Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 5
The Creating Hope Act: what is old is new again
Authors Tolbert J, Goldman J, Kauffman R, Abdel-Rahman S
Received 18 February 2014
Accepted for publication 29 March 2014
Published 3 June 2014 Volume 2014:5 Pages 49—57
DOI https://doi.org/10.2147/PHMT.S46069
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Jaszianne A Tolbert,1,2 Jennifer L Goldman,2-4 Ralph E Kauffman,2,4 Susan M Abdel-Rahman2,4
1Division of Hematology/Oncology, 2Division of Clinical Pharmacology and Therapeutic Innovation, 3Division of Infectious Diseases, Children's Mercy Hospital, Kansas City, MO, USA; 4Department of Pediatrics, University of Missouri-Kansas City, School of Medicine, Kansas City, MO, USA
Abstract: The Creating Hope Act, passed as part of the Food and Drug Administration Safety and Innovation Act of 2012, is among the newest laws intended to foster drug development for rare and neglected diseases in children. The act expands the priority review voucher incentive that first appeared in the Food and Drug Administration Amendments Act of 2007 and was intended to stimulate the development of products for the prevention and treatment of tropical diseases. Notably, legislative and regulatory initiatives aimed at enhancing drug development both for use in children and for rare diseases have intermittently emerged over the past 3 decades. This manuscript provides an overview of related legislation that has preceded the Creating Hope Act and examines the potential impact of the new act in the context of the outcomes that have been observed with the earlier initiatives.
Keywords: orphan drug, rare disease, pediatric, drug development, priority review voucher
Introduction
Recently, the Creating Hope Act (CHA) was signed into law as part of the Food and Drug Administration Safety and Innovation Act (FDASIA).1 The bill was proposed to foster drug development for rare pediatric diseases including childhood cancer. Not surprisingly, the link to pediatric cancer has precipitated strongly positive opinions in the media as associations with kids and cancer are wont to do. The CHA has been heralded as “a milestone”, “historic”, and “fundamentally transformative”. However, initiatives aimed at enhancing drug development, both for use in children and for rare and neglected diseases, are not new. Numerous legislative initiatives have surfaced over the past several decades and, while the proponents and opponents change with each new initiative, the fundamental intent is largely the same. So, what voids does the new Creating Hope Act address and which does it neglect to fill? This manuscript will examine the new act in the context of the legislations for rare (orphan) diseases, tropical diseases, and diseases of childhood that have preceded it.
Drug development for rare diseases
Rare disease populations were among the first to benefit from legislation that offered financial and regulatory incentives to stimulate drug development.2 Prior to the Orphan Drug Act (ODA), there was a critical lack of medications to address the needs of an estimated 25 million individuals who are afflicted with one of countless diseases that occur in fewer than 200,000 individuals in the US. In an attempt to address these unmet medical needs, the US federal government enacted legislation that provided some commanding incentives for companies willing to undertake the development of drugs and biologics used in the diagnosis, treatment, or prevention of these conditions (Table 1). The ODA provided tax credits to offset the cost of drug development, waived the user fees that typically accompany a marketing application to the US Food and Drug Administration (FDA), and offered 7 years of marketing exclusivity for products receiving approval.2
