Back to Journals » Therapeutics and Clinical Risk Management » Volume 15
The clinical significance of statins-macrolides interaction: comprehensive review of in vivo studies, case reports, and population studies
Authors Abu Mellal A ![]() , Hussain N
, Hussain N ![]() , Said ASA
, Said ASA ![]()
Received 31 May 2019
Accepted for publication 8 July 2019
Published 23 July 2019 Volume 2019:15 Pages 921—936
DOI https://doi.org/10.2147/TCRM.S214938
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Abdallah Abu Mellal,1 Nadia Hussain,2 Amira SA Said2
1College of Health and Human Sciences, Charles Darwin University, Darwin, Northern Territory, Australia; 2College of Pharmacy, Al Ain University of Science and Technology, Al Ain, UAE
Abstract: The objectives of this article were to review the mechanism and clinical significance of statins-macrolides interaction, determine which combination has the highest risk for the interaction, and identify key patients’ risk factors for the interaction in relation to the development of muscle toxicity. A literature review was conducted in PubMed and Embase (1946 to December 2018) using combined terms: statins – as group and individual agents, macrolides – as group and individual agents, drug interaction, muscle toxicity, rhabdomyolysis, CYP3A4 inhibitors, and OAT1B inhibitors, with forward and backward citation tracking. Relevant English language in vivo studies in healthy volunteers, case reports, and population studies were included. The interaction between statins and macrolides depends on the type of statin and macrolide used. The mechanism of the interaction is due to macrolides’ inhibition of CYP3A4 isoenzyme and OAT1B transporter causing increased exposure to statins. The correlation of this increased statin’s exposure to the development of muscle toxicity could not be established, unless the patient had other risk factors such as advanced age, cardiovascular diseases, renal impairment, diabetes, and the concomitant use of other CYP3A4 inhibitors. Simvastatin, lovastatin, and to lesser extent atorvastatin are the statins most affected by this interaction. Rosuvastatin, fluvastatin, and pravastatin are not significantly affected by this interaction. Telithromycin, clarithromycin, and erythromycin are the most “offending” macrolides, while azithromycin appears to be safe to use with statins. This review presented a clear description of the clinical significance of this interaction in real practice. Also, it provided health care professionals with clear suggestions and recommendations on how to overcome this interaction. In conclusion, understanding the different characteristics of each statin and macrolide, as well as key patients’ risk factors, will enable health care providers to utilize both groups effectively without compromising patient safety.
Keywords: drug interaction, rhabdomyolysis, muscle toxicity, HMG-Co A reductase inhibitors, CYP3A4 inhibitors, OATP1B inhibitors
Introduction
Statins are a class of cholesterol-lowering drugs and currently considered the most prescribed lipid-modifying therapy.1 Statins’ effect on lipid profile is caused by their competitive inhibition of HMG-CoA reductase enzyme, which is responsible for controlling the rate-limiting step of hepatocyte cholesterol synthesis. This reduction in hepatic cholesterol synthesis is augmented by the induction of low-density lipoprotein (LDL) receptor expression which enhances the hepatic removal of the atherogenic LDL cholesterol from circulation.2 The widespread use of statins in clinical practice is due to their very well documented benefits in reducing morbidity and mortality related to cardiovascular diseases.3 Macrolide antibiotics are also widely used in treating different infections in community and hospital settings. Most of the information available about the interaction between statins and macrolides were found under the heading of “Cytochrome P450 subfamily 3A4 (CYP3A4) inhibitors – statins interaction”. CYP3A4 inhibitors comprise a large number of drugs that have different inhibition potencies as well as different effects on several other transporters. The aim of this study was to explore the clinical significance of the specific interaction between statins and macrolides and provide recommendations on how to deal with such interaction.
Literature review
A literature review was performed through PubMed and Embase (1946 to December 2018) using different MeSH terms as well as combined keywords. The following keywords were used: CYP3A4 inhibitors, pharmacokinetics, HMG-Co A reductase inhibitors, statins, drug interaction, rhabdomyolysis, macrolides, muscle toxicity, myalgia, organic anion transporting polypeptides 1B (OATP1B) inhibitors, simvastatin, lovastatin, atorvastatin, rosuvastatin, pravastatin, fluvastatin, pitavastatin, erythromycin, clarithromycin, roxithromycin, telithromycin, and azithromycin with forward and backward citation tracking. Relevant English language in vitro studies as well as in vivo studies in healthy volunteers, case reports, and population studies were included in the review.
The interacting drugs
Chemically, simvastatin, atorvastatin, fluvastatin, and lovastatin are considered lipophilic compared to rosuvastatin, pitavastatin, and pravastatin.4 All statins are administered in the active form except simvastatin and lovastatin which are administered as lactone pro-drugs. Despite the rapid absorption of statins, their systemic bioavailability is low due to significant first-pass effect. However, since the liver is their main site of action, efficient first-pass uptake by hepatocytes has greater importance than systemic bioavailability. The major mechanism of hepatocyte uptake of lipophilic statins is passive diffusion, while the more hydrophilic statins will be subjected to an extensive transporter-mediated process.1 All statins are significantly metabolized by hepatic CYP450 enzymes except pravastatin and rosuvastatin, both drugs are cleared from the body mainly unchanged in urine and feces. Rosuvastatin and pravastatin renal clearances occur mainly through active renal tubular secretion and accounts for 10% and 20% of the drug total clearance, respectively. Therefore, CYP3A4 inhibition has no significant impact on the pharmacokinetics of pravastatin and rosuvastatin.5,6 Pitavastatin is minimally metabolized by CYP2C9 and most of the dose is excreted unchanged in the feces.7 CYP3A4 isoenzyme; the most available CYP450 isoenzyme; is the major metabolizing enzyme in relation to simvastatin, lovastatin, and atorvastatin.8,9 Fluvastatin is extensively metabolized by CYP2C9; and to a lesser extent by CYP3A4 and CYP2C8; to three major metabolites, and only 5% of the administered dose is renally cleared.10 Table 1 represents a summary of important pharmacokinetic parameters of different statins.
