Back to Journals » Clinical Ophthalmology » Volume 9
The association between neovascular age-related macular degeneration and regulatory T cells in peripheral blood
Authors Madelung C, Krüger Falk M, Sørensen TL
Received 3 February 2015
Accepted for publication 17 March 2015
Published 25 June 2015 Volume 2015:9 Pages 1147—1154
DOI https://doi.org/10.2147/OPTH.S82116
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Scott Fraser
Christopher Fugl Madelung,1,2 Mads Krüger Falk,1,2 Torben Lykke Sørensen1,2
1Clinical Eye Research Unit, Department of Ophthalmology, Copenhagen University Hospital, Roskilde, Denmark; 2University of Copenhagen, Copenhagen, Denmark
Purpose: To investigate regulatory T cells (Tregs) and subsets of the Treg population in patients with neovascular age-related macular degeneration (AMD).
Patients and methods: Twenty-one neovascular AMD cases and 12 age-matched controls without retinal pathology were selected. Patients were recruited from our outpatient retinal clinic. Control individuals were typically spouses. The diagnosis of neovascular AMD was confirmed using fluorescein and indocyaningreen angiography. Fresh venous blood was analyzed by flow cytometry using fluorochrome-conjugated antibodies to the Treg surface antigens CD4, CD25, CD127, CD45RA, and CD31. Main outcome measures were the percentage of CD25highCD127low Tregs, the percentage of CD45RA+ naïve Tregs, and the percentage of CD31+ recent thymic emigrant Tregs.
Results: Comparing patients with neovascular AMD to controls, no significant differences were found in the percentages of CD4+ lymphocytes, CD25highCD127low Tregs, CD45RA+ naïve Tregs, or CD31+ recent thymic emigrant Tregs.
Conclusion: Our data does not indicate an altered state of systemic Treg cells in neovascular AMD.
Keywords: Tregs, macular degeneration, immunosenescence, CD45RA antigens, CD31 antigens
Introduction
Age-related macular degeneration (AMD) is the leading cause of visual impairment in aged individuals worldwide. While early stages of the disease (dry AMD) are mostly asymptomatic, late stages of the disease cause severe vision loss due to disruption of the retinal anatomy and ultimately photoreceptor degeneration. Late stages of AMD are characterized by the invasion of new choroidal blood vessels (choroidal neovascularizations) into the subretinal space causing edema and rapid vision loss – denoted neovascular AMD – or atrophy of the retinal pigment epithelium in the macula causing irreversible degeneration of photoreceptors – denoted geographic atrophy.1
The pathogenesis of AMD is not known in detail, but research strongly suggests an involvement of the immune system. The current belief is that dysregulation of the immune system leading to overactivation of inflammatory processes is involved in inducing and advancing the pathology found in AMD.2 A contribution of the innate immune responses, in particular the complement system, is well established. The Y402H polymorphism of complement factor H has been found to account for 43% of the disease heritability, and altered complement regulatory proteins have been described in the blood.3,4 Some studies indicate that systemic alterations in the B and T cells of the adaptive immune system might be involved. Patients with AMD have increased autoantibodies with affinity to components of the retina in their peripheral blood, and age-related changes in the T cell population in peripheral blood is associated with AMD.5–7 Risk factors for developing AMD include genes associated with the complement system as well as behavioral factors such as cigarette smoking, obesity, and sunlight exposure, but the major risk factor is age.3,8,9 In the immune system, the most drastic age-related change is the involution of the thymus. The age-associated decline in thymic function contributes to a general decline in the immune system of older individuals known as immunosenescence. This phenomenon is thought to lead to an increased susceptibility to infections, cancer, and autoimmune diseases.10 An important quality of the immune system is the ability to initiate an effective immune response to foreign antigens whilst tolerating the organism’s own antigens. This mechanism of self-tolerance is thought to be mediated by a lymphocyte of thymic origin called the regulatory T cell or Treg.11,12 The population of Tregs, found within the CD4+ T helper cell population, has been characterized by a high expression of CD25 (interleukin [IL]-2-receptor alpha unit) and a low expression of CD127 (IL-7-receptor alpha unit).14,15
Recent studies have found that Treg frequencies are increased in the elderly, suggesting an association between immunosenescence and Tregs.16,17 Further research into the different subsets of the Treg population has shown that although the total population of circulating Tregs is increased or at least preserved in aged individuals, the prevalence of naïve Tregs declines with age as a consequence of decreased thymic function.18,19 The population of Tregs can be divided into subsets based on different expressions of surface antigens. The CD45RA isoform of the protein tyrosine phosphatase CD45 is located on the surface of naïve T cells. This can be used to distinguish between naïve and memory subsets of Tregs. Tregs expressing high levels of CD45RA are naïve Tregs (nTregs), whereas those expressing low levels of CD45RA are memory or effector Tregs (mTregs).13 Naïve Tregs have been shown to proliferate both in and outside the thymus. CD31 (PECAM-1) can be used to identify the subset of naïve Tregs generated in the thymus, known as recent thymic emigrant Tregs (RTE-Tregs).20
We hypothesize that changes in the Tregs population due to age-related involution of the thymus contribute to the pathogenesis of neovascular AMD. Therefore, the aim of this study was to investigate the frequencies of circulating CD4+ T-cells, CD4+ CD25high CD127low Tregs, CD45RA+ naïve Tregs, and CD31+ RTE-Tregs in patients with neovascular AMD compared with healthy, age-matched controls.
