0 and cross-checked, respectively. The items assessed were as follows: 1) random sequence generation; 2) allocation concealment; 3) blinding of participants and personnel; 4) blinding of outcome assessment; 5) incomplete outcome data; 6) selective reporting; and 7) other sources of bias. Disagreements were solved by discussion with a third reviewer (PFS).
Data synthesis
The meta-analysis was conducted using RevMan 5.3 software. For each trial, we extracted HRs and the corresponding 95% CI of the effects of chemotherapy on OS and PFS directly from the trial reports. If not reported, they would be estimated from Kaplan–Meier curves. If insufficient data were available, supplementary data were sought directly from the trial investigators. We used a fixed-effect model (Mantel–Haenzsel) a priori. If significant heterogeneity was found, we assessed the results using a random-effect model. I2 statistic and chi-squared test were used to assess the heterogeneity among trials and I2 >50% or χ2 p-value <0.1 was considered as significant heterogeneity. We also planned subgroup analyses according to 1) whether chemotherapy was combined with ADT; 2) the drugs used for chemotherapy. Publication bias was assessed by Egger’s test and funnel plot created using RevMan 5.3 software.
Results
Characteristics of the included trials
A flowchart of the selection process of eligible trials is shown in Figure 1. In total, 4,007 patients in eight RCTs were included in the meta-analysis.15–22 Of the eight included RCTs, five15–19 were full articles and the other three20–22 were abstracts. The main characteristics of the included trials are summarized in Table 1.
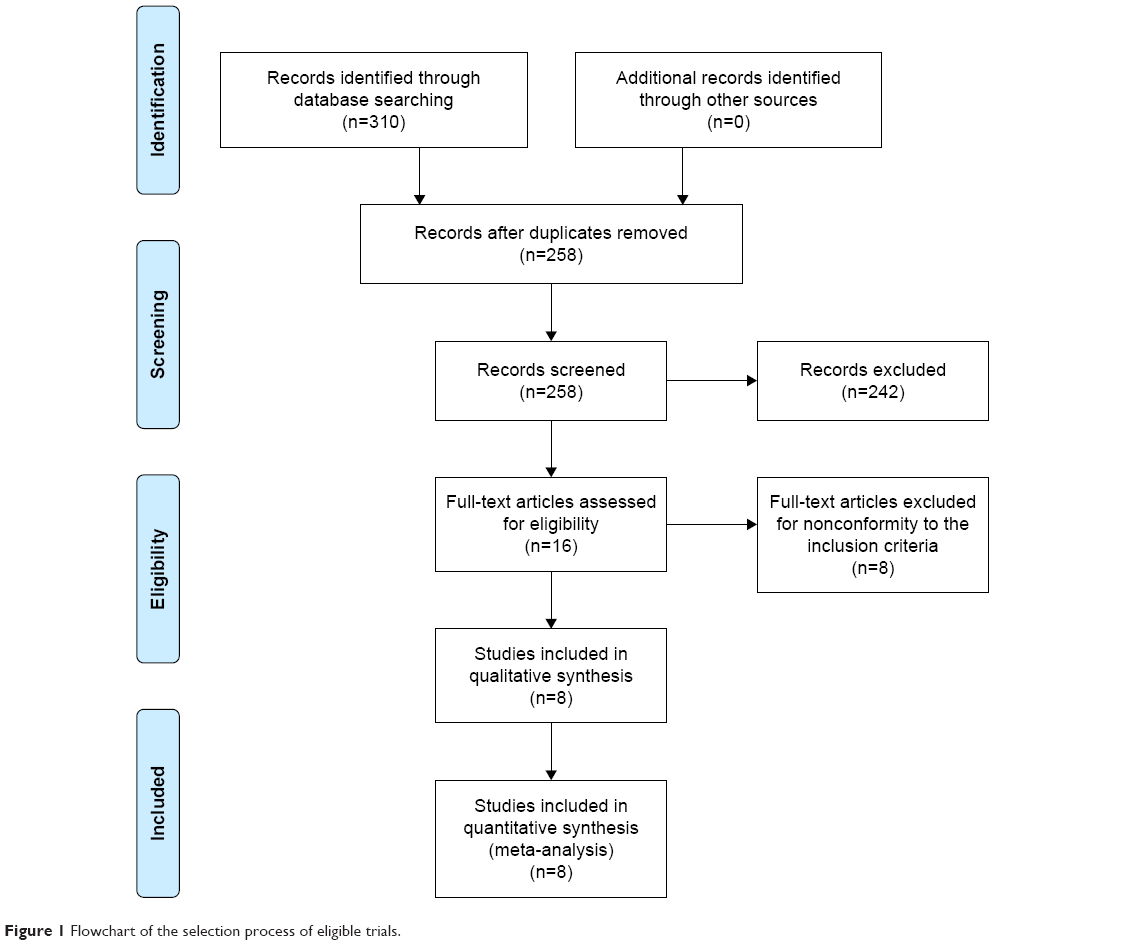 | Figure 1 Flowchart of the selection process of eligible trials. |
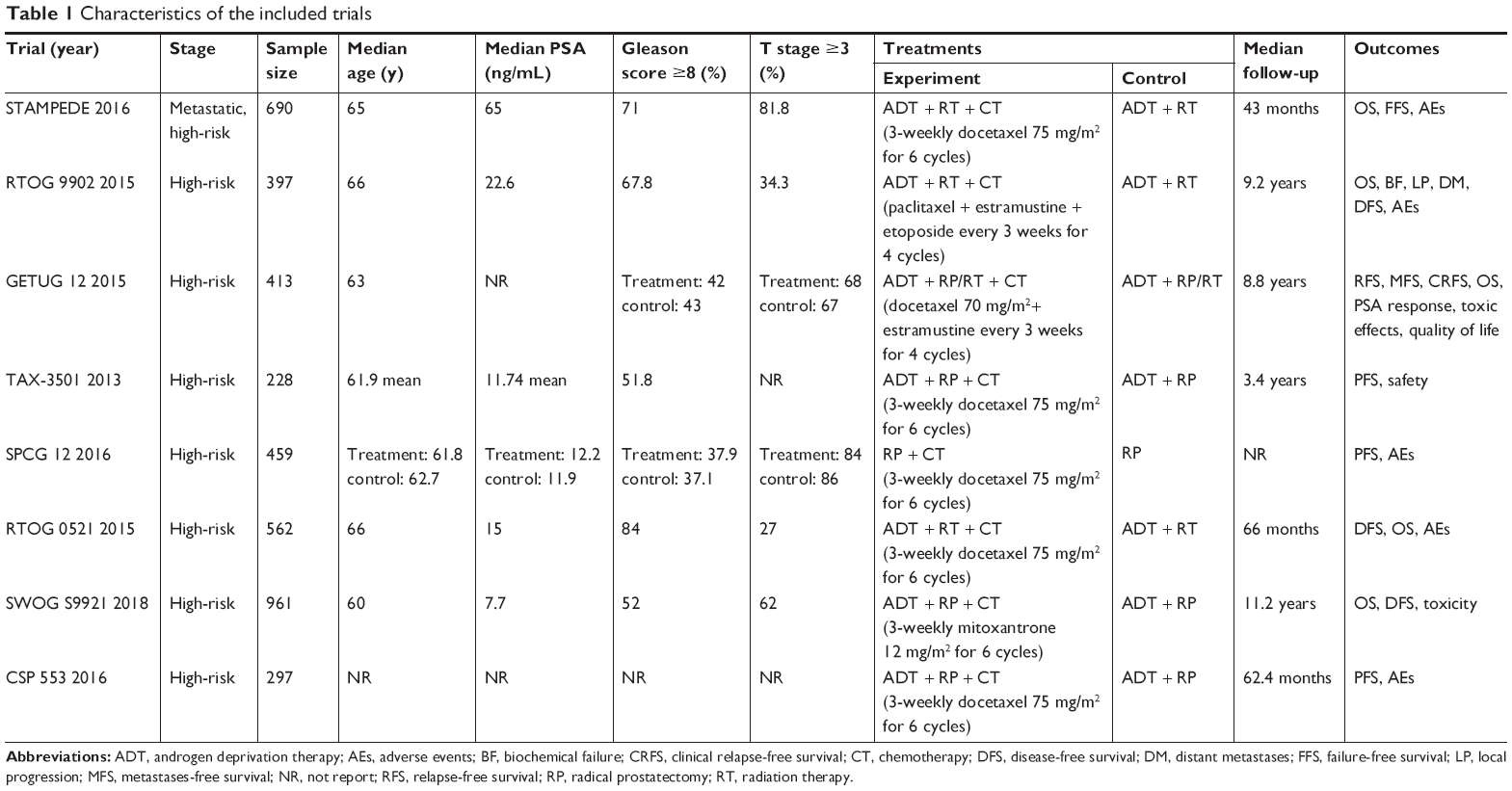 | Table 1 Characteristics of the included trials |