 |
Table 1 Summary of selected pharmacokinetic data of statins1,8,10,15,56,87 |
Macrolides is a group of antimicrobial drugs characterized by the presence of a macrocyclic lactone ring in their structures. Erythromycin is rapidly degraded in the stomach’s acidic environment to different compounds that are responsible for the gastrointestinal side effects of erythromycin. Clarithromycin, roxithromycin, and azithromycin are newer agents in this class and have been created semi-synthetically by modifications to erythromycin. The aim of these modifications is to create more acid-stable alternatives with longer half-life and extended spectrum of activity. The newer macrolides have an extended spectrum of activity toward certain species compared to erythromycin.11 Telithromycin is structurally related to macrolides (ketolides) designed to have dual binding to bacterial ribosomes in order to overcome the resistance of certain bacteria. The long half-life of telithromycin, azithromycin, and roxithromycin allows their use as a single daily dose. Erythromycin, clarithromycin, and telithromycin are extensively metabolized by CYP3A4 isoenzyme, roxithromycin undergoes limited metabolism, while azithromycin is not metabolized and mainly excreted as unchanged drug.11–13
The mechanisms of the interaction
Inhibition of CYP3A4
The most important site of interaction between statins and macrolides is CYP3A4 isoenzyme. The mechanism involves inhibiting CYP3A4 by certain macrolides, which will result in increased exposure to statins metabolized by the same isoenzyme. CYP3A4 is considered the most abundant CYP450 isoenzyme in the liver and intestine. It is involved in the metabolism of more than 50% of drugs currently available on the market. In the intestine, CYP3A4 is responsible for first-pass drug metabolism and contribute to drug clearance through the gut wall. Despite the fact that more than 20 allelic variants of CYP3A4 have been identified, the clinical significance of this variation has not been demonstrated in clinical practice.14 Inhibition of CYP3A4 isoenzyme is expected to have a significant impact on the blood level concentration of simvastatin, lovastatin, and atorvastatin. However, the magnitude of this inhibition will vary depending on the potency of the inhibition and the relative contribution of intestinal and hepatic CYP3A4 to the total bioavailability of the drug. Simvastatin and lovastatin’s bioavailability is more dependent on CYP3A4 isoenzyme than atorvastatin, therefore the inhibition of CYP3A4 isoenzyme has a larger effect on their exposure compared to atorvastatin.8,15
The affinity or the potency of inhibition of CYP3A4 isoenzyme by different macrolides has been explored in many in vitro studies. These studies concluded that erythromycin, clarithromycin, and telithromycin are the most potent inhibitors of CYP3A4 isoenzyme, followed by the weak inhibitor, roxithromycin, and finally azithromycin, which in some studies showed results comparable to placebo.16–19 The dual inhibition of intestinal and hepatic CYP3A4 does not occur at the same speed, intestinal CYP3A4 has a fast onset which peaks in about 2 days, while hepatic CYP3A4 may take a few days to reach maximum inhibition.20
The inhibition of CYP3A4 by macrolides is believed to be due to mechanism-based inhibition which results in the formation of a tight and irreversible metabolic intermediate (MI) complex which inactivates the isoenzyme. The N-demethylation of erythromycin, clarithromycin, troleandomycin, and oleandomycin forms reactive nitrosoalkanes which inactivate CYP3A4 by forming MI complex.21 Mechanism-based inhibition of CYP3A4 is more likely to cause significant drug-drug interaction than reversible inhibition, as the body will not overcome this inhibition until new CYP3A4 proteins are synthesized. Macrolides' inhibition of CYP3A4 through the formation of MI complex is expected to be time-dependent in its onset and more evident after multiple doses compared to a single dose.22
Inhibition of OATPs
Membrane transporters can either facilitate the uptake of their substrates into cells (influx) or facilitate the excretion of their substrates out of cells (efflux). Human OATPs are a membrane influx transporter family which consists of eleven proteins; OATP1B1, OATP1A2, OATP1B3, and OATP2B1 are the most characterized ones in relation to their influence on drug disposition.23 OATP1B1, 1B3, and 2B1 are expressed mainly in the sinusoidal membranes of hepatocytes and facilitate the entry of many endogenous and exogenous substances into liver cells. SLCO1B1 gene is responsible for the formation of OATP1B1 transporter and it has been found to be polymorphic with many single nucleotide polymorphisms (SNPs), and sequence variations have been identified. Two important SNPs that can form four distinct haplotypes are c.521T > C (p.Val174Ala) and c.388A > G (p.Asn130Asp). Haplotypes SLCO1B1*5 (c.388A-c.521C) and SLCO1B1*15 (c.388G-c.521C) have been associated with reduced transport activity.24
Simvastatin acid, the active form of simvastatin, is a substrate of OATP1B1. The contribution of this transporter to the hepatic uptake of simvastatin acid is estimated to be 75% of the hepatic extraction ratio.8 In a single dose study in healthy volunteers who carry SLCO1B1 c.521T > C variant, simvastatin acid exposure was increased by 40%.25 For atorvastatin, no human data available for the contribution of OATP1B1 in its hepatic uptake, but data from studies conducted on rats indicate more than 90% of the total hepatic uptake is mediated by OTAP1B1 transporter.26 In a single dose study in healthy volunteers (atorvastatin 40 mg), the administration of rifampin infusion, an OATP1B1 inhibitor, increased atorvastatin acid exposure by 6.8 fold.27 Pravastatin is mainly renally excreted and the rest will undergo hepatic metabolism. Since pravastatin is a hydrophilic compound, OATP1B1 transporter is needed for hepatic cellular uptake.8 In one study, a participant with SLCO1B1 c.521CC genotype showed 91% and 74% more pravastatin exposure compared to participants with c.521TT or c.521TC genotypes respectively, while fluvastatin exposure did not show any significant difference. In a rat model, when rifampicin (OATP1B1/1B3 inhibitor) was coadministered with fluvastatin, area under the serum concentration-time curve (AUC) increased by 2.5 fold. This supports the assumption that fluvastatin’s hepatic uptake is more dependent on OATP1B3 transporter.28,29
Metabolism of rosuvastatin is a minor route with most of the drug excreted unchanged in feces and urine. It is estimated that OATP1B1/B3 is responsible for 70% of rosuvastatin’s total hepatic uptake, the rest (about 30%) is transported via bile acid uptake transporter.8 The influence of SLCO1B1 c.521cc genotype was larger on atorvastatin exposure compared to rosuvastatin in healthy volunteers, which indicates that atorvastatin uptake is more dependent on OATP1B1 transporter.30 For pitavastatin, hepatic elimination as unchanged drug in bile is the major metabolic pathway. In vitro studies showed that pitavastatin is an OATP1B1/B3 substrate with 1B1 as the major contributor to its hepatic uptake. Increased exposure to pitavastatin has been reported in individuals with SLCO1B1 c.521CC genotype or when coadministered with rifampicin.31,32 Lovastatin is also a substrate of OATP1B1, but has not shown any affinity toward OATP1B3.33
In summary, possibly all statins are substrates of OATP1B1 but to different extents. Simvastatin, lovastatin, atorvastatin, and pitavastatin are the most affected statins when OATP1B1 inhibitor is used.
The magnitude of macrolides' inhibition of different transporters as well as the level of contribution of each transporter to total drug clearance is essential information to determine the significance of drug-drug interactions. The inhibitory effect of different macrolides on OATP1B1/1B3 transporters was evaluated in an in vitro model, the IC50 for each macrolide against OATP1B1 was 96 µM (clarithromycin), 121 µM (telithromycin), 153 µM (roxithromycin), and 217 µM (erythromycin). In relation to macrolides' affinity for OATP1B3 using the same model, telithromycin showed the strongest inhibition in vitro with IC50 of 11 µM, while clarithromycin, erythromycin, and roxithromycin showed slightly similar affinities with IC50 of 32 µM, 34 µM, and 37 µM respectively. Azithromycin did not show any inhibition of both transporters.34 Hirano et al calculated the inhibition constant for clarithromycin and erythromycin against OATP1B1 and found that clarithromycin has a stronger affinity for OATP1B1 than erythromycin (8.25 µM and 11.4 µM respectively) and may cause a significant clinical interaction with OATP1B1 substrates.35 These in vivo variations in inhibition values can be explained by the use of different expression systems and different substrates.
The effect on other transporters
P-glycoproteins (P-gp), which is a member of multidrug resistance proteins subfamily, is an efflux transporter that may play a role in the absorption of lovastatin, simvastatin acid, and atorvastatin acid, but not the lactone forms.36 However, an in vitro study using Caco-2 cells showed low affinity of these statins for P-gp and suggested that P-gp inhibition is unlikely to cause significant drug interactions.37 Clarithromycin, erythromycin, and azithromycin are considered weak P-gp inhibitors when coadministered with P-gp substrates such as digoxin and fexofenadine, they resulted in less than 2 fold increase in AUC.38 All other statins are not affected by P-gp inhibition.39
Breast cancer resistance protein (BCRP) is another efflux transporter that is located in the intestine, placenta, liver, and to a lesser extent in renal proximal tubules. In the intestine, BCRP has a similar role to P-gp in restricting the absorption of its substrates through the intestinal wall. An important SNP in ABCG2 gene, which encodes for BCRP, is ABCG2 c.421C>A, which is associated with reduced expression of the transporter, causing reduction in its efflux capacity.40 In healthy volunteers, c.421AA genotype was associated with 2.44 fold and 1.72 fold increase in the exposure to rosuvastatin and atorvastatin respectively.41 Also, similar results were found with fluvastatin (1.7 fold increase) and simvastatin lactone (2.11 fold increase). Pravastatin and pitavastatin exposure was not significantly altered in individuals with c.421AA genotype.42
Organic anion transporters (OATs) play a role in the renal clearance of pravastatin and rosuvastatin. More than 90% of the renal clearance of pravastatin and rosuvastatin is mediated by OAT3 transporter which is responsible for their active removal from circulation.43 Multidrug resistance protein 2 (MRP2) has a significant effect on the intestinal absorption of pravastatin.44 The sodium-dependent taurocholate co-transporting polypeptide (NTCP) is another transporter present exclusively in hepatocytes, and contributes to the homeostasis of bile salt in the body.45 NTCP contributes differentially to statins' hepatic uptake with other transporters such as OATP1B1/1B3. Rosuvastatin, pravastatin, pitavastatin, and fluvastatin are considered NTCP substrates. In vitro inhibition of NTCP by taurocholate affected mostly the hepatic uptake clearance of rosuvastatin and pitavastatin, but to a moderate extent.46 Despite the importance of all these transporters, there was no evidence in the literature that any of the macrolides were BCRP, OAT3, MRP2, and NTCP inhibitors.