Materials and methods
Study participants
During a period of 2 months, 39 individuals (patients and relatives) attending the outpatient retinal clinic at the Department of Ophthalmology, Copenhagen University Hospital, Roskilde, Denmark were asked to participate in this case–control study. We included non-smokers aged 70 years or more. For the case group, we included patients with a history of neovascular AMD. For the control group, we included patients attending the outpatient clinic, but without a history of retinal disease, and relatives, typically spouses of patients attending the outpatient clinic. The current study was approved by the Regional Committee of Ethics in Research of Region Zealand (SJ-142) and followed the tenets of the Declaration of Helsinki. Informed oral and written consent was obtained from all participants. After giving their informed consent, participants were interviewed with regard to their medical history, use of medications, alcohol use, prior smoking habits, weight, height, and physical activity. Individuals were excluded if they had a medical history with chronic inflammatory diseases such as rheumatoid arthritis or diabetes; were current smokers; were taking anti-inflammatory medications such as prednisolone; or had received intravitreal treatment with the anti-vascular endothelial growth factors ranibizumab (trade name Lucentis®; Novartis International AG, Basel, Switzerland) or aflibercept (trade name Eylea®; Bayer AG, Leverkusen, Germany) within the last 30 days. We used C-reactive protein as a marker of systemic inflammation and excluded patients with levels of 10 mg/L or more, as elevated levels probably reflect ongoing inflammatory processes other than AMD.
Visual acuity was assessed for both eyes using the method from the Early Treatment Diabetic Retinopathy Study (ETDRS),21 and weight and height was measured for participants unsure about their measurements. In order to thoroughly assess retinal pathology in the participants, we performed fundus photography (Carl Zeiss Meditec AG, Jena, Germany) and spectral domain-optical coherence tomography (SD-OCT) and fundus autofluorescence imaging (Spectralis® Heidelberg retinal angiography-OCT [HRA-OCT]; Heidelberg Engineering, Heidelberg, Germany), and graded our findings in accordance with the clinical age-related maculopathy grading system (CARMS).22 Participants were assigned to the case group if they received a CARMS grade of 5 (consistent with neovascular AMD) or to the control group if they received a CARMS grade of 1 (consistent with fewer than 10 drusen). The diagnosis of neovascular AMD was confirmed using fluorescein and indocyaningreen angiography. Participants with a CARMS grade between 2 and 4 were excluded. Sample size power calculation using a significance level of 0.05 and a power of 80% suggested that ten individuals from each group should be included.
Blood sample preparation
Venous blood samples were obtained from all participants between 8 am and 11 am. Two 4 mL tubes containing ethylenediaminetetraacetic acid were obtained for white blood cell (WBC) count and flow cytometry, and one 4 mL tube containing lithium-heparin was obtained for determining levels of C-reactive protein.
We determined the WBC count, lymphocyte count, and granulocyte count with a Sysmex® XE-5000 Hematology Analyzer (Sysmex Corporation, Kobe, Japan). A “volume” corresponding to 10×106 WBCs was transferred to a 50 mL tube containing a 10% solution of red blood cell lysis buffer (Novo Nordisk A/S, Bagsværd, Denmark) and was incubated for 10 minutes at room temperature in the dark. The WBCs were purified by centrifuge three times at 500 g for 5 minutes each time, and the pellets were transferred to a tube containing a buffer solution. A volume corresponding to 500.000 WBCs was incubated for 30 minutes at room temperature in the dark with the combination of fluorochrome-conjugated antibodies needed for flow cytometry. We also prepared tubes with a combination of fluorochrome-conjugated negative isotype controls to correct for unspecific binding. The solutions were then centrifuged and washed.
