Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
The Cerebroprotein Hydrolysate-I Plays a Neuroprotective Effect on Cerebral Ischemic Stroke by Inhibiting MEK/ERK1/2 Signaling Pathway in Rats
Authors Ren Y, Ma X, Wang T, Cheng B, Ren L, Dong Z, Liu H
Received 1 April 2021
Accepted for publication 15 June 2021
Published 6 July 2021 Volume 2021:17 Pages 2199—2208
DOI https://doi.org/10.2147/NDT.S313807
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Taro Kishi
Yuqian Ren,1 Xiaoqing Ma,1 Tingting Wang,1 Baohe Cheng,2 Leiming Ren,3 Zehua Dong,4 Hongling Liu5
1Institute of Cerebrovascular Disease, The Affiliated Hospital of Qingdao University, Qingdao, 266003, People’s Republic of China; 2Shandong Haoyun International Hospital of Stem Cells, Jinan, Shandong, 250001, People’s Republic of China; 3Institute of Chinese Integrative Medicine, Hebei Medical University, Shijiazhuang, 050017, People’s Republic of China; 4Department of Intensive Care Unit, The Affiliated Hospital of Qingdao University, Qingdao, 266003, People’s Republic of China; 5Department of Pharmacy, The Affiliated Hospital of Qingdao University, Qingdao, 266003, People’s Republic of China
Correspondence: Zehua Dong; Hongling Liu Tel +86-139 63958230
; +86-18661808612
Email [email protected]; [email protected]
Objective: To investigate the neuroprotective effect and mechanism of cerebroprotein hydrolysate-I (CH-I) on cerebral ischemia/reperfusion injury in rats.
Methods: A total of 100 adult healthy male SD rats were randomly divided into a sham group, model group, CH-I treated group, and cerebrolysin (CBL) positive group, consisting of 20 rats in each group. The middle cerebral artery occlusion/reperfusion (MCAO/R) model of rats was built by inserting a suture into the left external carotid artery (ECA) through the internal carotid artery (ICA). Treatment was performed by intraperitoneal injection of CH-I (20 mg/kg). The neurobehavioral function of rats was evaluated by modified neurological severity scores (mNSS). TTC staining was used to detect the cerebral infarction volume (CIV) of rats. The morphological and structural changes of nerve cells were observed by HE staining and the neuronal apoptosis was counted by TUNEL assay. Immunohistochemical (IHC) analysis was used to detect BDNF and pMEK1/2 expressions. The expressions of BDNF, pMEK1/2, pERK1/2, and pCREB were determined with Western blotting.
Results: After treatment with CH-I, the mNSS and CIV of rats were improved (P< 0.05). And the CH-I can reduce the degeneration and apoptosis of nerve cells in rats (P< 0.01). Western blotting showed that the expressions of pMEK1/2, pERK1/2, and pCREB in rats were increased, while the expression of BDNF was decreased after modeling (P< 0.05). After treatment, the expressions of pMEK1/2, pERK1/2, and pCREB in the CH-I group were decreased (P< 0.05), while the expression of BDNF was significantly increased (P< 0.05) compared with the model group. IHC showed that the expression of BDNF and pMEK1/2 was consistent with Western blotting.
Conclusion: It is suggested that the CH-I might play a neuroprotective role by inhibiting the expression of MEK-ERK-CREB and enhancing the expression of BDNF after cerebral ischemia/reperfusion injury, thus improving the neurobehavioral function of MCAO/R rats.
Keywords: cerebroprotein hydrolysate-I, cerebral ischemia/reperfusion injury, apoptosis, MAPK/ERK1/2 signaling pathway, rats
Introduction
Acute ischemic stroke is a common type of stroke.1,2 Ultra-early thrombolytic therapy is currently recognized as the main operative treatment, but the treatment time window of this therapy is only 3–6 hours, and there are many complications after thrombolytic therapy,2,3 which limits its clinical application. Neuronal damage in the ischemic penumbra or peri-infarct area is transient and reversible in the first few hours after ischemia.2 A series of studies have shown that the application of neuroprotective drugs as adjuvant therapy of thrombolytic therapy can contribute to improving the neurological function of patients with ischemic stroke.4–6 Cerebroprotein hydrolysate-I (CH-I), a mixture, was prepared from denatured proteins with a total nitrogen content of more than 120 mg/g.7 It contained 16 kinds of amino acids and 25–35% small molecule polypeptides (<10,000 Da).7 It also contained many nerve growth factors, which have similar pharmacodynamic characteristics with endogenous neurotrophic factors, and have neurotrophic and neuroprotective effects.8–11 It has been found that CH-I and cerebrolysin (CBL) have similar effects to brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF), and have neuroprotective effects such as reducing the inflammatory response, apoptosis, and encephaledema.8,9,11 BDNF is a protein synthesized in the brain, which is widespread in the central nervous system and can promote the recovery of function after stroke.12 It has been noted that the expression of BDNF is decreased after stroke, and the neurobehavioral function of cerebral ischemia rats can be improved by up-regulating BDNF expression.13 Mature BDNF dimer binds to tyrosine kinase receptor B (TrkB) to induce autophosphorylation of tyrosine residues and then regulate intracellular signaling pathways: Mitogen-activated protein kinase/Extracellular signal-regulated protein kinase (MAPK/ERK1/2), which leads to the phosphorylation and activation of downstream cAMP response element binding protein (CREB), and further mediates gene transcription of neurons.14,15 Ischemia/reperfusion (I/R) is a complex process, which refers to the interruption of blood flow to tissues and then recovery.16 Inflammation, oxidative stress, and apoptosis were important pathophysiological pathways after I/R injury.16–18 The MAPK signaling pathway is a major signaling pathway in the process of cerebral ischemia reperfusion injury (CIRI), and MAPK kinase (MEK)/ERK is an important part of the MAPK signaling pathway.19 CIRI can activate the MEK/ERK1/2 signaling pathway, which can reduce brain nerve cell death and improve prognosis by inhibiting the MEK/ERK1/2 signaling pathway.19,20 This study sought to investigate whether CH-I could play a neuroprotective effect against CIRI by intervening the MEK/ERK1/2 signaling pathway and downstream CREB pathway.
