Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
The direct and indirect effects of α-synuclein on microtubule stability in the pathogenesis of Parkinson’s disease
Authors Carnwath T, Mohammed R ![]() , Tsiang D
, Tsiang D
Received 23 February 2018
Accepted for publication 3 April 2018
Published 27 June 2018 Volume 2018:14 Pages 1685—1695
DOI https://doi.org/10.2147/NDT.S166322
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Roger Pinder
Tom Carnwath,1 Raihan Mohammed,2 Daniel Tsiang3
1Department of Zoology, University of Cambridge, Cambridge, UK; 2Faculty of Medicine, University of Cambridge, Cambridge, UK; 3Faculty of Engineering, Imperial College London, London, UK
Abstract: Despite decades of research, the mechanism of Parkinson’s disease pathogenesis remains unclear. Studies have focused heavily on the protein α-synuclein, which is the primary component of Lewy bodies, the pathologic inclusions that are the hallmark of Parkinson’s on the cellular level. While the roles of α-synuclein in causing mitochondrial dysfunction and disruptions to the proteasomal system have been well documented, recently, its role in effecting microtubule dynamics has been investigated as a potential source of pathogenicity. Here, we evaluate the evidence for and against the role of α-synuclein in destabilizing microtubules, causing axonal transport deficits and eventually neurodegeneration. We present evidence for a model where α-synuclein has both a direct and indirect effect on microtubule stability. Directly, it may act as a microtubule-associated protein, binding to microtubules and directly effecting their dynamics. Indirectly, it may promote the hyperphosphorylation of the microtubule stabilizing protein, tau, leading to tau aggregation with other microtubule stabilizing proteins, hence indirectly causing microtubule destabilization. This model provides insights into the function of α-synuclein and tau in Parkinson’s disease pathogenesis and raises the possibility that this role that may also be conserved in Alzheimer’s disease.
Keywords: tubulin, tau, axon, phosphorylation, Alzheimer’s disease
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease (AD) and it is estimated to affect over 6 million people worldwide. At the cellular level, Parkinson’s can be defined as the selective and progressive loss of dopaminergic neurons in particular areas of the brain. This reduction in dopamine transmission leads to the motor symptoms, characterized by a slowness of movement tremor and postural instability, that are currently the basis of diagnosis, as well as various non-motor symptoms such as depression. Currently, the only treatments for PD are symptomatic treatments that center on artificially restoring dopamine levels in the brain. However, while very effective in the short term, these drugs gradually lose their potency and often lead to devastating side effects in the long term, such as dyskinesia. Therefore, a therapy that targets the underlying cause of the depleted dopamine levels, by preventing the death of dopaminergic neurons in these areas of the brain, would be significant.
In order to develop such a therapy, knowledge of the pathogenesis of PD is important. PD is a multifactorial disease with both environmental and genetic factors. This is the basis for its clinical classification as either sporadic or familial PD. Reflecting the genetic and environmental origins of the disease, the primary method of generating PD experimental models in animals and cell cultures centers on using toxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and overexpressing PD-associated mutant genes such as α-synuclein. These models, as well as immunohistochemical studies on postmortem PD patient brain samples, have elucidated many of the processes involved in the early stages of the disease. These processes include mitochondrial dysfunction, α-synuclein aggregation and disruptions to the proteasomal degradation system, and the pathological changes in them seem to be conserved in both sporadic and familial PD, suggesting a common etiology to the disease. Recently, microtubules (MTs) have also been implicated in the pathogenesis of PD.
MTs are involved in many different cellular processes including mitosis, generation of cellular polarity and intracellular transport. The role of MTs in transporting proteins and organelles is particularly important in neurons due to the length of axons. This axonal transport is primarily mediated by MTs, both from the cell body to the synaptic bouton (anterograde transport) and vice versa (retrograde transport). Therefore, the dynamics of MTs and their level of stability are crucial for axonal transport and, hence, the correct functioning of neurons. Relating this to PD, a Wallerian-like mechanism of neurodegeneration is thought to take place, where the axon retreats into the cell soma before the cell dies, and this process is inherently linked with MT depolymerization. Indeed, the first visible sign of Wallerian degeneration is the fragmentation of MTs.1 Therefore, destabilization of MTs may represent an early, critical step in the loss of functionality of dopaminergic neurons in PD.
Many different cytosolic factors can affect MT stabilization. MT-associated proteins (MAPs) such as tau are the primary way by which the cell regulates MT dynamics; however, various chemicals can also have an effect. Both PD-inducing toxins (including MPTP, rotenone and 6-hydroxydopamine) and PD-associated proteins (including α-synuclein, tau, leucine-rich repeat kinase 2 [LRRK2] and parkin) seem to affect the stability of MTs.1 It, therefore, appears that MT destabilization could be a point of convergence that links the genetic and environmental factors involved in PD.
One MT-associated factor with a central role in PD pathology is α-synuclein. This is a 140-amino acid protein that has been strongly linked to PD via genome-wide association studies.1 Both mutations and duplications of α-synuclein have been shown to be associated with PD, suggesting overexpression alone is sufficient to have a pathogenic effect.2 Further highlighting α-synuclein as a central player in PD pathogenesis, aggregates of α-synuclein have been identified as the primary component of Lewy bodies (LBs), the characteristic aggregates of protein found in the cytoplasm of neurons in the PD brain and a hallmark of the disease.