Flow cytometry
Flow cytometry was performed less than 5 hours after venipuncture. A Beckman Coulter FC500 flow cytometer (Beckman Coulter Inc, Brea, CA, USA) was used to record 100.000 events from each sample. For determining the frequency of Tregs and the various subtypes within this population, we used the following fluorochrome-conjugated antibodies: anti-CD4 phycoerythrin-Texas red conjugate IgG1 (immunoglobulin G1), clone RTF-4G (AbCam, Cambridge, MA, USA), anti-CD25 phycoerythrin-cyanine 5 conjugate IgG2a, clone B1.49.9 (Beckman Coulter), anti-CD127 phycoerythrin conjugate IgG1, clone A019D5 (BioLegend, San Diego, CA, USA), anti-CD31 fluorescein isothiocyanate conjugate IgG1, clone WM59 (BioLegend), and anti-CD45RA phycoerythrin-cyanine 7 conjugate IgG2b, clone HI100 (BioLegend). Corresponding negative isotype- and fluorochrome-matched antibodies were used to adjust for unspecific binding during data analysis (Figure 1D–E). Kaluza® software (Beckman Coulter) was used for the analysis of the flow cytometry data.
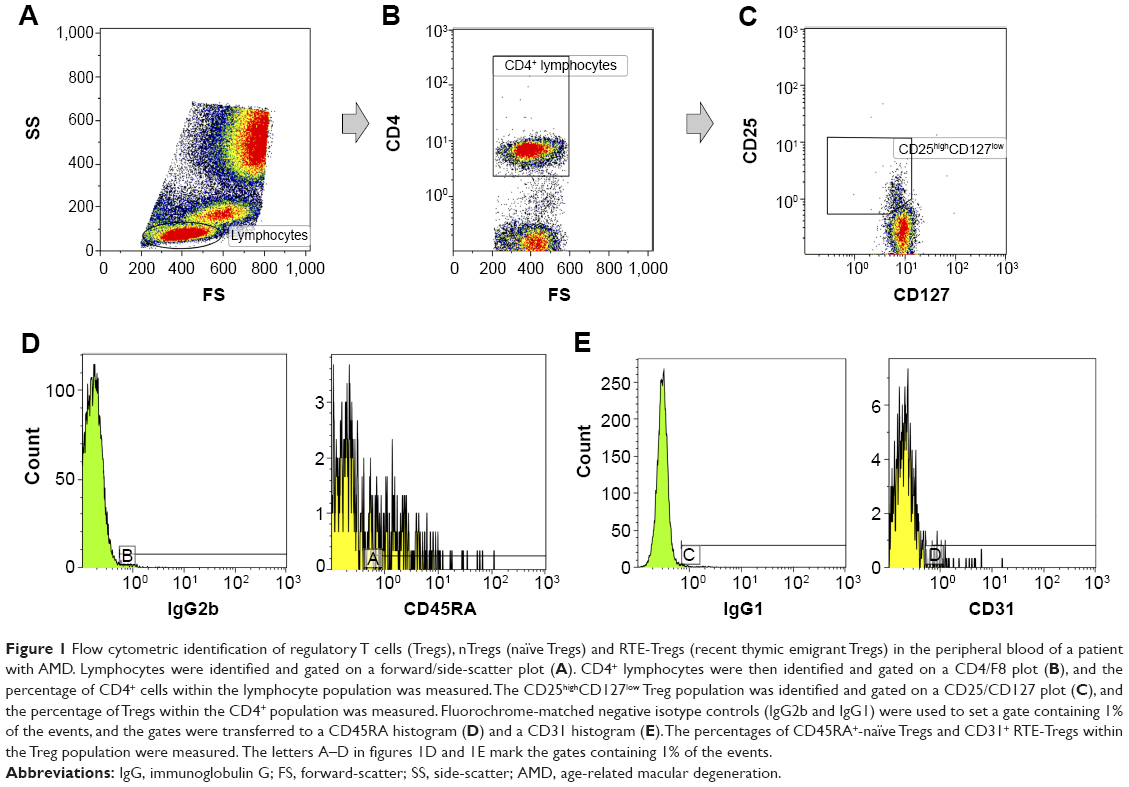 | Figure 1 Flow cytometric identification of regulatory T cells (Tregs), nTregs (naïve Tregs) and RTE-Tregs (recent thymic emigrant Tregs) in the peripheral blood of a patient with AMD. Lymphocytes were identified and gated on a forward/side-scatter plot (A). CD4+ lymphocytes were then identified and gated on a CD4/F8 plot (B), and the percentage of CD4+ cells within the lymphocyte population was measured. The CD25high CD127low Treg population was identified and gated on a CD25/CD127 plot (C), and the percentage of Tregs within the CD4+ population was measured. Fluorochrome-matched negative isotype controls (IgG2b and IgG1) were used to set a gate containing 1% of the events, and the gates were transferred to a CD45RA histogram (D) and a CD31 histogram (E). The percentages of CD45RA+-naïve Tregs and CD31+ RTE-Tregs within the Treg population were measured. The letters A–D in figures 1D and 1E mark the gates containing 1% of the events. |
A gating strategy was designed to identify the Tregs and the naïve Treg and RTE-Treg subsets (Figure 1). Events were first gated with an elliptical lymphocyte gate on the forward-scatter/side-scatter plot. Then, a CD4/forward-scatter plot was used to gate the CD4+ lymphocytes. Finally, a CD25/CD127 plot was used to identify the CD4+ CD25high CD127low Tregs. Within the population of Tregs, the percentage of the following subsets was measured: CD25high CD127low CD45RA+ nTregs and CD25high CD127low CD31+ RTE-Tregs.