Methods and Materials
Materials
CH-I, No. 0190501–1, Zhitong Biopharma Co. Ltd., China; CBL, No. 1–21380, EVER Neuro Pharma GmbH, Austria;2,3,5-Triphenyltetrazolium chloride (TTC), No. T8877, SIGMA, USA; Hematoxylin-Eosin (HE) staining kit, No. G1120, Beijing Solarbio Science & Technology Co. Ltd., China; Colorimetric TUNEL apoptosis assay kit (C1091), Beyotime Biotechnology Co. Ltd., China; Rabbit anti-IgG MEK1/2 (#380797), pMEK1/2 (Ser217/221, #310050), ERK1/2 (#343830), pERK1/2 (Thr202/Tyr204, #301245), CREB (#383983), pCREB (Ser133, #380697), ZEN-BIOSCIENCE, China; Rabbit anti-mouse polyclonal antibody BDNF (Ab-DF6387), Affinity Co. Ltd., USA.
Animal Models
A total of 100 healthy adult male SD rats, weight 230–260 g, SPF grade, was granted by the Experiment Animal Center of Shandong Drug Inspection Institute (SCXK(LU) 20190007). The rats were acclimatized to the environment in the laboratory for 1 week at room temperature (23±2)°C, with natural light, free eating, and drinking water. All animal procedures were approved by the Ethics Committee of the Affiliated Hospital of Qingdao University (Permit No. QYFY WZLL 26273) and followed the Guide for the Care and Use of Laboratory Animals.21 Twenty rats were randomly selected as the sham group, and the other 80 rats were established by the middle cerebral artery occlusion/reperfusion (MCAO/R) model. All rats were anesthetized by intraperitoneal injection of 10% chloral hydrate (0.3 mL/kg). The MCAO model was established by inserting a suture through the left external carotid artery (ECA)-internal carotid artery (ICA) until 18–22 mm from the left common carotid artery (CCA) bifurcation.22 After 2 hours of ischemia, the suture was removed to restore reperfusion, the preparation of MCAO/R model was completed. The anal temperature of the animals was kept at 36–37°C during the operation. The MCAO/R model was successfully determined by the peak of regional cerebral blood flow (rCBF) to 30% and below baseline (perflux 5000, Sweden), rCBF recovered to 80% or more after the suture was pulled out, and the modified neurologic severity score (mNSS)23 was 7–14 after the animal woke up. Rats in the sham group were immediately removed the suture from the ICA by 10 mm, and the remaining procedures were performed the same as the operation group. Twenty rats who had not recovered or died after 2 hours were removed. The other successful models were randomly divided into model group, treated group, and positive group, 20 in each group.
Intervention Measures
Treated group and positive group: the CH-I and CBL were prepared into 1 mL solution with 0.9% normal saline, and the rats were intraperitoneally injected with CH-I (20 mg/kg)11 and CBL (20 mg/kg) at 0, 3, 6, and 12 hours after reperfusion, respectively. The sham group and model group rats were given 1 mL normal saline simultaneously. After reperfusion at 24 hours, all the animals were killed, and then the corresponding indexes were detected (Figure 1).
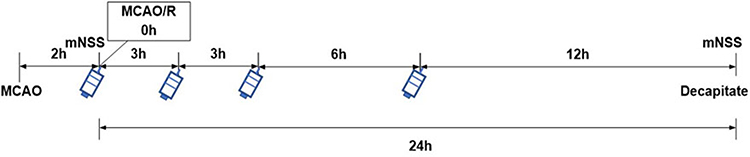 |
Figure 1 Schematic diagram of administration in rats. |
Evaluation Indexes
mNSS Scores
The mNSS includes motor function (tail lifting test and placing rats on floor), sensory tests (placing test and proprioceptive test), beam balance tests, reflex absence (pinna reflex, corneal reflex, and startle reflex), and abnormal movements (seizures, myoclonus, myodystony). The total score is 18 points. The higher the score, the more serious the neurological function damage. The test was repeated at 0 and 24 hours after MCAO/R.