However, despite extensive research, the physiological role of α-synuclein remains unclear. While there is some evidence suggesting that it may act as a chaperone for SNARE complexes, recently, an alternative function for α-synuclein as a MAP has been proposed. This is a controversial area, raising many questions including what the nature of the MT–α-synuclein interaction is, whether α-synuclein causes MT instability in pathogenic conditions and whether this happens directly by a pathogenic change in the interaction of α-synuclein and MTs, or if tau and the kinases – protein kinase A (PKA) and glycogen synthase kinase-3β (GSK-3β) – mediate an indirect effect of α-synuclein on MT stability. This review will discuss the evidence for and against the role of α-synuclein in interacting with MTs, effecting their dynamics and, under pathogenic conditions, destabilizing them, disrupting axonal transport and enhancing tau pathology as well as its own. We evaluate numerous studies within this field and categorize the evidence as either a direct or indirect effect on MT stability (Table 1). The understanding of this pathogenic pathway in PD provides opportunities for therapeutic intervention and highlights a possible conserved role for α-synuclein and tau pathology in Alzheimer’s pathogenesis.
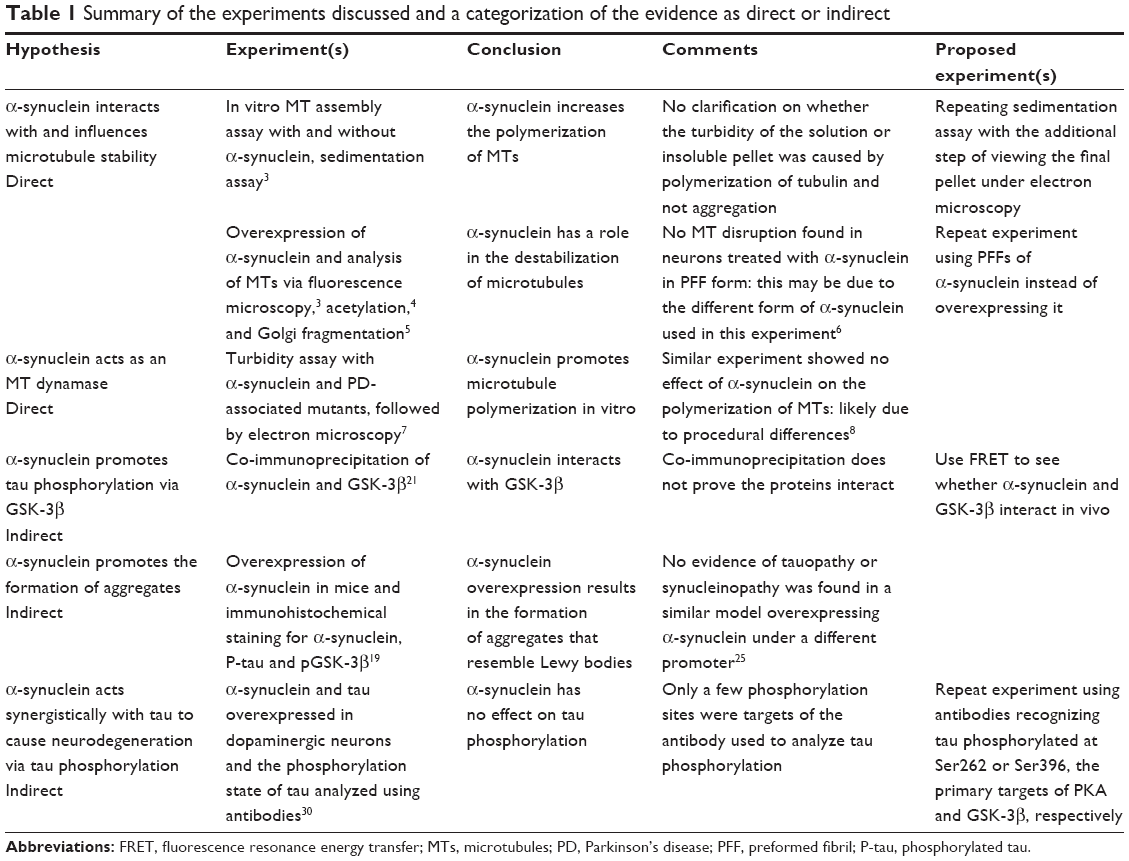 | Table 1 Summary of the experiments discussed and a categorization of the evidence as direct or indirect |
α-synuclein interacts with and influences MT stability
In order to first test the ability of α-synuclein to interact with MTs in vitro and in vivo, pull downs in hamster’s brain homogenate and immunostaining for α-synuclein in HeLa cells were done. The co-immunoprecipitation of α-synuclein with tubulin in the homogenate combined with the co-localization of α-synuclein and MTs in HeLa cells provides good evidence that α-synuclein interacts with MTs in vivo. To determine whether α-synuclein has any direct effects on MT stability, in vitro MT assembly assays in the presence of α-synuclein were also performed.3 First, various concentrations of recombinant α-synuclein or His-GST (control) were mixed with tubulin in a polymerization buffer and the turbidity of the solution was measured at certain time points. The authors concluded from this experiment that the turbidity of the solutions increases rapidly with α-synuclein in a dose-dependent manner, suggesting that this effect is due to the increased formation of MTs with α-synuclein. However, if α-synuclein really was increasing the polymerization of MTs, one would expect a steady increase in the turbidity of the solution with time, the gradient of which should be significantly greater than control. Instead, as soon as α-synuclein is added (time zero), the turbidity of the solution is instantly much greater than that of the control and as time progresses, the gradients of the slopes show minimal difference. Therefore α-synuclein could simply be aggregating with the tubulin and forming insoluble aggregates that instantly increase the turbidity of solution.
To confirm their results, a sedimentation assay was performed with samples containing tubulin and α-synuclein polymerized for a period before being centrifuged.3 Incubation of α-synuclein and tubulin together led to a much greater proportion of tubulin in the pellet (insoluble) fraction than when tubulin and GST (control) were incubated. The authors concluded that α-synuclein is promoting the assembly of insoluble MTs, which accumulate in the pellet fraction. However, there is nothing presented here to prove that MTs are being formed; the increase in the tubulin in the pellet fraction could just as well be insoluble aggregates as MTs. Therefore, these results suggest that α-synuclein either promotes MT polymerization or aggregates with tubulin. These possibilities could be distinguished by viewing the pellet fraction from the sedimentation assay under electron microscopy to reveal its ultrastructure (tubules or aggregates), but this was not done in this study.
