Back to Journals » Journal of Inflammation Research » Volume 19
The Crosstalk Between Efferocytosis and Macrophage Polarization in Diabetic Wounds: A Comprehensive Review
Authors He Y, Hu J, Ma A, Du P, Yang M, Xiong X, Deng Y
Received 17 September 2025
Accepted for publication 2 February 2026
Published 25 March 2026 Volume 2026:19 566523
DOI https://doi.org/10.2147/JIR.S566523
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Quan Zhang
Yuxin He,1,2 Jie Hu,1,2 Anqi Ma,1,2 Peiyang Du,1,2 Mengdie Yang,1,2 Xia Xiong,2 Yongqiong Deng1
1Department of Dermatology, Chengdu Integrated TCM and Western Medicine Hospital, Chengdu, Sichuan, 610041, People’s Republic of China; 2Department of Dermatology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China
Correspondence: Xia Xiong, Department of Dermatology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China, Email [email protected] Yongqiong Deng, Department of Dermatology, Chengdu Integrated TCM and Western Medicine Hospital, Chengdu, Sichuan, 610041, People’s Republic of China, Email [email protected]
Abstract: Chronic refractory cutaneous wounds are major complications of diabetes. A hallmark of this pathology is persistent inflammation, driven in part by the accumulation of apoptotic cells (ACs) due to impaired clearance. Efferocytosis, as the primary process for removing ACs, was disordered when triggered by the diabetic microenvironment, further leading to impaired macrophage polarization and consequently delaying wound healing. This review also focuses on the abnormal crosstalk between efferocytosis and macrophage polarization and its modulated signaling pathways governing this axis, and reviewing potential novel therapeutic strategies targeting the restoration of clearing ACs. This review may offer new insights and directions for overcoming current treatment drawbacks in diabetic wound healing.
Keywords: apoptotic cells, efferocytosis, macrophage polarization, diabetic wound healing
Introduction
Diabetes, a chronic metabolic disease, affected the health of 537 million individuals globally in 2022 and is projected to rise to 783 million by 2045.1 Chronic hyperglycemia induces progressive damage to multiple organ systems, such as the cardiovascular, cerebrovascular, renal, ophthalmic, and peripheral vascular systems. Among these complications, 19–34% of diabetic patients develop diabetic foot ulcers, imposing a heavy burden on patients’ quality of life and healthcare systems.2 Globally, diabetic wounds account for between 50% and 70% of all limb amputations, with statistics indicating that one leg is amputated every 30 seconds as a result.3 Furthermore, the annual healthcare costs associated with chronic wounds constitute approximately 6% of total medical expenditures in the developed countries.4 Current clinical approaches for managing diabetic wounds primarily include glycemic control, surgical debridement, negative pressure therapy, vascularized flap reconstruction, topical antibiotics, and wound dressing application.5 While these established methods may partially relieve symptoms, they do not reduced the amputation rate and fail to adequately target the underlying pathological mechanisms. This limitation highlights the urgent need for novel treatment strategies based on the pathogenesis of the disease.
The defining hallmark of the non-healing diabetic wound is a state of persistent, non-resolving inflammation.6 The diabetic wound microenvironment is locked in a chronic inflammatory response that actively impedes the transition into proliferation phase.7 Central to this dysregulation is the macrophage, a master regulator of immunity and repair. Macrophages undergo a timely phenotypic switch from pro-inflammatory (M1) state to anti-inflammatory (M2) state in normal wound healing. In diabetes, this critical transition fails, and macrophages remain pathologically arrested in a pro-inflammatory phenotype, perpetuating a cycle of tissue damage and impaired repair.8,9 Emerging evidence now found that the clearance of apoptotic cells (ACs)—a process known as efferocytosis—is closely related to macrophage polarization.10 Successful efferocytosis can drive the shift of macrophages from M1 to M2 phenotype. The transition stimulate the release of crucial anti-inflammatory mediators such as transforming growth factor-β (TGF-β) and interleukin-10 (IL-10).11,12 Thus, efferocytosis serves as a pivotal factor in regulating macrophage polarization, concluding the inflammatory response and launching subsequent tissue regeneration, acting as a core biological bridge connecting the inflammatory phase to the proliferative phase.13,14 In the diabetic wound microenvironment, however, this sophisticated regulatory axis is severely disrupted.15 Hyperglycemia, advanced glycation end products (AGEs), oxidative stress, hypoxia, biofilm, protease load, cellular senescence, neutrophil extracellular traps (NETs) and impaired angiogenesis converge to cripple efferocytosis by mainly disrupting both the recognition and engulfment phases: they suppress key bridging molecules and their receptors (MerTK), and inhibit essential intracellular machinery like Rac1 for phagocytic cup formation.16–20 Defective efferocytosis, in turn, starves macrophages of the signals required for phenotypic switching, locking them in a chronic inflammatory state and fueling a vicious cycle of impaired clearance and sustained inflammation.21–23
Therefore, this review discusses the central role of macrophage efferocytosis- polarization axis and its regulatory mechanisms in impaired diabetic wound healing, in order to provide a theoretical foundation and future perspectives for breaking the vicious cycle of inflammation in diabetic wounds and advancing clinical translation.
Molecular and Cellular Mechanisms in Efferocytosis
Efferocytosis, the process of phagocytic clearance of ACs, is a key biological mechanism for maintaining tissue homeostasis and regulating immune responses.14 Efferocytosis involve series of tightly regulated molecular events. Altogether, the processes of efferocytosis regulated by four steps.
Recruitment
Phagocytes do not randomly search for ACs but directionally along chemotactic gradients established by ACs-derived soluble factors termed “find-me” signals.24 These chemoattractants guide phagocytes to precisely and efficiently target ACs for clearance. At the same time, it also promotes the rearrangement of phagocyte skeleton, strengthens the expression and digestion mechanism of phagocytic receptors, and promotes the regeneration and anti-inflammatory properties of phagocytes.25 Currently, four well-characterized “find-me” signals have been characterized: lysophosphatidylcholine (LPC),26 sphingosine 1-phosphate (S1P),27 fractalkine (CX3CL1),28 and nucleotides (ATP and UTP) (Figure 1A). 29
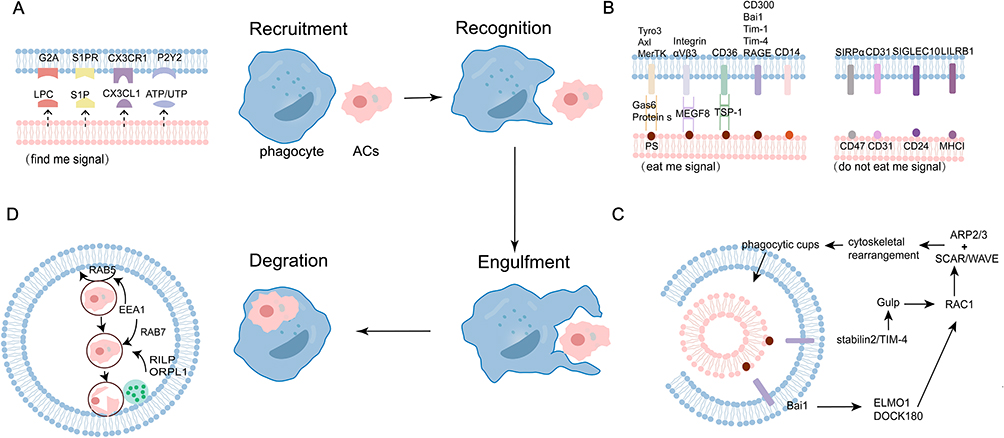 |
Figure 1 The processes of efferocytosis. (A) Apoptotic cells (ACs) first release chemotactic “find-me” signals—including lysophosphatidylcholine (LPC), sphingosine-1-phosphate (S1P), CX3CL1, and ATP/UTP—to attract phagocytes. (B) Upon arrival, these phagocytes discriminate ACs through balanced “eat-me” and “don’t-eat-me” signals, while further cell-surface “eat me” and “do not eat me” signals assist with the subsequent stage of cell engulfment. (C) Following recognition, phagocytes rapidly initiate cytoskeletal rearrangements and form a phagocytic cup to efficiently engulf the ACs—a process largely dependent on the Rho family of small GTPases, particularly Rac1. (D) Finally, the internalized ACs are then processed through a maturation sequence in which nascent phagosomes progress from early to late stages before fusing with lysosomes; Rab5-GTP and Rab7-GTP sequentially govern the transition to early and late phagosomes, respectively, ensuring complete degradation. |
LPC was the earliest identified “find-me” signal in ACs clearance.24 It was reported that LPC is primarily generated via hydrolysis of membrane phosphatidylcholine in ACs, a process mediated by caspase-3-dependent activation of phospholipase A2 (PLA2).30 Once release, LPC binds to and activates the G-protein-coupled receptor G2A, promoting the migration of phagocytes toward ACs. S1P is another critical lipid “find-me” signal.27 It exhibits high affinity for its cognate receptors S1PR1–5, which regulates key cellular processes such as macrophage migration, adhesion, and invasion.31 The binding of S1P by S1PR on macrophages not only inhibits apoptosis but also initiates an autocrine signaling cascade. This cascade involves peroxisome proliferator-activated receptor α (PPARα), which upregulates the expression of receptors for dying cells.32,33 Consequently, S1P has the potential to induce sustained efferocytosis. It is hypothesized that specific cell types, such as apoptotic B cells and neurons, may release soluble CX3CL1, which could then be sensed by CX3CR1 receptors on phagocytes and theoretically contribute to their recruitment. However, direct evidence placing CX3CL1 firmly within the established “find-me” signal paradigm of efferocytosis remains limited, and its expression appears restricted to specific contexts.14 Additionally, nucleotides such as ATP and UTP act as “find-me” signals in the process of efferocytosis. These molecules are recognized by purinergic receptors, particularly P2Y subtypes, on monocytes and macrophages.34
Recognition
After localizing ACs, the second phase of efferocytosis is “Recognition”, mediated by interactions between “eat-me” signals on ACs and corresponding receptors on phagocytes.
Current evidence identifies multiple recognition ligands including phosphatidylserine (PS), oxLDL, calreticulin, ICAM3, complement component 1q (C1q) and Annexin I (Figure 1B).14 Among these, PS represents the most fundamental and prominent “eat me” signal. 35 As a phospholipid, PS is mainly located in the inner leaflet of plasma membrane under normal conditions. This characteristic asymmetry in phospholipid distribution is actively sustained by flippase enzymes, which facilitate the ATP-dependent translocation of specific phospholipids from the outer to the inner leaflet.36,37 However, during cellular stress or apoptosis, the asymmetric distribution is disrupted, leading to PS externalization to the outer membrane leaflet where it becomes accessible for recognition by phagocytic receptors.38 PS can directly interact with phagocytes receptors, including CD300 family, Bai1, Tim-4, Tim-1, the receptor for advanced glycation end products (RAGE), etc. Alternatively, PS indirectly engages TAM families (Tyro3, Axl, MerTK), integrins, and CD36 via soluble bridging molecules,39,40 such as growth arrest-specific gene 6 (Gas6), milk fat globule-epidermal growth factor 8 (MFG-E8), protein S, CCN1 and thrombospondin-1.39 This recognition mechanism—whether mediated indirectly through bridging molecules or via direct contact with ACs—also extends to other engulfment receptors that are not specific to PS.