TTC Staining
Five rats were chosen from each group. All rats MCAO/R 24 hours later, and anesthetized intraperitoneally with chloral hydrate. After the normal saline was injected into the heart, the brain was taken completely. The brain was placed in the rat brain mold. The coronal sections were cut back and forth from the middle point of the junction between the forehead and the visual cross. The thickness was 2 mm, immersed in 1% of TTC at 37°C for 10 minutes in a dark incubator. The ischemia area showed pale white and the normal area showed bright red. The infarct area of each layer was calculated by Image J System, and the percentage of cerebral infarction volume (CIV) in the same hemisphere volume was calculated. CIV=sum of the infarct area of each section/sum of the area of each section.
HE Staining
In each group, five rats were given intraperitoneal injection of chloral hydrate, followed by perfusion 0.9% sodium chloride and 4% paraformaldehyde through the heart to reach the brain. Conventional gradient ethanol dehydration, xylene transparent, paraffin embedded, back continuous coronal section at the optic chiasm, thickness 5 µm, each three pieces extracted one, pasted on the poly lysine treatment section, 4°C preservation. The paraffin sections of the brain tissue were dewaxed with xylene, dehydrated with ethanol, stained with hematoxylin for 2 minutes, stained with eosin for 1 minute, and then routinely dehydrated, transparent, and sealed. Under the light microscope (Olympus IX-141, Japan), the nuclei of neurons were blue and the cytoplasm was red in varying degrees. At high magnification (×400), four visual fields were collected from the parietal cortex randomly, and the denatured cells were counted. And the degree of damage was expressed by the denatured cell index (DCI=number of denatured cells/total number of cells).
TUNEL Staining
According to the instructions of the TUNEL cell apoptosis detection kit, the above paraffin sections were taken and added to biotin labeled solution at 37°C for 60 minutes, PBS for 3-times, incubated with Streptavidin-HRP working solution at room temperature for 30 minutes, PBS 5 minutes for 3-times, developed DAB color in dark, and dehydrated, transparent, and sealed routinely. Negative control showed no positive staining without TdT. The positive apoptotic cells were brown under a microscope. Four fields of vision were randomly taken in the cortex area under high magnification (×400), and TUNEL positive cells were counted. Apoptotic cell index (ACI)=the number of apoptotic positive cells/the total number of cells.
Western Blotting
Five rats in each group received perfusion with 0.9% NaCl 250 mL through the heart. The whole brain was extracted, and the ischemic brain tissue of the parietal cortex was separated after the optic chiasma and placed into a 1.5 mL EP tube. Cell lysate was added and homogenized by ultrasound, centrifuged at 12,000 rpm for 20 minutes, and the supernatant was left after precipitation removal. The protein concentration was determined by BCA (BL521A, Biosharp Life Science Co. Ltd., USA) method. Added 1/4 × loading buffer, 95–97°C for 5 minutes, and stored at −80°C. The above protein samples were separated by 10% SDS-PAGE and then transferred onto a PVDF membrane by a wet method. The primary antibody (Rabbit anti-pMEK1/2, pERK1/2, pCREB, BDNF, 1:1,000) was incubated at 4°C overnight. TBST for 10 minutes in 3times-. The secondary antibody (1:10,000) was incubated at room temperature for 2 hours. Bio-Rad 2000 imaging systems were used to scan the membranes and Image J System was used to analyze gray values of each band. The relative value protein (RVP)=gray value of target protein/gray value of internal parameters was calculated, repeating the experiment three -times, and the mean value was taken.
Immunohistochemistry
The above paraffin sections were treated with conventional dewaxing and hydration, sodium citrate heat antigen repair at 96°C for 15 minutes, blocking endogenous peroxidase (HRP), sealing at room temperature for 40 minutes, pMEK1/2 (1:100) incubated at 4°C overnight, secondary antibody (1:200) incubated at 37°C for 30 minutes, stained with DAB, and re-stained with hematoxylin. Under light microscope, brown granules in the cytoplasm were positive cells. Negative control sections were stained with 0.1 mol/L PBS instead of primary antibody. Four non-overlapping fields were randomly observed in each section of a cortical area under a microscope (×400), and the average was taken. The expression intensity of the protein was denoted by positive cell index (PCI)=number of positive cells/total number of cells.
Statistical Analysis
SPSS 20.0 software was used for data analysis. The measurement data were expressed by mean±standard deviation (x±s). Multi-group comparison was performed by one-way ANOVA, and the pairwise comparison was performed by Bonferroni-corrected tests. The values were significant (P<0.05) and extremely significant (P<0.01).
Results
CH-I Can Improve the Neurobehavioral Function of Rats
The mNSS score of the sham group was zero. Compared with the sham group, the score of the model group, CH-I group, and CBL group was significantly increased (P<0.01). After treatment, the score of the CH-I group and CBL group were significantly lower than that of the model group (P<0.01), but there was no significant difference between the CH-I group and CBL group (P>0.05). The results indicated that CH-I and CBL could improve neurological behavioral function in MCAO/R rats (N=12) (Figure 2).
