Back to Journals » OncoTargets and Therapy » Volume 13
The CXCR4 Antagonist, AMD3100, Reverses Mesenchymal Stem Cell-Mediated Drug Resistance in Relapsed/Refractory Acute Lymphoblastic Leukemia
Authors Wang S, Wang X, Liu S, Zhang S, Wei X ![]() , Song Y, Yin Q
, Song Y, Yin Q
Received 12 February 2020
Accepted for publication 7 June 2020
Published 6 July 2020 Volume 2020:13 Pages 6583—6591
DOI https://doi.org/10.2147/OTT.S249425
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Shan Wang, Xiaojiao Wang, Sha Liu, Shengnan Zhang, Xudong Wei, Yongping Song, Qingsong Yin
Department of Hematology, The Affiliated Cancer Hospital of Zhengzhou University (Henan Cancer Hospital), Zhengzhou, People’s Republic of China
Correspondence: Yongping Song; Qingsong Yin Email [email protected]; [email protected]
Purpose: To investigate the role of the CXCR4/CXCL12 axis in chemotherapy resistance in refractory/relapsed (R/R) ALL patients.
Methods: CXCR4 expression on ALL cells from newly diagnosed or R/R ALL patients were detected using flow cytometry. The CXCR4/CXCL12 signaling pathway was blocked by the CXCR4 inhibitor AMD3100 in a co-culture model of primary drug-resistant ALL cells and umbilical cord mesenchymal stem cells (UCMSCs). Surface CXCR4 expression, apoptosis rate, and apoptosis-related protein expression in primary ALL cells under various treatments were detected.
Results: Of the 37 ALL patients examined, CXCR4 expression was higher in R/R patients than that in those with newly diagnosed disease. Similarly, in in vitro co-cultures of drug-resistant ALL cells with UCMSCs, the expression of CXCR4 was increased in the presence of vincristine (VCR), but reduced when VCR was combined with the CXCR4 antagonist AMD3100. Additionally, the supernatants of ALL-UCMSC co-cultures contained high CXCL12 concentrations, which were upregulated by VCR and significantly decreased by the combination of VCR plus AMD3100. Furthermore, the apoptosis rate of ALL cells significantly decreased, Bax expression was downregulated, and Bcl-2 was upregulated when ALL was co-cultured with UCMSCs compared with ALL cells alone. With the addition of VCR, the apoptosis rate mildly increased, Bax was upregulated, and Bcl-2 was downregulated. Nevertheless, the above results were further intensified, particularly Bax expression, when VCR was combined with AMD3100.
Conclusion: The CXCR4 antagonist could effectively reverse MSC-mediated drug resistance by blocking the CXCR4/CXCL12 axis and sensitizing leukemic cells from R/R ALL patients to chemotherapy drugs.
Keywords: acute lymphoblastic leukemia, relapsed/refractory, CXCR4/CXCL12 signaling, bone marrow microenvironment, drug resistance
Introduction
Although therapeutic regimens for acute lymphoblastic leukemia (ALL) have improved, only 30–40% of adult ALL patients can achieve long-term disease-free survival (DFS) when compared with 80% of children with ALL.1,2 Relapse and treatment failure in leukemia is mainly attributed to the presence of minimal residual diseases (MRD) and multidrug resistance (MDR). Patients with relapsed/refractory (R/R) ALL have poor prognosis, with a median overall survival (OS) ranging from 4.5 to 8.4 months.3
It has been found that interactions between ALL cells and the bone marrow microenvironment constitutively generate growth-promoting and anti-apoptotic signals, which are essential for drug resistance and relapse.4,5 CXC chemokine receptor-4 (CXCR4) is widely expressed on hematopoietic cells, and in the clinic, high CXCR4 expression has been linked to inferior outcome for B-ALL patients.6,7 The chemokine CXCL12, previously called stromal cell-derived factor-1 (SDF-1), is constitutively secreted by bone marrow stromal cells (BMSCs) and regulates the retention and migration of hematopoietic progenitor cells, mature hematopoietic cells, and various cancer cells, including B-ALL cells, and induces resistance.8 In addition to bone marrow derived stem cells (BMSC), there are also umbilical cord and placenta sourced MSCs. In this research, we chose umbilical cord mesenchymal stem cells (UC-MSCs), compared with BMSCs, UC-MSCs not only have convenience in collection, but also low risk of contamination and good proliferation ability. Most importantly, they having similar biological characteristics, and no significant differences in the ability of CXCL12 secretion.9 Moreover, CXCL12 has pro-survival and growth-promoting effects in normal and malignant B-cells. Once CXCL12 binds to CXCR4, the resulting activation of CXCR4/CXCL12 plays a key role in homing, survival, proliferation, and the extramedullary infiltration of ALL cells.10
Disrupting interactions between malignant hematopoietic cells and the bone marrow microenvironment is a promising therapeutic strategy that has attracted considerable attention. Various targets aiming at the interactions between these cells and the bone marrow microenvironment are currently being investigated.11,12 After blocking these interactions, leukemia cells can be mobilized from the protective niche in the bone marrow into peripheral blood (PB), as a result, which may enhance the efficacy of chemotherapy and reduce the occurrence of MDR and MRD.13,15 Many preclinical and clinical studies have revealed the potential clinical utility of targeting the CXCR4/CXCL12 axis in leukemia using CXCR4 antagonists, including small molecules, peptides, and monoclonal antibodies.16 AMD3100 (plerixafor) is a reversible small molecule inhibitor of CXCR4. It is currently used in combination with granulocyte colony-stimulating factor (G-CSF) as an HSC mobilizing agent and has been shown to effectively mobilize hematopoietic stem cells in patients with non-Hodgkin’s lymphoma in the clinic.17 Both preclinical18 and clinical studies19 have suggested that AMD3100 can effectively enhance sensitivity to anti-leukemia therapies and blast mobilization.
