Back to Journals » OncoTargets and Therapy » Volume 10
The combination astemizole–gefitinib as a potential therapy for human lung cancer
Authors Chávez-López MDG, Zúñiga-García V, Hernández-Gallegos E, Vera E, Chasiquiza-Anchatuña CA ![]() , Viteri-Yánez M, Sanchez-Ramos J, Garrido E, Camacho J
, Viteri-Yánez M, Sanchez-Ramos J, Garrido E, Camacho J ![]()
Received 21 June 2017
Accepted for publication 12 October 2017
Published 6 December 2017 Volume 2017:10 Pages 5795—5803
DOI https://doi.org/10.2147/OTT.S144506
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yao Dai
María de Guadalupe Chávez-López,1 Violeta Zúñiga-García,1 Elisabeth Hernández-Gallegos,1 Eunice Vera,1 Carmen Alexandra Chasiquiza-Anchatuña,1,2 Marco Viteri-Yánez,1,2 Janet Sanchez-Ramos,3 Efraín Garrido,3 Javier Camacho1
1Department of Pharmacology, Center for Research and Advanced Studies of the National Polytechnic Institute, Mexico City, Mexico; 2Department of Life Sciences and Agriculture, University of the Armed Forces ESPE, Sangolquí, Ecuador; 3Department of Genetics and Molecular Biology, Center for Research and Advanced Studies of the National Polytechnic Institute, Mexico City, Mexico
Abstract: Lung cancer is a major cause of cancer mortality. Thus, novel therapies are urgently needed. Repositioning of old drugs is gaining great interest in cancer treatment. Astemizole is an antihistamine proposed to be repositioned for cancer therapy. This drug targets several molecules involved in cancer including histamine receptors, ABC transporters and the potassium channels Eag1 and HERG. Astemizole inhibits the proliferation of different cancer cells including those from cervix, breast, leukemia and liver. Gefitinib is widely used to treat lung cancer; however, no response or drug resistance occurs in many cases. Here, we studied the combined effect of astemizole and gefitinib on the proliferation, survival, apoptosis and gene and protein expression of Eag1 channels in the human lung cancer cell lines A549 and NCI-H1975. Cell proliferation and survival were studied by the MTT method and the colony formation assay, respectively; apoptosis was investigated by flow cytometry. Gene expression was assessed by real-time polymerase chain reaction (RT-PCR), and protein expression was studied by Western blot analysis and immunocytochemistry. We obtained the inhibitory concentrations 20 and 50 (IC20 and IC50, respectively) values for each drug from the cell proliferation experiments. Drug combination at their IC20 had a superior effect by reducing cell proliferation and survival in up to 80% and 100%, respectively. The drugs alone did not affect apoptosis of H1975 cells, but the drug combination at their IC20 increased apoptosis roughly four times in comparison to the effect of the drugs alone. Eag1 mRNA levels and protein expression were decreased by the drug combination in A549 cells, and astemizole induced subcellular localization changes of the channel protein in these cells. Our in vitro studies strongly suggest that the combination astemizole–gefitinib may be a novel and promising therapy for lung cancer patients.
Keywords: astemizole, gefitinib, potassium channels, lung cancer
Introduction
Lung cancer is the major cause of cancer-related deaths worldwide.1,2 Most of the primary lung cancers are non-small-cell lung cancer (NSCLC).3,4 The most important oncogene drivers in NSCLC patients are mutations in the epidermal growth factor receptor (EGFR) gene; actually, activating mutations are a prime therapeutic target.5,6 The most common “sensitizing mutations” in EGFR (exon 19 deletions and exon 21 L858R mis-sense substitutions) result in constitutive activation of the receptor without ligand binding. In accordance, some of the EGFR tyrosine kinase inhibitors (TKIs) target mutant EGFRs.5,7–9 The first-generation EGFR inhibitors such as gefitinib were the first EGFR-targeted therapies to be registered and later approved by the US Food and Drug Administration (FDA) as a treatment for lung cancer.6,10 Unfortunately, although many patients initially respond to EGFR-targeted therapies, most of them eventually developed resistance and relapsed.11 One of the mechanisms of drug resistance involves the extrusion of TKIs by ATP-binding cassette (ABC) multidrug efflux pumps.12,13 ABCG2 (breast cancer resistance protein) transports gefitinib which also interacts with ABCB1 and ABCC1.14 Because drug resistance decreases the efficacy of the drug, it is necessary to find alternative therapeutic strategies for lung cancer patients.