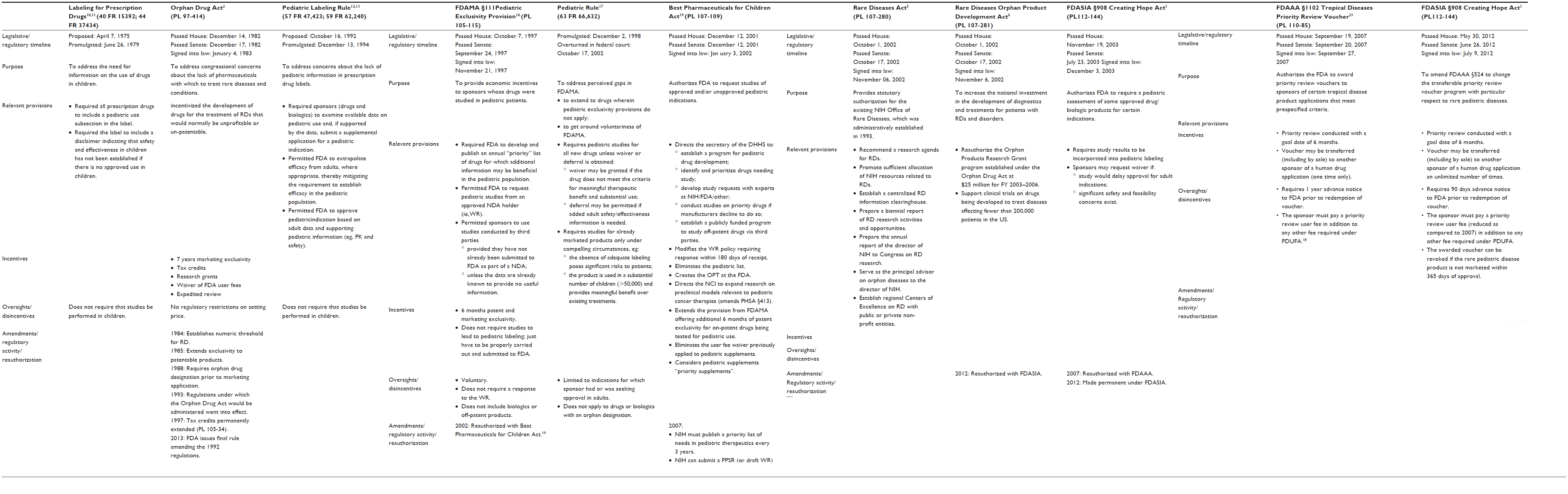 | Table 1 Legislative and regulatory initiatives related to drug development for RDs and children |
In the 30 years since this law went into effect, over 400 drugs have received an orphan drug designation with approval for marketing, representing, in recent years, nearly one-third of FDA approvals for new molecular entities.3 Over 200 rare diseases are addressed by these new medications, and cancer, along with pediatric conditions, features prominently in this list.4 This surge in development lay in stark contrast to the decade preceding the ODA, wherein ten drugs received marketing approval in as many years. These statistics leave little question as to the impactfulness of the incentives established by the ODA, evidenced further by the number of international regulatory agencies that have adopted similar legislation. The success of the ODA has also stimulated other legislative initiatives surrounding rare diseases. These include the Rare Diseases Act and Rare Diseases Orphan Product Development Act, which are intended to foster a research agenda around rare and neglected diseases (Table 1),5,6 and regulations that aim to accelerate the review and approval of drugs for serious and life-threatening conditions (Table 2).7
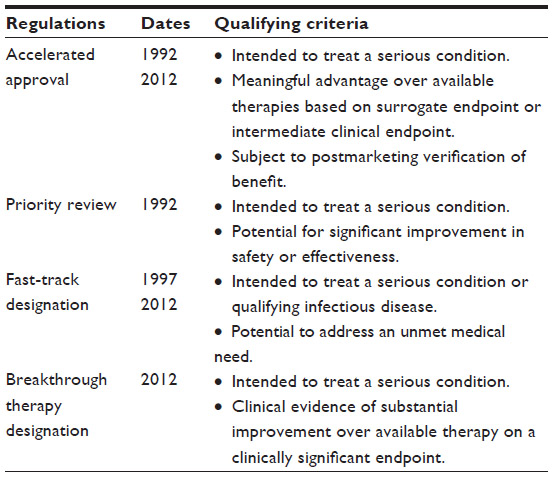 | Table 2 Expedited programs authorized by the US Food and Drug Administration |
It is important to recognize that biotechnology companies sponsor the majority of orphan-designated products.8 In fact, the rate of growth for orphan products has paralleled growth of the biotechnology sector since 1987.9 Among all of the incentives offered by the ODA, the promise of added exclusivity appears to be the principal driver that affords small, start-up companies the ability to secure the private capital needed to fund the drug development process.9 The putative value of other incentives (eg, priority review vouchers [PRVs]) for a company wherein the portfolio is small and the development pipeline is limited will be considered later in this manuscript.
Drug development for children
Recognition that drug use in children was occurring without pediatric safety and efficacy data stimulated regulatory activity as early as the mid-1970s (Table 1).10,11 Regulations promulgated at the end of that decade, and again 15 years later, required sponsors to address “pediatric use” in the product label.12,13 However, neither regulation required that studies be performed in children and, with no enforcement mechanism, their impact on pediatric drug development remained relatively limited.14 At the time, the major impediments to pediatric drug development included limited expertise in conducting pediatric pharmaceutical studies; perceived difficulties in recruiting pediatric participants; concerns related to liability and ethics surrounding investigations in a vulnerable population; and economic disincentives that accompany the relatively small pediatric market for most drugs.