In this study, focus was given to the CXCR4/CXCL12 axis and interactions between leukemia blast cells and bone marrow stromal cells. Human primary relapsed/refractory B-ALL cells and UCMSCs were co-cultured to imitate the bone marrow microenvironment. Then, changes in the surface CXCR4 expression, the apoptosis rate of leukemic blast cells, and expression of apoptosis-related proteins were detected under various treatment conditions, including UCMSCs, chemotherapy drugs, and CXCR4 antagonists.
Patients and Methods
Clinical Sample Collection
All the patients enrolled were informed with the study and signed the informed consent form before the study. In accordance with the Declaration of Helsinki for protocols reviewed and approved by the Institutional Review Board of the Affiliated Cancer Hospital of Zhengzhou University, bone marrow samples were obtained from patients that fulfilled the clinical and immunophenotypic criteria for B-cell ALL in the Hematology Department. Bone marrow mononuclear cells (BMMCs) were isolated by density gradient centrifugation over Ficoll-Paque (GE Healthcare) and used fresh for flow cytometry detection or culture, or they were viably frozen in fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific) plus 10% dimethylsulfoxide (DMSO, Sigma-Aldrich) in liquid nitrogen.
Co-Culture System
As mentioned above, the bone marrow samples of relapsed and refractory B-ALL patients (peripheral blood leukocyte number is more than 50 ×109/L at diagnosis, and the proportion of ALL cells in bone marrow were ≥95%) were collected and detected for next cell culture. Then, the isolated ALL cells were cultured in RPMI 1640 medium containing 20% FBS for less than 24 hours. The UCMSCs were purchased from Shandong Cell Tissue Bank and cultured in DMEM/F12 (1:1) medium containing 10% FBS. The medium was changed every 48–72 hours. When the cells grew and became dense, they were digested with trypsin containing 0.25% EDTA and passaged. Passages less than five generations were used in experiments.
To investigate the protective role of the bone marrow microenvironment on ALL cells, UCMSCs were co-cultured directly with ALL cells to mimic the ALL bone marrow microenvironment. Briefly, 1 × 105 of UCMSCs were seeded into each well of a 6-well culture plate where they were cultured for approximately 24 hours, and the unattached cells were washed away. Then, 1 × 106 B-ALL cells per well were added to create the co-culture system. The groups were set as follows: primary ALL cells alone; primary ALL cells plus UCMSC co-culture; primary ALL cells plus UCMSCs plus 5 μg/mL AMD3100 (plerixafor); primary ALL cells plus UCMSC co-culture plus 1 μmol/L VCR; primary ALL cells plus UCMSC co-culture plus 1 μmol/L VCR and 10μmol/L (5 μg/mL) AMD3100. We chose VCR because it is most commonly used and has a high efficacy against ALL in the clinic. The concentrations of AMD310020 and VCR21 were determined according to the literature.
Detection of Surface CXCR4 Expression on Primary ALL Cells by Flow Cytometry
Surface CXCR4 expression was determined by flow cytometry after gating on CD45+CD19+cells. Briefly, mononuclear cells isolated from bone marrow samples or cells from each group cultured for 24 hours were stained with a saturating concentration of anti-CD45-perCP, anti-CXCR4-PE, and anti-CD19-APC, and respective isotype control antibodies for 30 minutes in the dark at room temperature. After staining, the cells were washed twice with RPMI 1640 containing 0.5% bovine serum albumin (BSA) and measured using a FACScan flow cytometer (BD Biosciences) followed by analysis with FCS Express 4 Plus Research Edition. Monoclonal antibodies (mAbs), including CD45-PerCP-Cy, CD19-APC, and CD184-PE (CXC chemokine receptor CXCR4) and relevant isotype control mAbs, were purchased from BD Biosciences. The expression of CXCR4 was shown as mean fluorescence intensity, ie, the fluorescence intensity of CXCR4 was divided by the fluorescence intensity of its isotype control.
Detection of CXCL12 in Supernatant by Enzyme-Linked Immunosorbent Assay (ELISA)
Supernatants from each group were collected, and the concentration of CXCL12 was detected using a human CXCL12 ELISA kit (R&D Systems), and the CXCL12 level in the supernatant was measured using a Quantikine kit within 30 minutes according to the manufacturer’s instructions (R&D Systems). High intra-assay and inter-assay precision was established by the manufacturer. The absorbance was recorded using a microplate reader (ELx808, Bio-Tek Instruments), and the data collection and analysis were performed using Gen5 software Version 1.08 (Bio-Tek Instruments).