The consequences of the interaction
Assuming that macrolides inhibit the metabolism/clearance of statins, this may lead to increased patient exposure to statins. This in turn can lead to one of four possibilities: first: no symptoms at all or no changes in markers of muscle toxicity (creatine kinase (CK)), the second possibility is development of mild muscle pain or myalgia without any increase in blood markers of muscle toxicity, the third possibility is development of myalgia with increase in CK (<10 times upper limit of normal), the final possibility is development of rhabdomyolysis, which usually starts with progressive muscle pain and weakness that is followed by severe increase in CK (10× > upper limit of normal) and/or increase in myoglobin level which can be detected in urine and which contributes to acute kidney injury.47 Rhabdomyolysis is a life-threatening condition that has a significant impact on patients’ health and quality of life. The aim of this review was to help health care professionals avoid the development of rhabdomyolysis by understanding the nature of macrolides-statins interaction and risk factors that predispose patients to such adverse events.
Studies in healthy volunteers
In order to explore the extent as well as the significance of statins' and macrolides' interaction, a few studies have been conducted in healthy volunteers and are summarized in Table 2. The effects of erythromycin, clarithromycin, and telithromycin on simvastatin exposure were variable and highly significant, however the comparison between them was inappropriate as one of them was a single dose study while the other two studies followed a multiple dose approach.48–51 In relation to atorvastatin, the increase in AUC after the administration of macrolides was dose-dependent. With atorvastatin 10 mg dosing, the increase in AUC was less than 2 fold, while with the 80 mg dosing the increase in AUC was more than 4 fold.48,52–54 Clarithromycin, erythromycin, and telithromycin inhibited both intestinal/hepatic CYP3A4 and OATP1B1, which resulted in this large increase in the exposure to atorvastatin and simvastatin. Azithromycin 500 mg daily for 3 days did not cause a significant change in atorvastatin exposure, which is in agreement with a molecular experiment that showed the clearance of triazolam through CYP3A4 did not change with the coadministration of azithromycin.17 The coadministration of clarithromycin with pravastatin doubled the subjects' exposure to pravastatin despite the fact that pravastatin is not metabolized by CYP3A4 isoenzyme.48 In fact, pravastatin’s hepatic clearance is estimated to account for 0.53 of drug total clearance; when considering that OATP1B1 is responsible of 95% of hepatic extraction, inhibiting OATP1B1 by clarithromycin theoretically will double pravastatin exposure.8 Erythromycin caused a slight decrease in AUC of rosuvastatin, which could be due to increased gut motility causing a slight decrease in rosuvastatin absorption. This result confirm that rosuvastatin clearance is not dependent on CYP3A4 activity.55 Pitavastatin AUC was increased by 2.8 fold when coadministered with erythromycin, which was more than expected (1.45 fold AUC reduction) and this could be due to underestimation of hepatic extraction ratio.56 All the above-mentioned trials did not take into consideration racial background or genotype testing of subjects, also, there was significant inter-individual variability in the extent of the statins' interaction with macrolides. Furthermore, these studies described the interaction in terms of increased exposure to statins, but none of these studies reported any signs of muscle toxicity.
 |
Table 2 Summary of studies of the administration of different statins with different macrolides in healthy volunteers |
Case reports from the literature
We identified 13 case reports of rhabdomyolysis suspected to be induced by addition of macrolides to long-term statin therapy; a summary has been provided in Table 3. Rhabdomyolysis is a severe skeletal muscle condition, usually starting with muscle pain and weakness due to the disruption of the muscle cells' integrity leading to a large release of creatine kinase and other intracellular components such as myoglobin which may cause dark urine and induce acute renal injury.57 Statin monotherapy rarely causes rhabdomyolysis, with estimated incidence of 0.3–13.5 cases per one million statin prescriptions.58 Case reports are confirmed cases of the outcome, which is rhabdomyolysis in this context, but could not necessarily establish causal relationship. On the other hand, analysis of these reports may help in identifying key factors that need to be considered to avoid such severe adverse events.
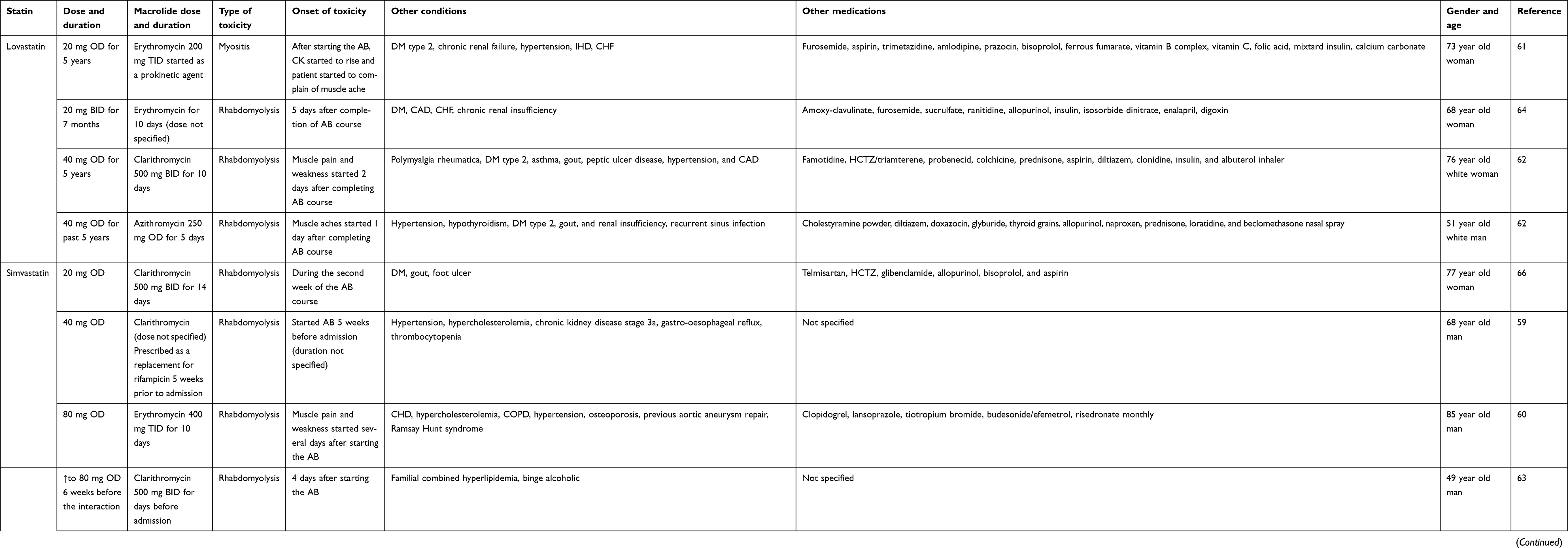 |
Table 3 Summary of case reports from literature implicating macrolides-statins interaction |
Lovastatin (four cases) and simvastatin (nine cases) were the only statins used in these reported cases.59–69 About 85% of the patients were ≥64 years old, of those about 54% were ≥75 years of age. In the literature, advanced age (65 years or older) is a risk factor for developing rhabdomyolysis with statin monotherapy.70 This risk will increase when combining statin with an interacting drug such as clarithromycin. Most of simvastatin users (78%) used a high dose of 80 mg daily, while lovastatin users used a medium dose of 40 mg daily. In another review of case reports of statin-induced rhabdomyolysis, most cases of simvastatin-induced rhabdomyolysis occurred at a dose of 40 mg.71 Also, Nguyen et al found that the OR of developing simvastatin-induced rhabdomyolysis was significantly increased when the simvastatin dose was larger than 20 mg.70 Clarithromycin was the most suspected offending agent in more than 50% of the cases, followed by erythromycin, while azithromycin was implicated in only one case. The onset of symptoms varied significantly between cases, but can be summarized into symptoms appearing after a few days from starting the macrolide or a few days after completing the macrolide course. In two cases, a longer period of about 2 weeks after the start/completion of the antibiotic was reported for the onset of symptoms. In almost all cases, muscle pain and weakness were the first complaints reported by patients.