The Recognition of “eat me” signal is a prerequisite for efferocytosis. In contrast, “don’t-eat-me” signals—such as CD31, CD47, CD24, PD-L1, and MHC I—protect healthy cells from phagocytosis when they are within the range of phagocytes attack.41 While viable cells may co-express both types of signals, ACs predominantly exhibit “eat-me” signals, leading to their targeted engulfment.24
Engulfment
In this stage, receptor-mediated signaling during efferocytosis promotes the actin polymerization and cytoskeletal rearrangement, allowing the plasma membrane to penetrate and localize and forming the “phagocytic cup”, that subsequently engulfs the ACs (Figure 1C). 42
The small GTPase Rac1, a key member of the Rho family, plays a central role in this stage.43 Rac1 activation occurs through two primary mechanisms: (1) the LRP1/GULP-dependent pathway, such as stabilin-2 and tim4;44–46 (2) the DOCK180/ELMO-dependent pathway, which requires the guanine nucleotide exchange factor (GEF) complex composed of DOCK180 and ELMO1.47 Brain-specific angiogenesis inhibitor 1 (BAI1) interacts with ELMO1 and functions as a bipartite GEF alongside DOCK180 to activate Rac1. BAI1 recognizes PS on ACs via its extracellular thrombospondin type 1 repeats, triggering the ELMO1–DOCK180–Rac1 signaling module and inducing actin cytoskeletal rearrangement.48 Once activated through either way, RAC1 is subsequently activates nucleation-promoting factors of the WASP family, SCAR and WAVE. These proteins associate with the ARP2/3 complex to nucleate new actin filaments and facilitate the formation of “phagocytic cup”.36
Degradation
Degradation is the terminal phase of efferocytosis. Following “phagocytic cup” closure, the newly formed phagosome undergoes a maturation process characterized by sequential Rab GTPase recruitment (Rab5→Rab7) and progressive acidification (Figure 1D). This maturation culminates in fusion with lysosomes containing pH-sensitive degrading enzymes, enabling complete degradation of the apoptotic cell cargo. Concurrently, anti-inflammatory factors such as IL-10 are released to resolve the immune response. Finally, membrane components and efferocytosis receptors are recycled to restore cellular homeostasis and maintain efferocytosis capacity.49,50
Early phagosomes recruit RAB5, a key regulator of endocytic transport and phagosome maturation.51 This step facilitates the assembly and activation of effector proteins such as early endosomal antigen 1 (EEA1) and the fusion mediators MON1A and MON1B.52,53 As maturation proceeds, phagosomes transition from early to late stages, characterized by the exchange of RAB5 for RAB7.36 Central displacement of late phagosomes and partial formation of the phagosomes / lysosome (phagolysosome) are regulated by the dynactin–dynein complex in conjunction with GTP-bound RAB7.54,55 The formation and migration of phagolysosomes require tremendous attention to membrane fusion. Soluble N-ethylmaleimide-sensitive fusion factor attachment protein receptor (SNAREs) and N-ethylmaleimide-sensitive factor (NSF), which function as catalytic elements in mediating membrane merger. The membrane fusion process is driven by a dynamic cycle of SNARE complex assembly and disassembly. During the assembly phase, syntaxin undergoes conformational activation to form a Q-SNARE precomplex, followed by the initiation of SNARE complex formation that overcomes the repulsion and ultimately establishes a stable cis-SNARE complex. In the disassembly phase, SNAPs and NSF are recruited to the complex, and ATP-dependent hydrolysis enables the recycling of SNARE proteins.56 Alternatively, LC3-associated phagocytosis (LAP) enhances this process through LC3 lipidation, which accelerates phagosome-lysosome fusion and the breakdown of ACs.36
Failure to efficiently clear ACs leads to secondary necrosis and subsequent release of damage-associated molecular patterns (DAMPs), which potently initiate proinflammatory responses that exacerbate local tissue damage.57 DAMPs are metabolically diverse entities, including genomic and mitochondrial DNA, high-mobility group protein B (HMGB), histones, cytoplasmic proteins (S100), cytokines (IL-1α, IL-33, IL-36) and other small molecules (ATP, UTP, uric acid crystals).58
Diabetic Microenvironment Triggering Impaired Efferocytosis in Diabetic Wounds
Impaired Efferocytosis in Diabetic Wounds
Normal wound healing is a highly coordinated and time-dependent dynamic process, in which timely and effective resolution of inflammation serves as a critical turning point to facilitate a smooth transition into the proliferative phase.59 Efferocytosis plays a central role mainly during the inflammatory phase, but its impact goes far beyond that, creating a favorable microenvironment for subsequent repair and regeneration by efficiently clearing dead or senescent cells. This process not only prevents ACs from undergoing secondary necrosis and releasing their harmful contents, thereby avoiding exacerbating tissue damage and inflammatory responses, but also, by activating a series of downstream signaling pathways, actively promotes resolution of inflammation, angiogenesis, and extracellular matrix remodeling, ultimately leading to successful wound healing.60
One of the core pathophysiological characteristics of diabetic wounds is that the wound is in a chronic inflammatory state for a long time, which cannot smoothly transition from the inflammatory phase to the proliferative phase.61 Emerging clinical and experimental evidence establishes impaired efferocytosis is a key link leading to this pathological condition.62,63 Savita et al provided the first direct evidence of dysfunctional efferocytosis in diabetic wounds, correlating it with a significant accumulation of ACs in diabetic wounds.63 This failure to clear ACs is not merely a consequence but an active driver of pathology; experimental induction of apoptosis in diabetic mice confirmed that an increased apoptotic load directly impedes wound closure.63 It established that macrophages taken from the wounds of diabetic mice exhibit impaired efferocytosis, resulting in accumulated ACs (including neutrophils and endothelial cells) within the wound microenvironment.64 Besides, the greater number of neutrophils in the wound exudate of the diabetic mice was also correlated with an impaired efferocytosis.65 The resulting ACs accumulation propagates inflammation by shifting the cytokine balance, typically characterized by elevated levels of pro-inflammatory mediators like tumor necrosis factor-α (TNF-α) and IL-6 alongside a reduction in the anti-inflammatory IL-10.59,66
Diabetic Microenvironment as A Trigger for Efferocytosis Dysfunction
In the complex pathological microenvironment of diabetic wounds, efferocytosis is disrupted by multiple factors.36,63 Hyperglycemia is a central biochemical feature of diabetes. In vitro experiments demonstrate that macrophages cultured under high glucose conditions exhibit a significantly reduced capacity to clear ACs compared to those maintained in normal glucose concentrations. Similarly, primary macrophages isolated from the bone marrow of diabetic mice display a markedly impaired ability to clear apoptotic cardiomyocytes relative to macrophages from control mice. This direct suppression may be attributed to various factors induced by hyperglycemia, including cellular metabolic disturbances, insufficient energy supply, and altered membrane fluidity, collectively leading to decreased efficiency in the recognition and phagocytosis of apoptotic cells by macrophages. Furthermore, high glucose upregulates the expression of a disintegrin and metalloproteinase 9 (ADAM9), which subsequently cleaves and inactivates MerTK, thereby impairing its ability to effectively recognize and engulf ACs.67 Persistent hyperglycemia driven the accumulation of AGEs. The binding of AGEs to the receptor for AGEs (RAGE) on macrophages activates downstream signaling pathways such as NF-κB, which promotes a strong pro-inflammatory response and further suppresses efferocytosis.19 Additionally, the AGEs–RAGE interaction inhibits Rac1 activity within macrophages, thereby disrupting cytoskeletal reorganization and preventing the formation of functional phagocytic cups necessary for engulfing ACs.68 Diabetic wounds are often characterized by impaired local microcirculation and tissue hypoxia. Hypoxia downregulates the expression of MerTK, a key receptor in efferocytosis.69 In contrast, recent experiments have shown that macrophages display a stronger efferocytosis under long-term (chronic) physiological hypoxia, characterized by increased internalization and accelerated degradation of ACs.70 Moreover, multiple factors in diabetic wounds—including hyperglycemia, mitochondrial dysfunction, and inflammatory cell infiltration—contribute to excessive reactive oxygen species (ROS) production and a state of oxidative stress.71 Elevated ROS levels activate various stress-related signaling pathways, including the p38 MAPK pathway, and lead to a significant down-regulation of MERTK expression through a series of responses, further impairing macrophage function.20 Diabetic wounds commonly exhibit overgrowth of pathogens such as Staphylococcus aureus (S. aureus) and Pseudomonas aeruginosa (P. aeruginosa), forming biofilms. These biofilms induce sustained elevation of TNF-α, IL-1β, and IL-6 while reducing IL-10 and TGF-β, which impairs phagosome maturation, thereby inhibiting efferocytosis.72 Neutrophils accumulating within diabetic wounds release large amounts of proteases that hydrolyze the central hinge region of CCN1, cleaving it into non-functional fragments. These fragments retain the ability to bind PS on ACs surfaces but can no longer interact with macrophage receptors, thereby blocking efferocytosis and leading to the accumulation of ACs.73 High glucose induced oxidative stress accelerates the senescence of fibroblasts and endothelial cells. Upon apoptosis, these senescent cells retain surface “eat me” signals, yet the senescence-associated secretory phenotype (SASP) releases abundant proteases, IL-6, IL-8, TNF-α can cleave cathecolamines like CCN1, blocking the bridging between ACs and macrophages; it may also suppresses macrophage MerTK receptor expression, directly impairing recognition and efferocytosis.73,74 NETs accumulate extensively in diabetic wounds. NETs significantly suppress the PI3K/Rac1 signaling axis—a central pathway regulating cytoskeletal rearrangement and phagocytosis, physically block the connection between ACs and macrophages, thereby inhibiting efferocytosis.17,18 Insufficient angiogenesis leads to reduced local blood perfusion in diabetic wounds. This prevents key molecules secreted by macrophages and endothelial cells—such as MFG-E8, Gas6, and complement C1q—from effectively reaching sites of ACs accumulation. Consequently, a shortage of “bridging molecules” occurs, hindering the recognition and binding between ACs and macrophages.75 Concurrently, impaired angiogenesis exacerbates the wound microenvironment characterized by hypoxia, hyperglycemia, and ROS accumulation, all of which inhibit macrophage efferocytosis. These factors work together to cause the accumulation of ACs and secondary necrosis, thus maintaining the chronic inflammatory state of the wound and ultimately hindering the healing of diabetic wounds.
The critical role of specific efferocytosis-related molecules is further highlighted by gain- and loss-of-function studies. Exogenous administration of bridging molecules like MFG-E8 or CCN1 has been shown to accelerate diabetic wound closure by enhancing ACs clearance and suppressing pro-inflammatory cytokines, with CCN1 acting through the activation of Rac1 in macrophages.73,76,77 Conversely, genetic deficiencies in components like C1q or CD36 exacerbate inflammation and delay healing.78 Collectively, these findings pinpoint the efferocytosis pathway, from recognition to engulfment, as a foundation for therapeutic intervention.