Apoptosis Detection of Primary ALL Cells Using Annexin V-FITC/PI Double Staining
Cell viability was analyzed by Annexin V-FITC and propidium iodide (PI) staining. Briefly, at least 5 × 105 ALL cells were collected and processed according to the protocol, followed by flow cytometry analysis (FACS Calibur, BD Biosciences, San Jose, CA, USA) within 1 hour. The annexin V (-) PI (-) double negative area revealed living cells, the annexin V (+) PI (+) double-positive area revealed late apoptotic and dead cells, and the annexin V (+) PI (-) area revealed early apoptotic cells. The apoptotic rate was the sum of cells in the region to the right.
Detection of Apoptosis-Related Protein Expression in Primary ALL Cells by Immunoblotting
Protein extracts were obtained, and protein content was determined using a detergent-compatible protein assay kit according to the manufacturer’s instructions (Bio-Rad). Equivalent amounts of total cell protein were boiled with NuPAGE LDS sample buffer (Invitrogen). This mixture was then loaded in 4–12% gradient SDS-polyacrylamide gels (Invitrogen) and transferred onto nitrocellulose membranes (GE Osmonics Labstore). Then, Western blot analysis was conducted for Bcl-2 and Bax, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control housekeeping protein. All antibodies were purchased from Cell Signaling Technology. The blots were incubated with species-specific HRP-conjugated secondary antibody (1:5000) for one hour at room temperature followed by incubation with the Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific) for 1 or 2 minutes.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism version 6.00 for Mac (GraphPad Software, La Jolla, CA, 2013) and SPSS software version 24.0 (SPSS, Chicago, IL, USA). The clinical information of the patients was expressed as the mean ± standard error of the mean (SEM) or median and range as appropriate. For in vitro experiments, results are presented as the mean ± standard deviation (SD) of at least four independent experiments. For Western blotting, the data shown are representative of three independent experiments. Flow cytometry data were analyzed using FCS Express 4 Plus software. Comparisons of proportions and variables between different groups were performed using the Mann–Whitney or unpaired t-test as appropriate. P ≤ 0.05 was considered statistically significant.
Results
Surface CXCR4 Expression Significantly Increases with Disease Recurrence
CXCR4 and its ligand CXCL12 have emerged as key cellular and molecular players in the cross talk between hematopoietic malignancies and their microenvironment. To explore the relationship between CXCR4 expression and drug resistance, CXCR4 expression was detected before treatment and at various time points during treatment in newly diagnosed patients and before treatment in R/R patients. The BM samples were collected from a total of 37 patients with ALL, including 20 newly diagnosed patients and 17 R/R patients. The patient characteristics are summarized in Table 1. There were no statistically significant differences between the two groups in terms of age, gender, BM primary cells, peripheral blood leukocytes, hemoglobin, and thrombocytes. The MFI for CXCR4 in the R/R group was 588.54 ± 248.71, which was significantly higher than the 317.06 ± 165.32 in the untreated group (P = 0.0003). Furthermore, of the 20 untreated patients, 4 progressed from newly-diagnosed to the R/R stage, and it was found that the expression of CXCR4 increased with R/R disease (Figure 1A and B). The majority of patients achieved complete remission, and only six patients underwent a second CXCR4 expression test at two weeks of induction. Interestingly, CXCR4 levels decreased at two weeks of treatment compared with pretreatment in untreated patients (Figure 1C).