The definitions of “high-risk” varied in each trial, but they were basically consistent with D’Amico criteria.5 In these trials, patients were assigned either to an experimental arm and received additional chemotherapy or to a control arm. As one RCT (TAX-3501) investigated immediate or deferred hormonal therapy after RP with or without chemotherapy, we divided it into two parts as two separate trials for analysis.18 Patients received chemotherapy consisting of docetaxel in seven trials,15,17,18,20–22 paclitaxel + estramustine + etoposide (TEE) in RTOG 9902 trial16 and mitoxantrone in SWOG S9921 trial.19 Three trials compared RT plus ADT with or without chemotherapy in patients with high-risk PCa.16,17,20 Four trials investigated the effects of additional chemotherapy to ADT plus RP.18,19,21 One trial investigated the efficacy of additional chemotherapy to RP only.22 In GETUG 12 trial, RT or RP combined ADT were admitted according to the status of lymph nodes, and the effects of additional chemotherapy were also investigated.15 The TAX 3501 trial was terminated ~21 months after its activation due, in part, to enrollment challenges.18 The STAMPEDE trial is still recruiting.17
Assessment of risk of bias
The risk of bias of the included trials is summarized in Figure 2. Due to inadequate information, seven trials had an unclear risk of bias for allocation concealment.16,18–22 One trial (GETUG 12) had a high risk of bias for allocation concealment because neither patients nor investigators were masked to treatment allocation.15 Eight trials were considered at high risk of bias for blinding because of no masking.15–20,22 A funnel plot was used to assess the risk of publication bias (Figure S1). An Egger’s test P-value of 0.968 indicated that there was no significant publication bias.