Dysregulation of the Macrophage Efferocytosis-Polarization Axis in Diabetic Wounds
The Importance of Macrophage Polarization in Wound Healing
Macrophages are highly plastic cells with different phenotypes, depending on the surrounding microenvironment. Currently, macrophages are categorized into two primary phenotype: M1 (classically activated) and M2 (alternatively activated), a classification based on their activation modalities, cell surface markers, or functions.79 M1 macrophages differentiate in response to IFN-γ and LPS, which stimulate the release of multiple pro-inflammatory mediators and ROS. These molecules are crucial for antimicrobial defense, infection control, and the initiation of inflammatory responses.80 Whereas M2 macrophages are induced by IL-4 and IL-13 from adaptive immune cells, leading to the production of anti-inflammatory factors and repair-promoting factors. These factors are primarily contribute to pathogen clearance, resolution of infections, and promotion of tissue regeneration.8,81 In addition, some researchers have subdivided M2 macrophages into M2a type (activated by IL-4 and IL-13), M2b type (activated by immune complexes and TLR), M2c type (activated by IL-10, TGF-β and glucocorticoid) and M2d type (activated by TLR antagonist and adenosine) according to different inducing factors and functions.39
In the early stages of wound repair, around 85% of macrophages in the wound exhibit the M1 phenotype and secrete cytokines such as IL-6 and TNF-α. These cytokines remove infections, wound debris, and ACs while also promoting inflammation. Around 80–85% macrophages convert to M2 macrophages in the late stage of repair. They can produce growth factors like IL-10, Insulin Like Growth Factor 1 (IGF-1), vascular endothelial growth factor (VEGF) and TGF-β, which not only dampen inflammation but also promote the proliferation, migration, and differentiation of keratinocytes, fibroblasts, and endothelial cells. These processes facilitate angiogenesis and accelerate wound close.82–84 Macrophages are key cells in regulating skin wound healing, and the specific removal of macrophages severely impairs the healing process.85 Numerous studies have already demonstrated that promoting the transition of M1 to M2 can accelerate wound healing.
Dysregulated Macrophage Polarization in Diabetic Wound
In the specific pathological context of diabetic wounds, sustained inflammation, dysregulated glucose homeostasis, and tissue hypoxia collectively contribute to the arrest of macrophages in the M1 pro-inflammatory polarization state, thereby disrupting normal tissue regeneration and wound closure processes.86 The diabetic milieu stimulates an increased production of hematopoietic stem cells in the bone marrow, which then differentiate into a greater number of monocytes entering the peripheral blood. These monocytes recruited to the wound site, where they differentiate into M1 macrophages expressing characteristic markers such as IL-1β and TNF-α, thereby amplifying local inflammation and delaying healing.87 In experimental models, wounds in diabetic mice exhibit significantly elevated monocyte infiltration compared to non-diabetic controls.88 Macrophages further demonstrate a persistent upregulation of M1 markers—including inducible nitric oxide synthase (iNOS), TNF-α, IL-1β, and matrix metalloproteinase-9 (MMP9)—alongside downregulation of M2 markers such as Arginase-1, CD206. A parallel shift is observed in human diabetic foot ulcers.87
Mechanistically, the hyperglycemic environment activates pro-inflammatory pathways like NF-κB and elevates oxidative stress, which collectively inhibit the phenotypic transition from M1 to M2 macrophages.89 Hypoxia further stabilizes the M1 state via HIF-1α upregulation, which not only sustains expression of iNOS and IL-6 but also suppresses M2 marker transcription.90 Additionally, hyperglycemia, AGEs, and hypoxia synergistically promote ROS overproduction. ROS functions as a signaling intermediate that activates the NLRP3 inflammasome, driving IL-1β maturation and release. This in turn perpetuates NF-κB activation via autocrine signaling, reinforcing M1 polarization.91 Lipid metabolism disorders in diabetes lead to elevated levels of palmitic acid, which activates the innate immune system through Toll-like receptors and engages the NF-κB signaling pathway, further promoting macrophages toward an M1 phenotype and enhancing secretion of IL-6 and TNF-α.92 Prolonged M1 polarization impairs the functional transition to reparative M2 macrophages, hindering endothelial and fibroblast proliferation and migration. This disruption leads to deficient angiogenesis, reduced collagen deposition, and delayed wound healing. Furthermore, the dysregulation of macrophage behavior diminishes VEGF-A and VEGFR1 signaling, which further compromises vascularization and tissue repair in diabetic wounds.93,94
The Abnormal Crosstalk Between Macrophage Efferocytosis and Polarization Leading to Diabetic Wounds
As the most extensively studied phagocytes, macrophages capable of efficient efferocytosis play a vital role in maintaining tissue homeostasis under physiological conditions. Their main role is to engulf pathogens and dead cells, which helps repair tissue damage, as well as produce chemokines and cytokines to call on extra leukocytes to protect the organism when needed. Following skin injury, macrophages are among the foremost immune cells recruited to wound sites, where they perform pleiotropic functions to orchestrate the healing process across its distinct phases.58
The efferocytosis function and phenotypic change of macrophages are interconnected and mutually influential processes. Efferocytosis serves as a key mechanism underlying macrophage phenotypic conversion. Under physiological conditions, the timely clearance of ACs by macrophages is not merely a clean-up operation; it serves as an active instructional signal that shift macrophages from M1 to M2 state.95,96 This M2 polarization, in turn, enhances the macrophage’s capacity for further efferocytosis, creating a self-reinforcing cycle that efficiently resolves inflammation and initiates tissue repair.76,97 The cytokine shift from pro-inflammatory IL-1β, IL-8, IL-10, and TNF-α towards IL-6, TGF-β, prostaglandin E2 (PGE2), and platelet-activating factor (PAF) is both a cause and a consequence of this virtuous cycle.98,99
In the diabetic wound, this critical feedback loop is disrupt. Metabolic stressors like hyperglycemia downregulate key “eat-me” signal receptors such as CD36 on macrophages, blinding them to ACs.59 Concurrently, a hyperglycemia microenvironment can produce a variety of inflammatory and chemotactic molecules, such as AGEs, MCP-1, DAMPs, and IL-1β stimulate inflammatory pathways like NLRP3, which directly suppresses efferocytosis and locks macrophages into a persistent M1 state.16,100 Besides, high levels of ROS and AGEs, interferes with intracellular signaling cascades. For instance, oxidative stress can disrupt the Nrf2 pathway,101 while AGEs-RAGE can inhibit anti-inflammatory signaling.102 The transition to M2 could not be initiated normally even though some of the efferocytosis proceeds normally. As a result, pro-inflammatory mediators of resolution, such as TGF-β and PGE2, are blocked, inflammation is reduced to a persistent state, and tissue repair is impeded. In wound healing, neutrophils serve a critical function as the primary leukocytes during the early phases.103 Impaired efferocytosis leads to the accumulation of apoptotic neutrophils, which progress to secondary necrosis. These cells release their intracellular contents and more DAMPs, further fueling inflammation and M1 polarization.64,65,104 The failure to transition to the M2 phenotype results in a persistent dominance of M1 macrophages. The M1 phenotype is inherently less efficient at efferocytosis and secretes cytokines like TNF-α that actively suppress the process.100 Simultaneously, This dual impairment creates a vicious cycle: impaired efferocytosis prevents the polarization switch to M2, while the sustained M1 dominance further inhibits the clearance of ACs. The consequence is the pathological hallmark of diabetic wounds: the accumulation of secondary necrotic cells, perpetual inflammation, and a failure to transition to the proliferative phase of healing.
The Signaling Pathway Modulating Macrophage Efferocytosis- Polarization Axis in Diabetic Wounds Healing (Figure 2)
JAK/STAT Signaling Pathway
The Janus kinase/signal transduction and transcription activation (JAK/STAT) pathway serves as a crucial regulatory axis through which efferocytosis modulates macrophage activation and polarization. Four tyrosine (Tyr) kinases (JAK1–3 and Tyk2) make up the JAK family. Before activation, STATs (STAT1–4, STAT5A, STAT5B, STAT6) are localized in the cytosol. Upon stimulation by cytokines, they bind to phosphorylated activated receptors and undergo phosphorylation by JAKs.105 Among the major STAT family members in these receptor downstream pathways, STAT1/3 primarily promote macrophage inflammatory cytokine expression and M1 polarization, whereas STAT5/6 regulates multiple M2 marker genes and serves as the master transcriptional regulator driving M2 polarization.106,107 The antagonism between STAT5 (promoting MerTK stabilization and M2 polarization) and STAT3 (suppressing MerTK), for instance, suggests that the functional outcome depends not on pathway activation but on the specific STAT isoforms engaged.107 Proteins like SOCS1 and SOCS3, upregulated by PS-Gas6/PROS1 signaling during efferocytosis, act as crucial negative feedback balancers, ubiquitinating JAKs and degrading STAT1/3, which reduces inflammation and suppresses M1 polarization.108,109 Similarly, the activation of the SOCS3/STAT3 axis by MFG-E8 demonstrates a paradoxical mechanism, where a single pathway can be harnessed to both promote M2 polarization and attenuate inflammatory signaling.110
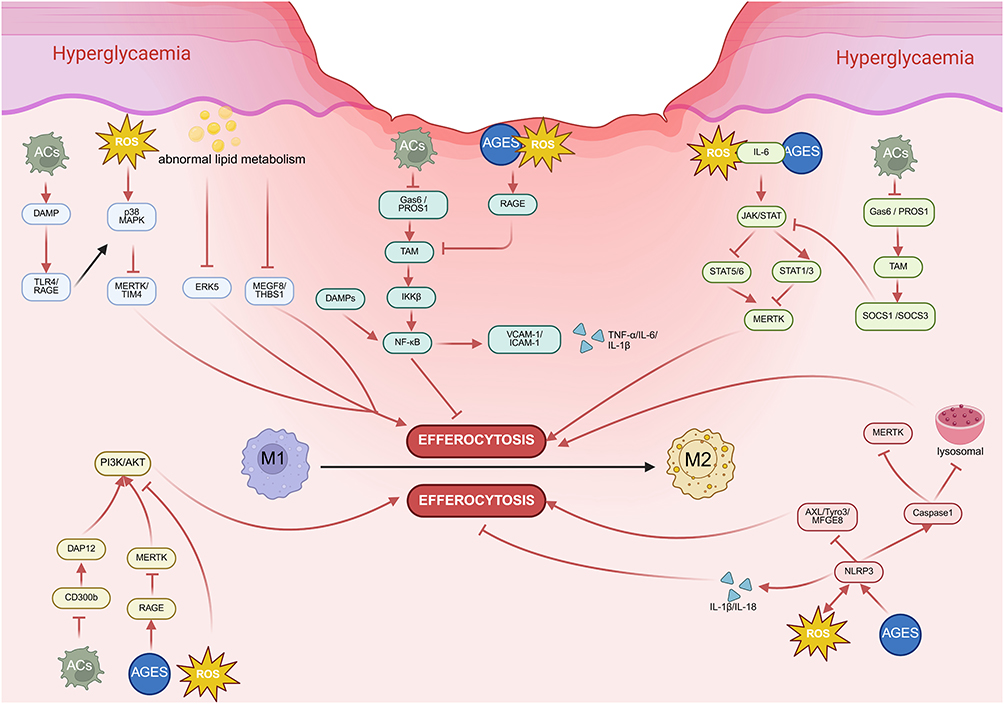 |
Figure 2 The vicious cycle of Impaired Efferocytosis and Sustained Inflammation in the diabetic wound microenvironment. MAPK: Hyperglycaemia/oxidative stress (ROS)/abnormal lipid metabolism activate MAPK signaling pathway (p38 MAPK, ERK5, which reduces expression of efferocytosis receptors (MerTK/TIM4) and bridging proteins (MFGE8/THBS1) via downregulation, thereby impairing efferocytosis and blocking M1-to-M2 phenotypic switching. NF-κB: TAM receptor (Tyro3, Axl, Mer)-Gas6/PROS axis suppresses NF-κB activation by blocking IκBα/β degradation and nuclear translocation, thereby inhibiting pro-inflammatory responses. Besides, hyperglycemia and AGEs activate NF-κB via RAGE engagement, triggering excessive production of pro-inflammatory cytokines (IL-6, IL-1β) and adhesion molecules (VCAM-1, ICAM-1) that trap the wound microenvironment in a chronic inflammatory state. PI3K/Akt: Hyperglycemia, ROS, and particularly the accumulation of AGEs, competitively engaging RAGE and disrupting CD300b/ MerTK activation, disrupting PI3K/AKT activation.NLRP3: Both ROS and AGEs can activate the NLRP3 inflammasome, subsequently sustaining inflammation by activating the AXL/Tyro3/MFGE8 pathway and driving caspase-1-dependent release of IL-1β and IL-18. JAK/STAT: hyperglycemia, AGEs, ROS and IL6PS-Gas6/PROS1 signaling upregulated SOCS1 and SOCS3, ubiquitinating JAKs and degrading STAT1/3, which reduces MerTK and suppresses efferocytosis. Figure created in BioRender. h, y. (2025) https://BioRender.com/xr9yuvz. |