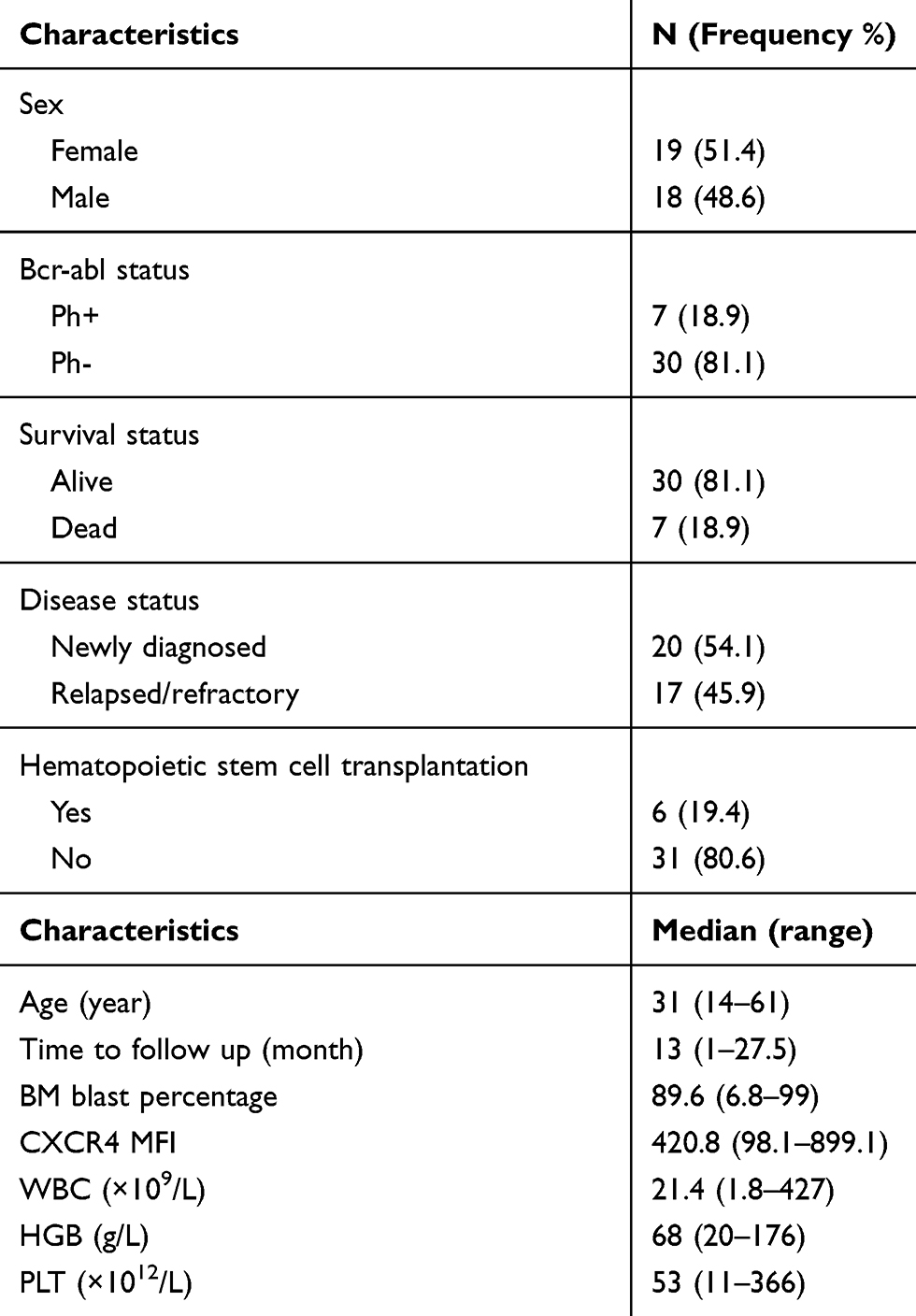 |
Table 1 Clinical Characteristics of ALL Patients (n = 37) |
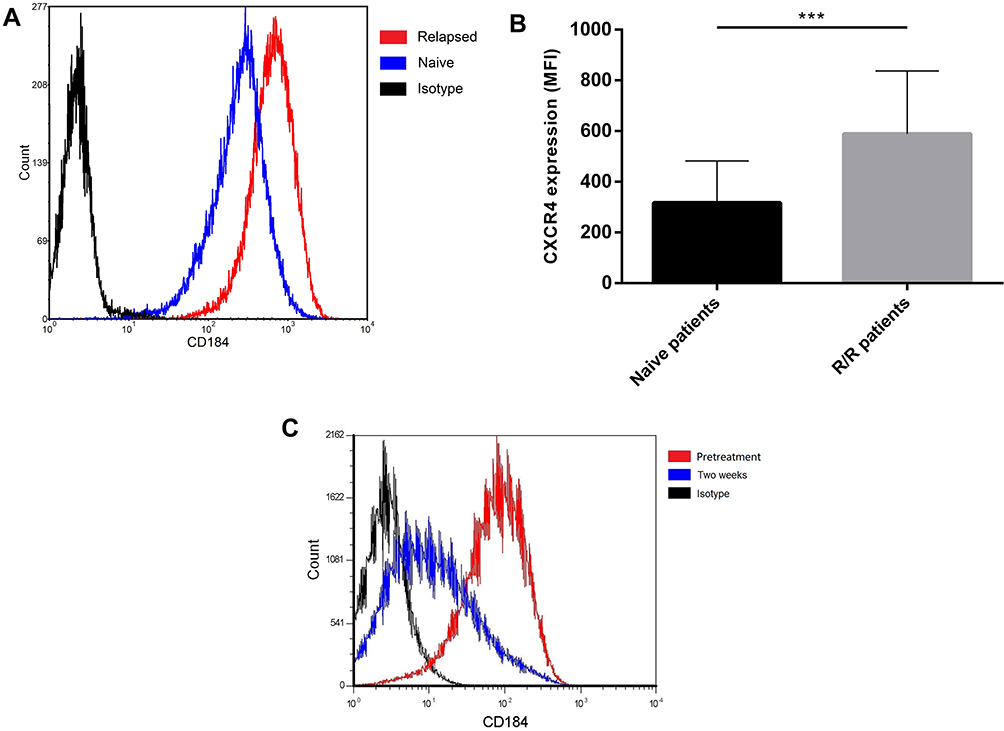 |
Figure 1 The increased surface CXCR4 expression is closely associated with recurrence in ALL patients. (A) One patient whose surface CXCR4 expression increased with disease recurrence. (B) The MFI of CXCR4 in the R/R group was significantly higher than that in untreated groups. (C) Surface CXCR4 levels decreased after two weeks of induction compared with pretreatment in untreated patients (***P < 0.001). |
Changes in the CXCL12 Concentration in Supernatants Under Different Treatment Conditions
Marrow stromal cells (MSCs) constitutively secrete the chemokine CXCL12, which is involved in the formation of complicated cytokine networks and mediates cross talk between ALL cells and the bone marrow microenvironment. UCMSCs also constitutively secrete the chemokine CXCL12, similar to MSCs. To investigate the protective role of the bone marrow microenvironment on ALL cells, UCMSCs were co-cultured with ALL cells to mimic the bone marrow microenvironment. It was found that supernatant from ALL-UCMSC co-cultures contained a high CXCL12 concentration after 24 hours (0.698 ± 0.088 ng), while the CXCL12 level was undetectable in the ALL alone group. In the co-cultured cells, there was no difference in CXCL12 secretion after the addition of AMD3100 (P = 0.532), but the CXCL12 concentration significantly increased when the culture was treated with VCR (1.351 ± 0.044 ng, P = 0.0079). However, the CXCL12 concentration was decreased when VCR was combined with AMD3100 compared with VCR alone (1.108 ± 0.041 ng, P = 0.0079, Figure 2A).