Statistical analysis
Statistical analysis was performed with IBM SPSS Statistics version 19 (IBM Corporation, Armonk, NY, USA). Demographics and clinical characteristics were compared using Mann–Whitney U-test or independent samples t-test for continuous variables and Pearson’s chi-square test for categorical variables.
We compared percentages of lymphocytes in the peripheral blood in the two groups. For normally distributed data, results were reported as mean ± standard deviation (SD), and the significance was tested with an independent samples t-test. For non-normally distributed data, results were reported as median ± interquartile range, and the significance was tested with the Mann–Whitney U-test.
Results
A total of 39 individuals were asked to participate in our study. Four subjects were excluded because their C-reactive protein levels exceeded the predefined cut-off value of 10 mg/L. One control subject was excluded on the basis of a CARMS grading of 3 consistent with dry AMD. One subject with neovascular AMD was excluded on the basis of extremely low values of Tregs (0.01% CD25high CD127low Tregs in the CD4+ lymphocyte population). Characteristics of the study population are summarized in Table 1. The study population (n=33) consisted of 21 patients with neovascular AMD and 12 healthy controls. The neovascular AMD group consisted of ten male and eleven female subjects with a median age of 82 years (interquartile range 78–86 years). The healthy control group consisted of six male and six female subjects with a median age of 75 years (interquartile range 72–79 years). Despite our effort to age-match case and control subjects, case subjects were significantly older than control subjects (P=0.012, Mann–Whitney U-test). To address the age difference, we performed a correlation analysis of the age and expression of Treg surface markers. No such correlation was found when we analyzed all individuals as a single group, as individual patients, and as control individuals (data not shown). This finding suggests that age in this age span does not significantly influence Treg cell populations.
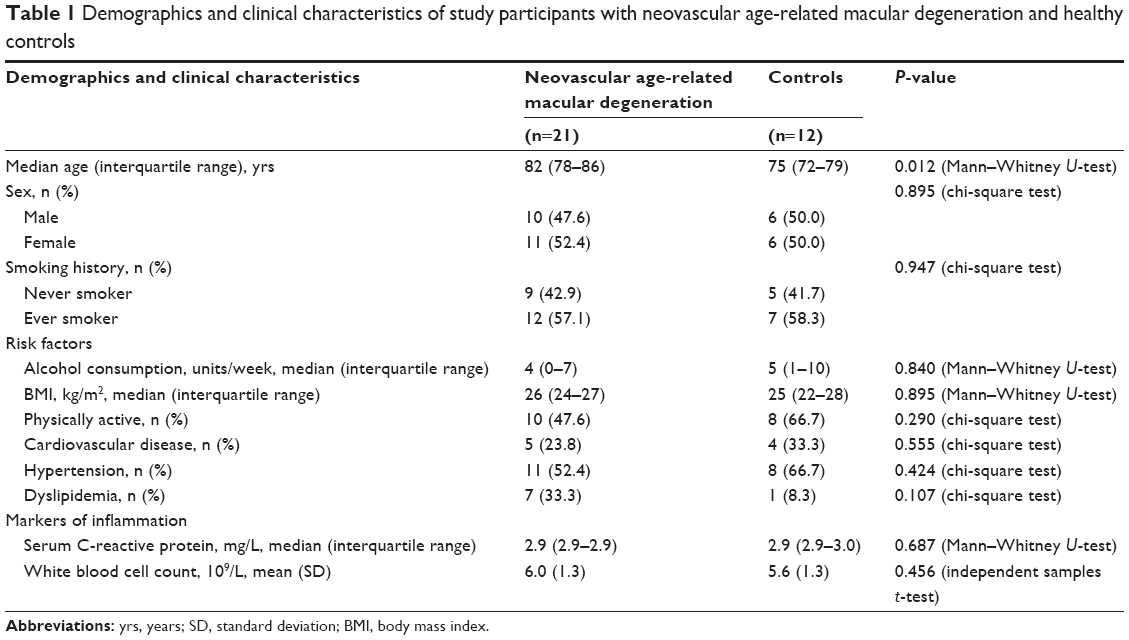 | Table 1 Demographics and clinical characteristics of study participants with neovascular age-related macular degeneration and healthy controls |
We found no significant differences in sex, prior smoking habits, alcohol consumption, body mass index, physical activity, cardiovascular disease, hypertension, or dyslipidemia. Biomarkers of inflammation – C-reactive protein level and leukocyte count, did not differ significantly between the two groups.
