Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
The association of neuron-derived orphan receptor 1 with pulmonary vascular remodeling in COPD patients
Authors Wang CG, Li C, Lei W ![]() , Jiang JH, Huang JA
, Jiang JH, Huang JA ![]() , Zeng DX
, Zeng DX ![]()
Received 16 September 2017
Accepted for publication 11 December 2017
Published 13 April 2018 Volume 2018:13 Pages 1177—1186
DOI https://doi.org/10.2147/COPD.S151820
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Chang-Guo Wang,1,* Chang Li,2,* Wei Lei,1 Jun-Hong Jiang,1 Jian-An Huang,1 Da-Xiong Zeng1
1Department of Respiratory and Critical Care Medicine, First Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China; 2Department of Thoracic Surgery, First Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China
*These authors contributed equally to this work
Introduction: Chronic hypoxia-induced pulmonary vascular remodeling is a feature of chronic obstructive pulmonary disease (COPD). Our previous reports indicate that neuron-derived orphan receptor 1 (NOR1) promoted pulmonary smooth muscle cell proliferation in vitro. But it remains unclear whether NOR1 participated into hypoxia-induced pulmonary vascular remodeling in COPD patients.
Patients and methods: For this study, we collected peripheral lung tissues of 26 male COPD patients with or without hypoxemia. We detected the pulmonary vascular remodeling in all the peripheral lung tissues. Primary human pulmonary arterial smooth muscle cells were also cultured in vitro and stimulated with hypoxia or normoxia. Cell proliferation and protein levels were detected.
Results: COPD patients with hypoxemia showed significantly enlarged pulmonary vessels wall thickness and increased protein levels of HIF-1α, smooth muscle actin, cyclin D1, and NOR1 when compared with those in normoxic patients. Moreover, hypoxia induced human pulmonary arterial smooth muscle cell proliferation and NOR1 overexpression in vitro. The plasmid-based NOR1 gene overexpression markedly promoted DNA synthesis and proliferation in hypoxia or normoxic cells. Human NOR1 gene-specific siRNA intensively suppressed DNA synthesis and proliferation. Transfection of NOR1 overexpression plasmid raised cyclin D1 protein levels, which could be significant inhibited by NOR1-specific siRNA or a CDK4/6 inhibitor PD0332991.
Conclusion: We concluded that NOR1 upregulation is associated with hypoxia-induced pulmonary vascular remodeling in COPD via promoting human pulmonary arterial smooth muscle cell proliferation.
Keywords: pulmonary arterial smooth muscle cells, neuron-derived orphan receptor 1, cyclin D1, hypoxia, pulmonary vascular remodeling
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive disease characterized by chronic airway inflammation and lung parenchyma destruction. As a common complication of COPD, pulmonary hypertension eventually leads to right ventricular hypertrophy and cardiac failure, which increases the risk of exacerbation and decreases survival.1 Vascular remodeling and hypoxic vasoconstriction in pulmonary small arteries were thought to be an important pathological basis of pulmonary hypertension in COPD patients.2 Pulmonary vascular remodeling is considered to result from chronic hypoxia, persistent inflammation, and loss of capillaries.1–3 Although many studies in patients or animal models have demonstrated the critical role of multiple growth factors or pathways (such as prostacyclin, nitric oxide, and endothelin) in pulmonary vascular remodeling, knowledge of the precise mechanism underlying this process in COPD remains limited.4–8
As a subfamily of the nuclear receptor superfamily, NR4A contains three kinds of mammalian nuclear receptors: NR4A1 (Nurr77), NR4A2 (Nurr1), and NR4A3 (neuron-derived orphan receptor 1[NOR1]).9 Acting as a kind of early-response gene, NOR1 could respond rapidly to various extracellular stimuli.10 NOR1 has been proven to promote nervous system development and metabolism.10,11 Recent reports demonstrated that NOR1 activation also played an important role in vascular diseases, such as atherosclerosis and vascular neointima formation after injury.12,13 However, it remained unclear whether NOR1 activation was involved in pulmonary vascular remodeling of COPD.