 | Figure 2 The risk of bias of the included trials. |
The effect of additional chemotherapy on OS
Four trials reported HRs of the effect of additional chemotherapy on OS,16,17,19,20 of which three compared RT + ADT plus additional chemotherapy with RT + ADT,16,17,20 and the other trial investigated RP + ADT with or without chemotherapy.19 The pooled analysis showed that additional chemotherapy could not significantly improve the OS (HR: 0.93; 95% CI: 0.79–1.09; P=0.37) (Figure 3). There was no statistically significant heterogeneity among the four trials (I2=31%, P=0.23).
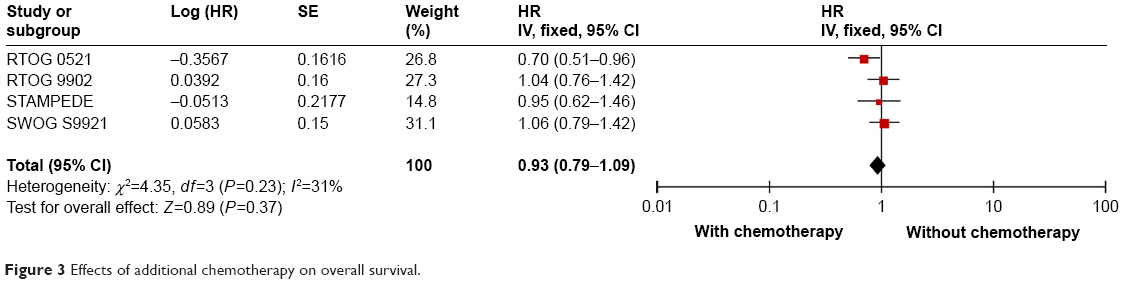 | Figure 3 Effects of additional chemotherapy on overall survival. |
The effect of additional chemotherapy on PFS
The data of PFS are available in all the included trials.15–22 The pooled analysis suggested a significantly longer PFS in high-risk PCa patients treated with additional chemotherapy (HR: 0.81; 95% CI: 0.74–0.90; P<0.0001) (Figure 4). No statistically significant heterogeneity was found among the nine trials (I2=32%, P=0.16). Eight trials reported the data of PFS and the results showed that additional chemotherapy to radical treatment plus ADT could significantly improve the PFS in high-risk PCa patients (HR: 0.82; 95% CI: 0.74–0.91; P=0.0002) (Figure 4). Two trials used non-docetaxel-based chemotherapy, one used TEE16 and the other used mitoxantrone.19 The meta-analysis showed no significantly prolonged PFS in high-risk PCa patients with non-docetaxel-based chemotherapy (HR: 0.97; 95% CI: 0.83–1.14; P=0.74) (Figure 5). Seven trials reported the PFS data in patients treated with docetaxel-based chemotherapy.15,17,18,20–22 The pooled results indicated that significant improvement in PFS was found in patients who had received docetaxel-based chemotherapy (HR: 0.73; 95% CI: 0.64–0.83; P<0.00001) (Figure 5).