Percentages of CD4+ lymphocytes in the lymphocyte population and CD25high CD127low Tregs in the CD4+ population were normally distributed. We found no significant difference in the percentage of CD4+ lymphocytes in the lymphocyte population between patients with neovascular AMD (mean 43.53, SD 13.37) and healthy controls (mean 39.7, SD 8.54) (P=0.379, independent samples t-test) (Table 2, Figure 2A). No significant difference was found in the percentage of CD25high CD127low Tregs in the CD4+ population between patients with neovascular AMD (mean 5.71, SD 1.80) and healthy controls (mean 6.09, SD 1.71) (P=0.554, independent samples t-test) (Figure 2B).
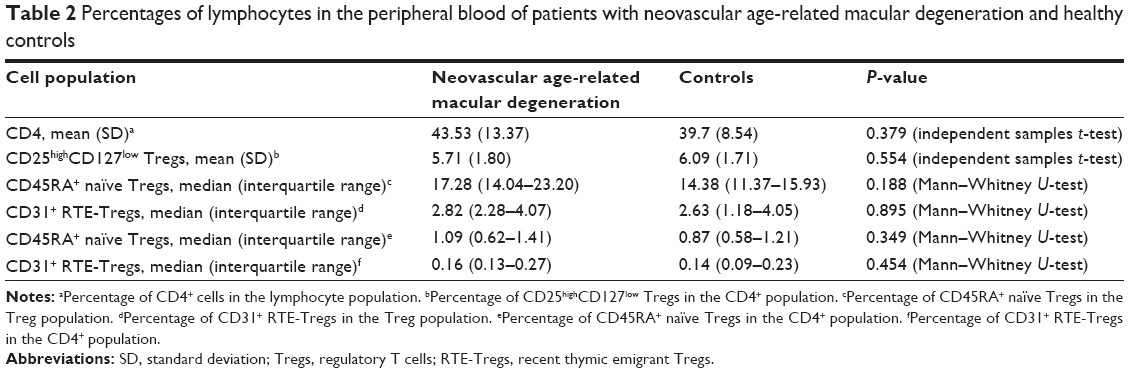 | Table 2 Percentages of lymphocytes in the peripheral blood of patients with neovascular age-related macular degeneration and healthy controls |
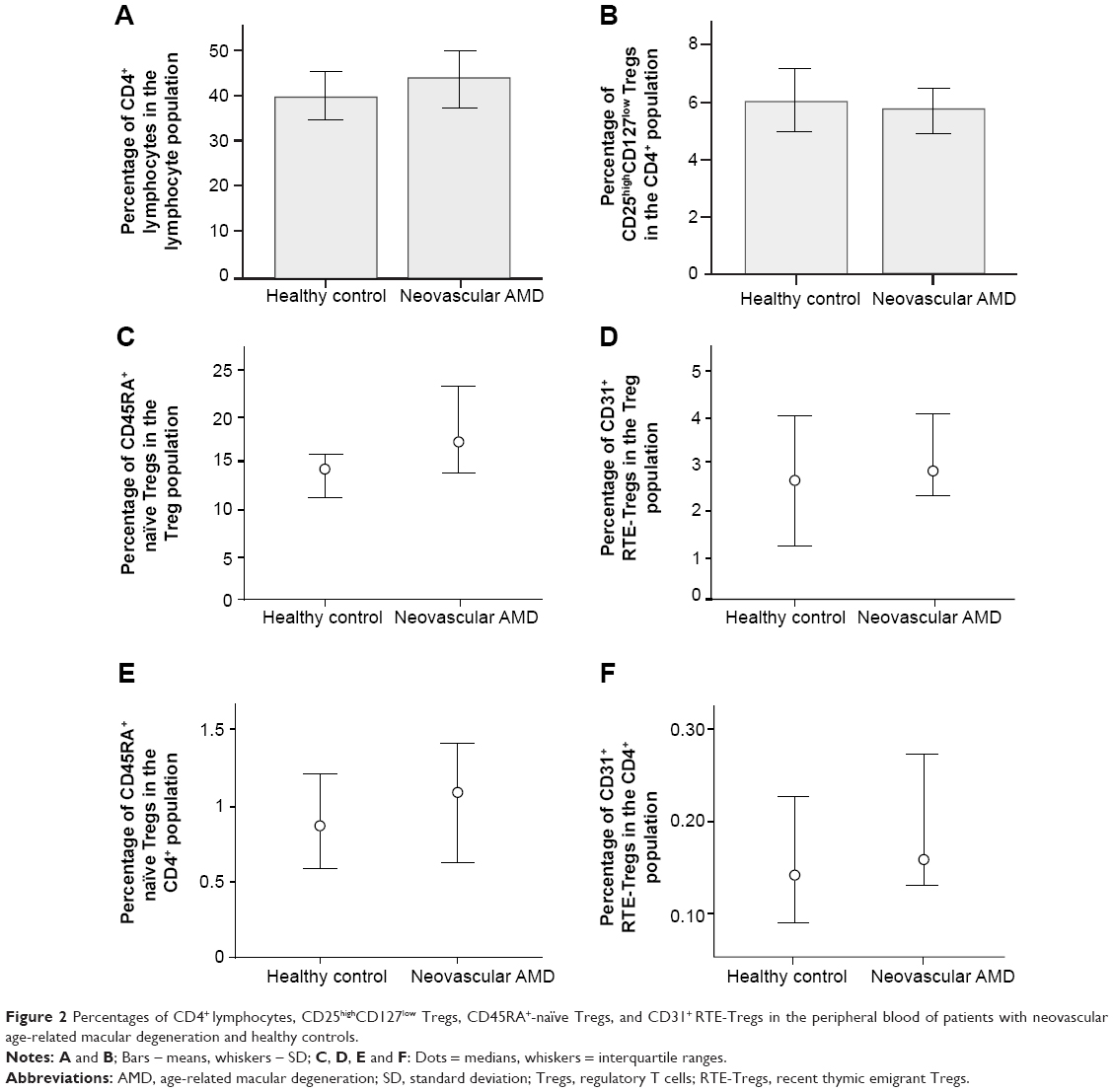 | Figure 2 Percentages of CD4+ lymphocytes, CD25high CD127low Tregs, CD45RA+-naïve Tregs, and CD31+ RTE-Tregs in the peripheral blood of patients with neovascular age-related macular degeneration and healthy controls. |
Percentages of CD45RA+-naïve Tregs and CD31+ RTE-Tregs in the Treg population were not normally distributed. No significant difference was found in the percentage of CD45RA+-naïve Tregs in the Treg population between patients with neovascular AMD (median 17.28, interquartile range 14.04–23.20) and healthy controls (median 14.38, interquartile range 11.37–15.93) (P=0.188, Mann–Whitney U-test) (Figure 2C). Comparing the percentages of CD31+ RTE-Tregs, we found no significant difference in the percentage of RTE-Tregs in the Treg population between patients with neovascular AMD (median 2.82, interquartile range 2.28–4.07) and healthy controls (median 2.63, interquartile range 1.18–4.05) (P=0.895, Mann–Whitney U-test) (Figure 2D).
Percentages of CD45RA+-naïve Tregs and CD31+ RTE-Tregs in the CD4+ population were not normally distributed. No significant difference was found in the percentage of CD45RA+-naïve Tregs in the CD4+ population between patients with neovascular AMD (median 1.09, interquartile range 0.62–1.41) and healthy controls (median 0.87, interquartile range 0.58–1.21) (P=0.349, Mann–Whitney U-test) (Figure 2E). Comparing the percentages of CD31+ RTE-Tregs, we found no significant difference in the percentage of RTE-Tregs in the CD4+ population between patients with neovascular AMD (median 0.16, interquartile range 0.13–0.27) and healthy controls (median 0.14, interquartile range 0.09–0.23) (P=0.454, Mann–Whitney U-test) (Figure 2F).
Discussion