 |
Figure 2 Surface CXCR4 expression and CXCL12 levels are increased by VCR, then are reversed by the CXCR4 antagonist. (A) CXCL12 concentration increased when the co-cultured cells were treated with VCR, then decreased when VCR was combined with AMD3100. (B) Showed one representative of experiment depicting the change of surface CXCR4 expression under different treatment conditions. (C) Surface CXCR4 level increased when the cells were treated with VCR, then decreased when VCR was combined with AMD3100 (Co: co-culture; V: VCR alone; AMD+V: the combination of AMD3100 and VCR) (*P < 0.05, **P < 0.01, ***P < 0.001). Abbreviation: ns, not significant. |
Changes in CXCR4 Expression on Primary ALL Cells Under Different Treatment Conditions
To understand the interaction between CXCR4 and the CXCL12 secreted by UCMSCs in the co-culture system, the MFI of CXCR4 on primary R/R B-ALL cells cultured with or without UCMSCs was examined (Figure 2B). The MFI of CXCR4 mildly decreased under co-culture conditions (261.87 ± 91.07) compared with ALL cells alone (335.20 ± 12.85; P = 0.0809), and it was significantly decreased after the addition of AMD3100 (134.71 ± 21.78) compared with the co-culture (P = 0.0174). Nevertheless, the CXCR4 level significantly increased when the cells were treated with VCR (426.00 ± 13.66; P = 0.0059). Interestingly, CXCR4 expression was significantly reduced again when AMD3100 was combined with VCR compared with VCR alone (275.78 ± 54.20; P = 0.0009) (Figure 2C).
UCMSC Co-Culture Overcomes the Drug-Induced Apoptosis of Primary B-ALL Cells and the CXCR4 Antagonist Reverses This Effect
To clarify the effects of UCMSCs on ALL cell drug resistance, the apoptosis rate of ALL cells was measured under different treatment conditions (Figure 3A). We found that the apoptosis rate of ALL cells decreased from 64.76 ± 2.37% in the ALL cell alone group to 57.06 ± 3.41% in the ALL-UCMSC co-culture group (P = 0.0079). The addition of AMD3100 or VCR resulted in similar increases in the apoptosis rate during co-culture (both P = 0.0079). Nevertheless, the apoptosis rate increased to 83.47 ± 2.43% when AMD3100 was combined with VCR (P < 0.0001), which is significantly higher than that found for VCR alone (74.60 ± 2.93) % (P = 0.0079, Figure 3B).