Polypharmacy and multiple comorbidities are obvious characteristics of this cohort of patients. Seventy-seven percent of patients suffered from cardiovascular diseases (hypertension 61.5%, coronary artery disease 53.8%, HF 23%), 53% suffered from diabetes, and 46.1% had a certain degree of renal impairment. Interestingly, 38.5% of patients represented with gout. Cardiovascular diseases, especially hypertension, diabetes mellitus (DM), and renal insufficiency are considered as risk factors for developing statin-induced muscle toxicity.70,72 All patients with DM had also reported renal impairment, except in one case, which possibly makes DM-induced renal impairment the highest risk, not just DM. Furthermore, infection, excessive alcohol consumption, and thyroid disorders are documented to increase the risk of rhabdomyolysis.57
Looking carefully at other medications used in each case, we could clearly identify many confounding agents that may at least have contributed to the development of rhabdomyolysis. Diltiazem, a very well-known CYP3A4 and P-gp inhibitor, was recognized as a major contributor to two cases, especially when combined with colchicine, a CYP3A4 and P-gp substrate. The interaction between colchicine and statins as well as colchicine and clarithromycin, can lead to colchicine toxicity which can cause rhabdomyolysis, especially if the patient is renally impaired.73,74 Amlodipine is considered a mild CYP3A4 inhibitor which can cause 30% increase in simvastatin acid exposure.75 Also, amlodipine is considered a potent BCRP inhibitor, as revealed by ligand-based virtual screening combined with in vitro testing.76 Furthermore, valsartan, telmisaratan digoxin, and glibenclamide have different inhibitory effects on OATP1B1. Lastly, all those patients suffered from infection, which can independently contribute to rhabdomyolysis.57 The complex interaction of advanced age, multiple comorbidities as well as polypharmacy in these case reports makes it difficult to come to a conclusion regarding a causal relationship.
Population studies from health records and databases
Electronic medical records or databases provide a great opportunity for researchers to monitor medication safety. Table 4 represents a short description of seven relevant population studies identified from the literature. Researchers conducted a screening of VigiBase (an adverse drug reaction database established by the World Health Organization) in July 2009 for rhabdomyolysis reports concerning macrolides and statins, and found that clarithromycin is the most commonly reported offender. Also, they reported 53 cases that involved azithromycin and statins, mostly simvastatin. However, in more than 30% of azithromycin cases, other well-established interacting agents were reported. Azithromycin does not inhibit CYP3A4 isoenzyme or is at least considered a very weak inhibitor, therefore the suspected interactions with statins could be a coincidental temporal relation. The nature of VigiBase spontaneous reporting system, as well as the lack of clinical details, limited the ability to establish a definite causal relationship.77
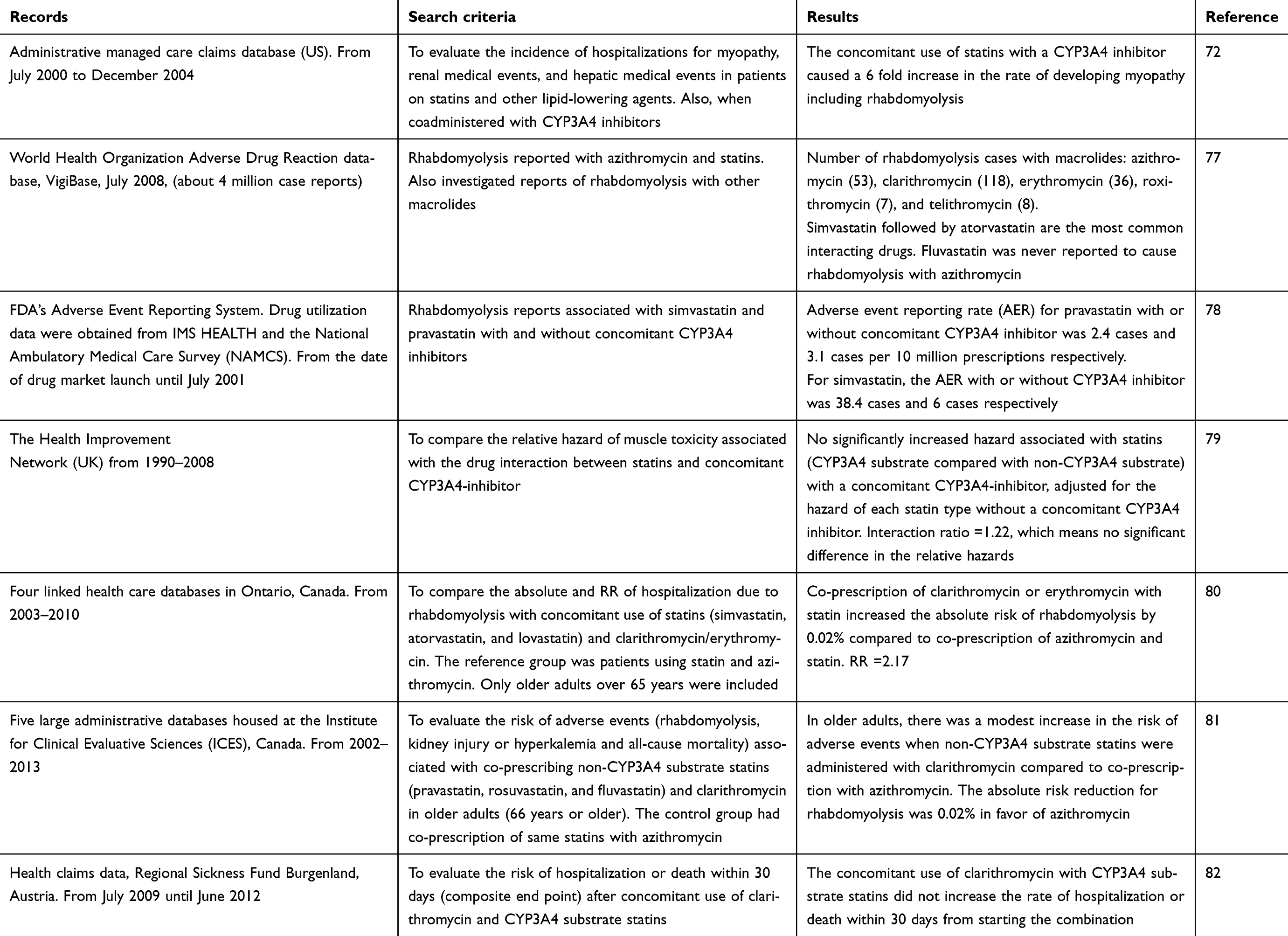 |
Table 4 Summary of population studies related to macrolides-statins interaction |
Rowan et al looked at the US Food and Drug Administration (FDA)’s Adverse Event Reporting System in order to compare the adverse event reporting rate (AER) for pravastatin and simvastatin with or without concomitant CYP3A4 inhibitor.78 With pravastatin, the AER was lower and did not change significantly with or without concomitant CYP3A4 inhibitor (2.4 and 3.1 cases per 10 million prescriptions respectively). While with simvastatin, the AER jumped from six cases without CYP3A4 inhibitor to 38.4 cases with CYP3A4 inhibitor per 10 million prescriptions. Clarithromycin and erythromycin contributed to more than 20% of simvastatin-induced rhabdomyolysis cases compared to none with pravastatin. The study concluded that concomitant use of simvastatin and a CYP3A4 inhibitor increased the risk for developing rhabdomyolysis. When looking at muscle toxicity as the investigated event – defined as myalgia, myopathy, and myositis – researchers screened The Health Improvement Network (THIN) database of primary care medical records in the UK from 1990–2008 to compare the relative hazard of muscle toxicity associated with the concomitant use of statins and CYP3A4 inhibitors.79 They stratified statins as CYP3A4 substrate (simvastatin and atorvastatin) and non-CYP3A4 substrate (fluvastatin, pravastatin, and rosuvastatin), clarithromycin and erythromycin were included in the analysis. There was no increase in the relative hazard of muscle toxicity between using statins alone and using statins with a CYP3A4 inhibitor. However, erythromycin and clarithromycin represented only 1.5% and 0.9% respectively of the total cases of concomitant use. Interestingly, no cases of muscle toxicity were reported with concomitant use of macrolides and non-CYP3A4 substrate statins (fluvastatin, pravastatin, and rosuvastatin), which supported the claim that the metabolism of these statins is not affected by CYP3A4 isoenzyme pathway. The results of this research questioned the clinical significance of increased statins’ exposure reported in healthy volunteers and its correlation to statins’ adverse effects in real practice.