Some previous reports demonstrated that NOR1 stimulated smooth muscle cell proliferation, endothelial cells survival, cells apoptosis, and adherence.14–19 By regulating downstream target gene transcription, NOR1 exerted distinct effects on the aforementioned cellular processes. As a key checkpoint of cell cycle, cyclin D1 has been proven to be a target of NOR1.12,15 Although our previous studies indicated the critical role of cyclin D1 in PASMC proliferation and pulmonary remodeling,20,21 we cannot confirm whether cyclin D1 was a downstream target gene of NOR1 in hypoxia-induced pulmonary vascular remodeling or PASMC proliferation.
So, in this study, we focused on the role of NOR1 upregulation in hypoxia-induced PASMC proliferation and pulmonary vascular remodeling in COPD patients.
Patients and methods
Reagents
Rabbit polyclonal antibody against GAPDH (sc-367714) human cyclin D1 (sc-718) or smooth muscle myosin heavy chain 11 (ab53219) and mouse monocloal antibody against smooth muscle actin (sc-53142) or BrdU (sc-32323) were all purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Rabbit polyclonal antibody against human NOR1 was purchased from Abcam (ab188752; Cambridge, MA, USA). Lipofactimine 200, TaqMan gene expression assays, and Trizol were purchased from Invitrogen Biotechnology, Inc. (Waltham, MA, USA). The specific siRNA targeted for human NOR1 gene (NOR1-siRNA) and negative-control siRNA (NC-siRNA) were synthesized by Shanghai Bio-Engineering Co. Ltd (Shanghai, People’s Republic of China).
Subjects
Human lung samples were obtained from 26 male patients who underwent pneumonectomy for lung volume reduction or lung carcinoma from January 2012 to December 2013 in our hospital. Lung tissue samples were obtained from normal appearing areas of the pulmonary parenchyma as far as possible from the tumor (at least 2 cm). Pulmonary function tests were performed before surgery in all patients. The arterial blood gas analysis was checked at least twice (at admission and before operation) with a blood gas analyzer (ABL-90; Radiometer Medical Aps, Copenhagen, Denmark). This study was approved by the research ethics committee of the First Affiliated Hospital of Soochow University, and written informed consents were obtained from all subjects.
Tissue preparation and morphometric analysis
Peripheral lung samples were treated with 4% (w/v) paraformaldehyde before embedding in paraffin. Samples were cut into multiple 5-μm-thick sections and then stained with hematoxylin and eosin (HE) for the morphological analysis under an optical microscope. The measurement was limited to medium and small arteries (≤500 μm diameter) with complete circumferential smooth muscle layer. As described previously,21 the vessel wall thickness was expressed as a percentage of the external diameter [(external diameter - internal diameter)/external diameter × 100%].
Immunohistochemistry
Sections were deparaffinized, rehydrated, and then soaked for antigen retrieval with citrate buffer. Primary antibody was added and incubated at 4°C overnight. Mouse monocloal antibody against α-smooth muscle actin (α-SMA, 1:500) and smooth muscle myosin heavy chain 11 (ab53219) were used as primary antibodies. Relative IgG was used as negative control instead of primary antibody. Then, the sections were incubated with secondary antibody with a commercial SP kit (Zhongshan Golden Bridge Biotech Co., Beijing, People’s Republic of China). Immunoreactivity was visualized with diaminobenzidine.