Drug repositioning involves the identification of novel indications for already existing, well-characterized and well-tolerated drugs reducing costs and bypassing safety concerns; this reposition strategy has emerged as an attractive alternative for cancer treatment.15–18 Astemizole is an anti-histamine that has gained enormous interest to be repositioned for cancer treatment because it targets several molecules involved in cancer including H1 histamine receptors, P-glycoproteins and members of the potassium channel family ether à-go-go.19–28 Human ether à go-go-1 (Eag1, Kcnh1 and Kv10.1) is a voltage-gated channel that displays oncogenic properties and has gained great interest in cancer research.25,26,29,30 The distribution of Eag1 is very restricted in normal tissues. It is mainly expressed in the brain, but low amounts can be detected in placenta, testes and adrenal glands and transiently in myoblasts.29,31,32 Conversely, Eag1 is overexpressed in most human tumors, including liver, cervical, lung, breast, colon and prostate cancer.28,32–35 Astemizole decreases tumor cell proliferation in vitro in breast, liver, cervical and lung cancer cell lines as well as in in vivo breast and liver cancer models.16,24,28,36 In addition, astemizole potentiates the effect of other anticancer drugs in leukemia and breast cancer cells and downregulates Eag1 mRNA expression in breast cancer cells; in addition, astemizole binds to chromatine.36–39 Interestingly, the use of astemizole and loratadine was associated with reduced mortality from different cancers, and astemizole induced sensitization to chemotherapy and reversion of multidrug resistance in NSCLC cells.16
Therefore, because of the high mortality-to-incidence ratio of lung cancer and the potential use of anti-histamines in cancer treatment as well as due to the multitarget properties of astemizole and its synergism with anticancer drugs, here we investigated whether the combination astemizole–gefitinib may be a potential anticancer treatment for lung cancer cells.
Materials and methods
Cells lines and reagents
The human NSCLC cell lines A549 and NCI-H1975 were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured according to the manufacturer’s instructions. Astemizole and DMSO were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Gefitinib (Iressa®, ZD1839) was kindly provided by AstraZeneca plc (Cambridge, UK). The anti-Eag1 antibody was purchased from Novus International (Littleton, CO, USA) and the anti-actin antibody from Sigma-Aldrich Co.
Metabolic activity
Cell proliferation (assessed by metabolic activity) was assayed by a colorimetric method with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as described previously.28 Briefly, 3×103 cells per well were seeded in 96-well plates and incubated for 72 hours in culture medium either alone or in the presence of astemizole, gefitinib or DMSO as vehicle. MTT (0.5 mg/mL) was added 4 hours before completing the whole incubation time. Absorbance data were obtained with a microplate photometer (Sunrise Touchscreen).
Colony formation assay
Cell survival was studied with the colony formation assay. Briefly, 2×102 A549 and 5×102 NCI-H1975 cells were cultured in 60 mm Petri dishes to allow the growth of colonies from single separated cells. Twenty-four hours after plating, the cells were incubated for 72 hours in culture medium alone or in the presence of DMSO or the drugs. Afterward, A549 and NCI-H1975 cells were left to grow for 6 and 11 days more, respectively, in the absence of the drugs. Then, cells were fixed in ethanol (absolute grade) for 15 minutes, stained with crystal violet (1%) for 15 minutes and then rinsed four times with water, observed with a microscope and counted.