Despite some flaws, a significant disruption of MTs in HeLa cells was observed in the presence of α-synuclein compared to a GST control. These observations were supported by the finding of a significant reduction in acetylated tubulin (associated with stable MTs) in a human dopaminergic neuronal cell line overexpressing α-synuclein compared to those expressing green fluorescent protein (GFP), as well as significantly decreased kinesin levels and fewer mitochondria in the neurites.4 Further, α-synuclein overexpression was reported to cause Golgi fragmentation in neuroblastoma cells, characteristic of MT dysfunction.5 Thus, there is reasonably strong evidence for α-synuclein having a role in the destabilization of MTs.
Disagreeing with these results, electron micrographs in neurons treated with α-synuclein preformed fibrils (PFFs), which seed the recruitment of endogenous α-synuclein into LB-like inclusions, revealed no visible sign of MT disruption. Additionally, the live imaging of various organelle markers in hippocampal cultures treated with PFFs revealed no significant difference in anterograde and retrograde transport of mitochondria compared to control cells treated with PBS.6 These contradictory results are likely due to the different ways in which α-synuclein pathology was induced in different models (simple overexpression vs treatment with PFFs). This is a notable issue throughout the literature and is discussed further in the “Discussion” section below.
Considering the evidence that α-synuclein promotes MT destabilization in pathogenic conditions, we now discuss the data exploring the mechanism by which α-synuclein interacts with MTs to affect their dynamics, first directly, then indirectly via tau.
α-synuclein directly regulates MT dynamics and may aggregate with tubulin in pathogenic conditions
Recently, evidence has come to light suggesting that wild-type (WT) α-synuclein has an alternative role, distinct from its proposed function as a chaperone, as an MT dynamase, regulating the polymerization dynamics of MTs. PD-associated α-synuclein mutants may lose this ability and instead aggregate with tubulin. α-synuclein has been found to interact with tubulin in vitro and it can be seen to co-localize with α-synuclein in LBs in PD brain sections. Additionally, it was shown via a turbidity assay combined with electron microscopy that WT α-synuclein can stimulate the polymerization of tubulin into MTs, while PD-associated A30P and A53T mutants form aggregates with tubulin instead.7 This result seems to confirm that α-synuclein can promote MT polymerization in vitro.
To test these conclusions, a similar experiment involving a turbidity assay and sedimentation test was performed.8 However, this indicated that oligomeric α-synuclein has no effect on the polymerization of MTs. The discrepancy of these results may be explained by the different sources of the tubulin used or more likely by the folding of α-synuclein: recently, circular dichroism spectroscopy was used to reveal that α-synuclein forms α-helical structures on binding to tubulin tetramers (α2β2).9 It was demonstrated that giving α-synuclein time to fold by preincubating it with tubulin for 10 minutes before analysis made the difference between α-synuclein influencing tubulin polymerization (with preincubation) or not (without preincubation). This suggests that the contradictory results of the above experiments may have been down to procedural differences in the time allowed between mixing of α-synuclein and tubulin, and analysis. It was also shown that various PD-associated mutants of α-synuclein do not display the same level of increase in α-helical content on binding tubulin tetramers as WT. Moreover, the MTs formed in the presence of the α-synuclein mutants were surrounded by aggregates of tubulin and α-synuclein, and the extent to which these aggregates formed in the different mutants inversely correlated with their ability to form tetramers. These results strongly suggest that WT forms of α-synuclein form α-helical secondary structures when interacting with tubulin to promote polymerization, whereas PD-associated mutant α-synuclein loses this ability and instead aggregates with tubulin.
In order to further probe the mechanism by which α-synuclein effects MT dynamics, the kinetics of MT assembly were analyzed in the presence of α-synuclein or controls and the products analyzed via electron microscopy. This revealed that there are more, shorter MTs in the presence of α-synuclein compared to control, suggesting that α-synuclein promotes nucleation of MTs and increases the frequency of catastrophe. This analysis was reinforced by the discovery that a fragment of α-synuclein (residues 43–63) containing four of the five PD-associated mutations shows sequence similarity to the tubulin-interacting domain of the stathmin family of MT depolymerizing proteins, which are also naturally unfolded proteins that adopt an α-helix secondary structure upon binding tubulin. It was, therefore, proposed that α-synuclein could act as a “dynamase”, a recent term used to describe kinesin-8, which is able to regulate both MT nucleation and catastrophe in Schizosaccharomyces pombe. While this is an extremely recent advance in the field (and hence, more experimental evidence is needed), it reinforces the idea that α-synuclein may have a direct effect on MT dynamics. However, recent evidence has also pointed to α-synuclein having an indirect effect on MT stability via the MAP, tau.
α-synuclein indirectly effects MT stability through tau
Tau is one of the major MAPs expressed in the neurons of the brain. It interacts with tubulin, promoting its polymerization, and binds to MTs to promote their stabilization. Similar to α-synuclein, pathogenic forms of tau are found in the brains of AD patients and have also been implicated in disrupting axonal transport.10 Over the past few decades, tau has primarily been associated with AD due to its propensity to self-assemble, when hyperphosphorylated, into paired helical filaments (PHFs) that make up the neurofibrillary tangles (NFTs) found in AD patient brains. However, the aggregations of tau (tauopathy) and its propensity to self-assemble, which are commonly associated with AD and PD, respectively, actually display a broad overlap between the diseases. For example, the appearance of NFTs in PD patient brains is much more frequent than an age-matched population and LBs can be found in around 60% of sporadic cases of AD.11 Therefore, the interaction between tau and α-synuclein may be important in both PD and AD pathogenesis. Focusing on PD, the MAPT locus, from which tau is spliced, is consistently identified in genome-wide association study as being second only to α-synuclein in its linkage to PD, with a p-value of <6.1×10−49.2,12 LBs that are immunoreactive for tau have been found in PD brains.13 Furthermore, it has been well documented that tau and α-synuclein promote the fibrillogenesis of each other and the A53T mutant of α-synuclein seems to enhance this effect. All this suggests that tau has a role in the pathology of α-synuclein aggregates and, considering its main function as an MT stabilizer, it has the potential to mediate and interact with some of the aforementioned effects of α-synuclein on MTs.