Cell cultures
As our previous report described,20 primary human PASMCs were separated from control subject and cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM). The distal pulmonary arteries were separated from the lung tissue of control subject and incubated in Hanks’ solution containing collagenase (1.5 mg/mL) for 15 min. After the adventitia layer and endothelium were carefully removed, the smooth muscle explants were digested with elastase (0.5 mg/mL) and collagenase (2.0 mg/mL) for 40 min at 37°C. The smooth muscle explants and digestive liquid were centrifuged at 1,000 rpm. The supernatants were removed and the sediments were washed twice with DMEM. All the sediments were diluted with DMEM and seeded into a 25–50 mL bottle. Cells were grown in DMEM containing 10% fetal bovine serum (FBS), streptomycin (100 mg/mL), and penicillin (100 U/mL) in a humidified incubator at 37°C (5% CO2, 21% O2). Passage 3–10 cells were used for all experiments. Before different treatment, cells were starved in serum-free media for 24 h.
Cell treatment with siRNA transfection or CDK4/6 inhibitor
For the transfection of human NOR1-specific siRNA (NOR1-siRNA), cells were grown to 70% confluence. The siRNA was mixed with Lipofectine 2000 (Invitrogen) according to the manufacturer’s instructions and then added to human PASMCs. Efficiency of transfection was assessed by monitoring the uptake of siRNA labeled with 6-carboxyfluorescein. Six hours after siRNA transfection, cells were cultured in hypoxia or normoxia with DMEM containing 2% FBS and then used for subsequent experiments. Hypoxia was achieved using a chamber equilibrated with a water-saturated gas mixture of 5% O2, 5% CO2, and 90% N2 at 37°C. The specific siRNA sequences targeting human NOR1 gene have been described as previous report:21 sense 5′-GCGUACAGAUAGUCUGAAATT-3′ and antisense 5′UUUCAGACUAUCUGUACGCTT3′. For preventing the function of cyclin D1, a CDK4/6-specific inhibitor PD0332991 was used as a blocker of cyclin D1-CDK4/6 complex. Cells were treated with PD0332991 (1 μmol/L) for 24 h and then cultured in hypoxia or normoxia with DMEM containing 2% FBS before subsequent experiments.
Western blot
After different treatments, whole protein was extracted from human PASMCs. The protein content was determined using Bradford reagent. A total of 50 μg protein was subjected to SDS-PAGE. NOR1 or cyclin D1 proteins were detected by immunobloting with antibodies against human NOR1 (1:200; Abcam [ab188752]) or cyclin D1 (1:300; Santa Cruz). The horseradish peroxidase-conjugated goat anti-rabbit IgG was used as the secondary antibody (1:1,000; Santa Cruz). Immunoreactivity was detected using an enhanced chemiluminescence kit according to the manufacturer’s instructions. The values of optical density of the visualized blots were analyzed with a gel imaging analysis system. GAPDH was used as control. The values of the relative density of target protein brand were normalized to the relative density of GAPDH.
Cell count and BrdU incorporation assay
Cells were plated into 6-well plates at a concentration of 4×104 cells/well. After treatment, cells were collected by trypsin digestion and total cell numbers were calculated with a hemocytometer (Cellometer Auto T4; Nexcelom Bioscience, Lawrence, MA, USA) after trypan blue exclusion.
For BrdU incorporation assays, cells were seeded into 24-well plates at a density of 1×104 cells/well. After different treatments, cells were incubated with 50 mM BrdU (Sigma-Aldrich, St Louis, MO, USA) for 4 h at 37°C and then fixed with cold acetone. Cells were permeabilized with 0.5% Triton X-100 and subsequently incubated with primary antibody (mouse anti-BrdU, 1:200; SantaCruz) and secondary antibody (goat anti-IgG, 1:1,000; SantaCruz) for 1 h at 37°C. Nuclei were stained using DAB. PBS instead of the primary antibodies was used as negative control. BrdU incorporation was expressed as the percentage of BrdU-positive cells.
Statistical analysis
All data are expressed as mean ± standard deviation and analyzed by t-test or one-way analysis of variance and followed by q-test using the computer software SPSS version 12.0 (SPSS Inc., Chicago, IL, USA). For all tests, P<0.05 was considered statistically significant.