Apoptosis
Apoptosis was studied by flow cytometry as described previously.28 Briefly, 4×104 cells were seeded in culture plates and incubated during 72 hours in culture medium alone or in the presence of astemizole, gefitinib or vehicle (DMSO). Camptothecin (apoptosis inductor) and methanol (necrosis inductor) were used as controls. Apoptosis was determined with the Annexin V-FITC kit (Thermo Fisher Scientific, Waltham, MA, USA) binding to phosphatidylserine and DNA staining by propidium iodide (PI). Experiments were performed by flow cytometry (CYAN ADP; Dako, Glostrup, Denmark). Percentages of viable (FITC-negative and PI-negative), apoptotic (FITC-positive and PI-negative) and late apoptotic (FITC-positive and PI-positive) cells were obtained by quadrant analysis using the Summit 4.3 software.
Immunocytochemistry
Cell lines were grown on charged glass slides and boiled for antigen retrieval, then blocked with endogenous peroxidase blocker (Bio SB, Santa Barbara, CA, USA) for 10 minutes and then incubated in the presence of 1:500 anti-Eag1 antibody overnight at 4°C. The slides were then incubated with secondary biotin antibody (Bio SB) for 15 minutes and then incubated with streptavidin polymer (Bio SB) for 15 minutes. The specific staining reaction was completed by incubating the slides in the presence of diaminobenzidine in buffer reaction solution (Bio SB) and observed as a brown staining. Sections were counterstained with hematoxylin (Dako). The slides were observed in an Olympus IX51 microscope, Olympus DP70 camera (Tokyo, Japan).
Real-time polymerase chain reaction (RT-PCR)
Total RNA was extracted from cell cultures with TRIzol reagent. Five micrograms of total RNA was reverse transcribed using the Moloney Murine Leukemia Virus Reverse transcriptase (M-MuLV) (New England BioLabs Inc., Ipswich, MA, USA). RT-PCR was performed with 1 μL of cDNA using the TaqMan™ detection system (Thermo Fisher Scientific) and the Universal PCR Master Mix reagents kit (Thermo Fisher Scientific). Probes previously developed from TaqMan were used to study Eag1 (ID: Hs00924320_m1) and Gusb (ID: Hs00939627_m1, as a constitutive gene) expression. The PCR reaction protocol was 95°C for 15 seconds and 60°C for 1 minute (40 cycles). Data were analyzed with the 2−ΔΔCt method.
Western blot
Cells were washed, scrapped and centrifuged, and the obtained pellet was resuspended in lysis buffer supplemented with protease inhibitors. Lysis was completed with the freezing–thawing process; the lysate was centrifuged and the supernatant collected. Forty micrograms of protein was separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis (10%), transferred to a nitrocellulose membrane and incubated with either the anti-Eag1 antibody (1:750) or the anti-actin antibody (1:100,000). The relative protein quantification was performed with the ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
Statistical analysis was performed with analysis of variance (ANOVA) followed by the Tukey–Kramer test using GraphPad Prism software version 5.0 (La Jolla, CA, USA). The differences between the drug combination groups and either the drug-alone groups at the corresponding concentration or the control or the vehicle are shown. The analysis of Western blot data was performed with the Student’s t-test. P-values <0.05 were considered to be statistically significant.
Results
Concentration-dependent effect of astemizole and gefitinib on the proliferation of lung cancer cells
We first investigated the effect of astemizole and gefitinib alone on the proliferation of A549 and NCI-H1975 lung cancer cells, assessed by the metabolic activity. The effect of gefitinib is well known in both cell lines,40,41 and the effect of astemizole has been previously reported in A549 cells.16 As expected, both drugs decreased the metabolic activity in a concentration-dependent manner in both cell lines (Figure 1). From these experiments, we obtained the IC20 and IC50 values for each drug in each cell line (Table 1).