The aim of our study was to investigate whether any change in frequencies of different subsets of Tregs could be found in patients with neovascular AMD. There are no previous reports on the potential involvement of Tregs in neovascular AMD pathogenesis, and there is limited knowledge of the role of Tregs in eye disease in general. However, Tregs have been implicated in the maintenance of the immune privilege of the eye, and Tregs have been reported to be influenced by the retinal pigment epithelium in vitro.23,24 Tregs induced by retinal pigment epithelium cells effectively suppress inflammatory cells (monocytes, B cells, and T cells), and intraocular T cells from the vitreous fluid of patients with acute uveitis, acute retinal necrosis, and cytomegalovirus retinitis.25 Tregs have been implicated in several neurodegenerative diseases. In animal models of Parkinson’s disease, Tregs have been shown to mediate neuroprotection through suppression of microglial stimulation by aggregated nitrated α-synuclein, the misfolded protein found in Parkinson’s disease Lewy bodies.26 Interestingly, Tregs were later found to have nigrostriatal neuroprotective properties when administered to mice with an animal model of Parkinson’s disease.27 In humans, Tregs from patients with Parkinson’s disease have been found to be dysfunctional, as their ability to suppress effector T cell function is impaired.28 In Alzheimer’s disease and mild cognitive impairment, the frequency of Tregs is increased.29 A shift from naïve to memory phenotype similar to the previously described age-related changes within subsets in the Treg population has been found in patients with Alzheimer’s disease.30 The role of Tregs has also been studied in multiple sclerosis, where the decrease in RTE-Treg numbers and the suppressive function of the Tregs is more pronounced in patients with relapse-remitting multiple sclerosis compared to age-matched controls.20,31 The changes in Tregs numbers and function found in Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis support the theory that dysfunction of the Treg population might be attributed to age-related involution of the thymus and subsequent decline in naïve Tregs and compensatory increase in memory Tregs. Our hypothesis for the role of Tregs in AMD pathogenesis was based on the idea that since the retina is a highly differentiated neuro-ectodermal tissue related to the central nervous system, changes in Tregs, similar to those found in neurodegenerative diseases, could lead to dysregulation of the immune system, causing overactivation of inflammatory processes in the retina.