Results
Clinical data
The baseline characteristics of all subjects are shown in Table 1. All subjects were male. There were no differences between the two groups with regard to age, body mass index, and smoking index. As expected from the selected criteria, the partial pressure of arterial oxygen of COPD patients with hypoxemia was markedly lower compared with those with normoxia (68.5±9.4 mmHg vs 86.3±11.6 mmHg, P<0.05). Although the forced expiratory volume in 1 second/fixed vital capacity ratio was similar, the forced expiratory volume in 1 second (% predicted) in hypoxemia patients was significantly lower than in normoxic patients (43.3±6.9 vs 75.7%±10.4%, P<0.05).
 | Table 1 Characteristics of recruited COPD patients |
Pulmonary vascular remodeling and NOR1 overexpression in COPD patients with hypoxemia
The lung tissue that was HE stained in each patient was focused on intermediate vessels (50–500 μm) for evaluating pulmonary vascular remodeling. As expected, the pulmonary vascular wall thickness and muscularized vessels ratio in COPD patient with hypoxemia were both significantly higher than in those with COPD with normoxia (Figure 1A, C, and D). Similar to this, the α-SMA protein expression in lung tissue of the hypoxemia patient was markedly higher than in normoxic patients (Figure 1B and E). Furthermore, we detected HIF-1α, NOR1, and cyclin D1 protein expression in explants of pulmonary artery of all subjects. As shown in Figure 1E and F, HIF-1α, NOR1, and cyclin D1 protein levels in explants of pulmonary artery in hypoxemia patients were higher than in normoxic patients. These results demonstrate the vascular remodeling and NOR1 upregulation in pulmonary vessels of COPD patients with hypoxemia.
 | Figure 1 Hypoxemia induced NOR1 upregulation and pulmonary vascular remodeling. |
Hypoxia induced cell proliferation and NOR1 overexpression
To explore the role of hypoxia in PASMC proliferation in COPD patients, we separated human PASMCs (Figure 2A) and stimulated it under hypoxia. As shown in Figure 2B, hypoxia (5% O2) stimulated cell proliferation in a time-dependent manner. Hypoxia also upregulated HIF-1α and NOR1 protein expression significantly (Figure 2C and D). Furthermore, hypoxia significantly promoted DNA synthesis (evaluated by BrdU incorporation) when compared with normoxia (Figure 2E and F). These results showed that hypoxia not only induced NOR1 overexpression but also enhanced cell proliferation and DNA synthesis.
 | Figure 2 Hypoxia induced NOR1 upregulation and human PASMC proliferation. |
Specific siRNA prevented hypoxia-induced NOR1 overexpression and cell proliferation
A specific siRNA of human NOR1 gene was used for the loss-of-function analysis. Although hypoxia (5% O2, 48 h) induced NOR1 upregulation in human PASMCs, NOR1-specific siRNA significantly suppressed hypoxia-induced NOR1 upregulation by about 70% (Figure 3A and B). But NC-siRNA did not affect NOR1 protein levels in cells subjected to hypoxia.
 | Figure 3 NOR1-specific siRNA inhibited proliferation in hypoxic cells. |
Additionally, hypoxia induced higher BrdU incorporation ratio and cell count compared with normoxia (Figure 3C–E). However, NOR1-specific siRNA markedly reduced BrdU incorporation and cell count in hypoxic cells (Figure 3C–E). NC-siRNA did not affect BrdU incorporation or cell count in hypoxic cells. These results showed that a specific siRNA not only successfully knocked down NOR1 protein expression but also suppressed DNA synthesis and proliferation in hypoxic cells.
Exodus NOR1 overexpression promoted hypoxia-induced cell proliferation and DNA synthesis
For gain-of-function analysis, we constructed a NOR1 overexpression plasmid and transfected human PASMCs. The transfection of this plasmid significantly elevated NOR1 protein levels not only in normoxic cells but also in hypoxic cells (Figure 4A and B). The cell count and BrdU incorporation in NOR1-overexpressed cells were both significantly higher than that of controls before and after hypoxia stimulation (Figure 4C–F). These results showed that exodus NOR1 overexpression promoted proliferation and DNA synthesis in both normoxic and hypoxic cells.