On the other hand, the previous results were not in agreement with results extracted from administrative managed care claims database in the US, where concomitant use of lipid-lowering medications and a CYP3A4 inhibitor increased the rate of developing myopathy requiring hospitalization by 6 fold.72 It is noteworthy to mention that the two studies had different definition codes for myopathy, also the later study had several limitations, it used aggregated data for all lipid-lowering drugs (statins and non-statins), also cerivastatin, which has been withdrawn from the market, significantly influenced the results (incidence rate per 10,000 person-years was three and four times that of simvastatin and atorvastatin respectively). Finally, the statistical analysis included all CYP3A4 inhibitors listed as one group without stratification, which makes it difficult to identify which agents significantly influenced the result.
In another attempt to explore the difference in the risk of developing rhabdomyolysis when statins are co-prescribed with different macrolides, Patel et al screened four linked health care databases in Ontario, Canada to compare the risk of developing rhabdomyolysis when statins metabolized by CYP3A4 (simvastatin, atorvastatin, and lovastatin) are co-prescribed with clarithromycin or erythromycin compared to statins co-prescribed with azithromycin as the reference group. A small absolute risk reduction of 0.02% was observed in favor of azithromycin group. Indeed, the total number of rhabdomyolysis cases was 34 out of more than 144,000 co-prescriptions, also, the study was limited only to patients older than 65 years.80
In addition to inhibiting CYP3A4 isoenzyme, clarithromycin can inhibit the transporters OATP1B1 and OATP1B3. Pravastatin and rosuvastatin’s hepatic uptake is controlled mainly by these transporters. To evaluate the effect of concomitant use of clarithromycin with non-CYP3A4 substrate statins (pravastatin, rosuvastatin, and fluvastatin), five large administrative databases housed at the Institute for Clinical Evaluative Sciences (ICES) were screened. The measured end points were hospitalization due to rhabdomyolysis, acute kidney injury or hyperkalemia, and all-cause mortality due to concomitant use of clarithromycin with non-CYP3A4 substrate statins. The control group was selected as those with concomitant use of same statin and azithromycin. The results showed a modest increase in the risk of outcomes (for rhabdomyolysis, the absolute risk reduction was 0.02%).81 To understand the clinical significance of clarithromycin-statins interaction, researchers from Austria screened a large health claims database for the composite end point of hospitalization or death within 30 days due to concomitant use of clarithromycin and CYP3A4 substrate statins. After calculating the adjusted risk, the concomitant use of clarithromycin and CYP3A4 substrate statins was not associated with increasing the risk of hospitalization or death.82 One of the limitations of this study was that they considered pravastatin a CYP3A4 substrate statin.
Discussion
The evidence behind macrolides-statins interaction varies significantly between the different types of drugs in each group. In relation to statins, most of this interaction was associated with simvastatin and lovastatin in case reports, while in healthy volunteers, simvastatin showed the highest level of increased exposure followed by atorvastatin 80 mg and pitavastatin 4 mg. In relation to macrolides, it is very obvious from this review that the risk of interaction is higher with telithromycin, erythromycin, and clarithromycin, while roxithromycin has less potential for the interaction and azithromycin appears to be safe to use. It is noteworthy to mention that from the 53 cases of rhabdomyolysis suspected to be due to azithromycin and statins combination mentioned by Strandell et al, in only three cases the reporters explicitly indicated that azithromycin was most likely to be the offending agent.77 Pharmacogenetic polymorphisms in CYP3A4 gene, SLCO1B1, and ABCG2 may have an influence on an individual’s susceptibility to this interaction. However, currently in clinical settings, genetic testing for polymorphisms in these genes is not available, and it is not likely to be available in the near future, unless it is proven to be cost-effective. The case reports presented in this review identified very well-known risk factors for rhabdomyolysis suspected to be due to macrolides-statins combination. Advanced age, cardiovascular diseases, renal impairment, DM, and concomitant interacting drugs are all considered risk factors for the development of statin-induced rhabdomyolysis with or without concomitant macrolides.57,70,71,83
The increased exposure of simvastatin, lovastatin, and atorvastatin when coadministered with clarithromycin, telithromycin, and erythromycin is evident, but the significance of this increased exposure to the development of muscle toxicity could not be established in real practice. In Japan, a retrospective cohort study using a claims database could not find a statistically significant difference between the incidence of muscle toxicity between statin users who took interacting drugs (as per product information; including clarithromycin, erythromycin, and telithromycin) compared to statin users not taking interacting drugs. The number of cases identified in this cohort was very small (three cases), so no stratification analysis against the type of interacting drug was performed.84 In another population study in Austria, the increased risk of hospitalization or death due to concomitant use of clarithromycin and statins was perfectly neutralized after adjusting for age, cardiovascular diseases, diabetes, and utilization of other antibiotics.82 Similar results have been shown in the UK, where the relative hazard of muscle toxicity, hepatic and renal dysfunction did not differ between statins (3A4 substrate/non-3A4 substrate) combined with CYP3A4 inhibitor compared to statins alone.79 On the other hand, rhabdomyolysis adverse event rate increased from six cases to 38.4 cases per 10 million prescriptions when simvastatin was coadministered with CYP3A4 inhibitor compared to simvastatin alone. However, this rate was based on domestic spontaneous reporting of adverse events, also clarithromycin/erythromycin represented 21% of the CYP3A4 inhibitors used in this cohort.78
Assuming that all CYP3A4 inhibitors are similar and including them as a group in the analysis is not the most appropriate approach to establish a causal relationship. Many of these inhibitors have a different affinity for CYP3A4 isoenzyme as well as the fact that many of them has different inhibitory effects on other transporters, eg, cyclosporine is a potent inhibitor of CYP3A4, P-gp, BCRP, and OATP1B1/1B3. Also, the proportion of each inhibitor in the group varied significantly, which makes it difficult to predict which inhibitor drove up the results. Perhaps the small amount of muscle toxicity detected in such studies makes it difficult to stratify the results as per the interacting agent.