Interaction with tau underlies some of the MT destabilizing properties of α-synuclein
The interaction between tau and α-synuclein has the potential to be a major avenue by which α-synuclein affects MT dynamics, given tau’s MT stabilizing properties. This is also an intriguing area as it could pertain to both AD and PD pathogenesis, as discussed later. It was found that α-synuclein oligomers and multimer protofilaments had no effect on tubulin polymerization in vitro unless tau was added to the MT assembly mixture.4 In the presence of tau, α-synuclein inhibited MT polymerization dose dependently and this inhibition could be alleviated by increased concentrations of tau. This suggests that α-synuclein may inhibit tau from stabilizing MTs, leading to MT depolymerization. Indeed, α-synuclein binds within the MT-binding domain of tau and this interaction is inhibited by tubulin.14 Interestingly, the repeat MT-binding domain of tau forms the core of PHFs in Alzheimer’s15 and fragments of tau derived from this domain have a much greater propensity to aggregate in vitro than full-length tau.16 This strongly suggests that exposure of the MT-binding domain of tau by releasing it from MTs might precipitate its aggregation; almost all the compounds known to cause tau aggregation, such as heparin, DNA and fatty acids, do so by competing with MTs to bind to the MT-binding domain of tau.17
Tau is a phosphoprotein, and this is central to its interaction with α-synuclein and, indeed, its role in pathogenesis: tau isolated from PHFs in AD is found to be hyperphosphorylated,18 and hyperphosphorylated tau is a component of LBs in PD.19 Tau can be phosphorylated by PKA and GSK-3β. Crucially, the phosphorylation of tau on certain residues, in particular, serine 262 (S262) and serine 396 (S396), reduces its affinity for binding MTs.20 This raises the possibility that α-synuclein may in part destabilize MTs via the phosphorylation of tau.
α-synuclein promotes tau phosphorylation via PKA and GSK-3β, leading to MT destabilization
α-synuclein has been shown to affect the phosphorylation state of tau via PKA and GSK-3β using overexpression, and expression of PD-associated GSK-3β lies within the MT-binding domain of tau. First, it was shown that α-synuclein enhances PKA-mediated tau phosphorylation at S262 in vitro. A monoclonal antibody was used for phosphorylated S262 and S356 of tau, and addition of α-synuclein to a mixture including tau and PKA led to a dose-dependent increase in phosphorylated tau, as indicated by an increase in antibody binding.14
Second, a toxin-based model showed that α-synuclein also promotes GSK-3β-mediated tau phosphorylation on different residues.21 This is particularly interesting because GSK-3β is heavily implicated in hyperphosphorylating tau to generate PHFs (as demonstrated in both cell culture and rodent models), promoting the fibrillogenesis of tau and regulating apoptosis. Cultured neuroblastoma cells transfected with α-synuclein were exposed to the commonly used toxin 1-methyl-4-phenylpyridinium (MPP) (which reliably induces Parkinsonism in humans and rodents). GSK-3β activity is negatively regulated by serine 9 (Ser9) phosphorylation and positively regulated by tyrosine 216 (Tyr216) phosphorylation. Thus, the Western blots were probed for phosphorylated (activated) GSK-3β (pTyr216-GSK-3β) and phosphorylated tau. The quantification of these blots showed that MPP+ treatment led to the activation of GSK-3β, phosphorylation of tau and accumulation of α-synuclein. Moreover, inhibition of GSK-3β using lithium and a more specific inhibitor, thiadiazolidinone-8, reversed these effects, proving that GSK-3β is likely to be key in mediating the pathogenic effects of MPP+ on cells.21
Although commonly used, the mechanism by which MPTP induces parkinsonism may not accurately reflect the sporadic or familial pathogenesis normally found in humans. These results were, therefore, validated by overexpressing α-synuclein in mice, which showed that this alone is able to induce increased pTyr216-GSK-3β. Looking at humans, a significant increase in pTyr216-GSK-3β was seen relative to total GSK-3β levels in the striata of PD patients compared to age-matched controls. Together, this provides strong evidence supporting a mechanism in which α-synuclein promotes the phosphorylation of tau by activating GSK-3β. Co-immunoprecipitation studies suggest α-synuclein physically interacts with GSK-3β to activate it and forms complexes with it and tau. However, co-immunoprecipitation does not prove the proteins interact in normal cellular conditions as it involves significant perturbation to the cell, including cell lysis and incubation with various reagents. Thus, to clarify the nature of the interaction of α-synuclein, tau and GSK-3β, a fluorescence resonance energy transfer-based method could be used to visualize whether these proteins interact in the cell. This would involve transfecting neuroblast cells with pairs of these proteins fused to fluorescence resonance energy transfer–suitable fluorophores and using confocal fluorescence microscopy to analyze whether they interact. If these proteins are shown to interact in vivo, this would provide additional evidence to support α-synuclein directly activating GSK-3β, which in turn directly phosphorylates tau.