Dysregulation of the JAK/STAT pathway is associated with various diseases.111–113 Under diabetic conditions, hyperglycemia, AGEs, and ROS collectively activate the JAK/STAT signaling pathway. Elevated levels of upstream inflammatory cytokines such as IL-6 under hyperglycemic conditions amplify JAK/STAT activation, driving the production of pro-inflammatory mediators including TNF-α and IL-1β.114 This cascade promotes M1 macrophage polarization and sustains inflammatory responses, while simultaneously impairing efferocytosis.115 The therapeutic challenge, therefore, is not JAK-STAT inhibition but “signal reselection”—shifting the balance from detrimental to beneficial arms. The promising approach of inhibiting JAK1 while activating STAT3/6 exemplifies this strategy, aiming to reconstitute the natural pro-resolving program.116
PI3K/Akt Signaling Pathway
The phosphatidylinositol 3-hydroxy kinase/protein kinase B (PI3K/ Akt) represents a canonical anti-apoptotic and proliferative signaling axis that regulates essential cellular processes including growth, survival, proliferation, and metabolic homeostasis. As an intracellular phosphatidylinositol kinase, PI3K binds and facilitates Akt phosphorylation upon activation, with Akt serving as the central node of this pathway.117 It is reported that efferocytosis was associated with a rapid increase in PI3K/Akt phosphorylation, and inhibition of PI3K has resulted in a severe defect in efferocytosis.118 Its position downstream of key efferocytosis receptors like MerTK and CD300b.118,119 By binding to these receptors, PS on ACs triggers the PI3K/Akt signaling cascade, which in turn enhances efferocytosis and promotes M2 polarization.120–122 The resulting M2 macrophages then produce anti-inflammatory mediators like TGF-β and IL-10, which further stabilize the PI3K/Akt activation state, ensuring inflammation is efficiently terminated.123
A vicious cycle of chronic inflammation is maintained when the PI3K/Akt pathway is inhibited under hyperglycemic and oxidative stress conditions. Hyperglycemia, oxidative stress, and particularly the accumulation of AGEs, competitively engaging RAGE and disrupting MerTK activation, directly suppress pathway activity.124 Subsequently, it cripples both efferocytosis and M2 polarization, locking the system in a pro-inflammatory state.18,125 This creates a self-perpetuating cycle characterized by defective efferocytosis, persistent M1 polarization, and unresolved inflammation. Targeting the PI3K/Akt pathway simultaneously improves efferocytosis and macrophage polarization, though its tissue-specific effects and long-term safety require further investigation.
NLRP3 Signaling Pathway
NOD-like receptor family pyrin domain containing 3 (NLRP3), a member of the NOD-like receptor (NLR) family of pattern recognition receptors, plays a critical role in various inflammatory diseases, including cryopyrin-associated periodic syndromes, Alzheimer’s disease, diabetes, gout, autoinflammatory diseases, and atherosclerosis when dysregulated.126,127 By activating caspase-1 and directly cleaving the intracellular domains of phagocytic receptors like MerTK, the NLRP3 hinders their capacity to detect PS on ACs and interferes with phagosome-lysosome fusion, both of which severely limit efferocytosis.128,129 What’s more, IL-1β and IL-18 are the principal mature inflammatory cytokines generated upon NLRP3 inflammasome activation. They not only propagate inflammatory signaling through autocrine/paracrine actions that drive M1 polarization,130 but also directly inhibit phagocyte activity that further impairs efferocytosis.128 Notably, NLRP3 activation can be triggered by ROS, while its activation further amplifies ROS production, establishing a self-reinforcing positive feedback loop. This vicious cycle exacerbates inflammatory responses, ultimately disrupting macrophage polarization and impairing efferocytosis.131–136
Within the diabetic wound, NLRP3 is essential for maintaining persistent inflammation. Large volumes of pro-inflammatory cytokines and ROS can be released when the NLRP3 is excessively activated by the hyperglycemic environment and AGEs.137 These events simultaneously impair efferocytosis through MerTK receptor cleavage and lysosomal dysfunction,129,138 while promoting a pro-inflammatory state via enhanced cytokine secretion and PPARγ signaling suppression.139 The resulting accumulation of ACs further fuels the NLRP3 activation, completing a vicious cycle that is self-sustaining and highly resistant to resolution.140,141 Besides, when phagocytes engulf ACs, they will produce signaling molecules and anti-inflammatory substances including TGF-β and IL-10, which can prevent NLRP3 from being assembled and activated, reducing inflammation and encouraging tissue repair.142 Inhibition of NLRP3 has been shown to upregulate key efferocytosis receptors (AXL, Tyro3, MFGE8), thereby promoting the healing of diabetic wounds.143 The future of NLRP3 therapeutics lies in locally-targeted inhibitors that can disrupt the pathological change during the chronic phase of diabetic wounds without compromising innate immunity, thereby restoring the body’s own regenerative program.
NF-κB Signaling Pathway
Nuclear factor kappa-B (NF-κB) is a key transcriptional regulator governing innate and adaptive immunity, and is critically involved in diverse physiological and pathological processes including inflammation, tumorigenesis, and immune regulation.144 The NF-κB signaling pathway consists of canonical and noncanonical branches. The canonical pathway is activated by IKKβ, whereas the noncanonical pathway depends on NIK and IKKα.145 A key regulatory node lies in the TAM receptor (Tyro3, Axl, Mer)-Gas6/PROS axis, which suppresses NF-κB activation by blocking IκBα/β degradation and nuclear translocation, thereby inhibiting pro-inflammatory responses in DCs and macrophages.146–148 MerTK deficiency is uniquely linked to defective efferocytosis and persistent NF-κB activation via damage-associated molecular patterns (DAMPs) from uncleared ACs, trapping macrophages in a pro-inflammatory M1 state and exacerbating inflammation. While the roles of Tyro3 and Axl in this cascade are less well-defined.149
Beyond TAM signaling, a self-reinforcing pathological loop emerges in diabetic wounds, driven by NF-κB dysregulation, impaired efferocytosis, and defective macrophage polarization. Persistent hyperglycemia and AGEs activate NF-κB via RAGE engagement, triggering excessive production of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β) and adhesion molecules (VCAM-1, ICAM-1) that trap the wound microenvironment in a chronic inflammatory state.150 This milieu not only blocks the transition of macrophages from M1 to M2 phenotypes but also disrupts the critical switch from inflammatory to proliferative healing phases151,152—yet the temporal dynamics of NF-κB activation in shaping macrophage plasticity remain poorly characterized. For instance, while acute NF-κB activation is essential for initiating wound healing, chronic activation drives pathology.153 The downregulation of efferocytosis receptors (eg, MerTK) by chronic NF-κB activation further exacerbates the imbalance by reducing anti-inflammatory mediators (TGF-β, IL-10), impairing M2 polarization, and delaying healing.146 Furthermore, hyperglycemia and AGEs promote ROS generation, which both activate NF-κB and are upregulated by it, creating a self-amplifying inflammatory loop.154 Thus, the field urgently needs a refined conceptual framework that distinguishes between “pathological” NF-κB activation (driven by AGEs/ROS/DAMPs) and “physiological” NF-κB signaling (essential for early healing). Therapeutic strategies must therefore seek to fine-tune NF-κB signaling rather than achieve its complete suppression.
MAPK Signaling Pathway
The mitogen-activated protein kinase (MAPK) signaling pathway represents one of the most crucial signal transduction networks in eukaryotic organisms, serving as a key regulatory pathway for cell proliferation, differentiation, apoptosis, and stress responses under both physiological and pathological conditions.155 Extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK are the primary members of the MAPKs family. They correspond to four conventional MAPK signaling cascades and are one of the common intersecting paths of signal transduction.156 A foundational observation is that the p38/JNK pathway typically promotes pro-inflammatory M1 polarization, while ERK signaling generally enhances M2 polarization.157 However, this opinion overlooks a self-reinforcing inflammatory cycle: activation of the p38 MAPK pathway induces proteolytic cleavage and shedding of the critical efferocytosis receptor MerTK/TIM-4 from macrophage membranes, impairing efferocytosis and blocking M1-to-M2 phenotypic switching.158,159 In turn, accumulated ACs undergo secondary necrosis, releasing DAMPs such as HMGB1 that perpetuate MAPK activation via TLR4/RAGE signaling—creating a feedforward loop that prolonged pro-inflammatory cytokine secretion and unresolved inflammation.160,161
Hyperglycemia-induced ROS rapidly phosphorylate and activate p38 MAPK, which directly represses MerTK transcription; this reduces efferocytosis of apoptotic neutrophils, locks macrophages in an M1 state, and delays wound healing.162 While p38 MAPK inhibition is widely proposed as a therapeutic strategy for diabetic wounds, conflicting preclinical evidence challenges its universal efficacy: in non-diabetic wound healing, p38 MAPK actually promotes keratinocyte migration and re-epithelialization via autophagy activation,163 whereas in hyperglycemic environments, it suppresses these reparative processes.117 Meanwhile, diabetes-related abnormal lipid metabolism reduces the ERK5 activity in macrophages, and decreases the expression of bridging proteins (MFGE8, THBS1) and “eat-me/find-me” signals to further impair efferocytosis.164 Notably, MFGE8 deficiency recapitulates diabetic wound pathology (persistent inflammation, impaired angiogenesis, delayed closure), while recombinant MFGE8 restores efferocytosis and accelerates healing6—yet ERK5’s role in regulating MFGE8 expression remains poorly characterized, representing a critical research gap. These observations underscore that effective therapeutics must be contextually tailored to the diabetic microenvironment (p38 selective inhibitors, ERK5 activators, or MerTK/MFGE8 supplementation), rather than targeting MAPK pathways in isolation.