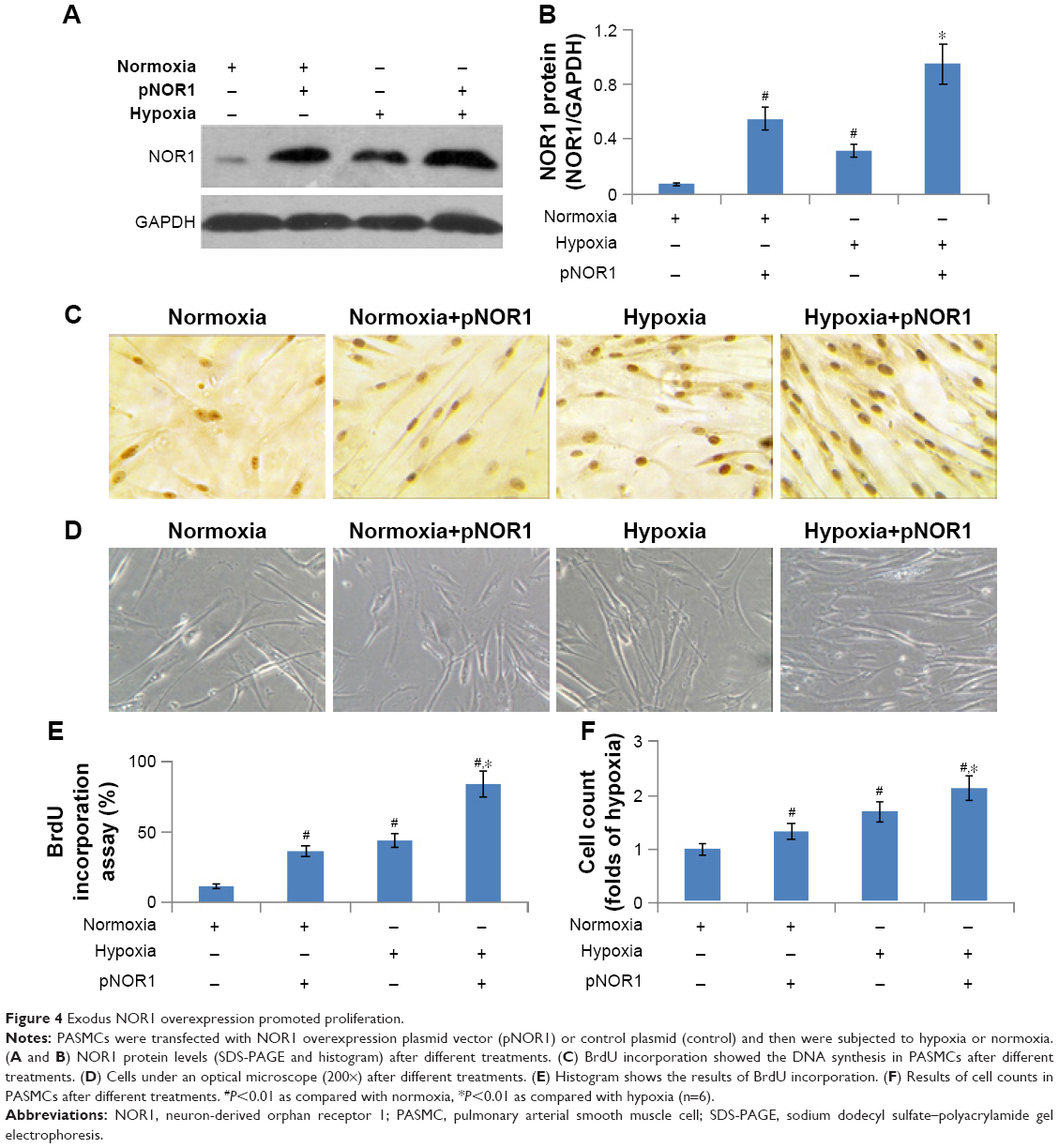 | Figure 4 Exodus NOR1 overexpression promoted proliferation. |
Cyclin D1 as target of NOR1 in hypoxia-induced cell proliferation
Cyclin D1, as a target gene of NOR1, has been demonstrated to promote cell proliferation. Based on this, we tried to uncover whether NOR1 regulated cyclin D1 expression in human PASMCs. As shown in Figure 5A and B, exodus NOR1 overexpression significantly raised cyclin D1 protein levels. Although hypoxia upregulated cylin D1 protein levels in human PASMCs, NOR1-specific siRNA significantly suppressed cyclin D1 protein expression by about 58% in hypoxic cells as compared with NC-siRNA (Figure 5C and D).