The connection between α-synuclein and tau phosphorylation is crucial because tau phosphorylation has been strongly linked to MT destabilization and may be a vital step in the pathogenesis of AD as well as PD. This was demonstrated by exposing PC12 cells to exogenous α-synuclein and measuring the levels of free and polymerized tubulin via sedimentation compared to unexposed control cells.22 This revealed that the cells exposed to α-synuclein had a significantly greater ratio of free to polymerized tubulin, indicating MT depolymerization. The addition of the specific GSK-3β inhibitor, SB-216763, fully reversed these effects, proving that the effects of α-synuclein on MT stability are GSK-3β mediated. The effect of GSK-3β on MT destabilization and cell viability was further investigated using rotenone, another toxin that is commonly used to model PD in cell cultures and animals, to show that it caused increased levels of pTyr216-GSK-3β, MT destabilization that was reversed by GSK-3β inhibition and cytotoxic effects that were ameliorated by taxol, an MT stabilizing drug.23 Together, these results highlight the link between GSK-3β and MT depolymerization and indicate that the well-known proapoptotic effects of GSK-3β may be, at least in part, due to its phosphorylation of tau and the resulting MT destabilization.
PD-associated α-synuclein mutations have also been used to show that this kind of mechanism for MT destabilization may also be relevant to familial PD cases and the PD-associated α-synuclein mutants. It was demonstrated that α-synuclein also activates PKA in vivo leading to the phosphorylation of S262 in tau and was shown that PD-associated mutants of α-synuclein promote PKA-mediated tau phosphorylation to a much greater extent than WT or an unspecific α-synuclein mutant in vitro.24 Furthermore, HEK293 cells were transfected with the various forms of α-synuclein and the effects on MT stability were analyzed via a sedimentation assay. Surprisingly, all the α-synuclein variants destabilized MTs by around 35% compared to vector-transfected cells (overall tubulin levels were unchanged) and there was no difference between cells expressing WT and mutated α-synuclein. Since HEK293 cells do not naturally express tau, this indicates that α-synuclein is able to destabilize MTs in the absence of tau, supporting the previously mentioned reports that α-synuclein has a direct, non-tau-mediated effect on MTs. Crucially, however, when tau was co-transfected along with the n-tau media variants, WT α-synuclein reduced MT stability by 11.9% compared to a tau and vector-transfected control, whereas the PD-associated mutants A30P, E46K and A53T dramatically decreased MT stability by 65.2%, 53.5% and 76.2%, respectively. This emphatically demonstrates the tau-mediated effect the PD-associated mutations in α-synuclein can have on MT stability. Overall, this supports a model where the PD-associated mutations of α-synuclein enhance its ability to phosphorylate tau residue S252 via PKA and, hence, indirectly destabilize MTs by reducing the ability of tau to stabilize them.
Finally, in order to directly test this model in mice, α-synuclein was overexpressed under the control of the platelet-derived growth factor promoter in transgenic mice.19 Analyzing the striata of these mice revealed a highly significant increase in α-synuclein, P-tau and pTyr216-GSK-3β, compared to non-transgenic control mice. These results were also shown to be relevant to sporadic PD in humans as (slightly less dramatic) increases in these proteins were found in postmortem PD brains compared to age-matched controls. Furthermore, each of these species was significantly enriched in the insoluble fraction (pellet) after treatment of the striatum with a detergent, hinting at the formation of insoluble aggregates. Immunohistochemical staining reinforces this point, showing these proteins co-localize with each other next to the nucleus in transgenic but not WT mice. Suggestively, these aggregates resemble LBs in size, shape and subcellular position. This provides strong evidence for the hypothesis that α-synuclein directly interacts with GSK-3β and tau and this leads to tau hyperphosphorylation and aggregation with α-synuclein. Connecting this to MT instability and neurodegeneration, it was revealed that α-tubulin in cytoskeleton-free fractions of the striata of the α-synuclein-overexpressing transgenic mice is significantly enriched compared to WT mice, while the dopaminergic neuron marker tyrosine hydroxylase is significantly reduced in this region (but not in the substantia nigra or basal ganglia). These results are particularly pertinent, given that α-synuclein gene duplications are known to be associated with PD. However, no evidence of tauopathy or synucleinopathy was found in a similar model of transgenic mice overexpressing α-synuclein under the murine 2′,3′-cyclic nucleotide 3′phosphodiesterase (CNP) promoter.25 These contradictory results may be due to the different promoters that α-synuclein was expressed under, the cells of the brain analyzed (oligodendricytes vs neurons of the striatum) or the method of staining (thioflavin-S vs tau). Nonetheless, overexpressing A53T mutant α-synuclein under the CNP promoter did result in abundant α-synuclein aggregates, and tau inclusions (which overlapped with the α-synuclein inclusions to some extent) were also found in half of the transgenic mice. Therefore, these results highlight the importance of the area of the brain and the form of α-synuclein on the development of pathogenic inclusions.
The above evidence suggests that α-synuclein can indirectly reduce MT stability by inhibiting the interaction of tau with MTs via PKA and GSK-3β, leading to the aggregation of α-synuclein, tau and GSK-3β and MT destabilization. This mechanism seems to be relevant to a diverse range of experimental models of PD, including rotenone, MPTP, α-synuclein overexpression and mutation-based studies, suggesting that it may be a conserved feature of both sporadic and familial PD. Precisely how hyperphosphorylated tau destabilizes MTs is more controversial. Simply, the synuclein-mediated dissociation of tau from MTs alone does not seem to be sufficient to induce significant MT destabilization, axonal transport deficits or, indeed, neurodegeneration since transgenic tau-knockout mice show only minor phenotypes and no significant effects on axonal transport or MTs in dopaminergic neurons.26 The increased susceptibility of hyperphosphorylated tau to form NFTs does not seem to be behind MT depolymerization as no correlation was found between the presence of NFTs and MT alterations in postmortem AD brains, despite there being significant reduction in MT length and density in AD brains compared to control.27
Instead, when phosphorylated on certain residues such as S262 and S396, tau may disrupt MT assembly and hyperphosphorylated tau sequesters other (stabilizing) MAPs as well as other tau molecules, forming aggregates and, hence, indirectly destabilizing MTs. This model is supported by the demonstration that very few MTs form in nocodazole and triton-treated cells supplemented with rat brain cytosol, in the presence of hyperphosphorylated tau isolated from AD patients’ brains28 or in the presence of PKA phosphorylated tau.29 Addition of the phosphatase PP2A largely restored MT assembly, highlighting the role of hyperphosphorylated tau in MT destabilization. AD-brain-isolated tau was also observed to co-stain with both endogenous tau and MAP2, suggesting sequestration of these proteins, and α-synuclein also binds MAP2 in vitro.4 Tau seems to overlap in function with MAP1B, another MT stabilizing protein found in neurons; so, redundancy between these two proteins could explain the lack of phenotype in tau-knockout mice. Overall, the sequestration of other MAPs may play an important role in the ability of hyperphosphorylated tau to cause MT instability and neurodegeneration.