The group analysis of statins as CYP3A4-substrate and non-CYP3A4-substrate also produced confusing results. Atorvastatin, simvastatin, and lovastatin are all included in the CYP3A4-substrate group despite the fact that inhibiting CYP3A4 by macrolides in healthy volunteers produced significant variation in exposure to these statins. This in turn, could overestimate the risk associated with atorvastatin compared to simvastatin and lovastatin. The population studies cited in this review which focused on CYP3A4-substrate statins did not stratify the outcomes measured by individual statins, except one study which found that the incidence of rhabdomyolysis with atorvastatin was significantly lower than simvastatin and lovastatin.72
In the two Canadian population studies discussed previously, the estimated incidence of rhabdomyolysis in older adults co-prescribed clarithromycin/erythromycin and statins was 0.03% regardless of the type of statin (3A4 or non-3A4 substrates).80,81 This similar incidence rate was not expected based on molecular and pharmacokinetic studies and raises a few issues related to the study type and design. The length of the macrolide course was not mentioned. Also, the events were recorded up to 30 days from the macrolide prescription date. This in turn could have overestimated the number of events, as in case reports most of the symptoms of muscle toxicity started a few days after the start of macrolides or a few days after the completion of the course. Also, the authors acknowledged the limitation of this type of study as well as the possibility of medical codes' insensitivity. Despite the fact that many researchers tried to account for all known variables, the type of infection was not taken into consideration, possibly due to the fact that most antibiotic treatments are empirical therapy, however, certain types of infection have been documented to be independent risk factors for developing rhabdomyolysis.47
Relevance to patient care and clinical practice
The interaction between macrolides and statins is more pronounced with simvastatin, lovastatin, and atorvastatin. In case reports, simvastatin 80 mg was the most commonly reported dose used by patients. In healthy volunteers, the largest increase in statin exposure with concomitant macrolides was with simvastatin 40 mg. Taking into consideration the FDA’s warning in relation to the increased risk of myopathy with simvastatin 80 mg,85 ceasing simvastatin therapy temporarily or using azithromycin is the most appropriate measure to avoid toxicity associated with concomitant use of macrolides, especially if there are other risk factors such as advanced age, diabetes, cardiovascular diseases, and using other interacting agents.86 Due to the similarity between metabolism of lovastatin and simvastatin, the same approach should be followed with lovastatin. It is important to mention that the FDA’s approved product information for lovastatin contraindicates the concomitant use of telithromycin, clarithromycin, and erythromycin with lovastatin.87
The risk of developing muscle toxicity with the coadministration of atorvastatin and macrolides is overestimated when it is included with simvastatin and lovastatin as “CYP3A4-substrate statins”. Concomitant administration of azithromycin with atorvastatin did not produce significant changes to atorvastatin AUC in healthy volunteers, which makes this macrolide a safe option for patients on atorvastatin therapy. Also, the increased exposure to atorvastatin (10–40 mg) when coadministered with clarithromycin/erythromycin is less likely to be of clinical significance in practice unless the patient has other risk factors for rhabdomyolysis. For higher doses of atorvastatin (80 mg), it is recommended to either use azithromycin or to temporarily cease atorvastatin until the end of the macrolide therapy. Fluvastatin exposure is not expected to significantly change with the coadministration of macrolide antibiotics.15 Pitavastatin AUC was increased by 2.8 fold when coadministered with erythromycin, therefore, the manufacturer suggests not to use this combination with pitavastatin dose above 1 mg.56 Since the proposed mechanism of this interaction is through inhibition of OATP1B1, the manufacturer’s suggestion could be extended to include clarithromycin and telithromycin. If higher doses of pitavastatin is used (>1 mg), we suggest using azithromycin or temporarily ceasing pitavastatin until the antibiotic course is completed. Pravastatin can be co-prescribed with clarithromycin with caution as long as the pravastatin dose does not exceed 40 mg.88 With pravastatin doses >40 mg, we suggest either reducing the dose or ceasing the medication temporarily or using azithromycin. Rosuvastatin is mainly excreted unchanged and can be safely coadministered with macrolide antibiotics.
Simvastatin, lovastatin, fluvastatin, and pravastatin have short half-lives of between 2–5 hours, while atorvastatin, rosuvastatin, and pitavastatin have longer half-lives of between 11–20 hours.1 Based on statins’ half-lives, after ceasing statin therapy temporarily, it will take 2–5 days to reach steady-state concentration after the re-commencement of treatment.89 Proper counseling is essential to reduce the risk of adverse drug events. All patients using concomitant statin-macrolide combinations should be advised to stop medication and refer to their doctors if they notice unusual muscle pain.
Conclusion
From this review, we can confidently suggest that macrolides-statins interaction can lead to increased exposure to statin therapy. The most affected statins are simvastatin, lovastatin, and to a lesser extent atorvastatin (80 mg). Rosuvastatin, fluvastatin, and pravastatin are not significantly affected by this interaction. In most patients, this increase in statin exposure will not have any clinical significance. The increased exposure to statin therapy in the presence of other risk factors such as advanced age, renal impairment, diabetes, cardiovascular diseases, and the use of other statin-interacting drugs may cause serious muscle toxicity. In relation to macrolides, clarithromycin, erythromycin, and telithromycin are the most offending agents, while roxithromycin has limited potential for this interaction and azithromycin appears to be safe to use with statins.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19(1):117–125. doi:10.1111/j.1472-8206.2004.00299.x
2. Stancu C, Sima A. Statins: mechanism of action and effects. J Cell Mol Med. 2001;5(4):378–387. doi:10.1111/j.1582-4934.2001.tb00172.x
3. Naci H, Brugts JJ, Fleurence R, Tsoi B, Toor H, Ades AE. Comparative benefits of statins in the primary and secondary prevention of major coronary events and all-cause mortality: a network meta-analysis of placebo-controlled and active-comparator trials. Eur J Prev Cardiol. 2013;20(4):641–657. doi:10.1177/2047487313480435
4. Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science (New York, NY). 2001;292(5519):1160–1164. doi:10.1126/science.1059344
5. Everett DW, Chando TJ, Didonato GC, Singhvi SM, Pan HY, Weinstein SH. Biotransformation of pravastatin sodium in humans. Drug Metab Dispos. 1991;19(4):740–748.
6. Martin PD, Warwick MJ, Dane AL, et al. Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin Ther. 2003;25(11):2822–2835.
7. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12(3):271–276. doi:10.1016/S1567-5688(11)70886-8
8. Elsby R, Hilgendorf C, Fenner K. Understanding the critical disposition pathways of statins to assess drug-drug interaction risk during drug development: it’s not just about OATP1B1. Clin Pharmacol Ther. 2012;92(5):584–598. doi:10.1038/clpt.2012.163
9. Henwood JM, Heel RC. Lovastatin. A preliminary review of its pharmacodynamic properties and therapeutic use in hyperlipidaemia. Drugs. 1988;36(4):429–454. doi:10.2165/00003495-198836040-00003
10. Igel M, Sudhop T, von Bergmann K. Metabolism and drug interactions of 3-hydroxy-3-methylglutaryl coenzyme A-reductase inhibitors (statins). Eur J Clin Pharmacol. 2001;57(5):357–364.
11. Beauduy CE, Winston LG. Tetracyclines, macrolides, clindamycin, chloramphenicol, streptogramins, & oxazolidinones. In: Katzung BG, editor. Basic & Clinical Pharmacology, 14e. New York, NY: McGraw-Hill Education; 2017: 815–825.
12. Shi J, Montay G, Bhargava VO. Clinical pharmacokinetics of telithromycin, the first ketolide antibacterial. Clin Pharmacokinet. 2005;44(9):915–934. doi:10.2165/00003088-200544090-00003
13. Zuckerman JM, Qamar F, Bono BR. Review of Macrolides (Azithromycin, Clarithromycin), Ketolids (Telithromycin) and Glycylcyclines (Tigecycline). Med Clin. 2011;95(4):761–791. doi:10.1016/j.mcna.2011.03.012
14. Parkinson A, Ogilvie BW, Buckley DB, Kazmi F, Czerwinski M, Parkinson O. Biotransformation of xenobiotics. In: Casarett and Doull’s Toxicology: The Basic Science of Poisons, 8e. Curtis D. Klaassen. New York, NY: McGraw-Hill Education; 2012: 185–366.