 | Figure 1 Effect of astemizole or gefitinib on the metabolic activity of lung cancer cells. |
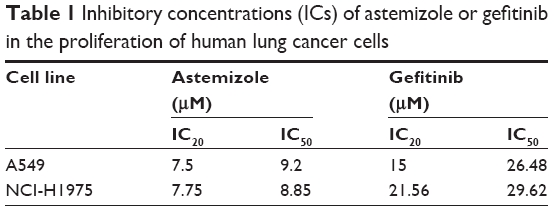 | Table 1 Inhibitory concentrations (ICs) of astemizole or gefitinib in the proliferation of human lung cancer cells |
Enhanced effect of the combination astemizole–gefitinib on the proliferation, survival and apoptosis of lung cancer cells
Cell proliferation experiments combining the drugs at their IC20 and IC50 concentrations were performed. The combination of the drugs had superior anti-proliferative effects in both cell lines. Astemizole-IC20 in combination with gefitinib-IC20 decreased metabolic activity by 80%, whereas the combination of astemizole-IC20 and gefitinib-IC50 decreased metabolic activity by 95% in A549 cells (Figure 2A). In the NCI-H1975 cells, the combinations of astemizol-IC20 plus gefitinib-IC20 and astemizole-IC20 plus gefitinib-IC50 showed a decrease of metabolic activity by 65% and 90%, respectively (Figure 2A). Based on these results, for the rest of the experiments, we decided to focus on the drug combination at the IC20 found in the cell proliferation assays (Table 1).
 | Figure 2 Enhanced effect of the combination astemizole–gefitinib on the metabolic activity, survival and apoptosis of lung cancer cells. |
Cell survival (assessed by the colony formation assay) was decreased in a very pronounced manner by the drug combination in comparison with the drugs alone in both cell lines (Figure 2B). In this assay, the combination decreased cell survival completely in both cell lines. Some representative images are shown in Figure 2B. Then, we studied apoptosis by flow cytometry. The drug combination decreased the percentage of viable cells more than the drugs alone in A549 cells, but the effect of the combination on apoptosis was not higher than that produced by astemizole alone (Figure 2C). However, while the drugs alone did not increase apoptosis in comparison with control experiments (in the absence of the drugs) in NCI-H1975 cells, the drug combination increased apoptosis almost four times in comparison with the effect of any of the drugs alone (Figure 2C).
The combination astemizole–gefitinib regulates Eag1 channel expression
Because Eag1 channel is one of the potential targets of astemizole, and channel expression is regulated by this drug in breast cancer cells,36 we wondered if the drug treatment might regulate Eag1 expression in both cell lines. The drug treatment did not modify Eag1 mRNA levels or protein expression in NCI-H1975 cells (data not shown). However, while the drugs alone did not affect Eag1 mRNA expression in A549 cells, the drug combination decreased Eag1 mRNA levels by 75% in comparison with the expression in either control experiments or the presence of any drug alone (Figure 3A). Western blot analysis also revealed that the drug treatment significantly decreased Eag1 protein expression in A549 cells (Figure 3B and C). Immunocytochemistry studies showed that control cells treated with either the vehicle or astemizole displayed strong brown immunostaining (Figure 3D–E). Despite that immunostaining was not that strong in the gefitinib-treated cells (Figure 3F), the weakest signal was observed in the cells treated with the drug combination (Figure 3G). Interestingly, astemizole induced subcellular accumulation of the channel in some parts of the cell adjacent to the nucleus (Figure 3E). This subcellular rearrangement of Eag1 after treatment was exhibited only in A549 cells.