The interaction of α-synuclein with tau precipitates axonal transport deficits
Although the pathogenic interaction between tau and α-synuclein is linked to MT destabilization, there is still a question as to how much of an effect this has on axonal transport. To answer this question, an enhancer trap was used to express α-synuclein and tau specifically in the motor neurons of drosophila and axonal transport defects were assayed for via the accumulation of the synaptic vesicle protein, DVGLUT (vesicular glutamate transporter), which is indicative of axon traffic jams.30 This revealed the combination of α-synuclein and tau to be synergistic as α-synuclein expression alone showed no DVGLUT accumulation, and tau alone showed reasonable accumulation; however, when expressed together, there was much greater DVGLUT accumulation.
This synergistic relationship on axonal transport and its relevance to neurodegeneration in PD were highlighted by the similar synergistic effects of α-synuclein and tau on dopaminergic cell loss in drosophila. Here, the GAL4 expression system was used to express α-synuclein and tau in dopaminergic cells, which were visualized by immunostaining for tyrosine hydroxylase. This showed that α-synuclein and tau expressed together promoted greater dopaminergic cell loss than α-synuclein or tau alone. In an attempt to understand the mechanism behind these findings, the authors also investigated tau phosphorylation; however, only the AT8 antibody against phosphorylated Ser202/Thr205 was used and no differences were found in tau phosphorylation between tau and α-synuclein expressing transgenic flies and WT. It would have been interesting to see if antibodies recognizing tau phosphorylated at Ser262 or Ser396 would have yielded the same results, given that these are the primary targets of PKA and GSK-3β, respectively. Nonetheless, although their relevance to humans in debatable, these experiments provide valuable evidence supporting the possibility that α-synuclein interacts with tau to promote MT instability, axonal transport disruption and eventually neurodegeneration.
Discussion
PD is marked by the loss of dopaminergic neurons in particular areas of the brain. Despite decades of research and the identification of many proteins and processes thought to play a role, the precise mechanisms that instigate neurodegeneration remain largely unknown. Here, we have reviewed the evidence for the role of α-synuclein in promoting dopaminergic cell loss via effecting MT stabilization. This may happen directly or indirectly. Directly, α-synuclein may have a physiologic role as an MT “dynamase”, regulating the dynamics of MTs, while pathogenic forms of α-synuclein interact differently, promoting aggregation with tubulin. These aggregates are associated with MT destabilization; however, the mechanism by which this happens is controversial. Indirectly, α-synuclein promotes the hyperphosphorylation of tau via the kinases GSK-3β and PKA, leading to its dissociation from MTs and aggregation. Again, the mechanism by which tauopathy and synucleinopathy disrupt MTs and axonal transport is currently unclear, although the sequestration of MAPs and normal tau may play a role.
Axonal transport deficits and the interaction between α-synuclein and tau are also thought to play a role in AD pathogenesis.31 Interestingly, a similar mechanism involving GSK-3β to disrupt axonal transport also seems to be applicable here, as it was demonstrated that tau is required for Aβ protein to activate GSK-3β and, hence, induce axonal transport disruptions (however, the role of tau in this case seemed to be independent of its interactions with tubulin).10 This highlights the importance of GSK-3β in particular and suggests that it could be a good target for therapies. Indeed, the GSK-3β inhibitor tideglusib has undergone a short-term Phase II clinical trial for AD treatment; however, no clinical benefit was found.32 It would be interesting to see if such a drug has any clinical benefit on PD, given its potency in animal models.33
It is becoming increasingly apparent that PD is a very heterogeneous disease: the physical and mental symptoms as well as the progression of the disease, the neuronal cell populations affected and the patient response to therapy all vary greatly in the PD population. It, therefore, follows that PD is likely to have many different etiologies, with different cellular processes being initially disrupted (eg, by age-related decline, environmental toxins, genetics or all of the above) in different people. Resonating with this, α-synucleinopathies have also been strongly implicated in disrupting other cellular pathways, most notably the autophagy-lysosomal pathway, the ubiquitin proteasome system and mitochondrial function.34 It is easy to imagine a mechanism whereby defects in any one of these processes could initiate pathogenic α-synuclein aggregation and, hence, start a deleterious positive feedback loop: mitochondrial dysfunction can promote α-synuclein aggregation via the production of reactive oxygen species, and axonal transport disruption as MT polymerization requires energy (guanosine triphosphate) and oxidative stress also inhibits axonal transport. Axonal transport disruption itself causes mitochondrial defects as their faulty distribution around the cell can derive some subcellular compartments of adenosine triphosphate, and it might contribute to α-synuclein accumulation. Proteasomal dysfunction causes α-synuclein aggregation because α-synuclein aggregates would normally be degraded by this system.