15. Hirota T, Ieiri I. Drug-drug interactions that interfere with statin metabolism. Expert Opin Drug Metab Toxicol. 2015;11(9):1435–1447. doi:10.1517/17425255.2015.1056149
16. Pinto AG, Wang YH, Chalasani N, et al. Inhibition of human intestinal wall metabolism by macrolide antibiotics: effect of clarithromycin on cytochrome P450 3A4/5 activity and expression. Clin Pharmacol Ther. 2005;77(3):178–188. doi:10.1016/j.clpt.2004.10.002
17. Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Inhibition of triazolam clearance by macrolide antimicrobial agents: in vitro correlates and dynamic consequences. Clin Pharmacol Ther. 1998;64(3):278–285. doi:10.1016/S0009-9236(98)90176-X
18. Ito K, Ogihara K, Kanamitsu S, Itoh T. Prediction of the in vivo interaction between midazolam and macrolides based on in vitro studies using human liver microsomes. Drug Metab Dispos. 2003;31(7):945–954. doi:10.1124/dmd.31.7.945
19. Polasek TM, Miners JO. Quantitative prediction of macrolide drug-drug interaction potential from in vitro studies using testosterone as the human cytochrome P4503A substrate. Eur J Clin Pharmacol. 2006;62(3):203–208. doi:10.1007/s00228-005-0091-x
20. Quinney SK, Malireddy SR, Vuppalanchi R, et al. Rate of onset of inhibition of gut-wall and hepatic CYP3A by clarithromycin. Eur J Clin Pharmacol. 2013;69(3):439–448. doi:10.1007/s00228-012-1339-x
21. Zhou S, Yung Chan S, Cher Goh B, et al. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet. 2005;44(3):279–304. doi:10.2165/00003088-200544030-00005
22. Lin JH, Lu AY. Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet. 1998;35(5):361–390. doi:10.2165/00003088-199835050-00003
23. Niemi M. Role of OATP transporters in the disposition of drugs. Pharmacogenomics. 2007;8(7):787–802. doi:10.2217/14622416.8.7.787
24. Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, Chiba K. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics. 2005;15(7):513–522.
25. Jiang F, Choi JY, Lee JH, et al. The influences of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of simvastatin, in relation to CYP3A4 inhibition. Pharmacogenomics. 2017;18(5):459–469. doi:10.2217/pgs-2016-0199
26. Menochet K, Kenworthy KE, Houston JB, Galetin A. Use of mechanistic modeling to assess interindividual variability and interspecies differences in active uptake in human and rat hepatocytes. Drug Metab Dispos. 2012;40(9):1744–1756. doi:10.1124/dmd.112.046193
27. Lau YY, Huang Y, Frassetto L, Benet LZ. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007;81(2):194–204. doi:10.1038/sj.clpt.6100038
28. Niemi M, Pasanen MK, Neuvonen PJ. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin Pharmacol Ther. 2006;80(4):356–366. doi:10.1016/j.clpt.2006.06.010
29. Chang JH, Zhang X, Messick K, et al. Unremarkable impact of Oatp inhibition on the liver concentration of fluvastatin, lovastatin and pitavastatin in wild-type and Oatp1a/1b knockout mouse. Xenobiotica. 2018;49:1–9.
30. Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2007;82(6):726–733. doi:10.1038/sj.clpt.6100220
31. Deng JW, Song IS, Shin HJ, et al. The effect of SLCO1B1*15 on the disposition of pravastatin and pitavastatin is substrate dependent: the contribution of transporting activity changes by SLCO1B1*15. Pharmacogenet Genomics. 2008;18(5):424–433. doi:10.1097/FPC.0b013e3282fb02a3
32. Prueksaritanont T, Chu X, Evers R, et al. Pitavastatin is a more sensitive and selective organic anion-transporting polypeptide 1B clinical probe than rosuvastatin. Br J Clin Pharmacol. 2014;78(3):587–598. doi:10.1111/bcp.12377
33. Kunze A, Huwyler J, Camenisch G, Poller B. Prediction of organic anion-transporting polypeptide 1B1- and 1B3-mediated hepatic uptake of statins based on transporter protein expression and activity data. Drug Metab Dispos. 2014;42(9):1514–1521. doi:10.1124/dmd.114.058412
34. Seithel A, Eberl S, Singer K, et al. The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab Dispos. 2007;35(5):779–786. doi:10.1124/dmd.106.014407
35. Hirano M, Maeda K, Shitara Y, Sugiyama Y. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos. 2006;34(7):1229–1236. doi:10.1124/dmd.106.009290
36. Keskitalo JE, Kurkinen KJ, Neuvoneni PJ, Niemi M. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin Pharmacol Ther. 2008;84(4):457–461. doi:10.1038/clpt.2008.25
37. Hochman JH, Pudvah N, Qiu J, et al. Interactions of human P-glycoprotein with simvastatin, simvastatin acid, and atorvastatin. Pharm Res. 2004;21(9):1686–1691.
38. Umeyama Y, Fujioka Y, Okuda T. Clarification of P-glycoprotein inhibition-related drug-drug interaction risks based on a literature search of the clinical information. Xenobiotica. 2014;44(12):1135–1144. doi:10.3109/00498254.2014.928958
39. Holtzman CW, Wiggins BS, Spinler SA. Role of P-glycoprotein in statin drug interactions. Pharmacotherapy. 2006;26(11):1601–1607. doi:10.1592/phco.26.11.1601
40. Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport–an update. Aaps J. 2014;17(1):65–82. doi:10.1208/s12248-014-9668-6
41. Keskitalo JE, Zolk O, Fromm MF, Kurkinen KJ, Neuvonen PJ, Niemi M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2009;86(2):197–203. doi:10.1038/clpt.2009.79
42. Keskitalo JE, Pasanen MK, Neuvonen PJ, Niemi M. Different effects of the ABCG2 c.421C>A SNP on the pharmacokinetics of fluvastatin, pravastatin and simvastatin. Pharmacogenomics. 2009;10(10):1617–1624. doi:10.2217/pgs.09.85
43. Watanabe T, Kusuhara H, Watanabe T, et al. Prediction of the overall renal tubular secretion and hepatic clearance of anionic drugs and a renal drug-drug interaction involving organic anion transporter 3 in humans by in vitro uptake experiments. Drug Metab Dispos. 2011;39(6):1031–1038. doi:10.1124/dmd.110.036129
44. Niemi M, Arnold KA, Backman JT, et al. Association of genetic polymorphism in ABCC2 with hepatic multidrug resistance-associated protein 2 expression and pravastatin pharmacokinetics. Pharmacogenet Genomics. 2006;16(11):801–808. doi:10.1097/01.fpc.0000230422.50962.91
45. Kullak-Ublick GA, Stieger B, Meier PJ. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology. 2004;126(1):322–342.
46. Bi YA, Qiu X, Rotter CJ, et al. Quantitative assessment of the contribution of sodium-dependent taurocholate co-transporting polypeptide (NTCP) to the hepatic uptake of rosuvastatin, pitavastatin and fluvastatin. Biopharm Drug Dispos. 2013;34(8):452–461. doi:10.1002/bdd.1861
47. Khan FY. Rhabdomyolysis: a review of the literature. Neth J Med. 2009;67(9):272–283.
48. Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol. 2004;94(9):1140–1146. doi:10.1016/j.amjcard.2004.07.080
49. Kantola T, Kivisto KT, Neuvonen PJ. Erythromycin and verapamil considerably increase serum simvastatin and simvastatin acid concentrations. Clin Pharmacol Ther. 1998;64(2):177–182. doi:10.1016/S0009-9236(98)90151-5
50. KETEX (telithromycin) tablets [Prescribing Information-drug Interactions]. Bridgewater, NJ 08807: Sanofi-aventis U.S. LLC; 2015.
51. Kaleem Z, Khan JA, Mushtaq Z, Altaf S, Javed I. Assessment of potential interaction between simvastatin and clarithromycin in healthy adult male subjects. Pak J Pharm Sci. 2018;31(3):801–806.
52. Siedlik PH, Olson SC, Yang BB, Stern RH. Erythromycin coadministration increases plasma atorvastatin concentrations. J Clin Pharmacol. 1999;39(5):501–504.
53. LIPITOR (atorvastatin) tablets [prescribing information]. NY, NY 10017: Parke-Davis-Pfizer Inc; 2009.
54. Amsden GW, Kuye O, Wei GC. A study of the interaction potential of azithromycin and clarithromycin with atorvastatin in healthy volunteers. J Clin Pharmacol. 2002;42(4):444–449.