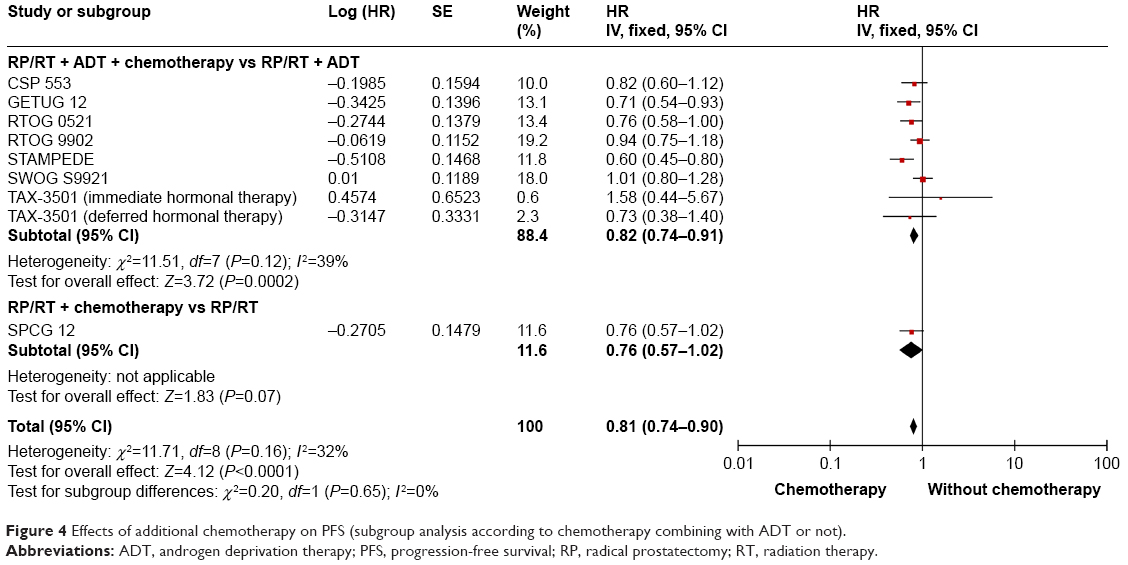 | Figure 4 Effects of additional chemotherapy on PFS (subgroup analysis according to chemotherapy combining with ADT or not). |
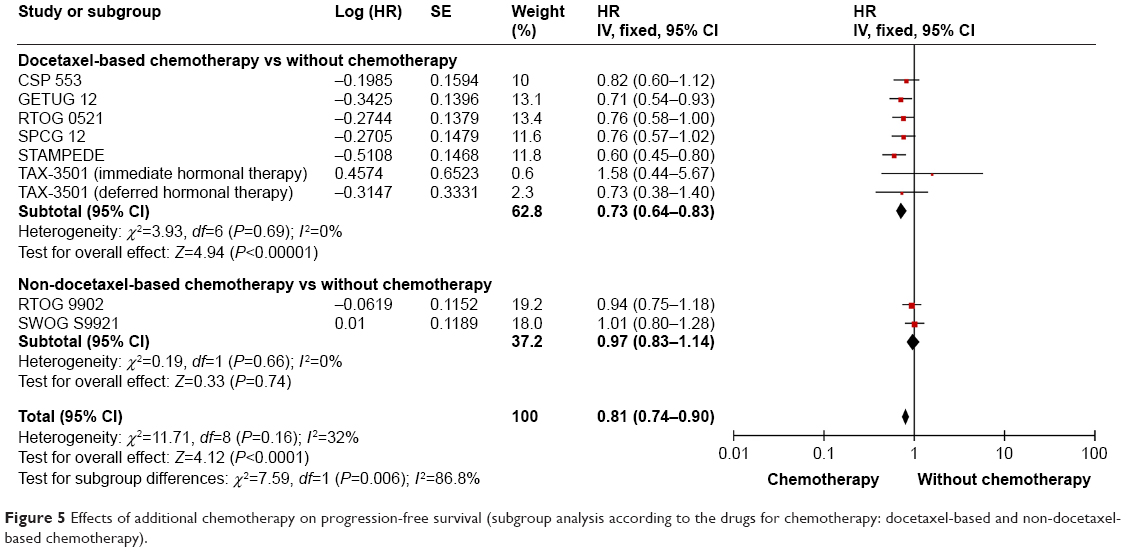 | Figure 5 Effects of additional chemotherapy on progression-free survival (subgroup analysis according to the drugs for chemotherapy: docetaxel-based and non-docetaxel-based chemotherapy). |
Outcomes of AEs
The most common AEs were neutropenia and febrile neutropenia. In the GETUG 12 trial, no toxicity-related death and grade 3–4 toxicity during the first 3 months were recorded, and the negative impact of chemotherapy on quality of life at 3 months disappeared at 1 year.23 In the RTOG 9902,16 the TAX-3501,18 and the SWOG S9921 trials,19 patients with chemotherapy had a higher incidence of ≥ grade 3 AEs than those without chemotherapy. As reported in the STAMPEDE17 and SPCG 12 trials,22 the most frequent ≥ grade 3 AE was febrile neutropenia (15% and 17.9%, respectively). Of all patients who received chemotherapy in the RTOG 0521, 65% suffered from ≥ grade 3 AEs definitely or probably related to treatment.20 In the CSP 553 trial, the most common ≥ grade 3 AEs related or possibly related to chemotherapy included neutropenia in 40%, hyperglycemia in 18%, and fatigue in 5%, with febrile neutropenia in 1.4%.21 Two patients died of infection/febrile neutropenia and one of acute myelogenous leukemia in the chemotherapy arm in the RTOG 9902 trial.16
Discussion
As is known, the development of PCa is dependent on androgen signaling and can be suppressed by ADT at the early stages.24 Despite the low testosterone concentration controlled by ADT, PCa still progresses. Two potential mechanisms have been proposed to explain this problem, which indicates that the resistance to ADT by PCa cells mainly may be the result of results from abnormal androgen receptor (AR) signaling and proliferation of androgen-independent cells.25,26 Several studies have reported that taxanes were able to impair the AR activities and interfere with the resistance mechanisms.27–29 Based on these preclinical data, RCTs investigating the effect of chemotherapy on PCa have emerged recently and verified its efficacy in mCRPC.12,13 However, the use of chemotherapy in non-metastatic PCa patients remains a matter of debate.30,31 Non-metastatic PCa patients represent a