Potentially Therapeutic Strategies by Targeting Efferocytosis
Small-Molecule Drugs
Small-molecule drugs targeting macrophage function have emerged as promising therapeutic strategies for modulating inflammation and tissue repair, with recent studies highlighting their diverse mechanisms of action. Ruxolitinib (JAK1/2 inhibitor) and MCC950 (NLRP3 inhibitor), although acting on distinct signaling pathways, both enhance macrophage efferocytosis. By inducing M2 polarization and upregulating MerTK, they effectively alleviate fibrosis and promote repair in models of systemic sclerosis and metabolic liver disease.129,165 Conversely, the PPAR γ inhibitor GW9662 exacerbates pathology by impairing efferocytosis.166,167 This contrasting evidence chain powerfully demonstrates the efficacy and sensitivity of efferocytosis as a therapeutic target. However, clinical data also show that pioglitazone, a PPARγ agonist, improves insulin sensitivity but increases heart failure risk.168
This raises a critical therapeutic dilemma: can PPARγ be considered a feasible target for promoting efferocytosis, or does its pleiotropy render it too risky for systemic use? The adverse effects also caution us that whether efferocytosis-enhancing benefits can be decoupled from metabolic side effects. Furthermore, the role of locally administered low-dose aspirin in diabetic wound healing reveals a “immune homeostasis reprogramming” strategy: it not only clears ACs by upregulating CD36 but also actively shifts arachidonic acid metabolism from pro-inflammatory leukotriene B4 (LTB4) to pro-resolving lipoxygenase A4 (LXA4).65 However, this approach raises its own set of evaluations: why does low-dose, local aspirin succeed where systemic NSAIDs often fail? This context-dependence—dose, route of administration, and disease microenvironment—remains understudied, representing a major gap in translating preclinical efferocytosis-enhancing strategies to humans. Effective drugs must prioritize enhancement of efferocytosis while minimizing disruption of other macrophage functions (phagocytosis of pathogens, metabolic homeostasis). Collectively, small-molecule drugs that target efferocytosis represent a promising class of therapeutics, but their full potential remains untapped due to unresolved controversies and descriptive rather than mechanistic preclinical studies.
Natural Medicines
Natural compounds offer a distinct therapeutic advantage over single-target synthetic drugs through their pharmacological activity, enabling the synergistic reprogramming of macrophage function to address the complexity of chronic inflammatory and metabolic diseases. For instance, both Baicalin and sulforaphane (SFN), despite their different origins, enhancing efferocytosis efficiency by relieving key endogenous inhibitory mechanisms—Baicalin inhibits the RhoA/ROCK pathway, while SFN activates the Nrf2/HO-1 axis to upregulate MerTK.12,169 This intervention on regulatory mechanisms allows them to effectively initiate inflammation resolution programs. Besides, Naoxintong (NXT), the Chinese patent medicine derived from natural ingredients, demonstrates a sophisticated multi-target mechanism: by suppressing JAK1 phosphorylation while selectively activating STAT3/STAT6, it not only drives the macrophage polarization from a pro-inflammatory M1 to a pro-repair M2 phenotype but also concurrently enhances their capacity to clear apoptotic neutrophils, thereby creating a positive feedback loop between polarization and efferocytosis in diabetic wounds.116 A critical unresolved question is whether the observed efficacy due to synergy among constituents, or is it predominantly driven by a single compound. Without deconvoluting the active ingredients, clinical translation is hampered by batch-to-batch variability and difficulty in establishing dose-response relationships. This also highlights a key translational challenge: identifying the definitive “active component” within multi-component formulations. Notably, ginsenoside Rg5 and isoliquiritigenin (ISL) collectively identify SLC7A11- a key mediator of ferroptosis and glutathione metabolism- as an emerging metabolic regulator. Through SLC7A11 inhibition, they alleviate its constraining effects on crucial metabolic pathways—including dendritic cell-mediated efferocytosis and macrophage glycolysis—thus establishing a crosstalk between immunoregulation and metabolic reprogramming.170–172 But preclinical studies of Rg5/ISL rarely evaluate ferroptosis markers in healthy tissues, focusing instead on therapeutic efficacy in diseased models. Systemic inhibition could induce ferroptosis in healthy tissues, leading to toxicity. In summary, multi-target pharmacology of natural compounds makes them ideal for addressing the network-level complexity of chronic inflammatory and metabolic diseases, but their full potential requires a shift from descriptive mechanism reporting to evaluative research.
Biomaterials
The emerging paradigm in biomaterial design for immune modulation moving beyond passive biocompatibility toward actively programmable systems that direct a coordinated sequence of immune responses to guide tissue regeneration. On one front, “resolution-accelerating” materials such as Ac2-26 peptide-modified surfaces or MerTK-enriched nanoparticles directly enhance the efferocytosis machinery and suppress M1 polarization, thereby driving established inflammation toward resolution.173–175 While this strategy is appealing for chronic inflammatory contexts, it raises a fundamental question: Does premature resolution of inflammation compromise tissue repair? Acute inflammation is a necessary step in regeneration, providing signals for cell recruitment and matrix remodeling; bypassing this phase could lead to incomplete tissue integration or functional deficits. Indeed, it’s crucial to achieve temporal precision (targeting only persistent, pathological inflammation) rather than global anti-inflammatory effects. A more nuanced approach involves “inflammatory priming”, where systems like the PDA-coated fiber matrix simulate acute-phase circumstances to reactivate macrophage efferocytosis, removing ACs while lowering oxidative stress and encouraging M2 polarization.176 Nevertheless, a critical translational question remains: preclinical studies typically use healthy animal models, would this strategy remain effective in animal or patients with pre-existing chronic inflammation, where the immune system is already hyperactivated? The most advanced concept of this paradigm is represented by integrated systems like Gel@fMLP/SiO2-FasL hydrogel—an autonomous programming platform that recruits neutrophils, orchestrates their timely apoptosis, and facilitates their subsequent removal, thereby executing a self-limited inflammatory cycle conducive to functional tissue repair.177 Despite its sophistication, the integrated system’s translational potential is limited by its lack of adaptability to patient-specific microenvironments. Additionally, both large-scale production and manufacturing, as well as long-term safety assessments, are issues that need to be addressed. In conclusion, the shift toward biomaterials represents a transformative advance in regenerative medicine, addressing the unmet need for coordinated inflammatory resolution and tissue repair, filling the gap in the field will mark a significant step forward in the treatment of diabetic wounds.
Conclusion
This review systematically emphasizes the pivotal role of macrophage efferocytosis-polarization axis in maintaining tissue homeostasis, and how their dysregulation in the pathological environment of diabetic wounds constitutes a common terminal pathway for healing failure. We have outlined that in the diabetic wound microenvironment, hyperglycemia-induced metabolic dysfunction and MerTK inactivation, AGE-RAGE-mediated inflammation and Rac1 inhibition, hypoxia, oxidative stress, biofilm, protease load, cellular senescence, NETs, and insufficient angiogenesis—which collectively impair macrophage recognition, engulfment, and clearance of ACs, leading to secondary necrosis, sustained inflammation, and ultimately disrupted efferocytosis. The impaired efferocytosis further causes dysfunction in the efferocytosis–polarization axis through key signaling pathways such as JAK-STAT, PI3K/Akt, NLRP3, NF-κB, and MAPK, trapping macrophages in a pro-inflammatory M1 state. This sustained M1 polarization further suppresses efferocytosis through the production of mediators such as TNF-α and ROS. The refined understanding mandates a paradigm shift in therapeutic strategy. Moving forward, the goal should evolve from simply suppressing inflammation to actively reprogramming the macrophage functional landscape. Currently, multiple drug-targeting studies have been conducted, including small molecules, natural medicines, and Biomaterials. However, due to challenges such as bioavailability, off-target effects, scalability, and long-term safety, there is still a long way to go before these therapies can be translated into clinical use. Embracing macrophage efferocytosis-polarization axis will be pivotal in transforming the stubborn challenge of diabetic wound healing into a tractable problem.
Data Sharing Statement
No datasets were generated or analysed during the current study.
Author Contributions
YH and JH are co-first authors. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. YH: writing–original draft, conceptualization, visualization. JH: writing–original draft, investigation. AM: writing–original draft, data curation. PD and MY: writing–review & editing, formal analysis. XX: writing–review & editing, conceptualization, supervision. YD: writing– review & editing, conceptualization.