 | Figure 3 The combination astemizole–gefitinib downregulates Eag1 expression in A549 cells. |
Discussion
Lung cancer is the main cause of cancer-related deaths worldwide, and thus, identifying new therapeutic strategies is urgently needed.1,2 Unfortunately, most of the patients either do not respond or develop resistance to one of the most common treatments for lung cancer, namely, gefitinib.11 Recently, the anti-histamine astemizole has gained great interest by its anticancer effects, either alone20,22,23,25,28,42 or in combination.16,20,36,37 In addition, epidemiological studies associated the use of astemizole and loratadine with reduced mortality from different cancers.16
The effect of gefitinib is well known in the A549 and NCI-H1975 lung cancer cells studied,40,41 whereas the effect of astemizole has been previously reported in A549 cells.16
Because astemizole has several targets involved in cancer,19–28,36,37,39 the possible mechanisms explaining the anti-proliferative effects here observed include blockage of oncogenic Eag1 potassium channels, decrease of Eag1 mRNA expression, inhibition of ABC multidrug transporters (which generate resistance to gefitinib)14 and antagonism of the histamine receptor H1. On the other hand, gefitinib may decrease cell proliferation by at least two possible mechanisms. A potential mechanism is the well-known EGFR-dependent pathway inhibition.6,10 The other is EGFR-independent by blocking H2-histamine and H4-histamine receptors, since gefitinib is able to antagonize these receptors and induce cytostasis and differentiation in leukemia cells.43
Here, we also observed that the combination astemizole–gefitinib at low concentrations had superior effects on the metabolic activity, survival and apoptosis of human lung cancer cells. The combination of the drugs at their IC20 decreased cell proliferation in up to 80%, whereas the combination of astemizole-IC20 and gefitinib-IC50 almost completely abolished proliferation in A549 cells. The drug combination also inhibited completely the survival of both cell lines. On the other hand, increased apoptosis was exclusively observed in NCI-H1975 cells only in the presence of the drug combination but not with the drugs alone. Some of the plausible mechanisms explaining the enhanced effects of the drug combination are the convergence on the histamine pathways (both can antagonize different histamine receptors), the increase in gefitinib concentration due to the blockage of ABC transporters by astemizole and/or the decrease in the expression of oncogenic Eag1 channels. Further studies are needed to elucidate the precise mechanism of the enhanced effects of the combination like testing the effect of other antihistamines, silencing or overexpressing the drug targets including histamine receptors, Eag1 potassium channels and ABC transporters, as well as testing the effect of other EGFR inhibitors.
In addition, we found that astemizole induced subcellular accumulation of Eag1 channels. More studies are needed to elucidate in which subcellular compartments this channel relocalization may be taking place and if this rearrangement may be associated with the anti-proliferative mechanism of astemizole. The cell lines displayed some differences in the drug responses. Despite that both cell lines were derived from biopsies of patients with NSCLC, the differences may reflect the heterogeneity of lung cancer cells observed in patients. However, the superior effect of the drug combination was maintained in both cell lines in several experimental approaches.
Astemizole is a nonsedating second-generation anti-histamine that does not cross the blood–brain barrier.44 This molecule was withdrawn from the market in several countries especially because severe cardiac side effects including prolongation of the Q-T segment and Torsade de Pointes were observed in cases of overdose.20,45–47 Our results show very strong effects when low concentrations of astemizole and gefitinib were combined. Then, astemizole may be safely administered at proper dose, and especially in combination with gefitinib, the dose may be even lowered. Despite that several studies explaining the precise mechanism of the combination effect are needed, these results suggest the combination astemizole–gefitinib as a novel therapeutic strategy for lung cancer that may help to decrease mortality from this disease.
Acknowledgments
This work was partially supported by AstraZeneca, Mexico (Project 07-1057 to JC). A similar abstract of this paper was presented at the 12th World Cancer Conference 2016 as a conference talk with interim findings. The abstract was published in “Scientific Tracks Abstracts” in Journal of Cancer Science & Therapy: https://www.omicsonline.org/proceedings/the-combination-astemizolegefitinib-as-a-novel-and-promising-therapy-for-human-lung-cancer-in-vitro-studies-54573.html. DOI:10.4172/1948-5956.C1.084.