Our findings do not show systemic alterations of Tregs in neovascular AMD, a finding that does not rule out a local ocular role of Tregs in AMD pathogenesis. However, studying Tregs is complicated by the challenge of identifying the “true” population of Tregs. The concept of Tregs was coined more than 30 years ago, when thymectomy in mice was discovered to lead to autoimmune diseases, and was confirmed when mice were selectively depleted of T cells with the surface markers CD4 and CD25 (the alpha unit of the [IL-2] receptor).32 In humans, however, these surface markers are not specific for Tregs alone. Peripheral blood contains up to 30% CD4+ CD25+ T cells, but only the 1%–2% cells with the highest CD25 expression have suppressive capabilities; the rest therefore cannot be considered regulatory. This led to the term CD4+ CD25high Tregs signifying the identification of Tregs by a high CD25 expression. The problem with this identification method is that a population of CD4+ T cells display a continuum of CD25 expression (Figure 1C), making the boundary between CD25high and CD25low highly arbitrary. A better marker was needed. The transcription factor FoxP3 was discovered to be essential for Tregs differentiation and function, and it was deemed the master regulator of Tregs.11 With FoxP3 being an intracellular marker, the need for a Treg-specific surface marker led to the discovery of the IL-7-receptor alpha unit, CD127, which by lack of surface expression identifies around 90% of FoxP3 Tregs in peripheral blood when combined with CD25high expression.13 T cells identified as CD4+, CD25high, and CD127low have excellent suppressive capabilities, which is why we found this method of identification useful for this study.15 It should be noted, however, that both FoxP3 and CD25high CD127low CD4+ T cells fail to exclude a non-regulatory population of unknown size.11 The search for the perfect combination of Treg markers continues, and when found, a consensus for the identification of Tregs will be a quantum leap for Treg research by allowing the comparison of results from different studies. These challenges make comparisons and conclusions from different studies using different cell type nomenclature difficult.
Our study has several strengths. The diagnosis was established using detailed imaging, enabling us to exclude control individuals with subretinal pathology suggestive of AMD. In addition, blood samples were carefully collected within a timespan of 3 hours, and flow cytometry was performed no more than 5 hours after venipuncture. Our exclusion criteria eliminated systemic inflammation as a source of potential bias, making the case and the control groups very comparable, but may also have made the sample less representable of AMD patients, as some AMD patients, like many aged individuals, have chronic diseases with systemic inflammation.
Despite our efforts to age-match case and control subjects, case subjects were significantly older than control subjects. This affects the validity of our findings, as aging affects the composition of the Tregs population.16–18 Therefore, the possibility exists that there are changes in the Treg population associated with neovascular AMD, which our study has been unable to detect. Better age-matching and a larger sample size are needed to fully answer the question.
Conclusion
We did not find any association between AMD and Tregs in peripheral blood. Since we studied systemic changes, local ocular changes involving Tregs cannot be ruled out.
Acknowledgments
We thank Yousif Subhi for his kind assistance with data analysis and Amardeep Singh for his help with the study design. This study was funded by Region Zealand’s Research Fund.
Author contributions
The following author contributions apply to the current study: study design and conduct (CFM, MKF, TLS), data collection (CFM, MKF), data management, analysis and interpretation (CFM, MKF, TLS), manuscript preparation, and review and approval (CFM, MKF, TLS).
Disclosure
The authors have no proprietary or commercial interests in any materials discussed in this article, and report no conflicts of interest in this work. The funding organization had no role in the design or conduct of this research.