large, heterogeneous population, whose biological features and disease stages change with cancer. Currently, non-metastatic PCa patients with high-risk features are more likely to develop recurrences and metastases even with ADT. Given the success of chemotherapy in CRPC population, patients with high-risk PCa may also benefit from additional chemotherapy.
To our knowledge, this was the first systematic review and meta-analysis to investigate the effect of additional chemotherapy on patients with high-risk PCa. The meta-analysis showed that additional chemotherapy in high-risk PCa patients had no impact on the OS but could significantly improve PFS.
The pooled HR of 0.93 indicated that additional chemotherapy might have a potential effect on improving the OS, but the result was not statistically significant. Several possible reasons may be responsible for the absence of significant differences in OS. First, the follow-ups in several trials are relatively short; the TAX 3501 trial was terminated early due to slow recruitment and the STAMPEDE trial is still recruiting patients. Thus, the available data for OS are not enough and less mature. Second, OS is affected by crossover treatments during the long follow-up. Patients may receive other active treatments, such as abiraterone and/or enzalutamide beyond progression. Unfortunately, the details of the post-progression treatments were not available in the included trials. Third, the chemotherapy regimens and number of cycles administered may have an effect. Four cycles of TEE were used in the RTOG 9902 trial16 and six cycles of docetaxel were used in the other three trials.17,19,20 Recently, a post hoc analysis of the mainsail study by de Morree et al has indicated that the total number of docetaxel cycles delivered was an independent and important contributor to the OS benefit provided by docetaxel chemotherapy and the median OS for patients receiving >7 cycles was significantly longer than for those receiving 5–7 cycles.32 Therefore, the difference in OS may emerge after increasing the cycles of docetaxel. Furthermore, the definition of “high-risk PCa” is not consistent in the included trials, and the differences in the baseline characteristics of patients, local treatments, and the use of chemotherapy can cause a clinical heterogeneity, which may have an influence on the result.