In 1990, the Institute of Medicine of the National Academy of Sciences, in conjunction with the National Institutes of Health (NIH), the American Academy of Pediatrics, the FDA, and representatives from the pharmaceutical industry, convened a workshop to address the problem. They concluded a need for all parties to prioritize and take ownership in pediatric drug development and proposed that: 1) the FDA explore mechanisms to facilitate the approval and labeling of drugs in children; 2) the US Congress develop legislation to address the economic disincentives; 3) pharmaceutical companies take a proactive stance in pediatric drug development; and 4) the NIH provide funding for pediatric drug studies, including the sponsorship of a collaborative pediatric research network to conduct the necessary studies.15 Over the ensuing decades, attempts were made by all parties to address these recommendations.
In 1997, the US Congress passed the Food and Drug Administration Modernization Act (FDAMA), which contained pioneering legislation for pediatric drug development in the US (Table 1).16 For the first time, there was economic incentive to develop drugs for children by means of an additional 6 months of exclusivity for new drugs or marketed drugs with protection still remaining. Under this statute, the FDA could submit a Written Request (WR) for pediatric studies. If the company voluntarily complied with the WR and completed the studies specified by the FDA, they were awarded 6 months of additional exclusivity for both the adult and pediatric indications, irrespective of whether data from the studies led to pediatric labeling. For drugs with a large adult market, additional exclusivity proved to be highly profitable. This incentive had an unprecedented impact on pediatric drug development, with hundreds of pediatric studies conducted (and drugs labeled for children) for medicines that had never previously been studied in children.
Despite its success, there were gaps in FDAMA. Principal among these was that the performance of pediatric studies was voluntary and there was no incentive to study drugs whose exclusivity had expired despite their value to children. Consequently, the FDA enacted the 1998 Pediatric Rule (Table 1).17 Under this regulation, the FDA could require pediatric studies of new drugs or biologicals if the product was likely to be used in a substantial number of pediatric patients (defined as >50,000). Notably, this rule applied only to the indications supported in the New Drug Application,17 which almost exclusively targeted adult conditions. Sponsors could not be required to conduct studies for unique pediatric indications. This rule was the first to mandate pediatric studies; however, it came under attack and was overturned by a US federal court in 2002 ruling that the FDA exceeded its statutory authority in promulgating the Pediatric Rule.
This court action became moot when Congress passed the Best Pharmaceuticals for Children Act (BPCA) in 2002 and the Pediatric Research Equity Act (PREA) in 2003 (Table 1).19,20 The BPCA extended the exclusivity provisions authorized under FDAMA and attempted to deal with shortcomings of the previous legislation.19 Under this new law, the FDA could conduct pediatric studies of drugs for which the manufacturer declined to respond to a WR. A mechanism by which to conduct pediatric studies of off-patent drugs was also addressed. Additionally, the FDA was required to establish: 1) the Office of Pediatric Therapeutics, which is responsible for facilitation and coordination of all FDA pediatric programs; 2) a Pediatric Pharmacology Advisory Committee, which focuses on issues surrounding pediatric pharmacology research (ethics included); and 3) a Subcommittee of the Oncologic Drugs Advisory Committee to help prioritize pediatric cancer drugs. PREA codified the requirements for pediatric studies embodied in the 1998 Pediatric Rule (Table 1).20 Under this new law, the FDA was given authority to require pediatric studies of drugs and biologics if the approved use was relevant to pediatrics. Unfortunately, sponsors were still permitted to request a waiver of the requirement for pediatric studies based on feasibility concerns, safety concerns, or putative delays in approval for the adult indication.