References
Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. Lancet. 2012;379(9827):1728–1738. | ||
Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. 2013;13(6):438–451. | ||
Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005; 308(5720):419–421. | ||
Singh A, Faber C, Falk M, Nissen MH, Hviid TVF, Sørensen TL. Altered expression of CD46 and CD59 on leukocytes in neovascular age-related macular degeneration. Am J Ophthalmol. 2012;154(1): 193–199.e2. | ||
Patel N, Ohbayashi M, Nugent AK, et al. Circulating anti-retinal antibodies as immune markers in age-related macular degeneration. Immunology. 2005;115(3):422–430. | ||
Faber C, Singh A, Krüger Falk M, Juel HB, Sørensen TL, Nissen MH. Age-related macular degeneration is associated with increased proportion of CD56(+) T cells in peripheral blood. Ophthalmology. 2013; 120(11):2310–2316. | ||
Morohoshi K, Ohbayashi M, Patel N, Chong V, Bird AC, Ono SJ. Identification of anti-retinal antibodies in patients with age-related macular degeneration. Exp Mol Pathol. 2012;93(2):193–199. | ||
Chakravarthy U, Wong TY, Fletcher A, et al. Clinical risk factors for age-related macular degeneration: a systematic review and meta-analysis. BMC Ophthalmol. 2010;10:31. | ||
Smith W, Assink J, Klein R, et al. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology. 2001;108(4):697–704. | ||
Gruver AL, Hudson LL, Sempowski GD. Immunosenescence of ageing. J Pathol. 2007;211(2):144–156. | ||
Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010; 10(7):490–500. | ||
Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med. 2007;13(3):108–116. | ||
Seddiki N, Santner-Nanan B, Martinson J, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203(7):1693–1700. | ||
Yu N, Li X, Song W, et al. CD4(+) CD25(+) CD127(low/-) T cells: a more specific Treg population in human peripheral blood. Inflammation. 2012;35(6):1773–1780. | ||
Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203(7):1701–1711. | ||
Rosenkranz D, Weyer S, Tolosa E, et al. Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J Neuroimmunol. 2007;188(1–2):117–127. | ||
Wang L, Xie Y, Zhu LJ, Chang TT, ing Mao YQ, Li J. An association between immunosenescence and CD4(+) CD25(+) regulatory T cells: a systematic review. Biomed Environ Sci. 2010;23(4):327–332. | ||
Santner-Nanan B, Seddiki N, Zhu E, et al. Accelerated age-dependent transition of human regulatory T cells to effector memory phenotype. Int Immunol. 2008;20(3):375–383. | ||
Valmori D, Merlo A, Souleimanian NE, Hesdorffer CS, Ayyoub M. A peripheral circulating compartment of natural naive CD4 Tregs. J Clin Invest. 2005;115(7):1953–1962. | ||
Haas J, Fritzsching B, Trübswetter P, et al. Prevalence of newly generated naive regulatory T cells (Treg) is critical for Treg suppressive function and determines Treg dysfunction in multiple sclerosis. J Immunol. 2007;179(2):1322–1330. | ||
Kaiser PK. Prospective Evaluation of Visual Acuity Assessment: A Comparison of Snellen Versus ETDRS Charts in Clinical Practice (An AOS Thesis). Transactions of the American Ophthalmological Society. 2009;107:311–324. | ||
Seddon JM, Sharma S, Adelman RA. Evaluation of the clinical age-related maculopathy staging system. Ophthalmology. 2006;113(2):260–266. | ||
Zhou R, Horai R, Silver PB, et al. The living eye “disarms” uncommitted autoreactive T cells by converting them to Foxp3(+) regulatory cells following local antigen recognition. J Immunol. 2012;188(4):1742–1750. | ||
Vega JL, Saban D, Carrier Y, Masli S, Weiner HL. Retinal pigment epithelial cells induce foxp3(+) regulatory T cells via membrane-bound TGF-β. Ocul Immunol Inflamm. 2010;18(6):459–469. | ||
Horie S, Sugita S, Futagami Y, Yamada Y, Mochizuki M. Human retinal pigment epithelium-induced CD4+CD25+ regulatory T cells suppress activation of intraocular effector T cells. Clin Immunol. 2010; 136(1):83–95. | ||
Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J Leukoc Biol. 2007;82(5):1083–1094. | ||
Reynolds AD, Stone DK, Hutter JAL, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol. 2010;184(5):2261–2271. | ||
Saunders JAH, Estes KA, Kosloski LM, et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J Neuroimmune Pharmacol. 2012;7(4):927–938. | ||
Saresella M, Calabrese E, Marventano I, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2010;21(3):927–938. | ||
Larbi A, Pawelec G, Witkowski JM, et al. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis. 2009;17(1):91–103. | ||
Lowther DE, Hafler DA. Regulatory T cells in the central nervous system. Immunol Rev. 2012;248(1):156–169. | ||
Sakaguchi S. Regulatory T cells: history and perspective. Methods Mol Biol. 2011;707:3–17. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.