The pooled analysis demonstrated that significant improvement in PFS was associated with additional chemotherapy in high-risk PCa patients. According to the results of subgroup analyses, ADT combined with additional chemotherapy was superior to ADT alone in high-risk PCa patients who received radical treatments, which may be the result of the synergistic effect of ADT and chemotherapy on AR signaling. Docetaxel-based chemotherapy could significantly improve PFS; however, non-docetaxel-based chemotherapy is not effective in delaying progression. Two reasons may be responsible for this result. First, the TEE regimen used in the RTOG 9902 trial would currently be regarded as obsolete because the systemic therapy options were limited at the time of study design.16 Second, mitoxanrone used in the SWOG S9921 may not be as effective as docetaxel. The TAX 327 trial has shown that in mCRPC, 3-weekly docetaxel led to superior survival and improved response in terms of pain, serum PSA level, and quality of life, when compared with mitoxantrone.33 The prolonged PFS in the current study indicated that additional chemotherapy could postpone the progression of high-risk PCa into metastatic diseases or CRPC and then potentially improve the OS. According to the SWOG S9346 trial,34 longer median OS was found in patients with metastatic hormone-sensitive prostate cancer who did not have PSA-progression within the first 7 months compared with those with PSA-progression (44 vs 10 months). The results of the TAX 327 trial also indicated that among the mCRPC patients receiving 3-weekly docetaxel, those with PSA response had a significantly longer OS.35 Thus, with the emergence of new trials and data, the improvement in PFS by additional chemotherapy may eventually translate into longer OS.
Not all PCa patients are suitable for additional chemotherapy. To refine individualized treatment strategies, it is necessary to determine the population that can receive maximal benefit from chemotherapy and those not suitable for additional chemotherapy. The subgroup analyses in the GETUG 12 and the SPCG 12 trials suggested patients with a Gleason score of <8 could gain more benefit from chemotherapy than those with a higher Gleason score, which might result from a differential effect of taxanes on AR trafficking according to Gleason score.15,22 Another two Phase III trials also reported that patients with a low Gleason score might derive greater benefit from chemotherapy.36,37 Lin et al found more benefits of chemotherapy in PFS for patients with tumor stage ≥ T3 b.21 Yu et al reckoned that PCa patients with a short PSA doubling time of <3 months would benefit from early introduction of docetaxel.30 In the latest advanced prostate cancer consensus conference (APCCC 2017), the panel voted on factors which they consider would render a man “unfit” for docetaxel.38 Of all, 96% voted for severe hepatic impairment, 82% for neuropathy grade ≥2 and 81% for platelets <50×109/L and/or neutrophils <1.0×109/L.
In total, additional chemotherapy was tolerated with life-threating AEs and severe toxicity occurring rarely. Neutropenia and febrile neutropenia were the most common AEs and in the RTOG 9902 trial,16 two patients were reported to have died of sepsis in the context of neutropenia. Therefore, intensive monitoring should be arranged for patients who are potentially immunocompromised to chemotherapy.39 Although the overall numbers of AEs in these clinical trials are acceptable, particular attention should be paid when chemotherapy is used in real-world population, especially when multiple therapies are used.
There are some limitations of the review. First, several trials have limited methodological quality and the risk of performance and detection bias is high. Second, the definition of “high-risk PCa” is not consistent in each trial and the baseline characteristics of the patients, the local treatments, drugs, doses, and cycles of chemotherapy varied among trials, which may lead to clinical heterogeneity. Third, the data for OS are not mature and the analyses of OS were restricted by limited number of trials.