Funding
This work was supported by Sichuan Provincial Department of Science and Technology Youth Fund Project (grant number 2024NSFSC1609) and Sichuan Medical Association General Program (grant number 2025YXKY0705).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Singh A, Shadangi S, PKr G, Rana S. Type 2 diabetes mellitus: a comprehensive review of pathophysiology, comorbidities, and emerging therapies. Compr Physiol. 2025;15(1):e70003. doi:10.1002/cph4.70003
2. Han C, Singla RK, Wang C. Application of biomaterials in diabetic wound healing: the recent advances and pathological aspects. Pharmaceutics. 2025;17(10):1295. doi:10.3390/pharmaceutics17101295
3. Vijayakumar V, Samal SK, Mohanty S, Nayak SK. Recent advancements in biopolymer and metal nanoparticle-based materials in diabetic wound healing management. Int J Biol Macromol. 2019;122:137–18. doi:10.1016/j.ijbiomac.2018.10.120
4. Powers JG, Higham C, Broussard K, Phillips TJ. Wound healing and treating wounds. J Am Acad Dermatol. 2016;74(4):607–625. doi:10.1016/j.jaad.2015.08.070
5. Jiang P, Li Q, Luo Y, et al. Current status and progress in research on dressing management for diabetic foot ulcer. Front Endocrinol. 2023;14:1221705. doi:10.3389/fendo.2023.1221705
6. Rong B, Jiang H, Zhu W, et al. Unraveling the role of macrophages in diabetes: Impaired phagocytic function and therapeutic prospects. Medicine. 2025;104(8):e41613. doi:10.1097/MD.0000000000041613
7. Patel S, Srivastava S, Singh MR, Singh D. Mechanistic insight into diabetic wounds: pathogenesis, molecular targets and treatment strategies to pace wound healing. Biomed Pharmacother. 2019;112:108615. doi:10.1016/j.biopha.2019.108615
8. Zheng Y, Ruan Z, Liu S, Yang X, Chen Z. Exosome-mediated macrophage polarization: pioneering pathways in diabetic wound healing. Int Immunopharmacol. 2025;161:115058. doi:10.1016/j.intimp.2025.115058
9. Liu Y, Xia G, Chen Y, et al. Purpurolide C-based microneedle promotes macrophage-mediated diabetic wound healing via inhibiting TLR4-MD2 dimerization and MYD88 phosphorylation. Acta Pharmaceutica Sinica B. 2023;13(12):5060–5073. doi:10.1016/j.apsb.2023.05.032
10. Liu H, Sun C, Jiang Y, et al. Eldecalcitol alleviates diabetic periodontitis by regulating macrophage efferocytosis and polarization via SOCE machinery. Int Immunopharmacol. 2025;146:113894. doi:10.1016/j.intimp.2024.113894
11. Cai Y, Chen K, Liu C, Qu X. Harnessing strategies for enhancing diabetic wound healing from the perspective of spatial inflammation patterns. Bioact Mater. 2023;28:243–254. doi:10.1016/j.bioactmat.2023.04.019
12. Huang Y, Wang B, Ma Z, et al. Sulforaphane promotes diabetic wound healing by regulating macrophage efferocytosis and polarization. Int Immunopharmacol. 2025;150:114243. doi:10.1016/j.intimp.2025.114243
13. Crunkhorn S. Promoting efferocytosis heals diabetic wounds. Nat Rev Drug Discov. 2022;21(7):491. doi:10.1038/d41573-022-00097-z
14. Zhang YQ, Nie R, Feng ZY, et al. Efferocytosis in tissue engineering: a comprehensive review of emerging therapeutic strategies for enhanced tissue repair and regeneration. Bioact Mater. 2025;52:155–181. doi:10.1016/j.bioactmat.2025.05.026
15. Yi J, Tang Q, Sun S, Xie H, Wang L, Yin X. Exosomes in diabetic wound healing: mechanisms, applications, and perspectives. DMSO. 2025;18:2955–2976. doi:10.2147/DMSO.S532885
16. Li K, Chen G, Luo H, et al. MRP8/14 mediates macrophage efferocytosis through RAGE and Gas6/MFG‐E8, and induces polarization via TLR4‐dependent pathway. J Cell Physiol. 2021;236(2):1375–1390. doi:10.1002/jcp.29944
17. Chen K, Murao A, Arif A, et al. Inhibition of efferocytosis by extracellular CIRP–induced neutrophil extracellular traps. J Immunol. 2021;206(4):797–806. doi:10.4049/jimmunol.2000091
18. Xie Y, Yang J, Zhu H, Yang R, Fan Y. The efferocytosis dilemma: how neutrophil extracellular traps and PI3K/Rac1 complicate diabetic wound healing. Cell Commun Signal. 2025;23(1):103. doi:10.1186/s12964-025-02092-4
19. Lin H, Yang Y, Wang X, et al. Targeting the AGEs-RAGE axis: pathogenic mechanisms and therapeutic interventions in diabetic wound healing. Front Med. 2025;12:1667620. doi:10.3389/fmed.2025.1667620
20. Zhu H, Wang W, Zhu J, et al. Methylglyoxal deteriorates macrophage efferocytosis in diabetic wounds through ROS-induced ubiquitination degradation of KLF4. Free Radic Biol Med. 2025;231:23–37. doi:10.1016/j.freeradbiomed.2025.02.030
21. Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. 2014;6(265):265sr6. doi:10.1126/scitranslmed.3009337
22. Wang M, Cao X, Shang Y, et al. Correlational analysis of PLIN1 with inflammation in diabetic foot ulcer wounds. Diabetes Res Clin Pract. 2024;209:111605. doi:10.1016/j.diabres.2024.111605
23. Jones RE, Foster DS, Longaker MT. Management of Chronic Wounds—2018. JAMA. 2018;320(14):1481. doi:10.1001/jama.2018.12426
24. Doran AC, Yurdagul A, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20(4):254–267. doi:10.1038/s41577-019-0240-6
25. Tajbakhsh A, Rezaee M, Kovanen PT, Sahebkar A. Efferocytosis in atherosclerotic lesions: malfunctioning regulatory pathways and control mechanisms. Pharmacol Ther. 2018;188:12–25. doi:10.1016/j.pharmthera.2018.02.003
26. Gude DR, Alvarez SE, Paugh SW, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine‐1‐phosphate as a “come‐and‐get‐me” signal. FASEB j. 2008;22(8):2629–2638. doi:10.1096/fj.08-107169
27. Naessens F, Demuynck R, Vershinina O, et al. CX3CL1 release during immunogenic apoptosis is associated with enhanced anti-tumour immunity. Front Immunol. 2024;15:1396349. doi:10.3389/fimmu.2024.1396349
28. Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–286. doi:10.1038/nature08296
29. Lauber K, Bohn E, Kröber SM, et al. Apoptotic cells induce migration of phagocytes via Caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113(6):717–730. doi:10.1016/S0092-8674(03)00422-7
30. Cartier A, Hla T. Sphingosine 1-phosphate: lipid signaling in pathology and therapy. Science. 2019;366(6463):eaar5551. doi:10.1126/science.aar5551
31. Luo B, Gan W, Liu Z, et al. Erythropoeitin signaling in macrophages promotes dying cell clearance and immune tolerance. Immunity. 2016;44(2):287–302. doi:10.1016/j.immuni.2016.01.002
32. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21(7):398–414. doi:10.1038/s41580-020-0232-1
33. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509(7500):310–317. doi:10.1038/nature13085
34. Segawa K, Nagata S. An apoptotic ‘Eat Me’ signal: phosphatidylserine exposure. Trends Cell Biol. 2015;25(11):639–650. doi:10.1016/j.tcb.2015.08.003
35. Balasubramanian K, Schroit AJ. Aminophospholipid asymmetry: a matter of life and death. Annu Rev Physiol. 2003;65:701–734. doi:10.1146/annurev.physiol.65.092101.142459
36. Liu X, Liu H, Deng Y. Efferocytosis: an emerging therapeutic strategy for type 2 diabetes mellitus and diabetes complications. JIR. 2023;16:2801–2815. doi:10.2147/JIR.S418334
37. Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148(7):2207–2216.
38. Bosurgi L, Brunelli S, Rigamonti E, Monno A, Manfredi AA, Rovere-Querini P. Vessel-associated myogenic precursors control macrophage activation and clearance of apoptotic cells. Clin Exp Immunol. 2015;179(1):62–67. doi:10.1111/cei.12356
39. Ma Y, Kemp SS, Yang X, Wu MH, Yuan SY. Cellular mechanisms underlying the impairment of macrophage efferocytosis. Immunol Lett. 2023;254:41–53. doi:10.1016/j.imlet.2023.02.001
40. Werfel TA, Cook RS. Efferocytosis in the tumor microenvironment. Semin Immunopathol. 2018;40(6):545–554. doi:10.1007/s00281-018-0698-5
41. Castellano F, Montcourrier P, Chavrier P. Membrane recruitment of Rac1 triggers phagocytosis. J Cell Sci. 2000;113(Pt 17):2955–2961. doi:10.1242/jcs.113.17.2955
42. Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998;17(23):6932–6941. doi:10.1093/emboj/17.23.6932
43. Ma Z, Thomas KS, Webb DJ, et al. Regulation of Rac1 activation by the low density lipoprotein receptor-related protein. J Cell Biol. 2002;159(6):1061–1070. doi:10.1083/jcb.200207070
44. Park SY, Kang KB, Thapa N, Kim SY, Lee SJ, Kim IS. Requirement of adaptor protein GULP during stabilin-2-mediated cell corpse engulfment. J Biol Chem. 2008;283(16):10593–10600. doi:10.1074/jbc.M709105200
45. Park D, Hochreiter-Hufford A, Ravichandran KS. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol. 2009;19(4):346–351. doi:10.1016/j.cub.2009.01.042
46. Gumienny TL, Brugnera E, Tosello-Trampont AC, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001;107(1):27–41. doi:10.1016/s0092-8674(01)00520-7
47. Das S, Owen KA, Ly KT, et al. Brain angiogenesis inhibitor 1 (BAI1) is a pattern recognition receptor that mediates macrophage binding and engulfment of Gram-negative bacteria. Proc Natl Acad Sci U S A. 2011;108(5):2136–2141. doi:10.1073/pnas.1014775108
48. Wang Y, Subramanian M, Yurdagul A, et al. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell. 2017;171(2):331–345.e22. doi:10.1016/j.cell.2017.08.041
49. Yin C, Argintaru D, Heit B. Rab17 mediates intermixing of phagocytosed apoptotic cells with recycling endosomes. Small GTPases. 2019;10(3):218–226. doi:10.1080/21541248.2017.1308852
50. Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62(2):317–329. doi:10.1016/0092-8674(90)90369-p
51. Flannagan RS, Jaumouillé V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. 2012;7:61–98. doi:10.1146/annurev-pathol-011811-132445
52. Rubino M, Miaczynska M, Lippé R, Zerial M. Selective membrane recruitment of EEA1 suggests a role in directional transport of clathrin-coated vesicles to early endosomes. J Biol Chem. 2000;275(6):3745–3748. doi:10.1074/jbc.275.6.3745
53. Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol. 2003;23(18):6494–6506. doi:10.1128/MCB.23.18.6494-6506.2003
54. Johansson M, Lehto M, Tanhuanpää K, Cover TL, Olkkonen VM. The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol Biol Cell. 2005;16(12):5480–5492. doi:10.1091/mbc.e05-03-0189
55. Han CZ, Ravichandran KS. Metabolic connections during apoptotic cell engulfment. Cell. 2011;147(7):1442–1445. doi:10.1016/j.cell.2011.12.006
56. Yoon TY, Munson M. SNARE complex assembly and disassembly. Curr Biol. 2018;28(8):R397–R401. doi:10.1016/j.cub.2018.01.005
57. Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi:10.1038/nature01991
58. Chen H, Shi R, Luo B, et al. Macrophage peroxisome proliferator-activated receptor γ deficiency delays skin wound healing through impairing apoptotic cell clearance in mice. Cell Death Dis. 2015;6(1):e1597. doi:10.1038/cddis.2014.544
59. Aitcheson SM, Frentiu FD, Hurn SE, Edwards K, Murray RZ. Skin wound healing: normal macrophage function and macrophage dysfunction in diabetic wounds. Molecules. 2021;26(16):4917. doi:10.3390/molecules26164917
60. Zhao Y, Li M, Mao J, et al. Immunomodulation of wound healing leading to efferocytosis. Smart Med. 2024;3(1):e20230036.