BPCA and PREA worked synergistically to enhance pediatric drug development. In 2007, both were reauthorized under the Food and Drug Administration Amendments Act (FDAAA).21 This renewal incorporated elements from both pieces of legislation and emphasized transparency, outcomes reporting, and pediatric formulation development. FDAAA also addressed the windfall profits that accompanied the minimal efforts of some manufacturers by requiring that applications for exclusivity be submitted at least 9 months prior to the expiration of the original patent. In 2012, BPCA and PREA were made permanent under FDASIA.22 As of January 2014, the FDA has received 842 proposed pediatric study requests, issued 439 WRs under the exclusivity incentives, and overseen the revision of 500 drug labels to incorporate pediatric information.23
Drug development for rare diseases in children
Conspicuously, only 15 of the 439 WRs issued by the FDA were related to oncology, and in the last 20 years, only two new drugs have been approved for use in childhood cancer. Thus, the perception that insufficient activity was taking place in the drug development arena for children with rare diseases persisted, despite the preexisting legislation detailed above. Motivated by the loss of her son to brain cancer, Nancy Goodman, and the “Kids v Cancer” nonprofit she founded, authored legislation that was subsequently signed into law (under FDASIA) as the CHA.24 The foundation of this act is a PRV, which is issued to a sponsor who successfully brings a product for a rare pediatric disease to market.25 This voucher entitles the holder to priority review and action by FDA within 6 months of a New Drug Application, irrespective of indication.
The PRV in and of itself is not novel to the CHA. These vouchers first appeared in FDAAA with the intent of stimulating the development of drugs for neglected tropical diseases (Table 1).26 The concept for this incentive program was proposed by academicians at Duke University (Durham, NC, USA) who argued that a voucher entitling the holder to a more rapid FDA review could be used to incentivize the development of drugs for neglected diseases in developing countries.27 Upon FDA approval of a drug developed for a neglected disease, the sponsor would receive a PRV that could, in turn: 1) be applied to the review of a drug that would not otherwise qualify for priority review; or 2) be transferred to another sponsor for the same intent. This shorter time to market could, in theory, generate hundreds of millions of dollars in added revenue for a “blockbuster” drug.27,28
Since the promulgation of the tropical diseases PRV program, only one voucher has been awarded. The decision to award this PRV in 2009 was met with criticism, because the subject of the approval, an antimalarial (Coartem™ [artemether/lumefantrine]; Novartis Pharma AG, Basel, Switzerland), had been readily available for nearly a decade. It had simply never been registered in the US.29 The sponsor subsequently used the PRV in 2011 on a medication for gouty arthritis (Ilaris™ [canakinumab]; Novartis Pharmaceuticals, Stein, Switzerland) that was denied approval, with the FDA requesting additional clinical data.30 With only a singular example in the 5 years that the PRV has been available, it is difficult to interpret the value of the preexisting legislation and predict the impact of its expansion with the CHA.
The CHA makes a few notable modifications to the earlier legislation.25 It expands the scope of diseases that are eligible for the incentive to include rare pediatric disease (eg, pediatric cancers). It also permits unlimited transfer of the voucher (including by sale). It shortens the notice of submission for the application from 1 year to 90 days; however, the PRV can also be revoked if the product for which the voucher was issued is not marketed within 1 year of approval. In conjunction with the expanded indications conferred by the CHA, there has also been a systematic decline in the nonrefundable user fee required of sponsors who redeem a PRV (Figure 1).31–34
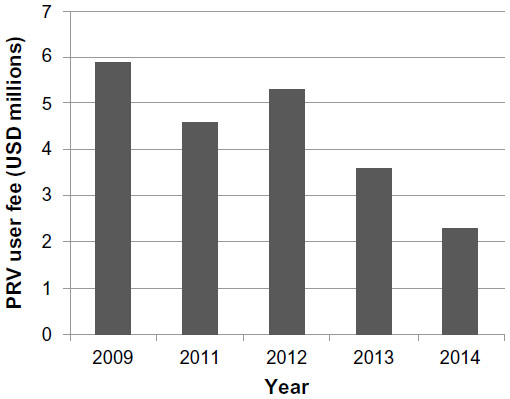 | Figure 1 PRV user fees as defined in the Federal Register. |
Missed opportunities
There remain shortcomings with the aforementioned legislation, which should be highlighted. The 6-month extended exclusivity incentive offered for pediatric drug development tends to select for drugs that have the greatest financial benefit for the sponsor in the adult market rather than strategically targeting high-priority pediatric needs. This oversight is not remedied under the FDASIA, wherein pediatric studies are mandated only for drugs with approved adult indications. Ostensibly, the ODA incentivizes drug development for rare pediatric diseases irrespective of adult indication; however, pediatric conditions that exceed the threshold criteria for a rare disease fall through the crack between FDASIA and the ODA. This could be remedied by incentivizing drug development for uniquely pediatric indications where prevalence estimates exceed the limits established by the 1984 amendment to the ODA.