Disclosure
CAC-A and MV-Y received financial support for undergraduate studies from Universidad de las Fuerzas Armadas, ESPE, Ecuador. The authors report no other conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Ervik M, et al. Cancer Incidence and Mortality Worldwide: IARC CancerBase No 11. Lyon, France: International Agency for Research on Cancer; 2013. GLOBOCAN 2012 v1. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Esposito L, Conti D, Ailavajhala R, Khalil N, Giordano A. Lung cancer: are we up to the challenge? Curr Genomics. 2010;11(7):513–518. | ||
de Groot P, Munden RF. Lung cancer epidemiology, risk factors, and prevention. Radiol Clin North Am. 2012;50(5):863–876. | ||
Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Trans Lung Cancer Res. 2015;4(1):36. | ||
Liu X, Wang P, Zhang C, Ma Z. Epidermal growth factor receptor (EGFR): a rising star in the era of precision medicine of lung cancer. Oncotarget. 2017;8(30):50209–50220. | ||
Zhu Q, Zhang S, Ding X, He B, Zhang H. Driver genes in non-small cell lung cancer: characteristics, detection methods, and targeted therapies. Oncotarget. 2017;8(34):57680–57692. | ||
Jackman DM, Yeap BY, Sequist LV, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non–small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12(13):3908–3914. | ||
Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361(10):958–967. | ||
Zhang H. Three generations of epidermal growth factor receptor tyrosine kinase inhibitors developed to revolutionize the therapy of lung cancer. Drug Des Devel Ther. 2016;10:3867. | ||
Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290(16):2149–2158. | ||
Hopper-Borge EA, Nasto RE, Ratushny V, Weiner LM, Golemis EA, Astsaturov I. Mechanisms of tumor resistance to EGFR-targeted therapies. Expert Opin Ther Targets. 2009;13(3):339–362. | ||
Özvegy-Laczka C, Cserepes J, Elkind NB, Sarkadi B. Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters. Drug Resist Updat. 2005;8(1):15–26. | ||
Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9(1):105–127. | ||
Lee H, Kang S, Kim W. Drug repositioning for cancer therapy based on large-scale drug-induced transcriptional signatures. PLoS One. 2016;11(3):e0150460. | ||
Ellegaard A-M, Dehlendorff C, Vind AC, et al. Repurposing cationic amphiphilic antihistamines for cancer treatment. EBioMedicine. 2016;9:130–139. | ||
Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3(8):673–683. | ||
Cheng F, Liu C, Jiang J, et al. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput Biol. 2012;8(5):e1002503. | ||
Parsons ME, Ganellin CR. Histamine and its receptors. Br J Pharmacol. 2006;147(S1):S127–S135. | ||
García-Quiroz J, Camacho J. Astemizole: an old anti-histamine as a new promising anti-cancer drug. Anticancer Agents Med Chem. 2011;11(3):307–314. | ||
Reynolds J, Akhter J, Morris D. In vitro effect of histamine and histamine H1 and H2 receptor antagonists on cellular proliferation of human malignant melanoma cell lines. Melanoma Res. 1996;6(2):95–100. | ||
García-Ferreiro RE, Kerschensteiner D, Major F, Monje F, Stühmer W, Pardo LA. Mechanism of block of hEag1 K+ channels by imipramine and astemizole. J Gen Physiol. 2004;124(4):301–317. | ||
Diaz L, Ceja-Ochoa I, Restrepo-Angulo I, et al. Estrogens and human papilloma virus oncogenes regulate human ether-a-go-go-1 potassium channel expression. Cancer Res. 2009;69(8):3300–3307. | ||
Downie BR, Sánchez A, Knötgen H, et al. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J Biol Chem. 2008;283(52):36234–36240. | ||
Pardo LA, Stühmer W. Eag1: an emerging oncological target. Cancer Res. 2008;68(6):1611–1613. | ||
Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8(12):982–1001. | ||
Avila E, Garcia-Becerra R, Rodríguez-Rasgado JA, et al. Calcitriol down-regulates human ether a go-go 1 potassium channel expression in cervical cancer cells. Anticancer Res. 2010;30(7):2667–2672. | ||
de Guadalupe Chávez-López M, Pérez-Carreón JI, Zuñiga-García V, et al. Astemizole-based anticancer therapy for hepatocellular carcinoma (HCC), and Eag1 channels as potential early-stage markers of HCC. Tumor Biol. 2015;36(8):6149–6158. | ||
Pardo LA, del Camino D, Sanchez A, et al. Oncogenic potential of EAG K(+) channels. EMBO J. 1999;18(20):5540–5547. | ||
Rodriguez-Rasgado JA, Acuna-Macias I, Camacho J. Eag1 channels as potential cancer biomarkers. Sensors. 2012;12(5):5986–5995. | ||
Occhiodoro T, Bernheim L, Liu J-H, et al. Cloning of a human ether-à-go-go potassium channel expressed in myoblasts at the onset of fusion. FEBS Lett. 1998;434(1–2):177–182. | ||
Hemmerlein B, Weseloh RM, de Queiroz FM, et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol Cancer. 2006;5(1):41. | ||
Farias LMB, Ocaña DB, Díaz L, et al. Ether a go-go potassium channels as human cervical cancer markers. Cancer Res. 2004;64(19):6996–7001. | ||
Ousingsawat J, Spitzner M, Puntheeranurak S, et al. Expression of voltage-gated potassium channels in human and mouse colonic carcinoma. Clin Cancer Res. 2007;13(3):824–831. | ||
Ortiz CS, Montante-Montes D, Saqui-Salces M, et al. Eag1 potassium channels as markers of cervical dysplasia. Oncol Rep. 2011;26(6):1377–1383. | ||
García-Quiroz J, García-Becerra R, Barrera D, et al. Astemizole synergizes calcitriol antiproliferative activity by inhibiting CYP24A1 and upregulating VDR: a novel approach for breast cancer therapy. PLoS One. 2012;7(9):e45063. | ||
Ishikawa M, Fujita R, Takayanagi M, Takayanagi Y, Sasaki K. Reversal of acquired resistance to doxorubicin in K562 human leukemia cells by astemizole. Biol Pharm Bull. 2000;23(1):112–115. | ||
García-Becerra R, Díaz L, Camacho J, et al. Calcitriol inhibits Ether-a go-go potassium channel expression and cell proliferation in human breast cancer cells. Exp Cell Res. 2010;316(3):433–442. | ||
Kong X, Chen L, Jiao L, et al. Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2. J Med Chem. 2014;57(22):9512–9521. | ||
Xu R, Shen H, Guo R, Sun J, Gao W, Shu Y. Combine therapy of gefitinib and fulvestrant enhances antitumor effects on NSCLC cell lines with acquired resistance to gefitinib. Biomed Pharmacother. 2012;66(5):384–389. | ||
Stabile LP, Lyker JS, Gubish CT, Zhang W, Grandis JR, Siegfried JM. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res. 2005;65(4):1459–1470. | ||
de Guadalupe Chávez-López M, Hernández-Gallegos E, Vázquez-Sánchez AY, Gariglio P, Camacho J. Antiproliferative and proapoptotic effects of astemizole on cervical cancer cells. Int J Gynecol Cancer. 2014;24(5):824–828. | ||
Yadav M, Singh AK, Kumar H, et al. Epidermal growth factor receptor inhibitor cancer drug gefitinib modulates cell growth and differentiation of acute myeloid leukemia cells via histamine receptors. Biochim Biophys Acta. 2016;1860(10):2178–2190. | ||
Nolen TM. Sedative effects of antihistamines: safety, performance, learning, and quality of life. Clin Ther. 1997;19:39–55. | ||
Kingswood JC, Routledge PA, Lazaris JH. A report of overdose with astemizole. Hum Toxicol. 1986;5:43–44. | ||
Bishop RO, Gaudry PL. Prolongated Q-T interval following astemizole overdose. Arch Emerg Med. 1989;6:63–65. | ||
Craft TM. Torsade de pointes after astemizole overdose. Br Med J. 1986;292:660. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.