61. Zhang W, Feng J, Ni Y, et al. The role of SLC7A11 in diabetic wound healing: novel insights and new therapeutic strategies. Front Immunol. 2024;15:1467531. doi:10.3389/fimmu.2024.1467531
62. Li Y, Ma F, Li H, et al. Dimethyl fumarate accelerates wound healing under diabetic conditions. J Mo Endocrinol. 2018;61(4):163–172. doi:10.1530/JME-18-0102
63. Khanna S, Biswas S, Shang Y, et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;5(3):e9539. doi:10.1371/journal.pone.0009539
64. Darby IA, Bisucci T, Hewitson TD, MacLellan DG. Apoptosis is increased in a model of diabetes-impaired wound healing in genetically diabetic mice. Int J Biochem Cell Biol. 1997;29(1):191–200. doi:10.1016/s1357-2725(96)00131-8
65. Dardenne C, Salon M, Authier H, et al. Topical aspirin administration improves cutaneous wound healing in diabetic mice through a phenotypic switch of wound macrophages toward an anti-inflammatory and proresolutive profile characterized by LXA4 release. Diabetes. 2022;71(10):2181–2196. doi:10.2337/db20-1245
66. Brem H, Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes. J Clin Invest. 2007;117(5):1219–1222. doi:10.1172/JCI32169
67. Suresh Babu S, Thandavarayan RA, Joladarashi D, et al. MicroRNA-126 overexpression rescues diabetes-induced impairment in efferocytosis of apoptotic cardiomyocytes. Sci Rep. 2016;6(1):36207. doi:10.1038/srep36207
68. Leerach N, Munesue S, Harashima A, et al. RAGE signaling antagonist suppresses mouse macrophage foam cell formation. Biochem Biophys Res Commun. 2021;555:74–80. doi:10.1016/j.bbrc.2021.03.139
69. Morey M, O’Gaora P, Pandit A, Hélary C. Hyperglycemia acts in synergy with hypoxia to maintain the pro-inflammatory phenotype of macrophages. PLoS One. 2019;14(8):e0220577. doi:10.1371/journal.pone.0220577
70. Wang YT, Trzeciak AJ, Rojas WS, et al. Metabolic adaptation supports enhanced macrophage efferocytosis in limited-oxygen environments. Cell Metab. 2023;35(2):316–331.e6. doi:10.1016/j.cmet.2022.12.005
71. Taha NA, Hussein AA, El-Hak HNG, El-Shenawy NS. A mini-review of nanoparticle therapeutics targeting oxidative stress and inflammation in diabetes. JoBAZ. 2025;86(1):29. doi:10.1186/s41936-025-00449-2
72. Lou J, Xiang Z, Zhu X, et al. Skin microbiota and diabetic foot ulcers. Front Microbiol. 2025;16:1575081. doi:10.3389/fmicb.2025.1575081
73. Jun JI, Kim KH, Lau LF. The matricellular protein CCN1 mediates neutrophil efferocytosis in cutaneous wound healing. Nat Commun. 2015;6(1):7386. doi:10.1038/ncomms8386
74. Kita A, Yamamoto S, Saito Y, Chikenji TS. Cellular senescence and wound healing in aged and diabetic skin. Front Physiol. 2024;15:1344116. doi:10.3389/fphys.2024.1344116
75. Cabrera JTO, Makino A. Efferocytosis of vascular cells in cardiovascular disease. Pharmacol Ther. 2022;229:107919. doi:10.1016/j.pharmthera.2021.107919
76. Das A, Ghatak S, Sinha M, et al. Correction of MFG-E8 resolves inflammation and promotes cutaneous wound healing in diabetes. J Immunol. 2016;196(12):5089–5100. doi:10.4049/jimmunol.1502270
77. Uchiyama A, Motegi SI, Sekiguchi A, et al. Mesenchymal stem cells-derived MFG-E8 accelerates diabetic cutaneous wound healing. J Dermatol Sci. 2017;86(3):187–197. doi:10.1016/j.jdermsci.2017.02.285
78. Bossi F, Tripodo C, Rizzi L, et al. C1q as a unique player in angiogenesis with therapeutic implication in wound healing. Proc Natl Acad Sci USA. 2014;111(11):4209–4214. doi:10.1073/pnas.1311968111
79. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi:10.12703/P6-13
80. Lei J, Shu Z, Zhu H, Zhao L. AMPK regulates M1 macrophage polarization through the JAK2/STAT3 signaling pathway to attenuate airway inflammation in obesity-related asthma. Inflammation. 2024;48(1):372–392. doi:10.1007/s10753-024-02070-x
81. M R-S, Teixeira da mota A, Jardim C, Serre K. Bringing macrophages to the frontline against cancer: Current immunotherapies targeting macrophages. Cells. 2021;10(9):2364. doi:10.3390/cells10092364
82. Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun. 2014;6(6):716–726. doi:10.1159/000364945
83. Fuchs AK, Syrovets T, Haas KA, et al. Carboxyl- and amino-functionalized polystyrene nanoparticles differentially affect the polarization profile of M1 and M2 macrophage subsets. Biomaterials. 2016;85:78–87. doi:10.1016/j.biomaterials.2016.01.064
84. Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol. 2013;93(6):875–881. doi:10.1189/jlb.1012512
85. Mirza R, DiPietro LA, Koh TJ. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol. 2009;175(6):2454–2462. doi:10.2353/ajpath.2009.090248
86. Wu X, He W, Mu X, et al. Macrophage polarization in diabetic wound healing. Burns Trauma. 2022;10:tkac051. doi:10.1093/burnst/tkac051
87. Song J, Wu Y, Chen Y, Sun X, Zhang Z. Epigenetic regulatory mechanism of macrophage polarization in diabetic wound healing (Review). Mol Med Rep. 2024;31(1):2. doi:10.3892/mmr.2024.13367
88. Barman PK, Urao N, Koh TJ. Diabetes induces myeloid bias in bone marrow progenitors associated with enhanced wound macrophage accumulation and impaired healing. J Pathol. 2019;249(4):435–446. doi:10.1002/path.5330
89. Peng X, He F, Mao Y, et al. miR-146a promotes M2 macrophage polarization and accelerates diabetic wound healing by inhibiting the TLR4/NF-κB axis. J Mol Endocrinol. 2022;69(2):315–327. doi:10.1530/JME-21-0019
90. Li Y, Tan J, Miao Y, Zhang Q. MicroRNA in extracellular vesicles regulates inflammation through macrophages under hypoxia. Cell Death Discov. 2021;7(1):285. doi:10.1038/s41420-021-00670-2
91. Yu W, Tao M, Zhao Y, Hu X, Wang M. 4′-Methoxyresveratrol Alleviated AGE-induced inflammation via RAGE-Mediated NF-κB and NLRP3 inflammasome pathway. Molecules. 2018;23(6):1447. doi:10.3390/molecules23061447
92. Korbecki J, Bajdak-Rusinek K. The effect of palmitic acid on inflammatory response in macrophages: an overview of molecular mechanisms. Inflamm Res. 2019;68(11):915–932. doi:10.1007/s00011-019-01273-5
93. Okonkwo UA, Chen L, Ma D, et al. Compromised angiogenesis and vascular integrity in impaired diabetic wound healing. PLoS One. 2020;15(4):e0231962. doi:10.1371/journal.pone.0231962
94. Gurevich DB, Severn CE, Twomey C, et al. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. EMBO J. 2018;37(13):e97786. doi:10.15252/embj.201797786
95. Hesketh M, Sahin KB, West ZE, Murray RZ. Regulate scar formation and chronic wound healing. IJMS. 2017;18(7):1545. doi:10.3390/ijms18071545
96. Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. J Leukocyte Biol. 2009;87(1):59–67. doi:10.1189/jlb.0409236
97. Kim SY, Nair MG. Macrophages in wound healing: activation and plasticity. Immunol Cell Biol. 2019;97(3):258–267. doi:10.1111/imcb.12236
98. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14(3):166–180. doi:10.1038/nri3607
99. Coulombe F, Jaworska J, Verway M, et al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of Type I interferon and apoptosis in macrophages. Immunity. 2014;40(4):554–568. doi:10.1016/j.immuni.2014.02.013
100. Barman PK, Koh TJ. Macrophage dysregulation and impaired skin wound healing in diabetes. Front Cell Dev Biol. 2020;8:528. doi:10.3389/fcell.2020.00528
101. Wang Y, Chen C, Li Y, et al. Kaempferol inhibits oxidative stress and reduces macrophage pyroptosis by activating the NRF2 signaling pathway. PLoS One. 2025;20(6):e0325189. doi:10.1371/journal.pone.0325189
102. Zhou Z, Tang Y, Jin X, et al. Metformin inhibits advanced glycation end products-induced inflammatory response in murine macrophages partly through AMPK activation and RAGE/NF κ B pathway suppression. J Diab Res. 2016;2016:1–10. doi:10.1155/2016/4847812
103. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. doi:10.1038/nri3399
104. PLOS ONE Editors. Expression of concern: topical insulin accelerates wound healing in diabetes by enhancing the AKT and ERK pathways: a double-blind placebo-controlled clinical trial. PLoS One. 2024;19(2):e0298558. doi:10.1371/journal.pone.0298558.
105. Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. 2016;12(1):25–36. doi:10.1038/nrrheum.2015.167
106. Czimmerer Z, Daniel B, Horvath A, et al. The transcription factor STAT6 mediates direct repression of inflammatory enhancers and limits activation of alternatively polarized macrophages. Immunity. 2018;48(1):75–90.e6. doi:10.1016/j.immuni.2017.12.010
107. Samarpita S, Srivastava S, Srikanth M, Miriam Jose A, Rithvik A, Rasool M. IL-17A/IL-17RA interaction blockade sensitizes synovial macrophages to efferocytosis and PD-L1 signaling via rewiring STAT-3/ADAM17/MERTK axis in rheumatoid arthritis animal model. Int Immunopharmacol. 2024;136:112343. doi:10.1016/j.intimp.2024.112343
108. Durham GA, Williams JJL, Nasim MT, Palmer TM. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol Sci. 2019;40(5):298–308. doi:10.1016/j.tips.2019.03.001
109. Jiang L, Chen XQ, Gao MJ, et al. The Pros1/Tyro3 axis protects against periodontitis by modulating STAT/SOCS signalling. J Cell Mol Medi. 2019;23(4):2769–2781. doi:10.1111/jcmm.14183
110. Ren J, Zhu B, Gu G, et al. Schwann cell-derived exosomes containing MFG-E8modify macrophage/microglial polarization for attenuating inflammation via the SOCS3/STAT3 pathway after spinal cord injury. Cell Death Dis. 2023;14(1):70. doi:10.1038/s41419-023-05607-4
111. Simon LS, Taylor PC, Choy EH, et al. The Jak/STAT pathway: a focus on pain in rheumatoid arthritis. Semin Arthritis Rheum. 2021;51(1):278–284. doi:10.1016/j.semarthrit.2020.10.008
112. Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108(5):1652–1660. doi:10.1182/blood-2006-02-002030
113. Gurzov EN, Stanley WJ, Pappas EG, Thomas HE, Gough DJ. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016;283(16):3002–3015. doi:10.1111/febs.13709
114. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6(1):402. doi:10.1038/s41392-021-00791-1
115. Soni B, Singh S. Synthetic perturbations in IL6 biological circuit induces dynamical cellular response. Molecules. 2021;27(1):124. doi:10.3390/molecules27010124
116. Fang L, Chen L, Song M, et al. Naoxintong accelerates diabetic wound healing by attenuating inflammatory response. Pharm Biol. 2021;59(1):250–259. doi:10.1080/13880209.2021.1877735
117. Zhou X, Guo Y, Yang K, Liu P, Wang J. The signaling pathways of traditional Chinese medicine in promoting diabetic wound healing. J Ethnopharmacol. 2022;282:114662. doi:10.1016/j.jep.2021.114662
118. Li S, Sun Y, Liang CP, et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circul Res. 2009;105(11):1072–1082. doi:10.1161/CIRCRESAHA.109.199570
119. Murakami Y, Tian L, Voss OH, Margulies DH, Krzewski K, Coligan JE. CD300b regulates the phagocytosis of apoptotic cells via phosphatidylserine recognition. Cell Death Differ. 2014;21(11):1746–1757. doi:10.1038/cdd.2014.86
120. Chen F, Li Y, Zhao L, et al. Anti-inflammatory effects of MerTK by inducing M2 macrophage polarization via PI3K/Akt/GSK-3β pathway in gout. Int Immunopharmacol. 2024;142(Pt A):112942. doi:10.1016/j.intimp.2024.112942
121. Schlegel J, Sambade MJ, Sather S, et al. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J Clin Invest. 2013;123(5):2257–2267. doi:10.1172/JCI67816
122. Tao H, Yancey PG, Babaev VR, et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res. 2015;56(8):1449–1460. doi:10.1194/jlr.M056689
123. Yang Y, Jia X, Qu M, et al. Exploring the potential of treating chronic liver disease targeting the PI3K/Akt pathway and polarization mechanism of macrophages. Heliyon. 2023;9(6):e17116. doi:10.1016/j.heliyon.2023.e17116
124. Jin Y, Huang Y, Zeng G, et al. Advanced glycation end products regulate macrophage apoptosis and influence the healing of diabetic foot wound through miR-361-3p/CSF1R and PI3K/AKT pathway. Heliyon. 2024;10(2):e24598. doi:10.1016/j.heliyon.2024.e24598
125. Yu T, Gao M, Yang P, et al. Insulin promotes macrophage phenotype transition through PI3K/Akt and PPAR-γ signaling during diabetic wound healing. J Cell Physiol. 2019;234(4):4217–4231. doi:10.1002/jcp.27185
126. Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol. 2011;166(1):1–15. doi:10.1111/j.1365-2249.2011.04440.x
127. Guo H, Callaway JB, Ting JPY. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–687. doi:10.1038/nm.3893
128. An Y, Zhang H, Wang C, et al. Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. 2019;33(11):12515–12527. doi:10.1096/fj.201802805RR
129. Wei S, Guan G, Luan X, et al. NLRP3 inflammasome constrains liver regeneration through impairing MerTK-mediated macrophage efferocytosis. Sci Adv. 2025;11(1):eadq5786. doi:10.1126/sciadv.adq5786
130. Hu H, Wang S, Chen C. Pathophysiological role and potential drug target of NLRP3 inflammasome in the metabolic disorders. Cell Signalling. 2024;122:111320. doi:10.1016/j.cellsig.2024.111320
131. Zhu X, Xu Y, Wang J, Xue Z, Qiu T, Chen J. Loss of NLRP3 reduces oxidative stress and polarizes intratumor macrophages to attenuate immune attack on endometrial cancer. Front Immunol. 2023;14:1165602. doi:10.3389/fimmu.2023.1165602
132. Tajbakhsh A, Bianconi V, Pirro M, Gheibi Hayat SM, Johnston TP, Sahebkar A. Efferocytosis and atherosclerosis: regulation of phagocyte function by MicroRNAs. Trends Endocrinol Metab. 2019;30(9):672–683. doi:10.1016/j.tem.2019.07.006
133. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. doi:10.1038/nri3520
134. Seimon TA, Nadolski MJ, Liao X, et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12(5):467–482. doi:10.1016/j.cmet.2010.09.010
135. Sun H, Sun Z, Xu X, et al. Blocking TRPV4 ameliorates osteoarthritis by inhibiting M1 macrophage polarization via the ROS/NLRP3 signaling pathway. Antioxidants. 2022;11(12):2315. doi:10.3390/antiox11122315
136. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. IJMS. 2019;20(13):3328. doi:10.3390/ijms20133328
137. Zhang X, Dai J, Li L, Chen H, Chai Y. NLRP3 inflammasome expression and signaling in human diabetic wounds and in high glucose induced macrophages. J Diab Res. 2017;2017:5281358. doi:10.1155/2017/5281358
138. Kinnunen K, Piippo N, Loukovaara S, Hytti M, Kaarniranta K, Kauppinen A. Lysosomal destabilization activates the NLRP3 inflammasome in human umbilical vein endothelial cells (HUVECs). J Cell Commun Signal. 2017;11(3):275–279. doi:10.1007/s12079-017-0396-4
139. Yang CC, Wu CH, Lin TC, et al. Inhibitory effect of PPARγ on NLRP3 inflammasome activation. Theranostics. 2021;11(5):2424–2441. doi:10.7150/thno.46873
140. Yan W, Ni T, Zhang Q, et al. MCC950 promotes diabetic wound healing through modulating macrophage polarization in an MDSC-dependent manner. Int Immunopharmacol. 2024;142(Pt A):112983. doi:10.1016/j.intimp.2024.112983
141. Qing L, Fu J, Wu P, Zhou Z, Yu F, Tang J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am J Transl Res. 2019;11(2):655–668.