Therefore, MT stabilizing drugs may be a promising area of future research in the treatment of PD, or perhaps specific subtypes of PD. Although the majority of MT stabilizing drugs are unable to cross the blood–brain barrier, and so do not represent valid therapeutic options, there are some that show promise. For example, epothilone D can cross the blood–brain barrier and reverse MT defects and stop nigrostriatal degeneration in mice treated with MPTP.35 This suggests that MT stabilization could be an efficacious treatment for PD.
Future perspectives
One of the key challenges in trying to elucidate the effects of pathogenic α-synuclein in cells is in replicating the exact nature of pathogenic α-synuclein as it occurs in sporadic PD. α-synuclein monomers can assimilate to form many different conformational, multimeric variants including oligomers, PFFs and fibrils. Differences in the precise form of α-synuclein generated in different models may be a key factor contributing to many of the contradictory results discussed above. Highlighting this point, α-synuclein PFFs alone can have distinctly different properties depending on how they are made in vitro. For example, PFFs generated by the repetitive seeded fibrillization of α-synuclein monomers in vitro have a much greater ability to seed not only the aggregation of α-synuclein, but that of tau as well, compared to PFFs formed de novo.36 Additionally, oligomers are now thought to be the most toxic form of α-synuclein, with fibrils being relatively inert.37 Therefore, specifically, the amount of oligomeric α-synuclein in the cell is likely to affect experimental results. Given that monomeric α-synuclein is thought to be in equilibrium with oligomeric α-synuclein, overexpression of α-synuclein may tip this balance, and hence, the strength of the promoter, temperature, endogenous α-synuclein or any other factor that might influence the equilibrium needs to be controlled for.
Although these different forms of α-synuclein may be hard to control for, and hence represent another difficulty in PD research, careful experimental design could exploit their different properties as useful controls. For example, the different abilities of the PFFs discussed above to cross-seed tau could be used to investigate whether α-synuclein has direct or indirect effects on MTs: one of the key unanswered questions raised above. Primary dopaminergic neurons isolated from transgenic mice containing a construct such as end binding protein 3 (EB3)-GFP would allow the visualization of MT dynamics as EB3 binds to the ends of MTs, tracking their growth. Live cell imaging would allow the tracking of many different MTs in these cells, and it could be possible to quantify any differences in the dynamics of MTs on addition of PFFs generated de novo, or by repetitive seeded fibrillization. If α-synuclein aggregates do cause MT depolymerization indirectly via tau, there should be a significant difference in the MT dynamics between the neurons treated with PFFs that cross-seed tau and those treated with PFFs generated de novo, that only seed α-synuclein. Thus, this would prove the role of tau sequestration in the α-synuclein-mediated depolymerization of MTs.
In order to prove the direct role of the kinases GSK-3β and PKA in tau phosphorylation and MT depolymerization, it would be interesting to observe the effects of their addition into an in vitro sedimentation assay. As described above, various authors have done sedimentation assays using lysed cells transfected with α-synuclein, tau and GSK-3β inhibitors; however, there could be many other proteins in the cell playing a role in the observed effects on MTs. Thus, performing an in vitro sedimentation assay with tubulin and various combinations of GST, α-synuclein, tau, GSK-3β and PKA, Western blotting the resulting pellet and supernatant fractions for tubulin, and viewing the part of the pellet fractions under an electron microscope would elucidate whether these components alone are able to significantly affect MT stability. According to the model proposed above, α-synuclein alone should cause MT polymerization and nucleation, leading to more tubulin being in the pellet fraction than with a GST control. Addition of both tau and α-synuclein should also result in more tubulin in the pellet fraction due to the additional MT stabilization. However, addition of α-synuclein, tau, GSK-3β and PKA together should lead to less tubulin in the pellet fraction, indicating free tubulin and, hence, MT depolymerization. It is also possible that more tubulin may appear in the pellet fraction due to its aggregation with α-synuclein, tau, GSK-3β and PKA. Therefore, electron microscopy of the pellet fraction could be used to confirm that this is the case (aggregates will be visible instead of tubules).
Finally, recent evidence has emerged demonstrating a strong analogy between the central nervous system (CNS) pathology described here and the peripheral nervous system (PNS) pathology concerning α-synuclein deposition. Skin biopsies from the leg, back and finger of PD and control patients were obtained and the levels of co-localized phosphorylated α-synuclein quantified.38 The results showed that peripheral nerve involvement is intrinsic to PD and reflective of known CNS pathologies, implying that skin biopsies could be a useful tool to study its pathogenesis. A more recent study using skin biopsies from only the leg showed that intraneural phosphorylated α-synuclein in the PNS is a reliable in vivo biomarker of idiopathic PD.39 The authors suggested that α-synuclein spreads from the CNS to the PNS along adrenergic skin nerves and phosphorylation causes a conformational change in the native protein. These findings may contribute to elucidating the pathogenesis of PD and this premortem histopathology may have greater diagnostic value than current methods. However, further studies are needed to determine the exact sensitivity and specificity of using skin biopsies as a diagnostic tool, since both studies also found differences in the innervation pattern and spatial distribution of phosphorylated α-synuclein in the PNS.
Overall, the addition of axonal transport disruption to the other dysfunctions that result from pathogenic α-synuclein highlights the interrelations between these processes and the need for a more integrative approach to PD pathogenesis. The application of systems biology may be useful in this regard, and relating the pathogenic interaction between tau, α-synuclein and MTs in PD to that in Alzheimer’s pathogenesis will be of interest.