55. Cooper KJ, Martin PD, Dane AL, Warwick MJ, Raza A, Schneck DW. The effect of erythromycin on the pharmacokinetics of rosuvastatin. Eur J Clin Pharmacol. 2003;59(1):51–56. doi:10.1007/s00228-003-0573-7
56. LIVALO (pitavastatin) prescribing information (drug interactions); 2016. Available from: https://www.kowapharma.com/documents/LIVALO_PI_CURRENT.pdf.
57. Torres PA, Helmstetter JA, Kaye AM, Kaye AD. Rhabdomyolysis: pathogenesis, diagnosis, and treatment. Ochsner J. 2015;15(1):58–69.
58. Davidson MH, Clark JA, Glass LM, Kanumalla A. Statin safety: an appraisal from the adverse event reporting system. Am J Cardiol. 2006;97(8a):32c–43c. doi:10.1016/j.amjcard.2005.12.008
59. Hill FJ, McCloskey SJ, Sheerin N. From a fish tank injury to hospital haemodialysis: the serious consequences of drug interactions. BMJ Case Rep. 2015;2015:bcr2015209961. doi:10.1136/bcr-2015-209961
60. Fallah A, Deep M, Smallwood D, Hughes P. Life-threatening rhabdomyolysis following the interaction of two commonly prescribed medications. Australas Med J. 2013;6(3):112–114. doi:10.4066/AMJ.2013.1616
61. Azmi HM, Fahad S, Kumar VA. Lovastatin-erythromycin induced myositis: case report and possible mechanism. J Pharm Pharm. 2015;1(1):30–32. doi:10.5530/jppcm.2015.1.7
62. Grunden JW, Fisher KA. Lovastatin-induced rhabdomyolysis possibly associated with clarithromycin and azithromycin. Ann Pharmacother. 1997;31(7–8):859–863. doi:10.1177/106002809703100710
63. Kahri AJ, Valkonen MM, Vuoristo MK, Pentikäinen PJ. Rhabdomyolysis associated with concomitant use of simvastatin and clarithromycin. Ann Pharmacother. 2004;38(4):719. doi:10.1345/aph.1D243
64. Spach DH, Bauwens JE, Clark CD, Burke WG. Rhabdomyolysis associated with lovastatin and erythromycin use. West J Med. 1991;154(2):213–215.
65. Alreja G, Inayatullah S, Goel S, Braden G. Rhabdomyolysis caused by an unusual interaction between azithromycin and simvastatin. J Cardiovasc Dis Res. 2012;3(4):319–322. doi:10.4103/0975-3583.102720
66. Wagner J, Suessmair C, Pfister HW. Rhabdomyolysis caused by co-medication with simvastatin and clarithromycin. J Neurol. 2009;256(7):1182–1183. doi:10.1007/s00415-009-5078-6
67. Page SR, Yee KC. Rhabdomyolysis in association with simvastatin and dosage increment in clarithromycin. Intern Med J. 2014;44(7):690–693. doi:10.1111/imj.12464
68. Lee AJ, Maddix DS. Rhabdomyolysis secondary to a drug interaction between simvastatin and clarithromycin. Ann Pharmacother. 2001;35(1):26–31. doi:10.1345/aph.10177
69. Molden E, Andersson KS. Simvastatin-associated rhabdomyolysis after coadministration of macrolide antibiotics in two patients. Pharmacotherapy. 2007;27(4):603–607. doi:10.1592/phco.27.4.603
70. Nguyen KA, Li L, Lu D, et al. A comprehensive review and meta-analysis of risk factors for statin-induced myopathy. Eur J Clin Pharmacol. 2018;74(9):1099–1109. doi:10.1007/s00228-018-2482-9
71. Mendes P, Robles PG, Mathur S. Statin-induced rhabdomyolysis: a comprehensive review of case reports. Physiother Can. 2014;66(2):124–132. doi:10.3138/ptc.2012-65
72. Cziraky MJ, Willey VJ, McKenney JM, et al. Statin safety: an assessment using an administrative claims database. Am J Cardiol. 2006;97(8a):61c–68c. doi:10.1016/j.amjcard.2005.12.011
73. Slobodnick A, Shah B, Pillinger MH, Krasnokutsky S. Colchicine: old and new. Am J Med. 2015;128(5):461–470. doi:10.1016/j.amjmed.2014.12.010
74. Davis MW, Wason S, Digiacinto JL. Colchicine-antimicrobial drug interactions: what pharmacists need to know in treating gout. Consult Pharm. 2013;28(3):176–183. doi:10.4140/TCP.n.2013.176
75. Nishio S, Watanabe H, Kosuge K, Uchida S, Hayashi H, Ohashi K. Interaction between amlodipine and simvastatin in patients with hypercholesterolemia and hypertension. Hypertens Res. 2005;28(3):223–227. doi:10.1291/hypres.28.223
76. Pan Y, Chothe PP, Swaan PW. Identification of novel Breast Cancer Resistance Protein (BCRP) Inhibitors by virtual screening. Mol Pharm. 2013;10(4):1236–1248. doi:10.1021/mp300547h
77. Strandell J, Bate A, Hagg S, Edwards IR. Rhabdomyolysis a result of azithromycin and statins: an unrecognized interaction. Br J Clin Pharmacol. 2009;68(3):427–434. doi:10.1111/j.1365-2125.2009.03473.x
78. Rowan C, Brinker AD, Nourjah P, et al. Rhabdomyolysis reports show interaction between simvastatin and CYP3A4 inhibitors. Pharmacoepidemiol Drug Saf. 2009;18(4):301–309. doi:10.1002/pds.1711
79. Rowan CG, Brunelli SM, Munson J, et al. Clinical importance of the drug interaction between statins and CYP3A4 inhibitors: a retrospective cohort study in the health improvement network. Pharmacoepidemiol Drug Saf. 2012;21(5):494–506. doi:10.1002/pds.3199
80. Patel AM, Shariff S, Bailey DG, et al. Statin toxicity from macrolide antibiotic coprescription: a population-based cohort study. Ann Intern Med. 2013;158(12):869–876. doi:10.7326/0003-4819-158-12-201306180-00004
81. Li DQ, Kim R, McArthur E, et al. Risk of adverse events among older adults following co-prescription of clarithromycin and statins not metabolized by cytochrome P450 3A4. Can Med Assoc J. 2015;187(3):174–180. doi:10.1503/cmaj.140950
82. Mesgarpour B, Gouya G, Herkner H, Reichardt B, Wolzt M. A population-based analysis of the risk of drug interaction between clarithromycin and statins for hospitalisation or death. Lipids Health Dis. 2015;14:131. doi:10.1186/s12944-015-0134-y
83. Kolovou V, Bilianou H, Kolovou G. Safe use of statins: focus on muscle toxicity. Clin Lipidol. 2016;11(1):16–22.
84. Chang C-H, Kusama M, Ono S, Sugiyama Y, Orii T, Akazawa M. Assessment of statin-associated muscle toxicity in Japan: a cohort study conducted using claims database and laboratory information. BMJ Open. 2013;3(4):e002040. doi:10.1136/bmjopen-2012-002040
85. New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. FDA Drug Safety Communication. August 6th, 2011. Available from: https://wayback.archive-it.org/7993/20170112031805/http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed December 11, 2018.
86. ZOCOR (simvastatin) tablets [prescribing information]. Cramlington, Northumberland, UK NE23 3JU: Merck Sharp & Dohme Ltd; 2010.
87. Lovastatin - FDA prescribing information, side effects and uses. Mylan Pharmaceuticals Inc., Morgantown, WV 26505 USA; 2018. Available from: https://www.drugs.com/pro/lovastatin.html. Accessed January 18, 2019.
88. PRAVACHOL (Pravastatin) tablets [Prescribing Information]. Princeton, New Jersey 08543 USA; Bristol-Myers Squibb Company; 2014.
89. Garcia MJ, Reinoso RF, Sanchez Navarro A, Prous JR. Clinical pharmacokinetics of statins. Methods Find Exp Clin Pharmacol. 2003;25(6):457–481.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.