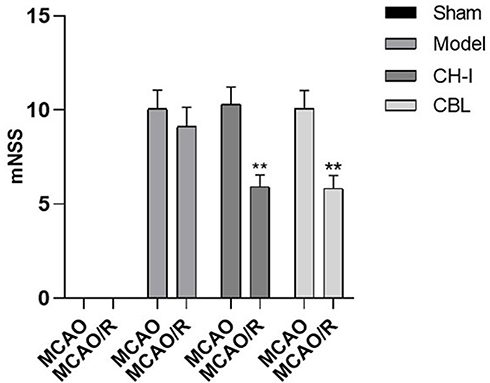 |
Figure 2 mNSS scores of MCAO/R rats before and after treatment. ** P<0.01 vs model group. |
CH-I Can Reduce Infarct Volume in Rats
There was no obvious infarct in the brain tissue of the sham group, which showed uniform red. And white infarct was discovered in the ischemic hemisphere of the model group, CH-I group, and CBL group (P<0.01). The range of cerebral infarction in the CH-I group and CBL group was markedly smaller than that in the model group (P<0.05), but there was no significant difference between the CH-I group and CBL group (P>0.05) (N=5) (Figure 3).
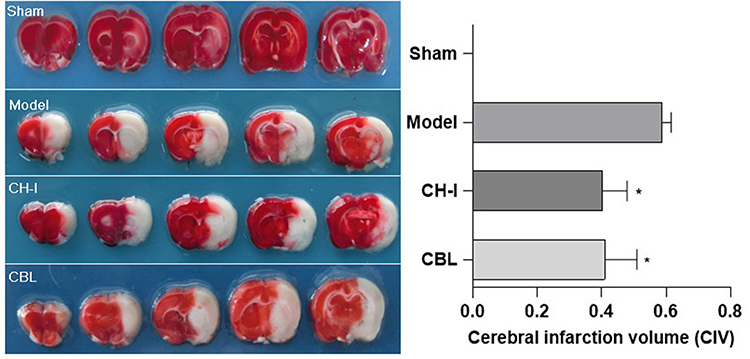 |
Figure 3 The cerebral infarction volume in rats, TTC staining. *P<0.05 vs Model. |
CH-I Can Improve the Pathological Structure of Nerve Cells in the Rat Brain
HE staining showed that the neurons in the sham group (DCI=0.4203±0.0832) were orderly arranged, with a complete structure and clear nucleus. The neurons in the model group (DCI=0.8559±0.0669) were arranged irregularly, with deeply colored and condensed nuclei, and the DCI value was much higher than that in the sham group (P<0.01). The degeneration of nerve cells in the CH-I group (DCI=0.6396±0.0717) and CBL group (DCI=0.6562±0.0738) were less than that in the model group (P<0.01). There was no significant difference in DCI between group CH-I and group CBL (P>0.05) (N=3) (Figure 4).
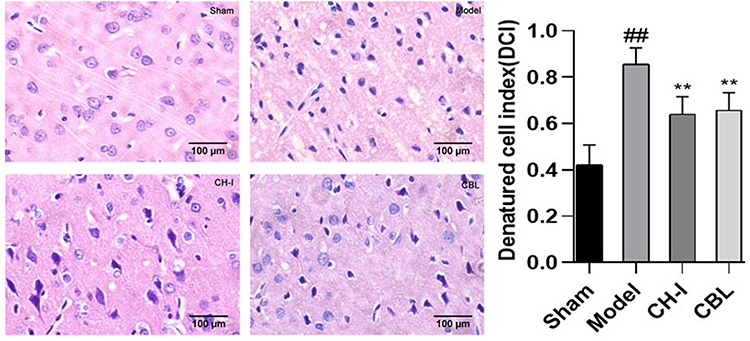 |
Figure 4 The morphology of neurons in the cortex of rats, HE staining (×400). ##P<0.01 vs Sham; **P<0.01 vs Model. |
CH-I Can Reduce the Number of Apoptotic Cells in Rat Brain Tissue
TUNEL staining showed that the neurons in the cerebral cortex in sham group (ACI= 0.0392±0.0140) were light blue, arranged orderly, with a complete structure, and a few brown apoptotic cells were observed. The apoptotic cells in the model group (ACI=0.6161±0.0655) were significantly increased compared to those in the sham group (P<0.01), and a large number of brown apoptotic cells were observed, with cell shrinkage and disordered arrangement. Compared with the model group, the apoptotic cells in the CH-I group (ACI=0.371±0.0655) and CBL group (ACI=0.4431±0.0794) were significantly improved (P<0.01), and the cell arrangement was more orderly. There was no notable difference between the CH-I group and CBL group (P>0.05). (N=3) (Figure 5).
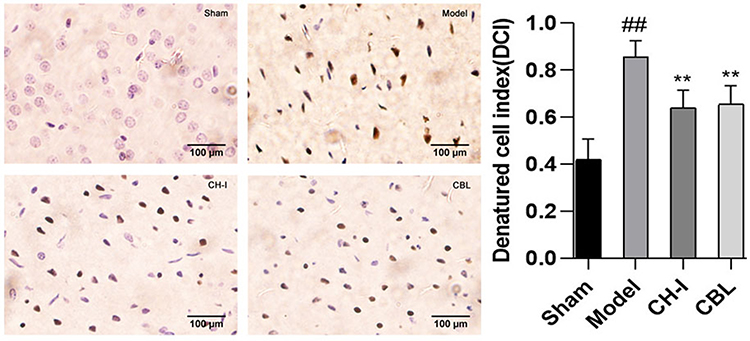 |
Figure 5 The morphology of neurons in the cortex of rats, TUNEL staining (×400). ##P<0.01 vs Sham; **P<0.01 vs Model. |
Effect of CH-I on Protein Expression of BDNF, pMEK1/2, pERK1/2, and pCREB
Western blotting was used to detect the expression of BDNF, pMEK1/2, pERK1/2, and pCREB proteins in the cortex ischemic area of rats after MCAO/R. The results were as in Figure 6 (N=3).