Conclusion
The meta-analysis demonstrates that significant improvement in PFS is associated with additional chemotherapy, especially docetaxel-based chemotherapy, in high-risk PCa patients. More trials and reliable data are needed to determine the effects of additional chemotherapy on OS in high-risk PCa population. Further investigations in PCa should also focus on the suitable patients for chemotherapy as well as the optimal use of chemotherapy.
Acknowledgments
This study was supported by grants from Natural Science Foundation of China (NSFC 81402110 and 81672547), Science and Technology Support Program of Sichuan Province (2015SZ0230-3) and 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Bolla M, van Tienhoven G, Warde P, et al. External irradiation with or without long-term androgen suppression for prostate cancer with high metastatic risk: 10-year results of an EORTC randomised study. Lancet Oncol. 2010;11(11):1066–1073. | ||
Rosenthal SA, Sandler HM. Treatment strategies for high-risk locally advanced prostate cancer. Nat Rev Urol. 2010;7(1):31–38. | ||
Boorjian SA, Karnes RJ, Rangel LJ, Bergstralh EJ, Blute ML. Mayo Clinic validation of the D’amico risk group classification for predicting survival following radical prostatectomy. J Urol. 2008;179(4):1354–1361. | ||
D’Amico AV, Whittington R, Malkowicz SB, et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA. 1998;280(11):969–974. | ||
Bastian PJ, Boorjian SA, Bossi A, et al. High-risk prostate cancer: from definition to contemporary management. Eur Urol. 2012;61(6):1096–1106. | ||
Granfors T, Modig H, Damber JE, Tomic R. Long-term followup of a randomized study of locally advanced prostate cancer treated with combined orchiectomy and external radiotherapy versus radiotherapy alone. J Urol. 2006;176(2):544–547. | ||
Pilepich MV, Winter K, Lawton CA, et al. Androgen suppression adjuvant to definitive radiotherapy in prostate carcinoma – long-term results of phase III RTOG 85-31. Int J Radiat Oncol Biol Phys. 2005;61(5):1285–1290. | ||
D’Amico AV, Chen MH, Renshaw AA, Loffredo M, Kantoff PW. Androgen suppression and radiation vs radiation alone for prostate cancer: a randomized trial. JAMA. 2008;299(3):289–295. | ||
Wirth MP, Weissbach L, Marx FJ, et al. Prospective randomized trial comparing flutamide as adjuvant treatment versus observation after radical prostatectomy for locally advanced, lymph node-negative prostate cancer. Eur Urol. 2004;45(3):267–270. | ||
Mcleod DG, Iversen P, See WA, et al. Bicalutamide 150 mg plus standard care vs standard care alone for early prostate cancer. BJU Int. 2006;97(2):247–254. | ||
de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–1154. | ||
Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. 2008;26(2):242–245. | ||
Vale CL, Burdett S, Rydzewska LHM, et al. Addition of docetaxel or bisphosphonates to standard of care in men with localised or metastatic, hormone-sensitive prostate cancer: a systematic review and meta-analyses of aggregate data. Lancet Oncol. 2016;17(2):243–256. | ||
Fizazi K, Faivre L, Lesaunier F, et al. Androgen deprivation therapy plus docetaxel and estramustine versus androgen deprivation therapy alone for high-risk localised prostate cancer (GETUG 12): a phase 3 randomised controlled trial. Lancet Oncol. 2015;16(7):787–794. | ||
Rosenthal SA, Hunt D, Sartor AO, et al. A phase 3 trial of 2 years of androgen suppression and radiation therapy with or without adjuvant chemotherapy for high-risk prostate cancer: final results of radiation therapy oncology group phase 3 randomized trial NRG oncology RTOG 9902. Int J Radiat Oncol Biol Phys. 2015;93(2):294–302. | ||
James ND, Sydes MR, Clarke NW, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. 2016;387(10024):1163–1177. | ||
Schweizer MT, Huang P, Kattan MW, et al. Adjuvant leuprolide with or without docetaxel in patients with high-risk prostate cancer after radical prostatectomy (TAX-3501): important lessons for future trials. Cancer. 2013;119(20):3610–3618. | ||