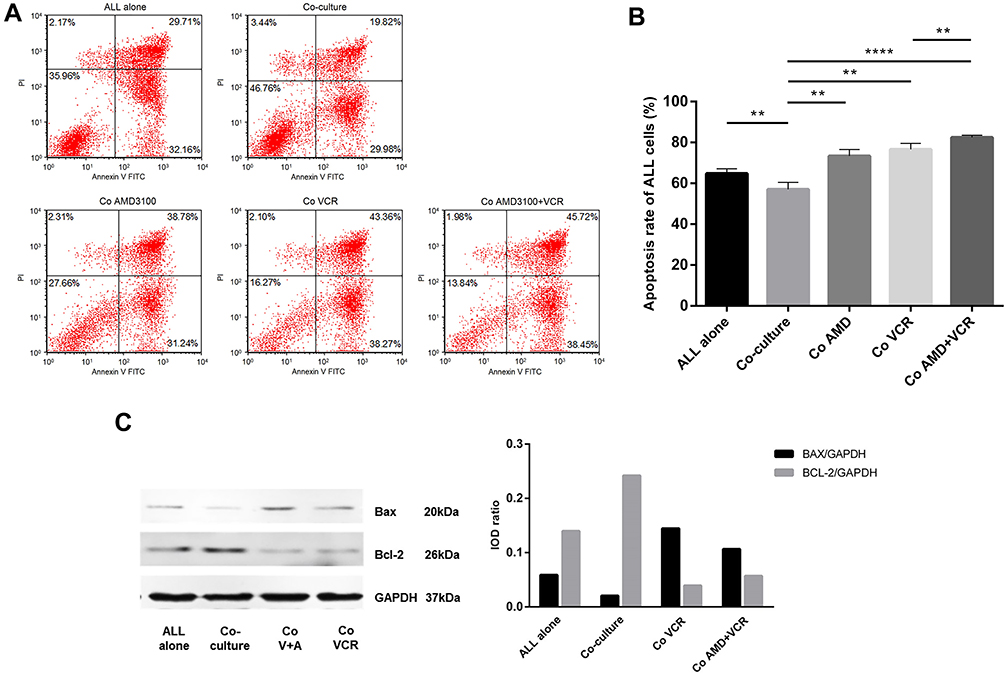 |
Figure 3 UCMSC co-culture overcomes the drug-induced apoptosis of primary B-ALL cells and the CXCR4 antagonist reverses this effect. (A) The dot plots showed a representative of experiment depicting change of the apoptosis rate of primary ALL cells under various treatment conditions. (B) The apoptosis rate of ALL cells decreased in ALL-UCMSC co-cultures, then increased with the addition of AMD3100 or VCR agent, and continued to increase when AMD3100 was combined with VCR. (C) UCMSCs reduce ALL apoptosis by upregulating Bcl-2 and downregulating Bax, and the CXCR4 antagonist sensitizes cells to VCR by upregulating Bax (Co: co-culture; V: VCR alone; AMD+V: the combination of AMD3100 and VCR) (**P < 0.01, ****P < 0.0001). Abbreviation: ns, not significant. |
UCMSCs Reduce ALL Apoptosis by Upregulating Bcl-2 and the CXCR4 Antagonist AMD3100 Sensitizes Cells to VCR by Upregulating Bax
To further understand the changes in apoptosis-related protein expression in primary ALL cells under different treatment conditions, the levels of the Bax and Bcl-2 proteins were detected. Bax was downregulated, and Bcl-2 was upregulated when ALL cells were co-cultured with UCMSCs. In contrast, Bax was upregulated, and Bcl-2 was downregulated after treatment with VCR. Furthermore, Bax continued to increase when AMD3100 was combined with VCR; however, the Bcl-2 level did not significantly change (Figure 3C). These results are representative of three independent experiments.
Discussion
There is compelling evidence for cross talk between ALL cells and stromal cells in the bone marrow microenvironment, which probably is one of the main causes for drug-resistance and recurrence in adult ALL patients. Disrupting this cross talk is an attractive therapeutic strategy. Nevertheless, the underlying mechanisms have not been fully characterized in R/R ALL. In this study, it was found that MSCs could protect primary ALL cells from apoptosis and VCR-induced cell death by downregulating CXCR4 and Bax expression and upregulating BCL-2. Furthermore, the CXCR4 antagonist AMD3100 inhibited the cross talk between chemo-resistant ALL cells and the microenvironment by blocking the CXCR4/CXCL12 axis, thereby reversing the protective effects of MSCs and sensitizing to VCR by upregulating Bax expression.
The CXCR4/CXCL12 axis plays a key role in homing hematopoietic stem cells and leukemic cells to the bone marrow microenvironment.8 Overexpression of CXCR4 grants leukemic blasts a higher capacity to seed into BM niches, thereby protecting leukemic cells from chemotherapy-induced apoptosis.22 It has been verified that CXCR4 overexpression on the surface of hematological tumors is strictly related to disease progression and poor prognosis.6,22,24 In addition, high expression of CXCR4 and phosphorylated CXCR4 is prone to extramedullary infiltration in ALL patients.7 In this study, we found that high CXCR4 expression is closely associated with recurrence in ALL patients, again confirming that it is a poor prognostic factor.
Of note, surface CXCR4 expression was induced to further increase on chemo-resistant leukemic cells by VCR, which is consistent with the increase of CXCR4 expression in the R/R ALL patients in this study and ALL cell lines reported in another study.25 In contrast, CXCR4 expression could be reduced on sensitive cells from untreated patients in accordance with a previous report in an ALL mouse model.25 More studies are needed to fully understand these phenomena. Simultaneously, the level of CXCL12 secreted by UCMSCs increased significantly when the drug-resistant cells were co-cultured with UCMSCs by VCR intervention. Furthermore, our data showed an interesting result that there is a bit difference between the AMD3100 and AMD3100+VCR in co-culture groups, we did not perform further experiments to elucidate this remaining question, but we assume that this may due to the strong effect of VCR. These findings confirm that activation of the CXCR4/CXCL12 axis is induced by VCR by increasing the CXCR4 and CXCL12 levels, which is then involved in ALL drug resistance.26 As shown in the ALL cells apoptosis, UCMSCs show direct protection to ALL cells apoptosis as well as VCR-induced cell death, which further demonstrates the protective effects of UCMSCs on ALL cells.