142. Mirza RE, Fang MM, Weinheimer-Haus EM, Ennis WJ, Koh TJ. Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes. 2014;63(3):1103–1114. doi:10.2337/db13-0927
143. Fan W, Li W, Lu H, Liu G. Exploring the mechanism of hematite ointment to reverse the macrophage efferocytosis escape of diabetic refractory wound through NLRP3 based on the theory of Weinong Zhangrou. Acta Chin Med Pharmacol. 2023;51(11):29–35. doi:10.19664/j.cnki.1002-2392.230233
144. Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545–558. doi:10.1038/nri.2017.52
145. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12(8):695–708. doi:10.1038/ni.2065
146. Rothlin CV, Ghosh S, Zuniga EI, Oldstone MBA, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124–1136. doi:10.1016/j.cell.2007.10.034
147. Wu X, Wang Z, Shi J, et al. Macrophage polarization toward M1 phenotype through NF-κB signaling in patients with Behçet’s disease. Arthritis Res Ther. 2022;24(1):249. doi:10.1186/s13075-022-02938-z
148. Jin X, Yao T, Zhou Z, et al. Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF-κB pathway. Biomed Res Int. 2015;2015:732450. doi:10.1155/2015/732450
149. Zhu Y, Ren S, Huang H, et al. Restoration of miR-299-3p promotes efferocytosis and ameliorates atherosclerosis via repressing CD47 in mice. FASEB J. 2024;38(15):e23857. doi:10.1096/fj.202400639R
150. Zhou M, Zhang Y, Shi L, et al. Activation and modulation of the AGEs-RAGE axis: implications for inflammatory pathologies and therapeutic interventions – a review. Pharmacol Res. 2024;206:107282. doi:10.1016/j.phrs.2024.107282
151. Kamal R, Awasthi A, Pundir M, Thakur S. Healing the diabetic wound: unlocking the secrets of genes and pathways. Eur J Pharmacol. 2024;975:176645. doi:10.1016/j.ejphar.2024.176645
152. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi:10.1101/cshperspect.a001651
153. Li J, Zhang Y, Kirsner RS. Angiogenesis in wound repair: angiogenic growth factors and the extracellular matrix. Microscopy Res Techn. 2003;60(1):107–114. doi:10.1002/jemt.10249
154. Morgan MJ, Gang LZ. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21(1):103–115. doi:10.1038/cr.2010.178
155. Papa S, Choy PM, Bubici C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene. 2019;38(13):2223–2240. doi:10.1038/s41388-018-0582-8
156. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-Activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83. doi:10.1128/MMBR.00031-10
157. Bai X, Guo YR, Zhao ZM, et al. Macrophage polarization in cancer and beyond: from inflammatory signaling pathways to potential therapeutic strategies. Cancer Lett. 2025;625:217772. doi:10.1016/j.canlet.2025.217772
158. Ou J, Li K, Yuan H, et al. Staphylococcus aureus vesicles impair cutaneous wound healing through p38 MAPK-MerTK cleavage-mediated inhibition of macrophage efferocytosis. Cell Commun Signal. 2025;23(1):14. doi:10.1186/s12964-024-01994-z
159. De Maeyer RPH, Van De Merwe RC, Louie R, et al. Blocking elevated p38 MAPK restores efferocytosis andinflammatory resolution in the elderly. Nat Immunol. 2020;21(6):615–625. doi:10.1038/s41590-020-0646-0
160. Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med. 2022;54(2):91–102. doi:10.1038/s12276-022-00736-w
161. Yang H, Wang H, Andersson U. Targeting Inflammation Driven by HMGB1. Front Immunol. 2020;11:484. doi:10.3389/fimmu.2020.00484
162. Li Q, Liu X, Du Y, et al. Protocatechuic acid boosts continual efferocytosis in macrophages by derepressing KLF4 to transcriptionally activate MerTK. Sci Signal. 2023;16(786):eabn1372. doi:10.1126/scisignal.abn1372
163. Ripszky Totan A, Imre MM, Parvu S, et al. Autophagy plays multiple roles in the soft-tissue healing and osseointegration in dental implant surgery—A narrative review. Materials. 2022;15(17):6041. doi:10.3390/ma15176041
164. Heo KS, Cushman HJ, Akaike M, et al. ERK5 activation in macrophages promotes efferocytosis and inhibits atherosclerosis. Circulation. 2014;130(2):180–191. doi:10.1161/CIRCULATIONAHA.113.005991
165. Bellamri N, Lelong M, Joannes A, et al. Effects of Ruxolitinib on fibrosis in preclinical models of systemic sclerosis. Int Immunopharmacol. 2023;116:109723. doi:10.1016/j.intimp.2023.109723
166. Yoon YS, Kim SY, Kim MJ, Lim JH, Cho MS, Kang JL. PPARγ activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol. 2015;8(5):1031–1046. doi:10.1038/mi.2014.130
167. Tang S, Lu C, Meng Z, et al. USP22 enhances atherosclerotic plaque stability and macrophage efferocytosis by stabilizing PPARγ. Commun Biol. 2025;8(1):678. doi:10.1038/s42003-025-08116-6
168. Sheikh IM, Hassan OA, Adam SM, et al. Association of pioglitazone with major adverse cardiovascular events, all-cause mortality, and heart failure hospitalizations: a systematic review. Cureus. 2023;15(10):e46911. doi:10.7759/cureus.46911
169. Cai X, Shi Y, Dai Y, Wang F, Chen X, Li X. Baicalin clears inflammation by enhancing macrophage efferocytosis via inhibition of RhoA/ROCK signaling pathway and regulating macrophage polarization. Int Immunopharmacol. 2022;105:108532. doi:10.1016/j.intimp.2022.108532
170. Xia W, Zhu Z, Xiang S, Yang Y. Ginsenoside Rg5 promotes wound healing in diabetes by reducing the negative regulation of SLC7A11 on the efferocytosis of dendritic cells. J Ginseng Res. 2023;47(6):784–794. doi:10.1016/j.jgr.2023.06.006
171. Feng SL, Luo HB, Cai L, et al. Ginsenoside Rg5 overcomes chemotherapeutic multidrug resistance mediated by ABCB1 transporter: in vitro and in vivo study. J Ginseng Res. 2020;44(2):247–257. doi:10.1016/j.jgr.2018.10.007
172. Gong X, Cai J, Zheng W, et al. Isoliquiritigenin alleviates SLC7A11-mediated efferocytosis inhibition to promote wounds healing in diabetes. Biomed Pharmacother. 2024;180:117578. doi:10.1016/j.biopha.2024.117578
173. Tang C, Wang H, Guo L, et al. CpG-conjugated silver nanoparticles as a multifunctional nanomedicine to promote macrophage efferocytosis and repolarization for atherosclerosis therapy. ACS Appl Mater Interfaces. 2023:
174. Qiu S, Liu J, Chen J, et al. Targeted delivery of MerTK protein via cell membrane engineered nanoparticle enhances efferocytosis and attenuates atherosclerosis in diabetic ApoE−/− Mice. J Nanobiotechnol. 2024;22(1):178. doi:10.1186/s12951-024-02463-y
175. Huang J, Xia C, Wei Y, et al. Annexin <sc>A1</sc> ‐derived peptide <sc>Ac2
176. Wang H, Zhang Y, Zhang Y, et al. Activating macrophage continual efferocytosis via microenvironment biomimetic short fibers for reversing inflammation in bone repair. Adv Mater. 2024;36(30):2402968. doi:10.1002/adma.202402968
177. Liu X, Dou G, Li Z, et al. Hybrid biomaterial initiates refractory wound healing via inducing transiently heightened inflammatory responses. Adv Sci. 2022;9(21):2105650. doi:10.1002/advs.202105650
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Bioactive Nanocomposite-Mediated Macrophage Metabolic Shift Orchestrates Pro-Regenerative Healing in the Diabetic Wound Microenvironment
Shen T, Xu X, Guo Z, Zhou W, Xiao X, Liao S, Huang J, Liu X, Fan Z
International Journal of Nanomedicine 2026, 21:617254
Published Date: 30 June 2026