 | Figure 5 NOR1 promoted proliferation via cyclin D1. |
In addition, we used CDK4/6-specific inhibitor PD0332991 as a blocker for cyclin D1 function. Although PD0332991 did not inhibit cyclin D1 protein expression, it still markedly lessened BrdU incorporation and cell count in hypoxic cells (Figure 5E–G). These results showed that CDK4/6-specific inhibitor PD0332991 could successfully prevent hypoxia-induced proliferation and DNA synthesis. Taken together, cyclin D1 might be a target gene of NOR1 to enhance proliferation in hypoxic PASMCs.
Discussion
Our study demonstrated the upregulation of NOR1 in pulmonary vascular remodeling in COPD patients with hypoxemia. Moreover, cyclin D1 might be the target of NOR1 in promoting proliferation. This was similar to recent reports which demonstrated that NOR1 also plays an important role in cardiovascular disease, such as neointima formation after vascular injury and atherosclerosis.12,13 Taken together, NOR1 might participate not only in systemic vascular diseases but also in pulmonary vascular diseases. However, it remains unclear whether the molecular mechanism underlying pulmonary vascular remodeling was similar to that in systemic vascular diseases.
As a transcriptional regulator, NOR1 is involved in cells proliferation, apoptosis, differentiation, and survival.13–16 NOR1 has been implicated in promoting systemic vascular SMC proliferation.11–16 But the role of NOR1 in pulmonary vascular diseases remains unknown. In this study, both hypoxia and exodus NOR1 overexpression enhanced human PASMC proliferation via NOR1 pathway. These revealed the role of NOR1 in augmenting PASMC proliferation, which was similar to previous reports in systemic vascular SMCs.12,14,17,18 So, we conclude that NOR1 induced SMC proliferation not only in systemic vessels but also in pulmonary vessels.
NOR1 could be activated rapidly in response to various extracellular stimuli, including serum, platelet-derived growth factor (PDGF), thrombin, insulin-like growth factor (IGF), and epidermal growth factor (EGF).10,11,14–16 Our previous study demonstrated that FBS induced NOR1 upregulation in human PASMCs in vitro.22 In this study, NOR1 could be upregulated in hypoxic conditions in human PASMCs. Although previous studies revealed that hypoxia stimulated NOR1 in survival response of endothelial cells,15 there was no report demonstrating the regulation of hypoxia in NOR1 in smooth muscle cells. This study was the first report showing hypoxia-stimulated NOR1 upregulation in human PASMCs. Moreover, this regulation induced proliferation and inhibited apoptosis. This mechanism might be involved in pulmonary vascular remodeling in COPD. Those results revealed the different role of NOR1 as a transcriptional regulator in different cells or tissue, even under the same stimuli.