Curiously, we also found that the surface expression of CXCR4 showed a decreasing trend when ALL cells were co-cultured with UCMSCs while the cell viability improved. These are seemingly contradictory results, which may be actually attributed to the increase in CXCL12 secreted by UCMSCs and then internalization of CXCR4 due to the binding of its ligand CXCL12. Nevertheless, subsequent activation of the intracellular signaling cascades induced by binding could maintain cell survival by downregulating the pro-apoptotic protein Bax and upregulating the anti-apoptotic protein Bcl-2, thereby significantly resisting VCR-induced apoptosis.
As previously reported, the small molecule CXCR4 antagonist AMD3100 (plerixafor) could mobilize AML cells from the protective BM microenvironment by blocking CXCL12 binding to CXCR4, resulting in susceptibility to chemotherapy.27,30 In this study, we found that the increased surface CXCR4 expression and CXCL12 secretion induced by VCR were downregulated due to the intervention of AMD3100. More importantly, as expected, AMD3100 could reverse the protective effects of UCMSCs and further improve the sensitivity of ALL cells to VCR to some degree by upregulating Bax, which is in concert with the literature where it was shown to significantly enhance the anti-leukemia effects of cytarabine,19 which more effectively impairs the protective effects of higher CXCR4 expression.25 Nevertheless, further experiments are needed to verify whether disturbing the CXCR4/CXCL12 axis will sensitize chemotherapy drugs in ALL xenograft models.
Conclusions
Together, these results confirmed that the bone marrow microenvironment did provided protections to the ALL cells through the CXCR4/CXCL12 axis, and they play an important role in drug resistance by activating the CXCR4/CXCL12 axis. Besides, these interactions can be cut off effectively, blocking CXCR4 by AMD3100 reverses its protective effects and sensitizes cells to chemotherapeutic drugs by downregulating CXCR4 and Bcl-2 and upregulating Bax. These findings provide experimental data for the clinical development of small molecule inhibitors in ALL patients.
Abbreviations
ALL, acute lymphoblastic leukemia; DFS, disease-free survival; MRD, minimal residual diseases; MDR, multidrug resistance; R/R, relapsed/refractory; OS, overall survival; BMSCs, bone marrow stromal cells; HPC, hematopoietic progenitor cells; G-CSF, granulocyte colony-stimulating factor; UCMSCs, umbilical cord mesenchymal stem cells.
Acknowledgments
The authors are grateful to Professor Yangqiu Li (Jinan University, Guangzhou) for outstanding manuscript edits and Central Lab (The Affiliated Cancer Hospital of Zhengzhou University) for assistance with data collection.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Steliarova-Foucher E, Colombet M, Ries LAG, et al. International incidence of childhood cancer, 2001–10: a population-based registry study. Lancet Oncol. 2017;18(6):719–731. doi:10.1016/S1470-2045(17)30186-9
2. Gaudichon J, Jakobczyk H, Debaize L, et al. Mechanisms of extramedullary relapse in acute lymphoblastic leukemia: reconciling biological concepts and clinical issues. Blood Rev. 2019;36:40–56. doi:10.1016/j.blre.2019.04.003
3. Gökbuget N, Stanze D, Beck J, et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120(10):2032–2041. doi:10.1182/blood-2011-12-399287
4. Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia. 2009;23(12):2233–2241. doi:10.1038/leu.2009.175
5. Shain KH, Tao J. The B-cell receptor orchestrates environment-mediated lymphoma survival and drug resistance in B-cell malignancies. Oncogene. 2014;33(32):4107–4113. doi:10.1038/onc.2013.379
6. Möhle R, Schittenhelm M, Failenschmid C, et al. Functional response of leukaemic blasts to stromal cell-derived factor-1 correlates with preferential expression of the chemokine receptor CXCR4 in acute myelomonocytic and lymphoblastic leukaemia. Br J Haematol. 2000;110(3):563–572. doi:10.1046/j.1365-2141.2000.02157.x
7. Konoplev S, Jorgensen JL, Thomas DA, et al. Phosphorylated CXCR4 is associated with poor survival in adults with B-acute lymphoblastic leukemia. Cancer. 2011;117(20):4689–4695. doi:10.1002/cncr.26113
8. Richardson SM, Kalamegam G, Pushparaj PN, et al. Mesenchymal stem cells in regenerative medicine: focus on articular cartilage and intervertebral disc regeneration. Methods. 2016;99:69–80. doi:10.1016/j.ymeth.2015.09.015
9. Baksh D, Yao R, Tuan RS. Comparison of proliferative and multilineage differentiation potential of human mesenchymal stem cells derived from umbilical cord and bone marrow. Stem Cells (Dayton, Ohio). 2007;25(6):1384–1392. doi:10.1634/stemcells.2006-0709