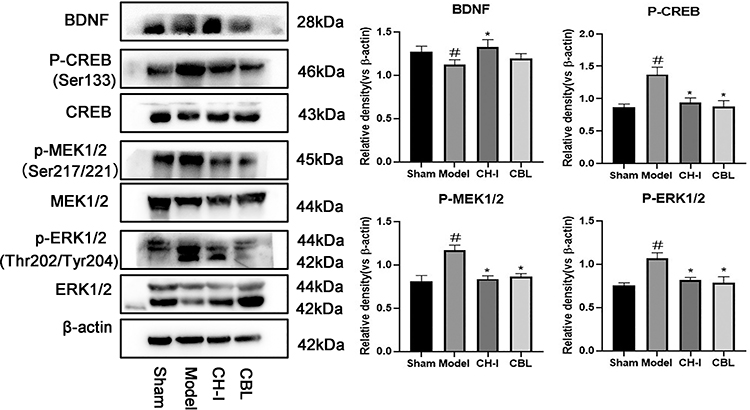 |
Figure 6 The protein expression level of BDNF/pMEK1/2/pERK1/2/pCREB. #P<0.05 vs Sham; *P<0.05 vs Model. |
BDNF Expression
In the model group (RVP=1.1265±0.0458), the expression of BDNF was weaker than that in the sham group (RVP=1.2717±0.0548) (P<0.05). The protein expression of the CH-I group (RVP=1.3262±0.0745) was clearly lower than that in the model group (P<0.05), however there was no significant difference between the CBL group (RVP=1.1959±0.0473) and model group (P>0.05), and there was no statistical difference between the CH-I and CBL group (P>0.05).
pMEK1/2 Expression
The pMEK1/2 content in rats brain of the model group (RVP=1.1683±0.0487) was enhanced compared to the sham group (RVP=0.8130±0.0544) (P<0.05). After the intervention with CH-I and CBL, the expression of pMEK1/2 in both two groups was lower than that in the model group (P<0.05). There was no significant difference between group CH-I (RVP=0.8387±0.0289) and group CBL (RVP=0.8643±0.0283) (P>0.05).
pERK1/2 Expression
The expression of pERK1/2 in the model group (RVP=1.0662±0.0548) was higher than that in the sham group (RVP=0.7529±0.0281) (P<0.05). After treatment, the protein expression of the CH-I group (RVP=0.8192±0.0246) and CBL group (RVP=0.7838±0.0594) were clearly weaker than the model group (P<0.05). However, there was no significant difference between the CH-I group and CBL group (P>0.05).
pCREB Expression
In the model group (RVP=1.3743±0.0914), the pCREB content in cerebral ischemic of rats was increased compared to the sham group (RVP=0.8661±0.0387) (P<0.05). The protein expression in the CH-I group (RVP=0.9405±0.0576) and CBL group (RVP=0.8791±0.0753) were much lower than that in the model group (P<0.05). There was no significant difference between group CH-I and group CBL (P>0.05).
Effect of CH-I on Expression of BDNF and pMEK1/2 Proteins Detected by Immunohistochemistry
The expression of BDNF in the model group (RVP=0.1895±0.0336) was clearly weaker that in the sham group (RVP=0.3239±0.0302) (P<0.01). After treatment, the protein expression of the CH-I group (RVP=0.3817±0.0454) and CBL group (RVP=0.3497±0.0496) were appreciably increased compared to the model group (P<0.05). But there was no statistical difference among CH-I group, CBL group, and sham group (P>0.05) (N=3) (Figure 7).
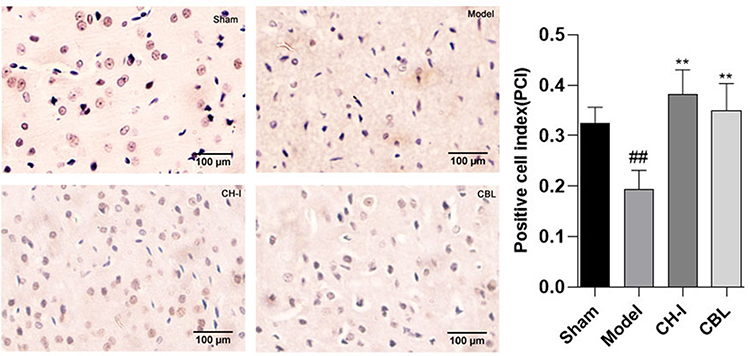 |
Figure 7 The expressions of BDNF in rats by IHC (×400). ##P<0.01 vs Sham; **P<0.01 vs Model. |
IHC staining showed that only a few neurons in the sham group (PCI=0.1675±0.0528) had positive expression of pMEK1/2. The expression of pMEK1/2 in the model group (PCI=0.4096±0.0769) was markedly higher than that in the sham group (P<0.01). The expression of pMEK1/2 in the CH-I group (PCI=0.2204±0.0470) and CBL group (PCI=0.2627±0.0592) was clearly lower than that in the model group (CH-I group, P<0.01; CBL group, P<0.05), but there was no significant difference between the two groups (N=3) (Figure 8).