NOR1 plays a role in different cellular processes via regulating the transcription of downstream target genes.9,10,15,19,23 Our previous study indicated that cyclin D1 might be a potential downstream target gene of NOR1 in serum-stimulated PASMCs.23 In this study, cyclin D1 was upregulated by exodus NOR1 overexpression and downregulated by NOR1-specific siRNA in PASMCs. This showed that NOR1 might upregulate cyclin D1 expression in hypoxic PASMCs. The result was similar to our previous study in serum-stimulated PASMCs and previous reports in endothelial cells.15,19,23 In response to mitogenic signals, NOR1 could bind as monomers to the nerve growth factor-induced clone B response element (NBRE) site in the promoter of target genes in SMC or endothelial cells. In this way, NOR1 might also augment cyclin D1 gene expression in hypoxic or serum-stimulated PASMCs.
Although NOR1-specific siRNA or cyclin D1-CDK4/6-specific inhibitor significantly prevented hypoxia-induced PASMC proliferation, neither of them suppressed cell proliferation completely. This might be due to two reasons. First, a specific siRNA was designed that binds to target RNA and does not integrate into the genome. So, siRNA only showed a transient effect on gene expression and did not suppress target genes completely or persistently. Second, some other genes or transcriptional factors besides NOR1 might be also involved in hypoxia-induced PASMC proliferation. Furthermore, cyclin D1 was not the only target gene of NOR1 in this process. Other genes might be also regulated by NOR1 in PASMCs, such as S-phase kinase-associated protein 2.24 Thus, more studies are needed to explore the mechanism of NOR1 in hypoxia-induced PASMC proliferation and pulmonary vascular remodeling.
There are some weaknesses in our study. First, we only examined NOR1 in pulmonary vascular remodeling in vitro. Further research in hypoxic animal models might benefit in uncovering the precise mechanism underlying hypoxia-induced pulmonary hypertension. Second, we used RNA interference technique (siRNA) for NOR1 gene knockdown in cells. The NOR1 gene knockout transgenic animals would be more helpful to reinforce our conclusion.
Conclusion
In conclusion, our study indicated that NOR1 upregulation was associated with hypoxia-induced pulmonary vascular remodeling in COPD patients. Cyclin D1 was a downstream target of NOR1 that regulates proliferation in hypoxic PASMCs. This pathway might play a role in hypoxia-induced pulmonary vascular remodeling in COPD. But the precise molecular mechanism underlying this process remains unclear. Further in vivo study is needed before NOR1 can become a potential target of pulmonary vascular remodeling therapy in COPD.
Acknowledgment
This study was supported by the Suzhou Municipal Application Basic Research Project (SYS201530), Program of Key Talents of Medical Science in Jiangsu Province (QNRC2016745), the Clinical Key Speciality Project of China, the Clinical Medicine Center of Suzhou (number Szzx201502), and the Suzhou Key Laboratory for Respiratory Medicine (number SZS201617).
Disclosure
The authors report no conflicts of interest in this work.
References
Wrobel JP, Thompson BR, Williams TJ. Mechanisms of pulmonary hypertension in chronic obstructive pulmonary disease: a pathophysiologic review. J Heart Lung Transplant. 2012;31(6):557–564. | ||
Sakao S, Voelkel NF, Tatsumi K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. Eur Respir Rev. 2014;23(133):350–355. | ||
Rowan SC, Keane MP, Gaine S, McLoughlin P. Hypoxic pulmonary hypertension in chronic lung diseases: novel vasoconstrictor pathways. Lancet Respir Med. 2016;4(3):225–236. | ||
Morrell NW, Adnot S, Archer SL, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(Suppl 1):S20–S31. | ||