Author contributions
All authors contributed to the conception, design, drafting and writing of the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the accuracy and integrity of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Cartelli D, Cappelletti G. Microtubule destabilization paves the way to Parkinson’s disease. Mol Neurobiol. 2016;54(9):6762–6774. | ||
Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. | ||
Zhou R, Huang Y, Li X, et al. Molecular interaction of α-synuclein with tubulin influences on the polymerization of microtubule in vitro and structure of microtubule in cells. Mol Biol Rep. 2009;37(7):3183–3192. | ||
Prots I, Veber V, Brey S, et al. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J Biol Chem. 2013;288(30):21742–21754. | ||
Lee H, Khoshaghideh F, Lee S, Lee S. Impairment of microtubule-dependent trafficking by overexpression of α-synuclein. Eur J Neurosci. 2006;24(11):3153–3162. | ||
Volpicelli-Daley L, Gamble K, Schultheiss C, Riddle D, West A, Lee V. Formation of -synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell. 2014;25(25):4010–4023. | ||
Alim M, Hossain M, Arima K, et al. Tubulin seeds α-synuclein fibril formation. J Biol Chem. 2001;277(3):2112–2117. | ||
Chen L, Jin J, Davis J, et al. Oligomeric α-synuclein inhibits tubulin polymerization. Biochem Biophys Res Commun. 2007;356(3):548–553. | ||
Cartelli D, Aliverti A, Barbiroli A, et al. α-Synuclein is a Novel Microtubule Dynamase. Sci Rep. 2016;6:33289. | ||
Vossel KA, Xu JC, Fomenko V, et al. Tau reduction prevents Aß-induced axonal transport deficits by blocking activation of GSK3ß. J Cell Biol. 2015;11;209(3):419–433. | ||
Geddes J. α-Synuclein: a potent inducer of tau pathology. Exp Neurol. 2005;192(2):244–250. | ||
Volpicelli-Daley L. Effects of α-synuclein on axonal transport. Neurobiol Dis. 2017;105:321–327. | ||
Ishizawa T, Mattila P, Davies P, Wang D, Dickson D. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol. 2003;62(4):389–397. | ||
Jensen P, Hager H, Nielsen M, Højrup P, Gliemann J, Jakes R. α-Synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem. 1999;274(36):25481–25489. | ||
Wischik C, Novak M, Edwards P, Klug A, Tichelaar W, Crowther R. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85(13):4884–4888. | ||
Kadavath H, Hofele R, Biernat J, et al. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A. 2015;112(24):7501–7506. | ||
Mukrasch M, Biernat J, von Bergen M, Griesinger C, Mandelkow E, Zweckstetter M. Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions. J Biol Chem. 2005;280(26):24978–24986. | ||
Morishima-Kawashima M, Hasegawa M, Takio K, et al. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270(2):823–829. | ||
Haggerty T, Credle J, Rodriguez O, et al. Hyperphosphorylated Tau in an α-synuclein-overexpressing transgenic model of Parkinson’s disease. Eur J Neurosci. 2011;33(9):1598–1610. | ||
Biernat J, Gustke N, Drewes G, Mandelkow E, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11(1):153–163. | ||
Duka T, Duka V, Joyce J, Sidhu A. α-Synuclein contributes to GSK-3β-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009;23(9):2820–2830. | ||
Gąssowska M, Czapski G, Pająk B, Cieślik M, Lenkiewicz A, Adamczyk A. Extracellular α-synuclein leads to microtubule destabilization via GSK-3β-dependent tau phosphorylation in PC12 cells. PLoS One. 2014;9(4):e94259. | ||
Hongo H, Kihara T, Kume T, et al. Glycogen synthase kinase-3β activation mediates rotenone-induced cytotoxicity with the involvement of microtubule destabilization. Biochem Biophys Res Commun. 2012;426(1):94–99. | ||
Qureshi H, Paudel H. Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and α-synuclein mutations promote tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J Biol Chem. 2010;286(7):5055–5068. | ||
Giasson B. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300(5619):636–640. | ||
Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70(3):410–426. | ||
Cash A, Aliev G, Siedlak S, et al. Microtubule reduction in Alzheimer’s disease and aging is independent of τ filament formation. Am J Pathol. 2003;162(5):1623–1627. | ||
Li B, Chohan M, Grundke-Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 2007;113(5):501–511. | ||
Liu F, Li B, Tung E, Grundke-Iqbal I, Iqbal K, Gong C. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci. 2007;26(12):3429–3436. | ||
Roy B, Jackson G. Interactions between Tau and α-synuclein augment neurotoxicity in a Drosophila model of Parkinson’s disease. Hum Mol Genet. 2014;23(11):3008–3023. | ||
Stokin G, Goldstein L. Axonal transport and Alzheimer’s disease. Annu Rev Biochemistry. 2006;75(1):607–627. | ||
del Ser T, Lovestone S, Boada-Rovira M, et al. A phase II randomized, double-blind, parallel group, 26-week study of GSK-3 inhibitor tideglusib in Alzheimer’s disease (ARGO trial). Alzheimer’s & Dementia. 2013;9(4):P689–P690. | ||
Morales-Garcia J, Palomo V, Redondo M, et al. Crosstalk between phosphodiesterase 7 and glycogen synthase kinase-3: two relevant therapeutic targets for neurological disorders. ACS Chem Neurosci. 2014;5(3):194–204. | ||
Villar-Piqué A, Lopes da Fonseca T, Outeiro T. Structure, function and toxicity of alpha-synuclein: the Bermuda triangle in synucleinopathies. J Neurochem. 2015;139:240–255. | ||
Cartelli D, Casagrande F, Busceti C, et al. Microtubule alterations occur early in experimental parkinsonism and the microtubule stabilizer epothilone D is neuroprotective. Sci Rep. 2013;3:1837. | ||
Guo J, Covell D, Daniels J, et al. Distinct α-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell. 2013;154(1):103–117. | ||
Lashuel H, Overk C, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14(1):38–48. | ||
Doppler K, Ebert S, Üçeyler N, et al. Cutaneous neuropathy in Parkinson’s disease: a window into brain pathology. Acta Neuropathol. 2014;128(1):99–109. | ||
Donadio V, Incensi A, Piccinini C, et al. Skin nerve misfolded α-synuclein in pure autonomic failure and Parkinson disease. Ann Neurol. 2016;79(2):306–316. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.