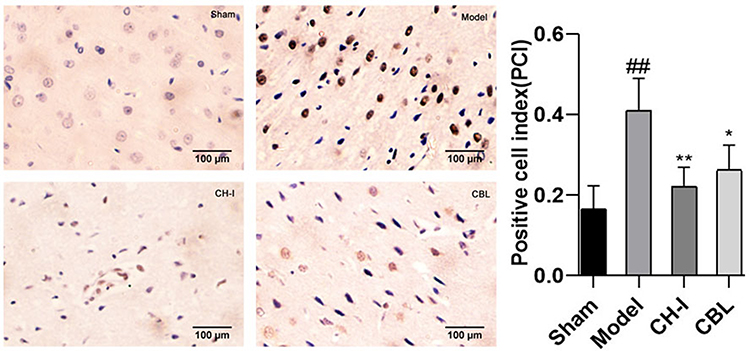 |
Figure 8 The expressions of pMEK1/2 in rats by IHC (×400). ##P<0.01 vs Sham; *P<0.05 vs Model; **P<0.01 vs Model. |
Discussion
Stroke is a common clinical cerebrovascular disease, which is divided into ischemic stroke and hemorrhagic stroke, of which about 85% are ischemic stroke.24 The present study investigated that treatment with neuroprotective drugs is beneficial to the early recovery of peripheral nerve function in the ischemic cortical after stroke.5,6,12 CH-I is a bioactive substance isolated and purified from fresh pig brain tissue.7 The free amino acids of 50–80% can enter brain neurons through the blood brain barrier (BBB) and strengthen protein synthesis in the brain.10,11,24 It can simulate similar pharmacodynamic characteristics of neurotrophic factors (NTFs), such as BDNF, NGF, and serum insulin-like growth factor-1 (IGF-1).8,10,24 In the current experiment, it has been found that CH-I can promote the repair of neurons and subsequently improve the dementia symptoms and delay the aggravation of dementia lesions of vascular dementia (VaD) rats by elevating the level of IGF-1 in VaD rats.14 Cao et al11 have found that continuous intraperitoneal injection of CH-I 14d to MCAO mice can improve the mNSS score, promote neuronal survival, and significantly improve infarct size in mice. BDNF is among the most abundant NTFs in brain tissue, mainly expressed in the synapse of the hypothalamus, cortex, and the basal part of the forebrain. BDNF can promote nerve growth and survival, promote angiogenesis and functional recovery after stroke, and play a key role in post-stroke rehabilitation.12,13,25,26 Ni et al13 confirmed that increasing the expression of BDNF could improve neurological function and sensory motor recovery in ischemic rats. TrkB, the specific receptor of BDNF, is a transmembrane protein on the nerve cell membrane. Phosphorylated TrkB activates three main intracellular signal transduction pathways: MAPK/ERK pathway leads to cell growth and differentiation; the phosphatidylinositol 3 kinase (PI3K)–Akt pathway induces cell survival; Phospholipase C gamma (PLCγ)-Ca2+ pathway changes synaptic plasticity.14,15 In vivo and in vitro studies have shown that PI3K–Akt and ERK1/2 are involved in autophagy after cerebral ischemia.1,27
After thrombolytic therapy, blood flow reperfusion will further aggravate brain damage, resulting in nerve injury and physical function defect or disability in patients.28 Thus, CIRI is a major pathological mechanism after stroke. Some evidence has shown that CIRI can activate the MAPK/ERK1/2 signal pathway, and then activate downstream mediators, leading to an inflammatory reaction, apoptosis, and other pathological processes.20,29 MAPK is a group of ubiquitous serine and threonine protein kinases in cells, and its expression level is relatively high in the central nervous system. Rat sarcoma/Rapidly accelerated fibrosarcoma (Ras/Raf)/MEK/ERK is the most classical signaling pathway of MAPK, and under the action of various growth factors and external stimuli, Raf can bind to MEK1/2 and phosphorylate it after Ras activation, which activates MEK1/2 and activated MEK1/2 can transfer phosphoric acid to the threonine and tyrosine residues of downstream kinase ERK1/2 to initiate the downstream ERK1/2 pathway.30,31 The most widely characterized feature of MAPK is ERK1/2, whose primary role is its catalytic role as a serine and threonine kinase.32 After ERK1/2 activation, the signal can be transduced from the cell membrane surface receptor to the nucleus and phosphorylated in the nucleus, then regulate various transcription factors, ultimately leading to the changes of gene expression.4,15 Some earlier studies have found that the level of phosphorylated MAPK changes in the brain tissue after stroke, and the phosphorylated MEK activates the downstream kinase ERK1/2, which in turn initiates the downstream pathway to regulate cell growth, differentiation, and other physiological and pathological processes by activating CREB and subsequently modifying BDNF.12,20,29 Burguete et al20 confirmed that ischemia/reperfusion enhanced the activity of the MAPK/ERK1/2 signaling pathway and markedly increased the phosphorylation of ERK1/2. Wang et al29 and other studies also indicate that the MEK-ERK1/2 pathway could be activated after cerebral ischemia in rats, and by intervening the MEK-ERK1/2 pathway could alleviate the apoptosis and inflammatory reaction of ischemic rats.