In addition, older drugs, which are important for pediatric therapeutics but no longer have marketing exclusivity protection, also remain inadequately addressed. Currently, there is no economic incentive for pharmaceutical sponsors to conduct studies of these drugs in children. Despite the provisions of FDASIA that authorize the FDA to issue a WR to the NIH to perform such studies, no specific funding has been appropriated for these activities, and the NIH Foundation has not provided funding as was originally envisioned. The few studies that have been conducted under this mechanism were supported by re-allocating funds from within the NIH budget. As of September 2013, 27 WRs for studies of 25 different drugs have been issued to the NIH (approximately one-third for rare and oncologic diseases); 12 drugs have been or are being studied; and one drug was relabeled for a pediatric indication.35–37 Specific appropriations to adequately fund pediatric studies of drugs with no marketing exclusivity that have been prioritized by the NIH (as mandated under FDASIA) would substantially enhance the success of this provision.
Conclusion
Legislative and regulatory initiatives during the past 3 decades have had a profound impact on the number of drugs studied and labeled for orphan indications as well as for general pediatric use. Particularly effective have been the economic incentives combined with mandates for pediatric studies now incorporated in FDASIA. However, it is difficult to predict whether the CHA will substantively influence the future trajectory of drug development for rare and neglected pediatric conditions. Whether sponsors will value the economic incentive offered by the newly authorized PRV in the same light as the extended exclusivity provisions offered under the earlier legislation remains to be determined. Furthermore, a potential deterrent is the substantial user fee to invoke a voucher-driven priority review without any guarantee that the review will occur within the target 6-month time frame and no assurance that the review will be followed by approval.
As written (ie, with a status review by the US Government Accountability Office after the third voucher has been issued),25 the short-term impact of the CHA will likely be a modest, incremental (ie, single-digit) increase in the development of drugs for rare diseases in children as opposed to the double- and triple-digit impact generated by the ODA, BPCA, and PREA. If the trend for PRVs awarded under the CHA mirrors that observed following the initial enactment of this incentive under FDAAA, the act will likely fail to meet the expectations of the individuals and groups anticipating an uptick in drug development for susceptible pediatric populations. Future amendments to the CHA legislation (if it is to be renewed) should consider increasing the number of available vouchers, eliminating the additional user fees, and guaranteeing a maximum time for completion of the review to increase the value of the PRV to the sponsor.
Disclosure
The authors report no conflicts of interest in this work.
References
The Food and Drug Administration Safety and Innovation Act (2012). United States Public Law No 112–144. | |
The Orphan Drug Act (1983). United States Public Law No 97–414. | |
Goodman JL. Working together to address the challenges of rare and neglected diseases [webpage on the Internet]. Statement before the US House of Representatives, Subcommittee on Africa, Global Health, Global Human Rights, and International Organizations Committee on Foreign Affairs. Silver Spring, MD: US Food and Drug Administration; 2013. Available from: http://www.fda.gov/NewsEvents/Testimony/ucm358834.htm. Accessed December 27, 2013. | |
Braun MM, Farag-El-Massah S, Xu K, Coté TR. Emergence of orphan drugs in the United States: a quantitative assessment of the first 25 years. Nat Rev Drug Discov. 2010;9:519–522. | |
The Rare Diseases Act (2002). United States Public Law No 107–280. | |
The Rare Diseases Orphan Product Development Act (2002). United States Public Law No 107–281. | |
US Department of Health and Human Services-Food and Drug Administration. Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics. Silver Spring, MD: US Food and Drug Administration; 2013 [June]. Available from: http://www.fda.gov/regulatoryinformation/guidances/. Accessed June 2013. | |
Wellman-Labadie O, Zhou Y. The US Orphan Drug Act: rare disease research stimulator or commercial opportunity? Health Policy. 2010;95:216–228. | |
The Orphan Drug Act. Implementation and Impact. OEI-09-00-00380. Department of Health and Human Services Office of Inspector General; 2001. Available from: http://oig.hhs.gov/oei/reports/oei-09-00-00380.pdf. Accessed December 27, 2013. | |