Wilkins MR, Wharton J, Gladwin MT. Update in pulmonary vascular diseases. Am J Respir Crit Care Med. 2013;188(1):23–28. | ||
Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122(12):4306–4313. | ||
Andersen CU, Mellemkjær S, Nielsen-Kudsk JE, Bendstrup E, Hilberg O, Simonsen U. Pulmonary hypertension in chronic obstructive and interstitial lung diseases. Int J Cardiol. 2013;168(3):1795–1804. | ||
Seeger W, Adir Y, Barberà JA, et al. Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol. 2013;62(Suppl 25):D109–D116. | ||
Pearen MA, Muscat GE. Minireview: nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol. 2010;24(10):1891–1903. | ||
Zhao Y, Bruemmer D. NR4A orphan nuclear receptors: transcriptional regulators of gene expression in metabolism and vascular biology. Arterioscler Thromb Vasc Biol. 2010;30(8):1535–1541. | ||
Martínez-González J, Badimon L. The NR4A subfamily of nuclear receptors: new early genes regulated by growth factors in vascular cells. Cardiovasc Res. 2005;65(3):609–618. | ||
Nomiyama T, Zhao Y, Gizard F, et al. Deficiency of the NR4A neuron-derived orphan receptor-1 attenuates neointima formation after vascular injury. Circulation. 2009;119(4):577–586. | ||
Zhao Y, Howatt DA, Gizard F, et al. Deficiency of the NR4A orphan nuclear receptor NOR1 decreases monocyte adhesion and atherosclerosis. Circ Res. 2010;107(4):501–511. | ||
Nomiyama T, Nakamachi T, Gizard F, et al. The NR4A orphan nuclear receptor NOR1 is induced by platelet-derived growth factor and mediates vascular smooth muscle cell proliferation. J Biol Chem. 2006;281(44):33467–33476. | ||
Martorell L, Gentile M, Rius J, et al. The hypoxia-inducible factor 1/NOR-1 axis regulates the survival response of endothelial cells to hypoxia. Mol Cell Biol. 2009;29(21):5828–5842. | ||
Medunjanin S, Daniel JM, Weinert S, et al. DNA-dependent protein kinase (DNA-PK) permits vascular smooth muscle cell proliferation through phosphorylation of the orphan nuclear receptor NOR1. Cardiovasc Res. 2015;106(3):488–497. | ||
Martínez-González J, Rius J, Castelló A, Cases-Langhoff C, Badimon L. Neuron-derived orphan receptor-1 (NOR-1) modulates vascular smooth muscle cell proliferation. Circ Res. 2003;92(1):96–103. | ||
Rodríguez-Calvo R, Guadall A, Calvayrac O, et al. Over-expression of neuron-derived orphan receptor-1 (NOR-1) exacerbates neointimal hyperplasia after vascular injury. Hum Mol Genet. 2013;22(10):1949–1959. | ||
Alonso J, Galán M, Martí-Pàmies I, et al. NOR-1/NR4A3 regulates the cellular inhibitor of apoptosis 2 (cIAP2) in vascular cells: role in the survival response to hypoxic stress. Sci Rep. 2016;6:34056. | ||
Zeng DX, Xu YJ, Liu XS, Wang R, Xiang M. Cigarette smoke extract induced rat pulmonary artery smooth muscle cells proliferation via PKCα-mediated cyclin D1 expression. J Cell Biochem. 2011;112(8):2082–2088. | ||
Zeng DX, Xu GP, Lei W, Wang R, Wang CG, Huang JA. Suppression of cyclin D1 by plasmid-based short hairpin RNA ameliorated experimental pulmonary vascular remodeling. Microvasc Res. 2013;90:144–149. | ||
Feng XJ, Gao H, Gao S, et al. The orphan receptor NOR1 participates in isoprenaline-induced cardiac hypertrophy by regulating PARP-1. Br J Pharmacol. 2015;172(11):2852–2863. | ||
Wang CG, Lei W, Li C, Zeng DX, Huang JA. Neuron-derived orphan receptor 1 promoted human pulmonary artery smooth muscle cells proliferation. Exp Lung Res. 2015;41(4):208–215. | ||
Gizard F, Zhao Y, Findeisen HM, et al. Transcriptional regulation of S phase kinase-associated protein 2 by NR4A orphan nuclear receptor NOR1 in vascular smooth muscle cells. J Biol Chem. 2011;286(41):35485–35493. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.