The results of this study showed that pMEK1/2, pERK1/2, and pCREB proteins were highly expressed in the ischemic cortex of rats in the model group, which was consistent with previous results.12,20,29,30 Compared with the sham group, the apoptosis of nerve cells and mNSS increased dramatically, and it prompts that CIRI can activate the MEK-ERK-CREB signaling pathway. After treatment with CH-I, the expression of pMEK1/2, pERK1/2, and pCREB were decreased, and the apoptosis and nerve function injury were alleviated compared with the model group. Western blotting tests and IHC staining showed that the expression of BDNF in model group was lower than that in the sham group, and the higher expression of BDNF was observed in the CH-I group. Immunohistochemistry showed that the expression of pMEK1/2 was in the line with that of Western blotting. These results suggested that CH-I may exert a protective effect by mediating the MEK-ERK-CREB pathway, increasing BDNF expression, and then improving neurological function and apoptosis of MCAO/R rats. In conclusion, CH-I may regularly ameliorate the neural function, apoptosis, and cerebral infarct volume in MCAO/R rats. This study only preliminarily discussed the protective role of CH-I in the treatment of cerebral ischemia-reperfusion. And whether it can play a brain protective role through other transcription factors downstream of MEK/ERK remains to be further studied. The pathological mechanism of stroke is sophisticated. From now on, it is necessary to further study the application effective and pathological mechanism of CH-I on stroke from more levels.
Acknowledgments
This work was supported by the National Natural Science Fund of China (81973501) and Hebei Zhitong Science Fund (2018ZD1901).
Disclosure
The authors declare no conflicts of interest.
References
1. Wang M, Liang X, Cheng M, et al. Homocysteine enhances neural stem cell autophagy in in vivo and in vitro model of ischemic stroke. Cell Death Dis. 2019;10(8):561. doi:10.1038/s41419-019-1798-4
2. Leung SW, Lai JH, Wu JC, et al. Neuroprotective effects of emodin against ischemia/reperfusion injury through activating ERK-1/2 signaling pathway. Int J Mol Sci. 2020;21(8):2899. doi:10.3390/ijms21082899
3. Prabhakaran S, Ruff I, Bernstein RA. Acute stroke intervention: a systematic review. JAMA. 2015;313(14):1451–1462. doi:10.1001/jama.2015.3058
4. Brainin M. Cerebrolysin: a multi-target drug for recovery after stroke. Expert Rev Neurother. 2018;18(8):681–687. doi:10.1080/14737175.2018.1500459
5. Zhang R, Liu C, Ji Y, Teng L, Guo Y. Neuregulin1β plays a neuroprotective role by inhibiting the Cdk5 signaling pathway after cerebral ischemia-reperfusion injury in rats. J Mol Neurosci. 2018;66(2):261–272. doi:10.1007/s12031-018-1166-3
6. Barfejani AH, Jafarvand M, Seyedsaadat SM, Rasekhi RT. Donepezil in the treatment of ischemic stroke: review and future perspective. Life Sci. 2020;263:118575. doi:10.1016/j.lfs.2020.118575
7. Hebei Zhitong Pharmaceutical Holding Group Co. Ltd. Denatured protein powder and brain protein hydrolysate prepared by this protein powder:CN201210235771.X[P]. 2012.
8. Zhang Y, Chopp M, Zhang ZG, et al. Cerebrolysin reduces astrogliosis and axonal injury and enhances neurogenesis in rats after closed head injury. Neurorehabil Neural Repair. 2019;33(1):15–26. doi:10.1177/1545968318809916
9. Jing XL, Hou YB, Ren LM, et al. Protective effects of Cerebroprotein Hydrolysate for injection (I) on vascular dementia in rats. Drug Eval Res. 2017;40(7):922–925. doi:10.7501/j.issn.1674-6376.2017.07.007
10. Ladurner G, Kalvach P, Moessler H, Cerebrolysin Study Group. Neuroprotective treatment with Cerebrolysin in patients with acute stroke: a randomised controlled trial. J Neural Transm (Vienna). 2005;112(3):415–428. doi:10.1007/s00702-004-0248-2.