Hussain M, Tangen CM, Thompson IM, et al. Phase III intergroup trial of adjuvant androgen deprivation with or without mitoxantrone plus prednisone in patients with high-risk prostate cancer after radical prostatectomy: SWOG S9921. J Clin Oncol. 2018;36(15):1498–1504. | ||
Sandler H, Hu C, Rosenthal S, Sartor O. Phase 3 trial (RTOG0521)-ADT plus RT versus AS plus RT followed by chemotherapy with docetaxel and prednisone for localized high risk PCa. J Clin Oncol. 2015;33(suppl; abstr LBA5002). | ||
Lin D, Garzotto M, Aronson W, et al. PI-LBA06 VA CSP#553 Chemotherapy after prostatectomy (CAP) for high risk prostate carcinoma: a phase III randomized study. J Urol. 2016;195(4):e1071. | ||
Ahlgren G, Flodgren P, Tammela TLJ, et al. A randomized phase III trial between adjuvant docetaxel and surveillance after radical prostatectomy for high risk prostate cancer: results of SPCG12. J Clin Oncol. 2016;34(15 Suppl):5001. | ||
Fizazi K, Lesaunier F, Delva R, et al. A phase III trial of docetaxel-estramustine in high-risk localised prostate cancer: a planned analysis of response, toxicity and quality of life in the GETUG 12 trial. Eur J Cancer. 2012;48(2):209–217. | ||
Agus DB, Cordon-Cardo C, Fox W, et al. Prostate cancer cell cycle regulators: response to androgen withdrawal and development of androgen independence. J Natl Cancer Inst. 1999;91(21):1869–1876. | ||
Tombal B. What is the pathophysiology of a hormone-resistant prostate tumour? Eur J Cancer. 2011;47(Suppl 3):S179–S188. | ||
Ahmed M, Li LC. Adaptation and clonal selection models of castration-resistant prostate cancer: current perspective. Int J Urol. 2013;20(4):362–371. | ||
Darshan MS, Loftus MS, Thadani-Mulero M, et al. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res. 2011;71(18):6019–6029. | ||
Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010;70(20):7992–8002. | ||
Gan L, Chen S, Wang Y, et al. Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res. 2009;69(21):8386–8394. | ||
Yu EY, Lin DW. Avoiding undertreatment of aggressive prostate cancer by early use of chemotherapy. JAMA Oncol. 2017;3(1):13–14. | ||
Graff JN, Alumkal JJ, Beer TM. Should chemotherapy be used in nonmetastatic prostate cancer? JAMA Oncol. 2017;3(1):11–12. | ||
de Morree ES, Vogelzang NJ, Petrylak DP, et al. Association of survival benefit with docetaxel in prostate cancer and total number of cycles administered: a post hoc analysis of the mainsail study. JAMA Oncol. 2017;3(1):68–75. | ||
Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–1512. | ||
Hussain M, Goldman B, Tangen C, et al. Prostate-specific antigen progression predicts overall survival in patients with metastatic prostate cancer: data from Southwest Oncology Group Trials 9346 (Intergroup Study 0162) and 9916. J Clin Oncol. 2009;27(15):2450–2456. | ||
Berthold DR, Pond GR, Roessner M, et al. Treatment of hormone-refractory prostate cancer with docetaxel or mitoxantrone: relationships between prostate-specific antigen, pain, and quality of life response and survival in the TAX-327 study. Clin Cancer Res. 2008;14(9):2763–2767. | ||
Sweeney C, Chen Y, Carducci M, et al. Chemohormonal therapy versus hormonal therapy for hormone naïve high volume newly metastatic prostate cancer (PRCA): ECOG LED Phase III randomized trial. Ann Oncol. 2014;25(Suppl 4):iv255–iv279 | ||
Gravis G, Fizazi K, Joly F, et al. Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(2):149–158. | ||
Gillessen S, Attard G, Beer TM, et al. Management of patients with advanced prostate cancer: the report of the advanced prostate cancer consensus conference APCCC 2017. Eur Urol. 2018;73(2):178–211. | ||
Rosenthal SA, Bae K, Pienta KJ, et al. Phase III multi-institutional trial of adjuvant chemotherapy with paclitaxel, estramustine, and oral etoposide combined with long-term androgen suppression therapy and radiotherapy versus long-term androgen suppression plus radiotherapy alone for high-risk prostate cancer: preliminary toxicity analysis of RTOG 99-02. Int J Radiat Oncol Biol Phys. 2009;73(3):672–678. |
Supplementary material
 | Figure S1 Funnel plot created by outcome of progression-free survival. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.