Labeling for prescription drugs used in man; proposed format for prescription drug advertisements. Fed Regist. 1975;40:15392. Available from: http://www.ncbi.nlm.nih.gov/nlmcatalog/ 7808722. | |
Labeling and prescription drug advertising; content and format for labeling for human prescription drugs. Fed Regist. 1979;44:37434. Available from: http://www.ncbi.nlm.nih.gov/nlmcatalog/7808722. | |
Specific requirements on content and format of labeling for human prescription drugs; proposed revision of “Pediatric use” subsection in the labeling. Fed Regist. 1992;57:47423. Available from: http://www.ncbi.nlm.nih.gov/nlmcatalog/7808722. | |
Specific requirements on content and format of labeling for human prescription drugs; revision of “Pediatric use” subsection in the labeling. Fed Regist. 1994;59:64240. Available from: http://www.ncbi.nlm.nih.gov/nlmcatalog/7808722. | |
Schreiner MS. Paediatric clinical trials: redressing the imbalance. Nat Rev Drug Discov. 2003;2(12):949–961. | |
Forum on Drug Development, Institute of Medicine, National Academy of Sciences. Report of a Workshop on Drug Development and the Pediatric Population. Washington, DC: National Academy Press; 1991. | |
Section 111 of the Food and Drug Administration Modernization Act (1997). United States Public Law No 105–115. | |
Food and Drug Administration. Regulations requiring manufacturers to assess the safety and effectiveness of new drugs and biological products; Final rule. Fed Regst. 1998;63:66631–66672. | |
Prescription Drug User Fee Act. [webpage on the Internet]. Silverspring: US Department of Health & Human Services. [updated May 6, 2014]. Available from: http://www.fda.gov/forindustry/userfees/prescriptiondruguserfee/. Accessed May 12, 2014. | |
Best Pharmaceuticals for Children Act (2002). United States Public Law No 107–109. | |
Pediatric Research Equity Act (2003). United States Public Law No 108–155. | |
Title IV and V of the Food and Drug Administration Amendments Act (2007). United States Public Law No 110–185. | |
Section 501 of the Food and Drug Administration Safety and Innovation Act (2012). United States Public Law No 112–144. | |
New Pediatric Labeling Information Database [webpage on the Internet]. Silver Spring, MD: US Food and Drug Administration;, 2013. Available from: http://www.accessdata.fda.gov/scripts/sda/sdNavigation.cfm?sd=labelingdatabase. Accessed April 28, 2014. | |
Kids V Cancer [homepage on the Internet]. Washington, DC: Kids v Cancer; 2014. Available from: http://www.kidsvcancer.org/. Accessed January 1, 2014. | |
Section 908 of the Food and Drug Administration Safety and Innovation Act (2012). United States Public Law 112–144. | |
Section 524 of the Food and Drug Administration Amendments Act (2007). Public Law 110–185. | |
Ridley DB, Grabowski HG, Moe JL. Developing drugs for developing countries. Health Aff (Millwood). 2006;25:313–324. | |
Noor W. Placing value on FDA’s priority review vouchers. In Vivo. 2009;27(8):1–8. | |
Anderson T. Novartis under fire for accepting new reward for old drug. Lancet. 2009;373:1414. | |
Novartis Media Release. FDA request additional clinical data on Novartis drug ACZ885 for the treatment of gouty arthritis. 2011; [August 29] Available from: http://www.novartis.com/newsroom/media-releases/en/2011/1541785.shtml. Accessed April 28, 2014. | |
Fee for using a priority review voucher in fiscal year 2014. Fed Regist. 2013;78:172:54656–54657. | |
Fee for using a priority review voucher in fiscal year 2013. Fed Regist. 2012;77:178:56649–56650. | |
Fee for using a priority review voucher in fiscal year 2012. Fed Regist. 2011;76:168:53910–53912. | |
Fee for using a priority review voucher in fiscal year 2011. Fed Regist. 2010;76:168:55589–55591. | |
Written Requests [webpage on the Internet] Eunice Kennedy Shriver National Institute of Child Health, Best Pharmaceuticals for Children Act. [updated September 22, 2013]. Available from: http://bpca.nichd.nih.gov/clinical/requests/Pages/index.aspx. Accessed January 13, 2014. | |
Zajicek A. Overview of Ongoing Overview of Ongoing BPCA-Sponsored Projects. [Presentation]. National Institute of Child Health & Human Development. Available from: http://bpca.nichd.nih.gov/clinical/documents/BPCA_mtg_110911_FINAL.pdf. Accessed January 13, 2014. | |
Guttmacher AE. Director’s Report, National Advisory Child Health and Human Development Council; 2014. Available from: https://www.nichd.nih.gov/about/advisory/nachhd/Documents/directors_report_201401.pdf. Accessed January 13, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.