11. Cao W, Zhang C, Chen R, et al. A novel cerebroprotein hydrolysate, CH1, ameliorates chronic focal cerebral ischemia injury by promoting white matter integrity via the Shh/Ptch-1/Gli-1 signaling pathway. Neuropsychiatr Dis Treat. 2020;16:3209–3224. doi:10.2147/NDT.S289990
12. Bu F, Min JW, Munshi Y, et al. Activation of endothelial ras-related C3 botulinum toxin substrate 1 (Rac1) improves post-stroke recovery and angiogenesis via activating Pak1 in mice. Exp Neurol. 2019;322:113059. doi:10.1016/j.expneurol.2019.113059
13. Ni GX, Liang C, Wang J, Duan CQ, Wang P, Wang YL. Astragaloside IV improves neurobehavior and promotes hippocampal neurogenesis in MCAO rats though BDNF-TrkB signaling pathway. Biomed Pharmacother. 2020;130:110353. doi:10.1016/j.biopha.2020.110353
14. von Bohlen Und Halbach O, von Bohlen Und Halbach V. BDNF effects on dendritic spine morphology and hippocampal function. Cell Tissue Res. 2018;373(3):729–741. doi:10.1007/s00441-017-2782-x
15. Kowiański P, Lietzau G, Czuba E, Waśkow M, Steliga A, Moryś J. BDNF:A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol. 2018;38(3):579–593. doi:10.1007/s10571-017-0510-4
16. Gul-Kahraman K, Yilmaz-Bozoglan M, Sahna E. Physiological and pharmacological effects of melatonin on remote ischemic perconditioning after myocardial ischemia-reperfusion injury in rats: role of Cybb, Fas, NfκB, Irisin signaling pathway. J Pineal Res. 2019;67(2):e12589. doi:10.1111/jpi.12589
17. Han D, Wang Y, Chen J, et al. Activation of melatonin receptor 2 but not melatonin receptor 1 mediates melatonin-conferred cardioprotection against myocardial ischemia/reperfusion injury. J Pineal Res. 2019;67(1):e12571. doi:10.1111/jpi.12571
18. Yu LM, Dong X, Xue XD, et al. Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: role of SIRT6. J Pineal Res. 2021;70(1):e12698. doi:10.1111/jpi.12698
19. Sun J, Nan G. The mitogen-activated protein kinase (MAPK) signaling pathway as a discovery target in stroke. J Mol Neurosci. 2016;59(1):90–98. doi:10.1007/s12031-016-0717-8
20. Burguete MC, Jover-Mengual T, López-Morales MA, et al. The selective oestrogen receptor modulator, bazedoxifene, mimics the neuroprotective effect of 17β-oestradiol in diabetic ischaemic stroke by modulating oestrogen receptor expression and the MAPK/ERK1/2 signalling pathway. J Neuroendocrinol. 2019;31(8):e12751. doi:10.1111/jne.12751
21. National Research Council. Guide for the Care and Use of Laboratory Animals: Eighth Edition. Washington, DC: The National Academies Press; 2011. doi:10.17226/12910
22. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi:10.1161/01.str.20.1.84
23. Chen J, Sanberg PR, Li Y, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32(11):2682–2688. doi:10.1161/hs1101.098367
24. Muresanu DF, Florian S, Hömberg V, et al. Efficacy and safety of cerebrolysin in neurorecovery after moderate-severe traumatic brain injury: results from the CAPTAIN II trial. Neurol Sci. 2020;41(5):1171–1181. doi:10.1007/s10072-019-04181-y
25. Yang J, Yan H, Li S, Zhang M. Berberine Ameliorates MCAO induced cerebral ischemia/reperfusion injury via activation of the BDNF-TrkB-PI3K/Akt signaling pathway. Neurochem Res. 2018;43(3):702–710. doi:10.1007/s11064-018-2472-4
26. Clarkson AN, Parker K, Nilsson M, Walker FR, Gowing EK. Combined ampakine and BDNF treatments enhance poststroke functional recovery in aged mice via AKT-CREB signaling. J Cereb Blood Flow Metab. 2015;35(8):1272–1279. doi:10.1038/jcbfm
27. Zhang D, Han S, Wang S, Luo Y, Zhao L, Li J. cPKCγ-mediated down-regulation of UCHL1 alleviates ischaemic neuronal injuries by decreasing autophagy via ERK-mTOR pathway. J Cell Mol Med. 2017;21(12):3641–3657. doi:10.1111/jcmm.13275
28. Zhai X, Chen X, Shi J, et al. Lactulose ameliorates cerebral ischemia-reperfusion injury in rats by inducing hydrogen by activating Nrf2 expression. Free Radical Biol Med. 2013;65(6):731–741. doi:10.1016/j.freeradbiomed
29. Wang T, Zhai L, Zhang H, Zhao L, Guo Y. Picroside II inhibits the MEK-ERK1/2-COX2 signal pathway to prevent cerebral ischemic injury in rats. J Mol Neurosci. 2015;57(8):335–351. doi:10.1007/s12031-015-0623-5
30. Wang ZQ, Wu DC, Huang FP, Yang GY. Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res. 2004;996(1):55–66. doi:10.1016/j.brainres
31. Chen M, Cecon E, Karamitri A, et al. Melatonin MT1 and MT2 receptor ERK signaling is differentially dependent on Gi/o and Gq/11 proteins. J Pineal Res. 2020;68(4):e12641. doi:10.1111/jpi.12641
32. Kovalska M, Kovalska L, Pavlikova M, et al. Intracellular signaling MAPK pathway after cerebral ischemia reperfusion injury. Neurochem Res. 2012;37(7):1568–1577. doi:10.1007/s11064-012-0752-y
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.