10. de Lourdes Perim A, Amarante MK, Guembarovski RL, de Oliveira CEC, Watanabe MAE. CXCL12/CXCR4 axis in the pathogenesis of acute lymphoblastic leukemia (ALL): a possible therapeutic target. Cell Mol Life Sci. 2015;72:1715–1723. doi:10.1007/s00018-014-1830-x
11. Bray LJ, Binner M, Körner Y, et al. A three-dimensional ex vivo tri-culture model mimics cell-cell interactions between acute myeloid leukemia and the vascular niche. Haematologica. 2017;102(7):1215–1226. doi:10.3324/haematol.2016.157883
12. Chen H, Sanchez E, Soof CM, et al. JAK1/2 pathway inhibition suppresses M2 polarization and overcomes resistance of myeloma to lenalidomide by reducing TRIB1, MUC1, CD44, CXCL12, and CXCR4 expression. Br J Haematol. 2019. doi:10.1111/bjh.16158
13. Passaro D, Quang CT, Ghysdael J. Microenvironmental cues for T‐cell acute lymphoblastic leukemia development. Immunol Rev. 2016;271:156–172. doi:10.1111/imr.12402
14. van den Berk LCJ, van der Veer A, Willemse ME, et al. Disturbed CXCR 4/CXCL 12 axis in paediatric precursor B‐cell acute lymphoblastic leukaemia. Br J Haematol. 2014;166:240–249. doi:10.1111/bjh.12883
15. Parameswaran R, Yu M, Lim M, Groffen J, Heisterkamp N. Combination of drug therapy in acute lymphoblastic leukemia with a CXCR4 antagonist. Leukemia. 2011;25(8):1314–1323. doi:10.1038/leu.2011.76
16. Cho B-S, Kim H-J, Konopleva M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: from bench to bedside. Korean J Intern Med. 2017;32:248–257. doi:10.3904/kjim.2016.244
17. Flomenberg N, Devine SM, DiPersio JF, et al. The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood. 2005;106(5):1867–1874. doi:10.1182/blood-2005-02-0468
18. Juarez J, Bradstock KF, Gottlieb DJ, Bendall LJ. Effects of inhibitors of the chemokine receptor CXCR4 on acute lymphoblastic leukemia cells in vitro. Leukemia. 2003;17(7):1294–1300.
19. Cooper TM, Sison EAR, Baker SD, et al. A Phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: a pediatric oncology experimental therapeutics investigators’ co.Pediatr Blood Cancer. 2017;64(8):e26414. doi:10.1002/pbc.26414
20. Pavlasova G, Borsky M, Seda V, et al. Ibrutinib inhibits CD20 upregulation on CLL B cells mediated by the CXCR4/SDF-1 axis. Blood. 2016;128(12):1609–1613. doi:10.1182/blood-2016-04-709519
21. Shang J, Chen Z, Wu W, Wei T, Chen W. Proliferation-inhibiting and multidrug-resistant reversing effect of bortezomib on human HL-60 cells. Zhonghua Xue Ye Xue Za Zhi. 2012;33(11):911–916.
22. Peled A, Klein S, Beider K, Burger JA, Abraham M. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine. 2018;109:11–16. doi:10.1016/j.cyto.2018.02.020
23. Du W, Lu C, Zhu X, et al. Prognostic significance of CXCR4 expression in acute myeloid leukemia. Cancer Med. 2019;8(15):6595–6603. doi:10.1002/cam4.2535
24. Vadillo E, Dorantes-Acosta E, Pelayo R, Schnoor M. T cell acute lymphoblastic leukemia (T-ALL): new insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018;32(1):36–51. doi:10.1016/j.blre.2017.08.006
25. Sison EAR, Magoon D, Li L, et al. Plerixafor as a chemosensitizing agent in pediatric acute lymphoblastic leukemia: efficacy and potential mechanisms of resistance to CXCR4 inhibition. Oncotarget. 2014;5(19):8947–8958. doi:10.18632/oncotarget.2407
26. Tesfai Y, Ford J, Carter KW, et al. Interactions between acute lymphoblastic leukemia and bone marrow stromal cells influence response to therapy. Leuk Res. 2012;36(3):299–306. doi:10.1016/j.leukres.2011.08.001
27. Stamatopoulos B, Meuleman N, De Bruyn C, et al. AMD3100 disrupts the cross-talk between chronic lymphocytic leukemia cells and a mesenchymal stromal or nurse-like cell-based microenvironment: pre-clinical evidence for its association with chronic lymphocytic leukemia treatments. Haematologica. 2012;97(4):608–615. doi:10.3324/haematol.2011.052779
28. Yu M, Gang EJ, Parameswaran R, et al. AMD3100 sensitizes acute lymphoblastic leukemia cells to chemotherapy in vivo. Blood Cancer J. 2011;1(4):e14. doi:10.1038/bcj.2011.13
29. Uy GL, Rettig MP, Motabi IH, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119(17):3917–3924. doi:10.1182/blood-2011-10-383406
30. Welschinger R, Liedtke F, Basnett J, et al. Plerixafor (AMD3100) induces prolonged mobilization of acute lymphoblastic leukemia cells and increases the proportion of cycling cells in the blood in mice. Exp Hematol. 2013;41(3):293–302.e1. doi:10.1016/j.exphem.2012.11.004
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.